")
Back to Journals » Open Access Rheumatology: Research and Reviews » Volume 11
Review of biologics, biosimilars, and intended copies in rheumatology, and current practice in Iraq
Authors Al Ani NA , Gorial FI, Al-Sulaitti S, Humadi JA , Awadh NI , Mounir M, El Dershaby Y, Jones H, Sunna N
Received 12 June 2018
Accepted for publication 28 September 2018
Published 24 December 2018 Volume 2019:11 Pages 1—9
DOI https://doi.org/10.2147/OARRR.S176965
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Chuan-Ju Liu
Nizar Abdulateef Al Ani,1 Faiq I Gorial,1 Saad Al-Sulaitti,2 Jasmine Abbas Humadi,3 Nabaa Ihsan Awadh,4 Mohamed Mounir,5 Yasser El Dershaby,5 Heather Jones,6 Nancy Sunna7
1Rheumatology Unit, Department of Medicine, College of Medicine, University of Baghdad, Baghdad, Iraq; 2Rheumatology Unit, Department of Internal Medicine, Al-Basra General Hospital, Al-Basra, Iraq; 3Rheumatology Unit, Department of Medicine, Al-Nahrain Medical College, Baghdad, Iraq; 4Rheumatology Unit, Department of Internal Medicine, Baghdad Teaching Hospital, Baghdad, Iraq; 5Inflammation & Immunology, Pfizer, Dubai, UAE; 6Inflammation & Immunology, Pfizer, Collegeville, PA, USA; 7Inflammation & Immunology, Pfizer, Amman, Jordan
Abstract: Biologic therapies are an important option in the treatment of patients with rheumatic disease. As the development of potential biosimilars increases, many countries are following the guidelines developed by the WHO, European Medicines Agency, or US Food and Drug Administration to create country-specific regulations for the review and approval of these products. Iraq does not yet have such regulations, and this presents a potential safety concern for patients. The analytical, nonclinical, and clinical data requirements for approval of a potential biosimilar are specific and scientifically rigorous. In some countries, products are available that have not met the stringent criteria for biosimilars; they are usually referred to as “intended copies”. Frequently, the available data are not sufficient to demonstrate that they are similar in efficacy and safety to the reference product. Thus, safety issues may arise once the product is in use, as was the case with Kikuzubam, an intended copy of rituximab that was withdrawn from the market in Mexico following reports of severe adverse reactions. It is important to implement scientific, evidence-based guidelines for the review, approval, therapeutic use, and monitoring of biosimilars, and to provide training on this topic to healthcare professionals and patients. In this review, we discuss issues related to the use and regulation of biosimilars, and the differences between biosimilars and intended copies. We also provide suggestions for including biosimilars as a treatment option in Iraq.
Keywords: biosimilar, intended copy, Iraq, rheumatology
Introduction
Guidelines for the treatment of rheumatoid arthritis (RA) and other rheumatic diseases focus on quickly achieving targeted endpoints in order to limit permanent joint damage and comorbidity, thereby enabling patients to maintain a high level of physical function.1–3 The European League Against Rheumatism recommends a target goal of 50% clinical improvement within 3 months and the achievement of remission or low disease activity within 6 months.2 The American College of Rheumatology (ACR) and the Asia Pacific League of Associations for Rheumatology (APLAR) also recommend implementing a treat-to-target strategy.4,5 Biologic therapies play a key role in treatment guidelines; however, their high cost has resulted in limited access and inequality in treatment options in some countries.2,6 This has created a demand for biosimilars as lower-cost alternatives.6 In order to protect patients, regulatory authorities should ensure that the safety and efficacy of potential biosimilars are comparable to those of the originator.7
A biosimilar is defined as a biologic therapy that is similar to an approved reference product with regard to quality, safety, and efficacy.8 Similarity must be demonstrated through comparisons of quality and biological activity, as well as nonclinical and clinical study data. Biosimilars are highly similar but not identical to the reference product.9
Conversely, products that do not meet the stringent criteria for biosimilars are usually referred to as intended copies or noncomparable biologics or copied biopharmaceutical products10,11 These products have not been adequately compared analytically or clinically to the originator, thus the data are not sufficient to demonstrate similar efficacy and safety.10,11 In some countries, intended copies were approved for use before rigorous biosimilar regulatory guidelines were developed.11 Differences in formulation, dosing, efficacy, or safety between the intended copy and the originator may exist, and this may pose a safety risk for patients.11 For example, Kikuzubam, an intended copy of rituximab, was withdrawn from the Mexican market in 2014 due to concerns of anaphylactic reactions and limited safety and efficacy data.12,13 In an example outside of rheumatology, intended copies of recombinant human erythropoietin in Thailand were found to differ substantially from the originator product, and they were also associated with increased immunogenicity.14,15
Currently, many countries worldwide are developing regulatory pathways for the approval of biosimilars, often based on the guidances developed by the WHO, European Medicines Agency (EMA), or US Food and Drug Administration (FDA).6,11 Iraq is one of the Middle East countries that still needs to develop its own guidelines for the approval of biosimilars.16
The healthcare system in Iraq has not yet recovered from years of war, political unrest, and sanctions.17 The Iraq Ministry of Health (MOH) is working to make access to health care more equitable; however, this is a complicated and challenging process.17 The MOH does provide health care, including medications, to all citizens and permanent residents of Iraq for free or for a very minimal fee.17,18 Although the health system is well funded, healthcare policies are outdated or lacking, including strategies for continued healthcare financing.17 For instance, biosimilars could provide a less expensive treatment option than originator biologics if a process existed for their review and approval.6,16
We searched the literature for information on the healthcare system in Iraq and the use of biosimilars in Iraq and the Middle East. In this review, we discuss challenges in the production, use, and regulation of biosimilars, as well as important issues related to the safety of biosimilars and intended copies. In addition, we provide author recommendations, as rheumatologists practicing in Iraq, for incorporating biosimilars into the healthcare system in Iraq.
Methods
We conducted a search of the published literature to investigate the manufacturing and regulatory requirements of biosimilars and the difference between biosimilars and intended copies. We also reviewed the literature to evaluate the healthcare system in Iraq and the status of biosimilar regulation in Iraq and the Middle East. As practicing rheumatologists in Iraq, we developed recommendations for biosimilar review and incorporation into treatment in Iraq, with patient safety as the most important consideration.
Manufacture of generic drugs versus biosimilars
A generic small molecule drug is chemically synthesized and equivalent to the reference drug with regard to chemical structure, quality, safety, and efficacy.19 Generic drugs have relatively simple chemical structures, thus an analytical demonstration of the molecular identity and a bioequivalence study in humans are enough to prove that the generic is equivalent to the originator.8,19
Conversely, biologic drugs are large molecules, most frequently proteins.8,19 The manufacture of a biologic product is a complex process that requires living cells; therefore, variability is a concern.20 Posttranslational modifications such as glycosylation and oxidation can affect the structure of the molecule.7 Additionally, manufacturing processes are usually proprietary, so companies interested in manufacturing a prospective biosimilar must develop the entire process de novo.20,21 Consequently, it is almost impossible to create a biologic that is identical to the originator.20 Even a slight variation in the manufacturing procedure can result in changes to the structure of the product, possibly altering its biological activity, safety, efficacy, and immunogenicity.7 Therefore, the manufacture of biologics must be closely monitored to ensure batch to batch consistency.
Regulatory requirements for biosimilar approval
Since the inherent small differences between an originator and a potential biosimilar can be important clinically, the WHO, EMA, and FDA have developed rigorous guidelines for the evaluation of biosimilars,8,21–23 which are much more complex than those for generic drugs.19 According to the WHO, a biosimilar, or a similar biotherapeutic product, has been demonstrated to be similar to an approved biologic with regard to quality, safety, and efficacy.8 The WHO, EMA, and FDA requirements specify that the primary amino acid sequence of the proposed biosimilar must be exactly the same as that of the reference drug.7,8 Additionally, any variations in molecular structure or posttranslational modifications must not affect safety, efficacy, or immunogenicity.7,8
The first step to demonstrating biosimilarity is to thoroughly analyze the structure and function of the potential biosimilar and compare these head-to-head with the reference product.8,11 This step is critical because similarity in structure and function provides the rationale to progress to the nonclinical and clinical studies. The nonclinical studies are conducted to provide evidence that the biosimilar affects physiologic processes in a manner similar to the reference product, with comparable toxicity. Then, clinical studies are conducted to demonstrate that the potential biosimilar and the reference product are similar with respect to pharmacokinetics, efficacy, and safety, including immunogenicity.11
The WHO guidelines for evaluation of a biosimilar require submission of the following documentation: 8,20
- Manufacturing follows Good Manufacturing Practices; documentation includes the known process information for the reference product.
- Physicochemical and biological characterization and a head-to-head preclinical comparison to the reference product.
- A repeat-dose preclinical toxicity study in an appropriate animal species.
- A single-dose head-to-head clinical pharmacokinetic study with the reference product.
- Phase III head-to-head clinical efficacy studies with the reference product.
- Clinical studies evaluating immunogenicity.
- A plan for conducting pharmacovigilance.
The clinical study requirements for biosimilars are not as extensive as for originator products. The FDA requires at least one efficacy trial that is adequately powered to show that it is equivalent to the reference product.7 According to the EMA, one comparative-dose pharmacodynamic study that is highly sensitive may be enough to demonstrate efficacy.7 For safety, both the EMA and FDA require at least one clinical trial that has enough patients to demonstrate equivalence.7 The FDA states that immunogenicity must be evaluated in one preapproval comparative trial and then in a postapproval trial, and the EMA requires evaluation of immunogenicity within the safety trial.7
The WHO, EMA, and FDA all require submission of a pharmacovigilance plan, so that benefit vs risk can be continually assessed.7,8 Postmarketing surveillance is critical for monitoring long-term safety and identifying any rare adverse events.6 Additionally, pharmacovigilance can monitor for adverse events that may occur when patients are switched between the reference product and the biosimilar. Once a biosimilar is approved, the manufacturer is responsible for conducting pharmacovigilance; however, the regulatory agency must monitor their compliance and address any safety concerns that may arise.8
Biosimilars in Europe and the USA
Table 1 lists the rheumatology biosimilars that have been approved by the EMA and the US FDA.24–29
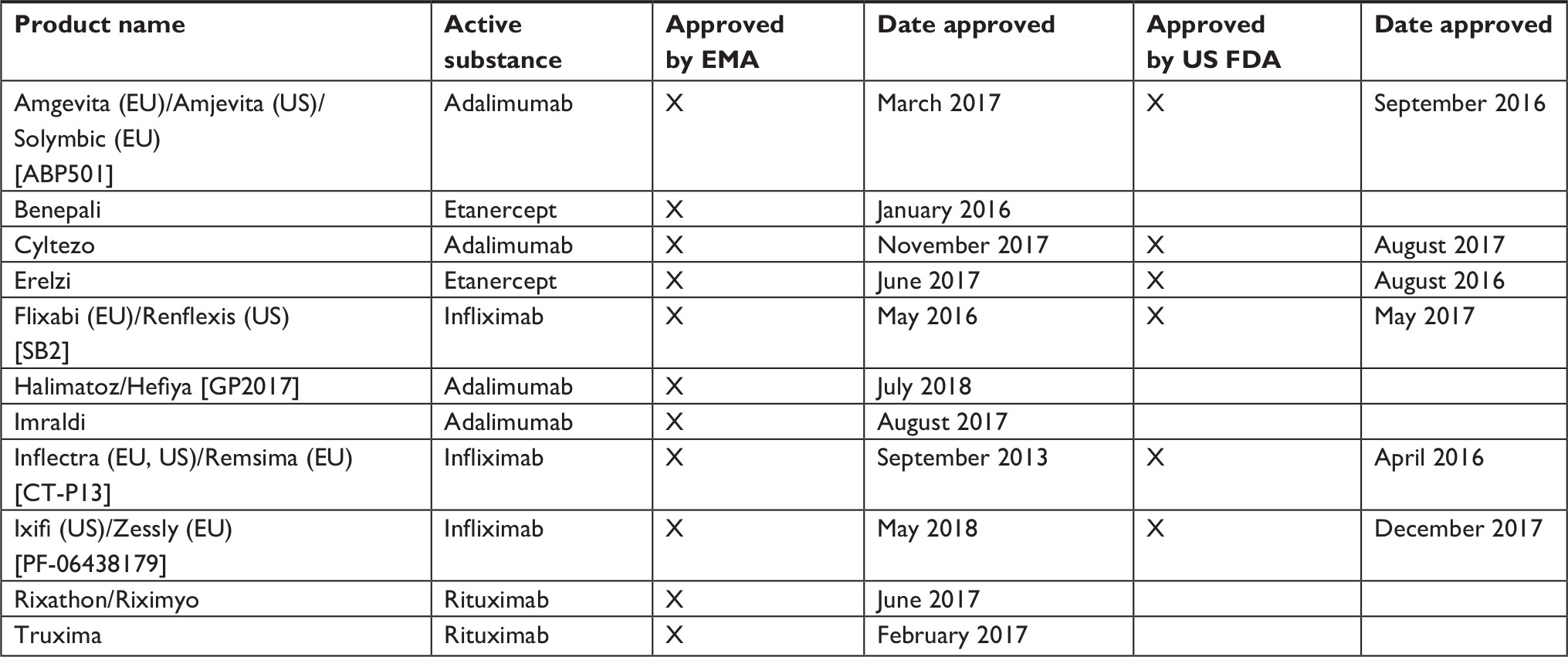 | Table 1 Biosimilars in rheumatology approved by the European Medicines Agency and the US Food and Drug Administration Abbreviations: EMA, European Medicines Agency; EU, European Union; US FDA, United States Food and Drug Administration. |
Terminology related to biosimilar use
Since biosimilars are required to be highly similar, but not identical, to an approved reference product, and since limited clinical trial data are available at approval,6,11 clinicians have raised practical questions about incorporating biosimilars into clinical practice. Many of the questions relate to the terms below.
Extrapolation
Extrapolation refers to the approval of a biosimilar for a particular indication of the reference product, even though a direct clinical comparison has not been conducted.30 Clinicians have questioned how regulators can be confident that a biosimilar will be safe and effective in the treatment of diseases for which it has no clinical data.13,31
Extrapolation occurs only after biosimilarity has been established. The WHO guidelines state that extrapolation may be considered if the following criteria are met: the model used for clinical testing was sensitive enough to identify any differences between the potential biosimilar and the reference product; the mechanism of action of the potential indications is the same; a thorough study of safety and immunogenicity has occurred; the efficacy results were acceptable and were evaluated using a noninferiority study.8 The FDA and EMA will allow extrapolation on a case-by-case basis.7,8
In order to justify extrapolation, it is important that countries first require rigorous analytical characterization of a proposed biosimilar, followed by studies of clinical pharmacokinetics, pharmacodynamics, safety, and efficacy that confirm biosimilarity.31 Once biosimilarity has been determined based on the totality of scientific evidence, then according to the principle that structure determines function, the biosimilar should perform the same as the reference product for all approved indications.31
Naming
There is no internationally recognized naming convention for biosimilars.11 The WHO is considering a proposal for all biologics, originators, and biosimilars to have a name with two parts, the International Nonproprietary Name and a unique identifier.11 However, some countries have developed their own naming conventions. This may create confusion for physicians and pharmacists when patients travel internationally or if they obtain medication from other countries.
Interchangeability and automatic substitution
Once a product has been approved as a biosimilar, then the prescriber can switch a patient from the reference product to the biosimilar and vice versa.13 Taking this a step further, in the USA, if the FDA were to designate a biosimilar as interchangeable with the reference product, then individual states can decide whether or not to allow pharmacists to automatically substitute it for the reference product, without consulting the prescribing clinician.13 The FDA has developed a draft guidance on the requirements for demonstrating interchangeability; no biosimilars have been approved as interchangeable with the reference product.13,32,33 Once the guidance is finalized, the policies required to implement it will be the responsibility of the individual states.
The definition of interchangeability provided by the European Commission is somewhat different, defining it as, “the medical practice of changing one medicine for another that is expected to achieve the same clinical effect in a given clinical setting and in any patient on the initiative, or with the agreement of the prescriber”.34 In the EU, the EMA does not designate interchangeability; it is a country-specific decision.13
The prospect of pharmacists being allowed to automatically substitute a biosimilar in place of the originator biologic without consulting the prescriber, as may be possible in the USA in the future, is concerning to some rheumatologists, for several reasons. The extent of pharmacovigilance varies across countries, and it may be difficult to monitor adverse events and track which biosimilar a patient is taking when an adverse event occurs.6,7 Additionally, if patients travel, the variability in naming conventions in different countries adds another challenge to tracking a patient’s particular therapy. Clinicians have also raised concerns about the risk of an immunogenic reaction after switching between biosimilars, since immunogenicity may lead to adverse events or a lack of efficacy.20,35
However, the evidence in the literature suggests that switching does not cause immunogenic reactions. Three large studies that have evaluated the safety of switching between an originator anti-tumor necrosis factor (TNF) agent and a biosimilar include EGALITY (switch between originator etanercept and GP2015), NOR-SWITCH (switch between originator infliximab and CT-P13), and a switch study with originator adalimumab and SB5.36–39 In all three studies, immunogenicity did not differ significantly between the originator biologic and the biosimilar, and switching did not affect immunogenicity.
Biosimilars in the Middle East
In 2007, experts in multiple sclerosis, endocrinology, and pharmacology from the Near and Middle East recommended the development of regional guidelines based on those used by the EMA.40 In 2015, a survey about biosimilars was conducted at the Pan-Arab Oncology Meeting in Beirut, Lebanon.41 A total of 117 questionnaires were completed by healthcare professionals within the disciplines of oncology (39%), hemato-oncology (14%), nursing (12%), surgery (9%), gynecology (5%), pharmacy (5%), urology (4%), radiology (3%), and other (9%). A majority of respondents were from Lebanon (62%); 3% were from Iraq. Seventy-seven of the 117 (66%) professionals were knowledgeable about biosimilars and 48 (41%) prescribed them. The primary rationale for prescribing biosimilars was their approval by the FDA or the EMA (69%); other reasons included the lower price vs the originator (65%), efficacy (48%), safety (42%), and positive manufacturer reputation (31%). According to these survey results, most healthcare professionals in the region are familiar with biosimilars; however, less than half prescribe them. Those who do prescribe biosimilars recognize the importance of having followed a rigorous, science-based review process.
Although many countries in the Middle East do not have biosimilar guidelines, several countries, including Iran, Lebanon, and Saudi Arabia, have developed guidelines.42 The Iran National Regulatory Authority used the WHO biosimilar guidelines as a resource; however, the Iranian guidelines are not as strict. Biosimilars manufactured by Iranian biopharmaceutical companies have been approved without undergoing a review as comprehensive as the WHO requires.43 The Ministry of Public Health in Lebanon follows the WHO biosimilar guidelines,44 and the biosimilar guidelines developed by the Saudi Food & Drug Authority are based on guidances from the EMA and the International Conference on Harmonisation.45,46
Regulation of drugs in Iraq
Drug evaluation and approval in Iraq are performed by the Drug Registration Committee within the MOH Directorate of Technical Affairs.47 Currently, ~8,600 trade drugs are registered with the MOH, and Iraq has 28 national pharmaceutical companies.47 However, there is no policy in place for the review and approval of biosimilars. According to the most recent information available, three biologic therapies that are used in rheumatology are approved in Iraq: the TNF inhibitors etanercept and infliximab, and the anti-CD20 monoclonal antibody, rituximab.48 Only the originator biologics are available; no biosimilars are approved in the country.16,42
Intended copies
Although no intended copies used in the treatment of rheumatic disease are approved in Iraq, two of them are manufactured in the neighboring country of Iran: Zytux, an intended copy of rituximab, and Altebrel, an intended copy of etanercept.42,49–51 Due to the proximity with Iran, these products may be accessible to some Iraqi patients. According to an analytical assessment of Altebrel, several attributes were not comparable to those of the originator: peptide sequence, distribution of charged species, and posttranslational modifications.52 Although the clinical impact of these differences is unknown, Altebrel should not be considered an etanercept biosimilar, and patients should not be switched between the two products. We are not aware of other countries in the Middle East with intended copies.42
Discussion
In Iraq, the healthcare system is trying to recover from years of war, and the process of making health care more equitable across the country is challenging and lengthy.17 Additionally, rheumatologists in Iraq face many challenges. For instance, inequalities in access to health care exist.17 Primary health care is provided to many poor patients at public clinics, approximately half of which do not have a medical doctor.17 Therefore, the diagnosis of RA and the referral of patients to a specialist may be delayed. Also, some patients may be reluctant to take methotrexate, because it is considered to be a chemotherapy drug.48 Further, the choice of medications for the treatment of rheumatic diseases is limited, and they are sometimes in short supply.17,48 The selection of approved anti-TNF agents is narrow, and other than rituximab, biologic drugs with alternative mechanisms of action, such as IL-6 antagonists, are not approved in Iraq.48
As healthcare costs increase, biosimilars can be an important option in the management of rheumatic diseases. Biosimilars are not only lower cost alternatives to existing biological therapies, they also introduce price competition. This may lead to a price decrease for the reference product and other products in the therapeutic area. Additionally, the cost savings for the healthcare system may result in increased access to treatment for patients. However, the decision to prescribe a particular therapy should not be based on cost alone.2,53,54 Patient safety, not cost, must be the highest priority.
Recommendations
In order to protect the safety of patients in Iraq, we believe that several steps must be taken to provide adequate resources (Table 2).
 | Table 2 Resources recommended by the authors to guide the safe use of biosimilars in Iraq |
Scientifically rigorous guidelines for the regulatory review of prospective biosimilars are needed in Iraq. The approval of biosimilars in Iraq should be the responsibility of an expert committee of scientists and clinicians that reviews all evidence of biosimilarity, including clinical comparability and switching studies. The regulatory requirements in Iraq should be stringent, similar to those of the WHO, FDA, or EMA, and suitably flexible to allow changes as necessary in the future. The lack of a scientifically based regulatory pathway for biosimilars may be a safety risk for Iraqi patients, because they may receive treatment with an intended copy that has been approved in another country but has not met the high standards required for a biosimilar.11 Regulations against the use of intended copies should be passed.
Nomenclature should be clear, precise, and consistent between countries. This is critical for conducting effective postapproval pharmacovigilance to monitor for immunogenicity or adverse events that may become apparent only with extended use by large numbers of patients.7,40
Multidisciplinary teams consisting of healthcare professionals, patients, manufacturers, policy makers, and regulatory authorities must work together to ensure the safe use of biosimilars, including their storage, administration, and pharmacovigilance. Healthcare professionals may face challenges related to pharmacovigilance, including 1) a lack of harmonized regulations between countries; 2) cultural variation in the practice of medicine; 3) priorities that compete with pharmacovigilance; 4) limited pharmacovigilance expertise; and 5) limited resources at the regulatory agencies.55 However, the importance and success of pharmacovigilance has been demonstrated by the withdrawal of Kikuzubam from the market in Mexico.12,13 This pharmacovigilance, in addition to postapproval studies and registry studies, enables the collection of real-world data that can help guide the decision-making process for clinicians.
Within Iraq, access to medications should be improved. In some areas of the country, community health centers are overwhelmed, and patients may choose a private clinic or hospital instead, even though many are unable to pay.56 The selection of rheumatology medications within the country is limited,48 and drug shortages occur due to complicated funding processes and long procurement times.17 One consideration, in situations that require a specific medication to treat a serious or life-threatening disease, may be self-financing by the health institution, thus making the necessary medication available at a suitable price and preventing patients from purchasing it from another country.
Additionally, guidelines are needed for incorporating biosimilars into clinical practice, to ensure that they are prescribed appropriately.54 Treatment choices should be based on discussion and shared decision-making between the patient and the physician.2 Only the treating rheumatologist understands the unique medical history and specific needs of a particular patient. Although automatic substitution of the biosimilar for the reference product may be allowed in the USA in the future, differences in the health care and legal systems between the USA and Iraq suggest that automatic substitution is not practical in Iraq. As stated by the ACR, the treating physician should be the only health care professional with the authority to switch a patient from the reference drug to a biosimilar.53 The decision to switch should be made on a case-by-case basis, with patient consent, and supported by scientific evidence.
For biosimilars to be successfully incorporated into treatment regimens in Iraq, it is necessary for health care providers and government decision makers to be highly knowledgeable about biosimilars. Education programs for health care providers should extend beyond physicians to include nurses and pharmacists. Pharmacists working at the MOH are closely involved in the drug review and approval process,47 and they must have a thorough understanding of biosimilar manufacturing and concepts such as switching between originators and biosimilars.
Additionally, educational materials are needed for patients and caregivers. Patients should have access to reliable information so they can better understand the available treatment choices.54 Together, prescribers and patients can consider the options and choose the therapy most appropriate for each patient.
Conclusion
Biosimilars can help decrease the cost of health care for governments and patients and increase access to biologic therapies. Although cost savings are desirable, the highest priority must be patient safety. Therefore, scientifically rigorous guidelines for the review, approval, therapeutic use, and monitoring of biosimilars are necessary, as well as educational materials for health care professionals and patients. Countries that do not have these resources, such as Iraq, should make it a priority to develop and implement them.
Compliance with ethical standards
The article does not contain clinical studies or patient data.
Acknowledgments
Medical writing support was provided by Jennica Lewis, PharmD, CMPP, of Engage Scientific Solutions and was funded by Pfizer.
Author contributions
All authors made substantial contributions to the conception or design of the work; or the acquisition, analysis, or interpretation of data. All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. All authors agree to be accountable for all aspects of the work.
Disclosure
Nizar Abdulateef Al Ani, Faiq I Gorial, Saad Al-Sulaitti, Jasmine Abbas Humadi, and Nabaa Ihsan Awadh report no conflicts of interest in this work. Yasser El Dershaby, Heather Jones, and Nancy Sunna are employees of Pfizer. Mohamed Mounir was an employee of Pfizer at the time the article was written.
References
Agarwal SK. Core management principles in rheumatoid arthritis to help guide managed care professionals. J Manag Care Pharm. 2011;17(9 Suppl B):S03–S08. | ||
Smolen JS, Landewé R, Bijlsma J, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann Rheum Dis. 2017;76(6):960–977. | ||
van der Heijde D, Ramiro S, Landewé R, et al. 2016 update of the ASAS-EULAR management recommendations for axial spondyloarthritis. Ann Rheum Dis. 2017;76(6):978–991. | ||
Singh JA, Saag KG, Bridges SL. 2015 American College of Rheumatology Guideline for the Treatment of Rheumatoid Arthritis. Arthritis Rheumatol. 2016;68(1):1–26. | ||
Lau CS, Chia F, Harrison A, et al. APLAR rheumatoid arthritis treatment recommendations. Int J Rheum Dis. 2015;18(7):685–713. | ||
Dörner T, Strand V, Cornes P, et al. The changing landscape of biosimilars in rheumatology. Ann Rheum Dis. 2016;75(6):974–982. | ||
Dörner T, Strand V, Castañeda-Hernández G, et al. The role of biosimilars in the treatment of rheumatic diseases. Ann Rheum Dis. 2013;72(3):322–328. | ||
World Health Organization, Expert Committee on Biological Standardization. Guidelines on evaluation of similar biotherapeutic products (SBPs). 2009. Available from: http://www.who.int/biologicals/areas/biological_therapeutics/BIOTHERAPEUTICS_FOR_WEB_22APRIL2010.pdf. Accessed June 29, 2017. | ||
European Medicines Agency. Biosimilar medicines. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/general/general_content_001832.jsp&mid=WC0b01ac0580bb8fda. Accessed September 17, 2018. | ||
Jacobs I, Petersel D, Shane LG, et al. Monoclonal antibody and fusion protein biosimilars across therapeutic areas: a systematic review of published evidence. BioDrugs. 2016;30(6):489–523. | ||
Mysler E, Pineda C, Horiuchi T, et al. Clinical and regulatory perspectives on biosimilar therapies and intended copies of biologics in rheumatology. Rheumatol Int. 2016;36(5):613–625. | ||
Castañeda-Hernández G, González-Ramírez R, Kay J, Scheinberg MA. Biosimilars in rheumatology: what the clinician should know. RMD Open. 2015;1(1):e000010. | ||
Dörner T, Kay J. Biosimilars in rheumatology: current perspectives and lessons learnt. Nat Rev Rheumatol. 2015;11(12):713–724. | ||
Halim LA, Brinks V, Jiskoot W, et al. How bio-questionable are the different recombinant human erythropoietin copy products in Thailand? Pharm Res. 2014;31(5):1210–1218. | ||
Praditpornsilpa K, Tiranathanagul K, Kupatawintu P, et al. Biosimilar recombinant human erythropoietin induces the production of neutralizing antibodies. Kidney Int. 2011;80(1):88–92. | ||
Center for Biologic Policy Evaluation. How robust are your country’s biosimilar medicine policies? Available from: https://biologicspolicy.com/#. Accessed August 24, 2017. | ||
Al Hilfi TK, Lafta R, Burnham G. Health services in Iraq. Lancet. 2013;381(9870):939–948. | ||
Ministry of Health of the Republic of Iraq, World Health Organization. Iraq Pharmaceutical Country Profile; 2011. Available from: http://www.who.int/medicines/areas/coordination/Iraq_PSCPNarrativeQuestionnaire_01022012.pdf. Accessed December 13, 2017. | ||
de Mora F. Biosimilar: what it is not. Br J Clin Pharmacol. 2015;80(5):949–956. | ||
Scheinberg MA, Kay J. The advent of biosimilar therapies in rheumatology—“O Brave New World”. J Nat Rev Rheumatol. 2012;8(7):430–436. | ||
Lee JF, Litten JB, Grampp G. Comparability and biosimilarity: considerations for the healthcare provider. Curr Med Res Opin. 2012;28(6):1053–1058. | ||
U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER). Scientific considerations in demonstrating biosimilarity to a reference product guidance for industry, April 2015. Biosimilarity; 2015. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf. Accessed May 17, 2016. | ||
European Medicines Agency, Committee for Medicinal Products for Human Use (CHMP).Guideline on similar biological medicinal products; 2014. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/10/WC500176768.pdf. Accessed August 31, 2017. | ||
European Medicines Agency. European public assessment reports. Available from: https://www.ema.europa.eu/en/medicines/download-medicine-data. Accessed August 30, 2018. | ||
US Food and Drug Administration, Department of Health and Human Services. Biosimilar Product Information, FDA-Approved Biosimilar Products. Available from: https://www.fda.gov/drugs/developmentapprovalprocess/howdrugsaredevelopedandapproved/approvalapplications/therapeuticbiologicapplications/biosimilars/ucm580432.htm. Accessed August 30, 2018. | ||
Generics and Biosimilars Initiative Journal. Biosimilars of adalimumab; 2014, updated 2018. Available from: http://www.gabionline.net/Biosimilars/General/Biosimilars-of-adalimumab. Accessed August 30, 2018. | ||
Generics and Biosimilars Initiative Journal. Biosimilars of infliximab; 2015, 2018. Available from: http://www.gabionline.net/Biosimilars/General/Biosimilars-of-infliximab. Accessed August 30, 2018. | ||
Generics and Biosimilars Initiative Journal. EMA approves adalimumab and trastuzumab biosimilars; 2018. Available from: http://www.gabionline.net/Biosimilars/News/EMA-approves-adalimumab-and-trastuzumab-biosimilars. Accessed August 30, 2018. | ||
Lamanna WC, Heller K, Schneider D, et al. The in-use stability of the rituximab biosimilar Rixathon®/Riximyo® upon preparation for intravenous infusion. J Oncol Pharm Pract. Epub 2017. | ||
Tesser JR, Furst DE, Jacobs I. Biosimilars and the extrapolation of indications for inflammatory conditions. Biologics. 2017;11:5–11. | ||
Generics and Biosimilars Initiative Journal. Biosimilars: extrapolation of clinical use to other indications; 2015. Available from: http://gabi-journal.net/biosimilars-extrapolation-of-clinical-use-to-other-indications.html?print. Accessed July 7, 2017. | ||
US Food and Drug Administration, Department of Health and Human Services. Center for Drug Evaluation and Research. List of Licensed Biological Products with (1) Reference Product Exclusivity and (2) Biosimilarity or Interchangeability Evaluations to Date, updated 8/8/18. Available from: https://www.fda.gov/drugs/developmentapprovalprocess/howdrugsaredevelopedandapproved/approvalapplications/therapeuticbiologicapplications/biosimilars/ucm411418.htm. Accessed August 24, 2018. | ||
US Food and Drug Administration, Department of Health and Human Services. Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER). Considerations in demonstrating interchangeability with a reference product, guidance for industry, Draft Guidance; 2017. Available from: https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM537135.pdf. Accessed August 24, 2018. | ||
European Commission. What you need to know about biosimilar medicinal products; 2013. Available from: http://www.medicinesforeurope.com/wp-content/uploads/2016/03/biosimilars_report_en.pdf. Accessed January 21, 2018. | ||
Ebbers HC, Chamberlain P. Interchangeability. An insurmountable fifth hurdle? GaBI J. 2014;3(2):88–93. | ||
Gerdes S, Thaçi D, Griffiths CEM, et al. Multiple switches between GP2015, an etanercept biosimilar, with originator product do not impact efficacy, safety and immunogenicity in patients with chronic plaque-type psoriasis: 30-week results from the phase 3, confirmatory EGALITY study. J Eur Acad Dermatol Venereol. 2018;32(3):420–427. | ||
Griffiths CEM, Thaçi D, Gerdes S, et al. The EGALITY study: a confirmatory, randomized, double-blind study comparing the efficacy, safety and immunogenicity of GP2015, a proposed etanercept biosimilar, vs. the originator product in patients with moderate-to-severe chronic plaque-type psoriasis. Br J Dermatol. 2017;176(4):928–938. | ||
Jørgensen KK, Olsen IC, Goll GL, et al. Switching from originator infliximab to biosimilar CT-P13 compared with maintained treatment with originator infliximab (NOR-SWITCH): a 52-week, randomised, double-blind, non-inferiority trial. Lancet. 2017;389(10086):2304–2316. | ||
Weinblatt ME, Baranauskaite A, Dokoupilova E. Switching from reference adalimumab to SB5 (adalimumab biosimilar) in patients with rheumatoid arthritis. Arthritis Rheumatol. 2018;70(6):832–840. | ||
Bohlega S, Al-Shammri S, Al Sharoqi I, et al. Biosimilars: opinion of an expert panel in the Middle East. Curr Med Res Opin. 2008;24(10):2897–2903. | ||
Farhat F, Othman A, El Karak F, Kattan J. Review and results of a survey about biosimilars prescription and challenges in the Middle East and North Africa region. Springerplus. 2016;5(1):2113. | ||
El Zorkany B, Al Ani N, Al Emadi S, et al. Biosimilars in rheumatology: recommendations for regulation and use in Middle Eastern countries. Clin Rheumatol. 2018;37(5):1143–1152. | ||
Cheraghali AM. Biosimilars; a unique opportunity for Iran national health sector and national pharmaceutical industry. Daru. 2012;20(1):35. | ||
Republic of Lebanon Ministry of Public Health. Guidance for registration of similar biological medicinal products (Biosimilars). Available from: http://www.moph.gov.lb/en/view/6917/guidance-and-list-of-requirements-for-registration-of-biosimilars-products. Accessed August 31, 2018. | ||
Heinemann L, Khatami H, Mckinnon R, Home P. An overview of current regulatory requirements for approval of biosimilar insulins. Diabetes Technol Ther. 2015;17(7):510–526. | ||
Saudi Food & Drug Authority. Guideline on biosimilar products, quality considerations; 2017. Available from: https://www.sfda.gov.sa/en/drug/drug_reg/Regulations/Guideline-on-biosimilar-products.pdf. Accessed December 1, 2017. | ||
Al-Jumaili AA, Hussain SA, Sorofman B. Pharmacy in Iraq: history, current status, and future directions. Am J Health Syst Pharm. 2013;70(4):368–372. | ||
World Health Organization. Essential medicines selection, Iraq, National Board for the Selection of Drugs, Essential Drugs List; 2010. Available from: http://www.who.int/selection_medicines/country_lists/irq/en/. Accessed December 20, 2017. | ||
Cheraghali AM. Current status of biopharmaceuticals in Iran’s pharmaceutical market. GaBI J. 2013;2(1):26–29. | ||
GaBI Online - Generics and Biosimilars Initiative. Biopharmaceutical products in Iran; 2013. Available from: http://www.gabionline.net/Biosimilars/Research/Biopharmaceutical-products-in-Iran. Accessed June 29, 2017. | ||
AryoGen Pharmed. Available from: http://www.aryogen.com/EN/index.html. Accessed October 26, 2017. | ||
Hassett B, Scheinberg M, Castañeda-Hernández G, et al. Variability of intended copies for etanercept (Enbrel®): data on multiple batches of seven products. MAbs. 2018;10(1):166–176. | ||
American College of Rheumatology, Committee on Rheumatologic Care. Biosimilars Position Statement; 2016. Available from: https://www.rheumatology.org/Portals/0/Files/Biosimilars-Position-Statement.pdf. Accessed August 24, 2017. | ||
Skingle D. Biosimilars: what do patients need to consider? RMD Open. 2015;1(1):e000141. | ||
Biswas P. Pharmacovigilance in Asia. J Pharmacol Pharmacother. 2013;4(Suppl 1):S7–S19. | ||
Al Hilfi TK. Toward a healthier Iraq. Yale J Biol Med. 2014;87(3):289–297. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.