")
Back to Journals » Drug Design, Development and Therapy » Volume 14
Reversible Small Molecule Inhibitors of MAO A and MAO B with Anilide Motifs
Authors Hagenow J, Hagenow S , Grau K, Khanfar M, Hefke L, Proschak E, Stark H
Received 30 October 2019
Accepted for publication 19 December 2019
Published 28 January 2020 Volume 2020:14 Pages 371—393
DOI https://doi.org/10.2147/DDDT.S236586
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Tin Wui Wong
Jens Hagenow,1 Stefanie Hagenow,1 Kathrin Grau,1 Mohammad Khanfar,1– 3 Lena Hefke,4 Ewgenij Proschak,4 Holger Stark1
1Heinrich Heine University Düsseldorf, Institute of Pharmaceutical and Medicinal Chemistry, Duesseldorf 40225, Germany; 2Faculty of Pharmacy, The University of Jordan, Amman 11942, Jordan; 3College of Pharmacy, Alfaisal University, Riyadh 11533, Saudi Arabia; 4Goethe University Frankfurt, Institute of Pharmaceutical Chemistry, Frankfurt 60438, Germany
Correspondence: Holger Stark
Heinrich Heine University Duesseldorf, Institute of Pharmaceutical and Medicinal Chemistry, Universitaetsstr. 1, Duesseldorf 40225, Germany
Tel +49 211 81-10478
Fax +49 211 81-13359
Email [email protected]
Background: Ligands consisting of two aryl moieties connected via a short spacer were shown to be potent inhibitors of monoamine oxidases (MAO) A and B, which are known as suitable targets in treatment of neurological diseases. Based on this general blueprint, we synthesized a series of 66 small aromatic amide derivatives as novel MAO A/B inhibitors.
Methods: The compounds were synthesized, purified and structurally confirmed by spectroscopic methods. Fluorimetric enzymological assays were performed to determine MAO A/B inhibition properties. Mode and reversibility of inhibition was determined for the most potent MAO B inhibitor. Docking poses and pharmacophore models were generated to confirm the in vitro results.
Results: N-(2,4-Dinitrophenyl)benzo[d][1,3]dioxole-5-carboxamide ( 55, ST-2043) was found to be a reversible competitive moderately selective MAO B inhibitor (IC50 = 56 nM, Ki = 6.3 nM), while N-(2,4-dinitrophenyl)benzamide ( 7, ST-2023) showed higher preference for MAO A (IC50 = 126 nM). Computational analysis confirmed in vitro binding properties, where the anilides examined possessed high surface complementarity to MAO A/B active sites.
Conclusion: The small molecule anilides with different substitution patterns were identified as potent MAO A/B inhibitors, which were active in nanomolar concentrations ranges. These small and easily accessible molecules are promising motifs, especially for newly designed multitargeted ligands taking advantage of these fragments.
Keywords: salicylic acid derivatives, molecular modeling, Parkinson’s disease, enzyme inhibitor, pharmacophore, structure-activity relationships
Plain Language Summary
Monoamine oxidases (MAO) A and B are neurotransmitter-catabolizing enzymes, which play a role in the pathophysiology of neurological diseases such as Parkinson’s disease, depression or schizophrenia. Small molecules consisting of two aryl moieties connected via a short spacer were shown to be potent MAO A/B inhibitors. In this study, aromatic amide derivatives with different structural variations and substitution pattern were demonstrated to have MAO A/B inhibition properties in a nanomolar concentration range. Compound 55 was found to be a reversible competitive MAO B preferring inhibitor (IC50 = 56 nM, Ki = 6.3 nM), while compound 7 showed a higher preference for MAO A (IC50 = 126 nM). Computational analysis confirmed in vitro binding properties as the respective anilides possessed high surface complementarity to MAO A/B active sites. These results suggest that the herein described anilides are small and easily accessible molecules, which may serve as promising precursors for the design of selective or multitargeting MAO A/B inhibitors.
Introduction
The neurotransmitter-catabolizing monoamine oxidases (MAO) are localized in the outer mitochondrial membrane and are classified into the A and B isoforms. MAO A is mainly involved in the degradation of serotonin, melatonin, norepinephrine, and epinephrine, and is expressed nearly ubiquitously in the human body. MAO B breaks down phenethylamine and benzylamine and it is highly expressed in the central nervous system (CNS). Dopamine, tyramine and tryptamine can be metabolized by both isoforms but with individual metabolic activity for each substrate. Consequently, inhibitors of both enzymes are established in the pharmacotherapy for neurological diseases.1–3 Moclobemide was the first marketed MAO inhibitor and entered the Swedish market in 1989.4 Slowing down the degradation of neurotransmitters like dopamine and norepinephrine via MAO A inhibition, this drug exhibits mood-lifting properties facilitating its use as an antidepressant.4 Since then the irreversible nonselective MAO inhibitor tranylcypromine as well as the irreversible MAO B inhibitors rasagiline and selegiline were approved for the treatment of depression and Parkinson’s disease (PD), respectively. Recently, safinamide as first reversible MAO B inhibitor in PD therapy was approved.5–7 As the imminent therapeutic benefits of MAO inhibitors are nowadays clear, multiple efforts have been made to develop new reversible and irreversible inhibitors. Due to long-term enzyme inactivation by irreversible MAO inhibitors, these can be associated with serious side effects or drug–drug interactions, eg leading to a serotonin syndrome.8 Reversible MAO inhibitors have a more favorable side effect profile; however, they can be displaced from the active site in case of high levels of the endogenous ligands. To maintain pharmacological efficacy, they are aimed to show a high affinity towards the MAOs and/or tight binding behavior.9 The design of such reversible and selective inhibitors of MAO A and B became a strongly researched area to identify suitable drug candidates for the treatment of neurological diseases, particularly of PD.
Having in mind that such diseases usually derive from multifactorial disorders, the development of sophisticated multitargeted ligands (MTLs) became of major exploration to treat these CNS disorders.10,11 The combination of different biological active moieties in one molecule is challenging since the affinity for each target needs to be preserved together with maintaining drug-likeness properties. Identification of small chemical entities with a promising target affinity are of great value in MTL drug design, as they might be easily combined or fused with other pharmacophores with minor influences on molecular size.
Accordingly, we synthesized a series of 66 anilides with diverse substitution patterns. The presented anilides are small-sized and have low molecular weights. The ligands show structural similarities to previously described potent MAO inhibitors assuring potential MAO inhibition activities. Multiple publications identified two aromatic moieties linked by a short bridging element as mutual MAO scaffold.8,12–16 Due to their high stability under physiological conditions, amides were used as linkers in our series. Aromatic moieties bearing different substituents were chosen to elucidate electronic and steric effects on the binding affinities towards both MAO isoforms. The presented ligands were characterized in vitro. For a better understanding of the binding modes, computational docking experiments were performed and a ligand-based pharmacophore-model was established.
Materials and Methods
Reagents and Instrumentation
Reagents and solvents for synthesis were purchased from Sigma-Aldrich, VWR Chemicals, Fisher Scientific, Panreac AppliChem, Alfa Aesar and Chemsolute and were used without further purifications (unless stated otherwise). 1H NMR and 13C NMR were recorded on a Bruker AMX spectrometer (Bruker, Germany) at 300 and 75 MHz respectively, where either CDCl3 or DMSO-d6 was used as a solvent. Tetramethylsilane was used as standard and chemical shifts are reported in parts per million (ppm). Elementary analyses (C, H, N) were measured on a CHN-Rapid (Heraeus, Germany) and were within 0.4% of the theoretical values for final compounds. LC-MS analysis was performed on a Bruker Elute SP; Column: Intensity Solo C18 RP; column dimensions: 100 x 2.1 mm; eluent: acetonitrile (1–40%) in water, 0.1% formic acid; flow: 0.3 mL/min) combined with mass spectrometric detection (Bruker amazon speed with ESI; detection: ion-trap; ion-polarity: positive; scan: 100–600 m/z). Data are given as retention time (tR), mass number ([M+H]+), and normalized peak area (%) as approximated purity. Melting points (m.p., uncorrected) were determined on a M-564 Büchi melting point apparatus (Büchi, Germany). Thin-layer chromatography (TLC) was carried out using pre-coated silica gel 60 with fluorescence indicator at UV 254 nm (Macherey-Nagel, Germany). The structure and purity of each compound were confirmed using 1H NMR, 13C NMR, LC-MS and/or elemental analysis.
Human recombinant monoamine oxidase A (MAO A, E.C. 1.4.3.4), human recombinant monoamine oxidase B (MAO B, E.C. 1.4.3.4), kynuramine dihydrobromide (KYN) and dimethylsulfoxide (DMSO) were purchased from Sigma Aldrich. For biological evaluation tests, compounds were dissolved in 100% DMSO (max. 10−3 M). Fluorescence intensity measurements were performed with an infinite M1000 Pro multimode reader (Tecan, Switzerland). Assay pipetting was partly automated using a Freedom EVO pipetting robot (Tecan, Switzerland).
Experimental Procedures
Using Method A the title compound was isolated as a white amorphous powder. Yield: 53%
1H NMR (300 MHz, DMSO-d6) δ 10.25 (s, 1H), 8.00–7.93 (m, 2H), 7.82–7.75 (m, 2H), 7.65–7.49 (m, 3H), 7.41–7.29 (m, 2H), 7.15–7.06 (m, 1H). 13C NMR (75 MHz, DMSO) δ 165.50, 139.13, 134.95, 131.49, 128.55, 128.33, 127.60, 123.60, 120.31.
Elemental Analysis: calc C 79.17%, H 5.62%, N 7.10%; found C 78.91%, H 5.53%, N 6.89%.
m.p.: 163°C (lit.163–164°C).35
Using Method C the title compound was isolated as a white amorphous powder. Yield: 83%
1H NMR (300 MHz, DMSO-d6) δ 11.95 (s, 1H), 8.51 (dt, J = 5.6, 1.3 Hz, 1H), 8.28–8.24 (m, 2H), 8.20–8.12 (m, 2H), 7.73–7.64 (m, 1H), 7.62–7.54 (m, 2H), 7.47 (td, J = 5.7, 2.7 Hz, 1H).
13C NMR (75 MHz, DMSO-d6) δ 166.89, 149.65, 143.30, 142.29, 132.94, 132.55, 128.60, 128.32, 120.34, 116.28.
LC-MS tR = 16.8 min; [M+H]+ = 198.9; 100%.
m.p.: 78°C (lit: 75–80°C).
Using Method A the title compound was isolated as a white solid. Yield: 57%
1H NMR (300 MHz, DMSO-d6) δ 10.65 (s, 1H), 8.06–7.92 (m, 4H), 7.87–7.78 (m, 2H), 7.68–7.49 (m, 3H).
13C NMR (75 MHz, DMSO) δ 166.15, 143.46, 134.34, 133.07, 131.99, 128.44, 127.80, 120.12, 119.04, 105.30.
Elemental Analysis: calc C 75.60%, H 4.54%, N 12.60%; found C 75.44%, H 4.24%, N 12.33%.
m.p.: 165°C (lit: 165–166°C).38
Using Method A the title compound was isolated as a white solid. Yield: 62%
1H NMR (300 MHz, DMSO-d6) δ 10.44 (s, 1H), 8.14–7.79 (m, 4H), 7.57 (ddd, J = 14.4, 7.8, 6.0 Hz, 3H), 7.37 (d, J = 8.6 Hz, 2H).
13C NMR (75 MHz, DMSO) δ 165.66, 143.82, 143.79, 138.36, 134.61, 131.68, 128.37, 127.65, 121.60, 121.41.
Elemental Analysis: calc C 59.79%, H 3.58%, N 4.98%; found C 59.76%, H 3.42%, N 4.90%.
m.p.: 179°C (lit: 176–178°C).40
Using Method D the title compound was isolated as a white solid. Yield 89%
1H NMR (300 MHz, DMSO-d6) δ 9.57 (s, 1H), 8.06–7.86 (m, 2H), 7.62–7.43 (m, 3H), 6.91 (d, J = 8.2 Hz, 1H), 6.47 (d, J = 2.5 Hz, 1H), 6.42 (dd, J = 8.3, 2.6 Hz, 1H), 2.08 (s, 3H).
13C NMR (75 MHz, DMSO) δ 165.29, 146.96, 134.81, 134.59, 131.13, 128.26, 127.78, 127.40, 124.87, 115.19, 111.43, 17.99.
Elemental Analysis: calc C 74.31%, H 6.05%, N 12.38%; found C 74.22%, H 6.05%, N 12.08%.
m.p.: 201°C (lit: 199–201°C).42
Using Method A the title compound was isolated as a faint yellow solid. 63%
1H NMR (300 MHz, DMSO-d6) δ 10.11 (s, 1H), 8.19 (dd, J = 2.6, 0.9 Hz, 1H), 8.12 (dd, J = 8.8, 2.8 Hz, 1H), 8.05–7.98 (m, 2H), 7.84 (d, J = 8.8 Hz, 1H), 7.68–7.52 (m, 3H), 2.41 (s, 3H).
13C NMR (75 MHz, DMSO) δ 165.59, 144.14, 143.00, 133.99, 133.88, 131.99, 128.47, 127.86, 125.57, 125.35, 121.49, 17.90.
Elemental Analysis: calc C 65.62%, H 4.72%, N 10.93%; found C 65.38%, H 4.42%, N 10.74%.
m.p.: 186°C (lit:181–186°C).42
Using Method A the title compound was isolated as a yellow solid. Yield 23%
1H NMR (300 MHz, DMSO-d6) δ 11.23 (s, 1H), 8.76 (d, J = 2.6 Hz, 1H), 8.59 (dd, J = 9.1, 2.7 Hz, 1H), 8.15 (d, J = 9.1 Hz, 1H), 8.05–7.91 (m, 2H), 7.78–7.53 (m, 3H).
13C NMR (75 MHz, DMSO) δ 164.49, 156.60, 141.19, 139.12, 137.12, 134.71, 131.32, 129.31, 123.56, 121.58, 119.84, 117.83, 116.95.
Elemental Analysis: calc C 54.36%, H 3.16%, N 14.63%; found C 54.21%, H 3.48%, N 14.80%.
m.p.: 199°C (lit: 199–201°C).44
Using Method D the title compound was isolated as an off-white solid. Yield 90%
1H NMR (300 MHz, DMSO-d6) δ 9.17 (s, 1H), 8.01–7.88 (m, 2H), 7.63–7.40 (m, 3H), 7.18 (d, J = 8.3 Hz, 1H), 6.31 (d, J = 2.3 Hz, 1H), 6.15 (dd, J = 8.4, 2.3 Hz, 1H), 5.08 (s, 2H), 3.71 (s, 3H).
13C NMR (75 MHz, DMSO) δ 153.40, 147.70, 134.82, 131.12, 128.28, 127.29, 126.68, 115.16, 105.13, 97.61, 55.12.
Elemental Analysis: calc C 69.41%, H 5.81%, N 11.56%; found C 69.49%, H 5.82%, N 11.38%.
m.p.: 115°C (lit: 112–113°C).
Using Method A the title compound was isolated as a yellow solid. Yield 60%
1H NMR (300 MHz, DMSO-d6) δ 9.63 (s, 1H), 8.31 (d, J = 8.9 Hz, 1H), 8.03–7.88 (m, 3H), 7.85 (d, J = 2.5 Hz, 1H), 7.69–7.49 (m, 3H), 4.00 (s, 3H).
13C NMR (75 MHz, DMSO) δ 165.32, 149.81, 143.47, 133.76, 132.16, 128.57, 127.62, 121.16, 116.53, 105.97, 56.55.
Elemental Analysis: calc C 61.76%, H 4.44%, N 10.29%; found C 61.71%, H 4.53%, N 10.33%.
m.p.: 149°C (lit: 148–151°C).
Using Method A the title compound was isolated as a pale-yellow solid. Yield 59%
1H NMR (300 MHz, DMSO-d6) δ 10.29 (s, 1H), 8.41 (d, J = 2.6 Hz, 1H), 8.27 (dd, J = 9.0, 2.6 Hz, 1H), 8.09 (d, J = 9.0 Hz, 1H), 8.05–7.95 (m, 2H), 7.73–7.50 (m, 3H).
13C NMR (75 MHz, DMSO) δ 165.49, 144.45, 141.31, 133.37, 132.36, 128.59, 128.01, 127.89, 126.58, 124.85, 122.89.
Elemental Analysis: calc C 56.44%, H 3.28%, N 10.13%; found C 56.14%, H 3.38%, N 10.18%.
m.p.: 161°C (lit: 160°C).
Using Method B the title compound was isolated as a white solid. Yield 75%
1H NMR (300 MHz, DMSO-d6) δ 10.80 (s, 1H), 8.75 (ddd, J = 4.8, 1.7, 0.9 Hz, 1H), 8.17 (dt, J = 7.8, 1.2 Hz, 1H), 8.07 (td, J = 7.7, 1.7 Hz, 1H), 7.96–7.88 (m, 2H), 7.69 (ddd, J = 7.5, 4.8, 1.4 Hz, 1H), 7.59–7.52 (m, 2H).
13C NMR (75 MHz, DMSO-d6) δ 162.65, 149.66, 148.41, 138.12, 137.77, 131.41, 127.00, 122.46, 122.24, 115.64.
Elemental Analysis: calc C 52.01%, H 3.27%, N 10.11%; found C 51.74%, H 3.06%, N 10.10%.
m.p.: 148°C (lit: 147–148°C).48
Using Method A the title compound was isolated as a white solid. Yield 61%
1H NMR (300 MHz, DMSO-d6) δ 11.83 (s, 1H), 10.40 (s, 1H), 7.99 (dd, J = 7.9, 1.7 Hz, 1H), 7.75–7.69 (m, 2H), 7.51–7.33 (m, 3H), 7.18–7.09 (m, 1H), 7.03–6.93 (m, 2H).
13C NMR (75 MHz, DMSO) δ 166.57, 158.48, 138.10, 133.62, 128.99, 128.69, 124.15, 120.96, 118.98, 117.42, 117.19.
Elemental Analysis: calc C 73.23%, H 5.20%, N 6.57%; found C 73.13%, H 5.21%, N 6.46%.
m.p.: 132°C (lit: 127–129°C).50
Using Method A the title compound was isolated as a white solid. Yield 47%
1H NMR (300 MHz, DMSO-d6) δ 11.41 (s, 1H), 10.66 (s, 1H), 7.94 (d, J = 8.8 Hz, 2H), 7.88 (dd, J = 7.9, 1.7 Hz, 1H), 7.83 (d, J = 8.8 Hz, 2H), 7.45 (ddd, J = 8.2, 7.2, 1.7 Hz, 1H), 7.05–6.92 (m, 2H).
13C NMR (75 MHz, DMSO) δ 166.45, 157.49, 142.68, 133.67, 133.15, 129.48, 120.40, 119.18, 118.96, 118.47, 117.05, 105.57.
Elemental Analysis: calc C 70.58%, H 4.23%, N 11.76%; found C 70.40%, H 4.11%, N 11.67%.
m.p.: 176°C (lit: 175–176.5°C).52
Using Method A the title compound was isolated as a yellow solid. Yield 22%
1H NMR (300 MHz, DMSO-d6) δ 12.41 (s, 1H), 11.96 (s, 1H), 9.01 (d, J = 9.4 Hz, 1H), 8.88 (d, J = 2.7 Hz, 1H), 8.59 (dd, J = 9.4, 2.8 Hz, 1H), 8.01 (dd, J = 8.0, 1.8 Hz, 1H), 7.50 (ddd, J = 8.6, 7.2, 1.8 Hz, 1H), 7.11–6.97 (m, 2H).
13C NMR (75 MHz, DMSO) δ 165.50, 142.73, 137.31, 132.90, 132.80, 128.81, 128.61, 127.91, 125.49, 121.16.
Elemental Analysis: calc C 51.49%, H 2.99%, N 13.86%; found C 51.40%, H 2.77%, N 13.97%.
m.p.: 212°C (lit: 213–214°C).54
Using Method A the title compound was isolated as a yellow solid. Yield 45%
1H NMR (300 MHz, DMSO-d6) δ 11.86 (s, 1H), 11.28 (s, 1H), 8.73 (d, J = 9.0 Hz, 1H), 8.03 (dd, J = 7.9, 1.8 Hz, 1H), 7.95 (dd, J = 9.0, 2.5 Hz, 1H), 7.85 (d, J = 2.5 Hz, 1H), 7.45 (ddd, J = 8.2, 7.2, 1.8 Hz, 1H), 7.09–6.96 (m, 2H), 4.04 (s, 3H).
13C NMR (75 MHz, DMSO) δ 163.54, 156.01, 147.85, 142.27, 134.60, 133.98, 131.09, 119.89, 118.33, 118.17, 117.34, 116.95, 105.64, 56.78.
Elemental Analysis: calc C 58.33%, H 4.20%, N 9.72%; found C 58.15%, H 4.05%, N 9.54%.
m.p.: 207°C (lit: 205–206°C).
Using Method A the title compound was isolated as a pale-yellow solid. Yield 55%
1H NMR (300 MHz, DMSO-d6) δ 12.18 (s, 1H), 11.42 (s, 1H), 8.89 (d, J = 9.3 Hz, 1H), 8.44 (d, J = 2.6 Hz, 1H), 8.31 (dd, J = 9.3, 2.7 Hz, 1H), 8.09 (dd, J = 7.9, 1.8 Hz, 1H), 7.53 (ddd, J = 8.2, 7.2, 1.8 Hz, 1H), 7.16–7.02 (m, 2H).
13C NMR (75 MHz, DMSO) δ 163.74, 156.15, 142.21, 141.45, 134.41, 131.14, 124.67, 123.78, 122.09, 120.45, 120.03, 117.82, 116.98.
Elemental Analysis: calc C 53.35%, H 3.10%, N 9.57%; found C 53.05%, H 3.32%, N 9.27%.
m.p.: 219°C (lit: 218–220°C).
Using Method A the title compound was isolated as a paleyellow solid. Yield 12%
1H NMR (300 MHz, DMSO-d6) δ 10.31 (s, 1H), 8.56 (dd, J = 8.7, 2.7 Hz, 1H), 8.52 (d, J = 2.6 Hz, 1H), 7.96 (d, J = 8.8 Hz, 1H), 7.92–7.86 (m, 2H), 7.38 (d, J = 7.9 Hz, 2H), 2.40 (s, 3H).
13C NMR (75 MHz, DMSO) δ 166.12, 145.09, 142.45, 141.88, 141.86, 141.84, 131.48, 130.41, 129.09, 127.87, 127.80, 126.04, 125.63, 124.29, 122.32, 122.25, 120.66, 20.99.
Elemental Analysis: calc C 55.56%, H 3.42%, N 8.64%; found C 55.46%, H 3.25%, N 8.39%.
m.p.: 144°C.
Using Method A the title compound was isolated as a white solid. Yield 63%
1H NMR (300 MHz, DMSO-d6) δ 10.32 (s, 1H), 8.04–7.95 (m, 2H), 7.83–7.74 (m, 2H), 7.65–7.55 (m, 2H), 7.36 (dd, J = 8.5, 7.3 Hz, 2H), 7.16–7.07 (m, 1H).
13C NMR (75 MHz, DMSO) δ 164.38, 138.92, 136.34, 133.61, 129.57, 128.58, 128.40, 123.77, 120.38.
Elemental Analysis: calc C 67.40%, H 4.35%, N 6.05%; found C 67.37%, H 4.21%, N 5.93%.
m.p.: 198°C (lit: 200–201°C).
Using Method C the title compound was isolated as a white solid. Yield 78%
1H NMR (300 MHz, DMSO-d6) δ 12.13 (s, 1H), 8.51 (ddd, J = 5.7, 1.7, 0.9 Hz, 1H), 8.36–8.14 (m, 4H), 7.73–7.60 (m, 2H), 7.47 (ddd, J = 7.1, 5.6, 1.5 Hz, 1H).
13C NMR (75 MHz, DMSO-d6) δ 165.86, 149.58, 143.24, 142.44, 137.80, 131.38, 130.33, 128.66, 120.45, 116.30.
LC-MS tR = 22.3 min; [M+H]+ = 232.9; 100%.
m.p.: 138°C (lit: 137–139°C).59
Using Method A the title compound was isolated as a white solid. Yield 49%
1H NMR (300 MHz, DMSO-d6) δ 10.67 (s, 1H), 8.00 (d, J = 1.6 Hz, 2H), 7.97 (d, J = 1.8 Hz, 2H), 7.81 (d, J = 8.8 Hz, 2H), 7.61 (d, J = 8.6 Hz, 2H).
13C NMR (75 MHz, DMSO) δ 164.97, 143.25, 136.87, 133.04, 132.97, 129.75, 128.49, 120.18, 118.98, 105.48.
Elemental Analysis: calc C 65.51%, H 3.53%, N 10.91%; found C 65.62%, H 3.51%, N 10.95%.
m.p.: 208°C.
Using Method A the title compound was isolated as a yellow solid. Yield 52%
1H NMR (300 MHz, DMSO-d6) δ 10.18 (s, 1H), 8.19 (d, J = 2.7 Hz, 1H), 8.11 (dd, J = 8.8, 2.7 Hz, 1H), 8.02 (d, J = 8.6 Hz, 2H), 7.81 (d, J = 8.8 Hz, 1H), 7.63 (d, J = 8.6 Hz, 2H), 2.40 (s, 3H).
13C NMR (75 MHz, DMSO) δ 164.57, 144.26, 142.76, 136.83, 134.00, 132.70, 129.81, 128.53, 125.72, 125.36, 121.48, 17.89.
Elemental Analysis: calc C 57.84%, H 3.81%, N 9.64%; found C 58.03%, H 3.99%, N 9.64%.
m.p.: 193°C.
Using Method A the title compound was isolated as a yellow solid. Yield 54%
1H NMR (300 MHz, DMSO-d6) δ 9.83 (s, 1H), 8.24 (d, J = 8.9 Hz, 1H), 8.03–7.91 (m, 3H), 7.88 (d, J = 2.5 Hz, 1H), 7.67–7.58 (m, 2H), 4.00 (s, 3H).
13C NMR (75 MHz, DMSO) δ 164.51, 150.30, 143.83, 136.97, 133.55, 132.59, 129.72, 128.60, 122.03, 116.45, 106.18, 56.56.
Elemental Analysis: calc C 54.83%, H 3.62%, N 9.13%; found C 54.67%, H 3.57%, N 8.86%.
m.p.: 200°C (decomposition).
Using Method A the title compound was isolated as a yellow solid. Yield 17%
1H NMR (300 MHz, DMSO-d6) δ 10.42 (s, 1H), 8.43 (d, J = 2.6 Hz, 1H), 8.27 (dd, J = 8.9, 2.6 Hz, 1H), 8.09–7.98 (m, 3H), 7.66 (d, J = 8.6 Hz, 2H).
13C NMR (75 MHz, DMSO) δ 164.56, 144.69, 141.12, 137.19, 132.13, 129.87, 128.69, 128.32, 127.00, 124.90, 122.89.
Elemental Analysis: calc C 50.19%, H 2.59%, N 9.00%; found C 50.44%, H 2.30%, N 8.96%.
m.p.: 219°C.
Using Method A the title compound was isolated as an off-white solid. Yield 20%
1H NMR (300 MHz, DMSO-d6) δ 10.41 (s, 1H), 8.41 (d, J = 2.6 Hz, 1H), 8.27 (dd, J = 9.0, 2.6 Hz, 1H), 8.04 (d, J = 9.0 Hz, 1H), 7.99–7.92 (m, 2H), 7.83–7.76 (m, 2H).
13C NMR (75 MHz, DMSO) δ 164.70, 144.65, 141.10, 132.49, 131.62, 130.00, 128.25, 126.92, 126.19, 124.88, 122.88.
Elemental Analysis: calc C 43.91%, H 2.27%, N 7.88%; found C 43.65%, H 1.90%, N 7.53%.
m.p.: 205°C.
Using Method D the title compound was isolated as an off-white solid. Yield 90%
1H NMR (300 MHz, DMSO-d6) δ 10.47 (s, 1H), 7.69 (dd, J = 8.3, 1.3 Hz, 2H), 7.36 (t, J = 7.9 Hz, 2H), 7.21–7.15 (m, 1H), 7.12 (d, J = 7.4 Hz, 1H), 6.73 (d, J = 2.0 Hz, 2H), 4.72 (s, 2H).
13C NMR (75 MHz, DMSO) δ 165.96, 148.64, 140.98, 138.50, 128.72, 123.71, 120.37, 120.29, 118.02, 117.50, 113.68.
Elemental Analysis: calc C 68.41%, H 5.30%, N 12.27%; found C 68.01%, H 4.96%, N 12.38%.
m.p.: 176°C (lit: 174–176°C).62
Using Method A the title compound was isolated as a white solid. Yield 61%
1H NMR (300 MHz, DMSO-d6) δ 12.78 (s, 1H), 10.57 (s, 1H), 8.79 (d, J = 2.9 Hz, 1H), 8.29 (dd, J = 9.1, 2.9 Hz, 1H), 7.72 (d, J = 8.0 Hz, 2H), 7.39 (t, J = 7.7 Hz, 2H), 7.16 (dd, J = 8.4, 5.5 Hz, 2H).
13C NMR (75 MHz, DMSO) δ 164.08, 163.27, 139.34, 137.89, 128.75, 128.33, 125.71, 124.39, 120.77, 119.33, 117.97.
Elemental Analysis: calc C 60.47%, H 3.90%, N 10.85%; found C 60.20%, H 3.85%, N 10.65%.
m.p.: 223°C (lit: 219–221°C).
Using Method A the title compound was isolated as a white solid. Yield 62%
1H NMR (300 MHz, DMSO-d6) δ 11.87 (s, 1H), 10.41 (s, 1H), 7.98 (d, J = 2.7 Hz, 1H), 7.77–7.64 (m, 2H), 7.47 (dd, J = 8.8, 2.7 Hz, 1H), 7.43–7.32 (m, 2H), 7.21–7.10 (m, 1H), 7.02 (d, J = 8.8 Hz, 1H).
13C NMR (75 MHz, DMSO) δ 164.96, 156.87, 137.95, 132.99, 128.72, 128.32, 124.29, 122.68, 120.81, 119.45, 119.04.
Elemental Analysis: calc C 63.04%, H 4.07%, N 5.66%; found C 62.83%, H 4.17%, N 5.55%.
m.p.: 209°C (lit: 211–212°C).63
Using Method A the title compound was isolated as a pale-yellow solid. Yield 46%
1H NMR (300 MHz, DMSO-d6) δ 12.26 (s, 1H), 10.88 (s, 1H), 8.40 (dd, J = 8.3, 1.6 Hz, 1H), 7.99 (d, J = 2.8 Hz, 1H), 7.54 (dd, J = 8.0, 1.5 Hz, 1H), 7.48 (dd, J = 8.7, 2.8 Hz, 1H), 7.43–7.34 (m, 1H), 7.17 (ddd, J = 8.0, 7.4, 1.6 Hz, 1H), 7.07 (d, J = 8.8 Hz, 1H).
13C NMR (75 MHz, DMSO-d6) δ 162.59, 155.35, 134.98, 133.33, 129.72, 129.29, 127.78, 125.32, 123.46, 122.76, 119.58, 118.99.
Elemental Analysis: calc C 55.35%, H 3.22%, N 4.96%; found C 55.35%, H 3.31%, N 4.75%.
m.p.: 189°C (lit:184–186°C).62
Using Method A the title compound was isolated as a pale-yellow solid. Yield 41%
1H NMR (300 MHz, DMSO-d6) δ 11.65 (s, 1H), 10.49 (s, 1H), 7.92 (t, J = 2.1 Hz, 1H), 7.90 (d, J = 2.7 Hz, 1H), 7.61 (ddd, J = 8.3, 2.1, 1.0 Hz, 1H), 7.47 (dd, J = 8.8, 2.7 Hz, 1H), 7.40 (t, J = 8.1 Hz, 1H), 7.20 (ddd, J = 8.0, 2.1, 1.0 Hz, 1H), 7.02 (d, J = 8.8 Hz, 1H).
13C NMR (75 MHz, DMSO-d6) δ 164.96, 156.41, 139.55, 133.01, 130.39, 128.44, 123.87, 122.72, 120.05, 119.94, 118.99.
Elemental Analysis: calc C 55.35%, H 3.22%, N 4.96%; found C 55.12%, H 3.25%, N 4.86%.
m.p.: 219°C (lit: 217–218°C).62
Using Method A the title compound was isolated as a pale-yellow solid. Yield 37%
1H NMR (300 MHz, DMSO-d6) δ 11.63 (s, 1H), 10.65 (s, 1H), 8.03–7.93 (m, 2H), 7.92–7.83 (m, 3H), 7.47 (dd, J = 8.8, 2.7 Hz, 1H), 7.03 (d, J = 8.8 Hz, 1H), 4.30 (q, J = 7.1 Hz, 2H), 1.32 (t, J = 7.1 Hz, 3H).
13C NMR (75 MHz, DMSO) δ 165.22, 164.87, 156.25, 142.53, 132.99, 130.11, 128.61, 125.00, 122.74, 120.28, 119.80, 118.96, 60.49, 14.17.
Elemental Analysis: calc C 60.10%, H 4.41%, N 4.38%; found C 60.46%, H 4.38%, N 4.32%.
m.p.: 214°C (lit: 212–214°C).
Using Method A the title compound was isolated as a pale-yellow solid. Yield 62%
1H NMR (300 MHz, DMSO-d6) δ 11.65 (s, 1H), 10.65 (s, 1H), 7.94 (d, J = 8.5 Hz, 2H), 7.90 (d, J = 2.7 Hz, 1H), 7.72 (dd, J = 8.7, 0.9 Hz, 2H), 7.46 (dd, J = 8.8, 2.7 Hz, 1H), 7.03 (d, J = 8.8 Hz, 1H).
13C NMR (75 MHz, DMSO) δ 165.02, 156.30, 141.75, 141.73, 133.02, 128.57, 125.97, 125.92, 122.78, 120.39, 120.07, 118.95.
Elemental Analysis: calc C 53.27%, H 2.87%, N 4.44%; found C 53.09%, H 2.63%, N 4.38%.
m.p.: 221°C (lit: 222–223°C).63
Using Method A the title compound was isolated as a yellow solid. Yield 9%
1H NMR (300 MHz, DMSO-d6) δ 11.46 (s, 1H), 10.84 (s, 1H), 8.28 (d, J = 9.2 Hz, 2H), 7.99 (d, J = 9.2 Hz, 2H), 7.84 (d, J = 2.8 Hz, 1H), 7.48 (dd, J = 8.8, 2.8 Hz, 1H), 7.04 (d, J = 8.8 Hz, 1H).
13C NMR (75 MHz, DMSO) δ 164.98, 155.83, 144.50, 142.70, 133.00, 128.73, 124.85, 122.77, 120.88, 120.01, 118.90.
Elemental Analysis: calc C 53.35%, H 3.10%, N 9.57%; found C 53.37%, H 3.02%, N 9.36%.
m.p.: 259°C (li: 260–262°C).62
Using Method A the title compound was isolated as a white solid. Yield 60%
1H NMR (300 MHz, DMSO-d6) δ 11.76 (s, 1H), 10.54 (s, 1H), 7.94 (d, J = 2.7 Hz, 1H), 7.83 (d, J = 9.0 Hz, 2H), 7.46 (dd, J = 8.8, 2.7 Hz, 1H), 7.41–7.30 (m, 2H), 7.02 (d, J = 8.8 Hz, 1H).
13C NMR (75 MHz, DMSO) δ 165.04, 156.73, 144.25 (m), 137.21, 133.02, 128.36, 122.71, 122.12, 121.46, 119.51, 118.99.
Elemental Analysis: calc C 50.70%, H 2.74%, N 4.22%; found C 50.30%, H 2.65%, N 4.06%.
m.p.: 200°C (lit: 200–202°C).
Using Method A the title compound was isolated as a yellow solid. Yield 55%
1H NMR (300 MHz, DMSO-d6) δ 11.75 (s, 1H), 7.93 (d, J = 2.7 Hz, 1H), 7.76–7.65 (m, 2H), 7.59–7.50 (m, 2H), 7.45 (dd, J = 8.8, 2.7 Hz, 1H), 7.01 (d, J = 8.8 Hz, 1H).
13C NMR (75 MHz, DMSO) δ 164.92, 156.63, 137.43, 133.00, 131.52, 128.38, 122.71, 122.57, 119.64, 119.00, 115.97.
Elemental Analysis: calc C 47.81%, H 2.78%, N 4.29%; found C 47.49%, H 2.47%, N 3.94%.
m.p.: 237°C (lit: 240–241.5°C).68
Using Method A the title compound was isolated as a yellow solid. Yield 16%
1H NMR (300 MHz, DMSO-d6) δ 12.33 (s, 1H), 10.73 (s, 1H), 8.52 (d, J = 9.0 Hz, 1H), 8.25–8.11 (m, 2H), 7.98 (d, J = 2.8 Hz, 1H), 7.53 (dd, J = 8.7, 2.8 Hz, 1H), 7.10 (d, J = 8.7 Hz, 1H), 2.45 (s, 3H).
13C NMR not performed due to poor solubility.
Elemental Analysis: calc C 54.83%, H 3.62%, N 9.13%; found C 54.78%, H 3.32%, N 8.96%.
m.p.: 229°C (lit: 225–227).
Using Method A the title compound was isolated as an off-white solid. Yield 51%
1H NMR (300 MHz, DMSO-d6) δ 11.42 (s, 1H), 10.76 (s, 1H), 8.36 (d, J = 1.8 Hz, 2H), 7.91–7.63 (m, 2H), 7.39 (dd, J = 8.8, 2.7 Hz, 1H), 6.96 (d, J = 8.8 Hz, 1H).
13C NMR (75 MHz, DMSO) δ 165.46, 156.39, 140.14, 133.21, 130.64 (q, J = 32.9 Hz), 128.47, 124.94, 122.73, 121.33, 120.22, 119.74, 118.97,116.70 (q, J = 7.8, 4.0 Hz).
Elemental Analysis: calc C 46.96%, H 2.10%, N 3.65%; found C 46.73%, H 1.70%, N 3.61%.
m.p.: 171°C.
Using Method A the title compound was isolated as a pale-yellow solid. Yield 63%
1H NMR (300 MHz, DMSO-d6) δ 12.51 (s, 1H), 11.24 (s, 1H), 8.77 (d, J = 9.2 Hz, 1H), 8.53 (dd, J = 9.2, 2.7 Hz, 1H), 8.43 (d, J = 2.7 Hz, 1H), 7.91 (d, J = 2.8 Hz, 1H), 7.51 (dd, J = 8.8, 2.8 Hz, 1H), 7.06 (d, J = 8.8 Hz, 1H).
13C NMR (75 MHz, DMSO) δ 162.50, 155.11, 142.28, 141.10, 134.09, 130.06, 128.68, 123.81, 123.74, 122.11, 122.02, 118.98, 118.95, 118.26.
Elemental Analysis: calc C 46.62%, H 2.24%, N 7.77%; found C 46.32%, H 1.94%, N 7.64%.
m.p.: 191°C.
Using Method A the title compound was isolated as an off-white solid. Yield 13%
1H NMR (300 MHz, DMSO-d6) δ 12.25 (s, 1H), 11.24 (s, 1H), 8.69 (d, J = 9.0 Hz, 1H), 8.04–7.82 (m, 3H), 7.50 (dd, J = 8.7, 2.9 Hz, 1H), 7.08 (d, J = 8.8 Hz, 1H), 4.04 (s, 3H).
13C NMR (75 MHz, DMSO) δ 162.21, 154.92, 147.93, 142.54, 134.19, 133.54, 130.00, 123.62, 119.85, 119.04, 118.34, 117.31, 105.72, 56.84.
Elemental Analysis: calc C 52.10%, H 3.28%, N 8.68%; found C 52.10%, H 3.28%, N 8.65%.
m.p.: 230°C (lit: 233–235°C).72
Using Method A the title compound was isolated as a pale-yellow solid. Yield 57%
1H NMR (300 MHz, DMSO-d6) δ 12.05 (s, 1H), 11.89 (s, 1H), 9.23 (d, J = 1.6 Hz, 1H), 8.09 (d, J = 8.3 Hz, 1H), 7.92 (d, J = 2.8 Hz, 1H), 7.77 (dd, J = 8.2, 1.7 Hz, 1H), 7.49 (dd, J = 8.8, 2.8 Hz, 1H), 7.06 (d, J = 8.8 Hz, 1H), 3.91 (s, 3H), 3.90 (s, 3H).
13C NMR (75 MHz, DMSO) δ 166.13, 165.35, 163.21, 155.43, 139.41, 133.74, 133.34, 131.12, 129.96, 123.60, 123.07, 122.67, 121.54, 120.19, 118.83, 52.78, 52.58.
Elemental Analysis: calc C 56.13%, H 3.88%, N 3.85%; found C 56.53%, H 3.64%, N 4.00%.
m.p.: 193°C.
Using Method A the title compound was isolated as a yellow solid. Yield 11%
1H NMR (300 MHz, DMSO-d6) δ 12.24 (s, 1H), 12.20 (s, 1H), 8.58 (dd, J = 11.9, 2.9 Hz, 1H), 8.30 (dd, J = 9.2, 5.8 Hz, 1H), 7.90 (d, J = 3.0 Hz, 1H), 7.49 (dd, J = 8.6, 2.9 Hz, 1H), 7.20 (ddd, J = 9.8, 7.3, 3.0 Hz, 1H), 7.05 (d, J = 8.7 Hz, 1H).
13C NMR (75 MHz, DMSO-d6) δ 164.87 (d, J = 252.5 Hz), 163.08, 155.34, 136.01 (d, J = 13.7 Hz), 134.52 (d, J = 2.7 Hz), 133.83, 130.12, 128.75 (d, J = 11.4 Hz), 123.36, 119.48, 118.92, 111.09 (d, J = 24.2 Hz), 109.40 (d, J = 29.8 Hz).
Elemental Analysis: calc C 50.26%, H 2.60%, N 9.02%; found C 50.19%, H 2.73%, N 9.02%.
m.p.: 178°C.
Using Method A the title compound was isolated as a brown solid. Yield 37%
1H NMR (300 MHz, DMSO-d6) δ 11.61 (s, 1H), 10.65 (s, 1H), 8.50 (d, J = 2.2 Hz, 1H), 7.89 (dd, J = 8.1, 2.5 Hz, 2H), 7.58–7.44 (m, 2H), 7.03 (d, J = 8.8 Hz, 1H), 3.33 (s, 3H).
13C NMR (75 MHz, DMSO) δ 165.18, 156.50, 148.51, 137.04, 133.10, 133.04, 128.39, 128.15, 125.13, 122.69, 119.80, 119.02, 115.79, 19.19.
Elemental Analysis: calc C 54.83%, H 3.62%, N 9.13%; found C 55.21%, H 3.50%, N 8.73%.
m.p.: 278°C.
Using Method A the title compound was isolated as a yellow solid. Yield 29%
1H NMR (300 MHz, DMSO-d6) δ 12.04 (s, 1H), 10.30 (s, 1H), 8.01 (d, J = 2.7 Hz, 1H), 7.46 (dd, J = 8.8, 2.7 Hz, 1H), 7.39 (d, J = 2.4 Hz, 1H), 7.25 (dd, J = 8.7, 2.4 Hz, 1H), 7.01 (d, J = 8.8 Hz, 1H), 6.95 (d, J = 8.7 Hz, 1H), 3.77 (s, 3H), 3.75 (s, 3H).
13C NMR (75 MHz, DMSO) δ 165.02, 157.36, 148.46, 145.70, 133.03, 131.21, 127.99, 122.58, 119.10, 118.81, 113.05, 111.77, 106.00, 55.63, 55.42.
Elemental Analysis: calc C 58.55%, H 4.59%, N 4.55%; found C 58.53%, H 4.68%, N 4.46%.
m.p.: 185°C.
Using Method A the title compound was isolated as a pale-yellow solid. Yield 56%
1H NMR (300 MHz, DMSO-d6) δ 11.75–11.27 (s, 1H), 10.56 (s, 1H), 7.90–7.76 (m, 2H), 7.47 (dd, J = 8.8, 2.7 Hz, 1H), 7.34 (t, J = 1.9 Hz, 1H), 7.03 (d, J = 8.8 Hz, 1H).
13C NMR (75 MHz, DMSO) δ 164.99, 156.11, 140.56, 133.99, 133.05, 128.53, 123.23, 122.74, 120.21, 118.93, 118.54.
Elemental Analysis: calc C 49.32%, H 2.55%, N 4.42%; found C 49.70%, H 2.20%, N 4.36%.
m.p.: 247 °C (lit: 247–249 °C).
Using Method A the title compound was isolated as an off-white solid. Yield 50%
1H NMR (300 MHz, DMSO-d6) δ 10.52 (s, 1H), 8.42 (d, J = 2.2 Hz, 1H), 8.32 (dd, J = 8.4, 2.2 Hz, 1H), 7.92 (d, J = 8.4 Hz, 1H), 7.74 (dd, J = 8.0, 1.7 Hz, 1H), 7.58 (dd, J = 8.0, 1.5 Hz, 1H), 7.43 (td, J = 7.6, 1.6 Hz, 1H), 7.32 (td, J = 7.7, 1.7 Hz, 1H).
13C NMR (75 MHz, DMSO-d6) δ 163.90, 148.38, 141.98, 133.87, 131.20, 130.11, 129.72, 128.35, 127.69, 127.56, 124.57, 122.40.
Elemental Analysis: calc C 50.19%, H 2.59%, N 9.00%; found C 50.11%, H 2.70%, N 8.81%.
m.p.: 171°C.
Using Method A the title compound was isolated as an off-white solid. Yield 51%
1H NMR (300 MHz, DMSO-d6) δ 10.95 (s, 1H), 8.44 (d, J = 2.2 Hz, 1H), 8.32 (dd, J = 8.4, 2.2 Hz, 1H), 7.99–7.87 (m, 2H), 7.58 (ddd, J = 8.3, 2.1, 1.0 Hz, 1H), 7.42 (t, J = 8.1 Hz, 1H), 7.22 (ddd, J = 8.0, 2.1, 1.0 Hz, 1H).
13C NMR (75 MHz, DMSO-d6) δ 163.57, 148.44, 141.85, 139.78, 133.18, 131.09, 130.62, 130.06, 124.63, 123.98, 122.49, 119.14, 118.09.
Elemental Analysis: calc C 50.19%, H 2.59%, N 9.00%; found C 50.26%, H 2.64%, N 8.92%.
m.p.: 163°C.
Using Method A the title compound was isolated as a pale-yellow solid. Yield 39%
1H NMR (300 MHz, DMSO-d6) δ 11.36 (s, 1H), 8.45 (d, J = 2.2 Hz, 1H), 8.36–8.25 (m, 3H), 7.97 (dd, J = 8.7, 5.3 Hz, 3H).
13C NMR (75 MHz, DMSO-d6) δ 164.04, 148.58, 144.35, 142.96, 141.45, 131.09, 130.15, 125.03, 124.67, 122.54, 119.54.
Elemental Analysis: calc C 48.54%, H 2.51%, N 13.06%; found C 48.16%, H 2.63%, N 12.81%.
m.p.: 197°C (lit: 196–198°C).
Using Method B the title compound was isolated as a yellow solid. Yield 20%
1H NMR (300 MHz, DMSO-d6) δ 10.26 (s, 1H), 10.11 (s, 1H), 8.38 (d, J = 2.2 Hz, 1H), 8.27 (dd, J = 8.4, 2.2 Hz, 1H), 8.01 (d, J = 2.6 Hz, 1H), 7.85 (d, J = 8.4 Hz, 1H), 7.08 (dd, J = 8.6, 2.6 Hz, 1H), 6.92 (d, J = 8.6 Hz, 1H).
13C NMR (75 MHz, DMSO-d6) δ 163.88, 148.22, 147.47, 142.16, 131.16, 130.13, 126.51, 124.96, 124.35, 122.24, 121.98, 116.59.
Elemental Analysis: calc C 47.79%, H 2.47%, N 8.56%; found C 47.68%, H 2.48%, N 8.51%.
m.p.: 223°C.
Using Method A the title compound was isolated as a white solid. Yield 66%
1H NMR (300 MHz, DMSO-d6) δ 10.22 (s, 1H), 7.95–7.81 (m, 2H), 7.60 (dd, J = 8.4, 2.1 Hz, 1H), 7.53 (d, J = 2.1 Hz, 1H), 7.41–7.30 (m, 2H), 7.08 (d, J = 8.5 Hz, 1H), 4.11 (qd, J = 7.0, 1.7 Hz, 4H), 1.36 (td, J = 6.9, 1.1 Hz, 6H).
13C NMR (75 MHz, DMSO) δ 165.01, 151.23, 147.59, 143.67, 143.64, 138.50, 126.46, 121.63, 121.35, 121.16, 112.52, 111.99, 63.96, 63.82, 14.67, 14.55.
Elemental Analysis: calc C 58.54%, H 4.91%, N 3.79%; found C 58.64%, H 5.16%, N 3.66%.
m.p.: 173°C.
Using Method A the title compound was isolated as a white solid. Yield 57%
1H NMR (300 MHz, DMSO-d6) δ 10.16 (s, 1H), 7.79–7.71 (m, 2H), 7.59 (dd, J = 8.4, 2.1 Hz, 1H), 7.56–7.49 (m, 3H), 7.07 (d, J = 8.5 Hz, 1H), 4.11 (qd, J = 7.0, 1.3 Hz, 4H), 1.36 (td, J = 7.0, 1.1 Hz, 6H).
13C NMR (75 MHz, DMSO) δ 164.96, 151.19, 147.57, 138.67, 131.31, 126.53, 122.22, 121.14, 115.03, 112.48, 111.99, 63.96, 63.82, 14.69, 14.57.
Elemental Analysis: calc C 56.06%, H 4.98%, N 3.85%; found C 56.19%, H 5.26%, N 3.77%.
m.p.: 198°C.
Using Method A the title compound was isolated as a white solid. Yield 64%
1H NMR (300 MHz, DMSO-d6) δ 10.20 (s, 1H), 8.56 (dd, J = 8.7, 2.7 Hz, 1H), 8.52 (d, J = 2.7 Hz, 1H), 7.93 (d, J = 8.8 Hz, 1H), 7.60 (dd, J = 8.4, 2.1 Hz, 1H), 7.53 (d, J = 2.1 Hz, 1H), 7.12 (d, J = 8.5 Hz, 1H), 4.24–4.01 (m, 4H), 1.37 (t, J = 6.9 Hz, 6H).
13C NMR (75 MHz, DMSO) δ 165.66, 151.64, 147.68, 145.02, 142.03, 131.43, 127.87, 125.97, 125.56, 125.10, 122.36, 121.35, 112.38, 112.13, 63.92, 63.88, 14.63, 14.53.
Elemental Analysis: calc C 54.27%, H 4.30%, N 7.03%; found C 54.18%, H 4.22%, N 6.96%.
m.p.: 158°C.
Using Method A the title compound was isolated as an off-white solid. Yield 65%
1H NMR (300 MHz, DMSO-d6) δ 10.08 (s, 1H), 8.41 (d, J = 2.6 Hz, 1H), 8.26 (dd, J = 9.0, 2.6 Hz, 1H), 8.04 (d, J = 9.0 Hz, 1H), 7.64 (dd, J = 8.4, 2.1 Hz, 1H), 7.56 (d, J = 2.1 Hz, 1H), 7.11 (d, J = 8.5 Hz, 1H), 4.12 (qd, J = 6.9, 5.4 Hz, 4H), 1.37 (t, J = 6.9 Hz, 6H).
13C NMR (75 MHz, DMSO) δ 164.82, 151.71, 147.70, 144.28, 141.56, 127.89, 126.54, 125.22, 124.84, 122.86, 121.51, 112.43, 112.10, 63.94, 63.89, 14.65, 14.54.
Elemental Analysis: calc C 55.98%, H 4.70%, N 7.68%; found C 56.00%, H 4.69%, N 7.54%.
m.p.: 168°C.
Using Method A the title compound was isolated as a white solid. Yield 64%
1H NMR (300 MHz, DMSO-d6) δ 10.06 (s, 1H), 7.83–7.72 (m, 2H), 7.59 (dd, J = 8.2, 1.8 Hz, 1H), 7.53 (d, J = 1.8 Hz, 1H), 7.34 (dd, J = 8.5, 7.3 Hz, 2H), 7.15–7.02 (m, 2H), 6.14 (s, 2H).
13C NMR (75 MHz, DMSO) δ 164.44, 149.98, 147.32, 139.18, 128.70, 128.50, 123.46, 122.79, 120.31, 107.87, 107.68, 101.76.
Elemental Analysis: calc C 69.70%, H 4.60%, N 5.81%; found C 69.54%, H 4.65%, N 5.77%.
m.p.: 142°C (lit: 138–139°C).76
Using Method A the title compound was isolated as a white solid. Yield 48%
1H NMR (300 MHz, DMSO-d6) δ 10.44 (s, 1H), 7.97 (d, J = 8.8 Hz, 2H), 7.80 (d, J = 8.7 Hz, 2H), 7.59 (dd, J = 8.1, 1.8 Hz, 1H), 7.52 (d, J = 1.8 Hz, 1H), 7.08 (d, J = 8.1 Hz, 1H), 6.15 (s, 2H).
13C NMR (75 MHz, DMSO) δ 165.02, 150.42, 147.41, 143.54, 133.02, 128.01, 123.21, 120.06, 119.06, 107.96, 107.80, 105.10, 101.91.
Elemental Analysis: calc C 67.67%, H 3.79%, N 10.52%; found C 67.62%, H 3.87%, N 10.67%.
m.p.: 193°C.
Using Method A the title compound was isolated as an off-white solid. Yield 18%
1H NMR (300 MHz, DMSO-d6) δ 9.92 (s, 1H), 8.19 (d, J = 2.7 Hz, 1H), 8.10 (dd, J = 8.8, 2.8 Hz, 1H), 7.78 (d, J = 8.8 Hz, 1H), 7.61 (dd, J = 8.1, 1.8 Hz, 1H), 7.53 (d, J = 1.8 Hz, 1H), 7.09 (d, J = 8.2 Hz, 1H), 6.15 (s, 2H), 2.39 (s, 3H).
13C NMR (75 MHz, DMSO) δ 164.56, 150.40, 147.43, 144.05, 143.11, 133.85, 127.74, 125.59, 125.33, 123.20, 121.47, 108.02, 107.83, 101.88, 17.89.
Elemental Analysis: calc C 60.00%, H 4.03%, N 9.33%; found C 59.85%, H 3.76%, N 9.22%.
m.p.: 230°C.
Using Method A the title compound was isolated as a yellow solid. Yield 10%
1H NMR (300 MHz, DMSO-d6) δ 11.04 (s, 1H), 8.74 (d, J = 2.6 Hz, 1H), 8.57 (dd, J = 9.0, 2.7 Hz, 1H), 8.12 (d, J = 9.1 Hz, 1H), 7.59 (dd, J = 8.2, 1.8 Hz, 1H), 7.47 (d, J = 1.8 Hz, 1H), 7.12 (d, J = 8.2 Hz, 1H), 6.17 (s, 2H).
13C NMR (75 MHz, DMSO) δ 164.49, 151.20, 147.73, 142.52, 140.60, 137.48, 128.56, 126.54, 125.30, 123.62, 121.14, 108.26, 107.68, 102.18.
Elemental Analysis: calc C 50.77%, H 2.74%, N 12.69%; found C 50.84%, H 2.46%, N 12.68%.
m.p.: 200°C.
Using Method A the title compound was isolated as a yellow solid. Yield 33%
1H NMR (300 MHz, DMSO-d6) δ 9.53 (s, 1H), 8.24 (d, J = 8.9 Hz, 1H), 7.94 (dd, J = 8.9, 2.5 Hz, 1H), 7.87 (d, J = 2.5 Hz, 1H), 7.59 (dd, J = 8.2, 1.8 Hz, 1H), 7.52 (d, J = 1.8 Hz, 1H), 7.08 (d, J = 8.1 Hz, 1H), 6.15 (s, 2H), 4.01 (s, 3H).
13C NMR (75 MHz, DMSO) δ 164.40, 150.58, 150.01, 147.55, 143.50, 133.87, 127.62, 123.09, 121.56, 116.53, 108.09, 107.67, 106.07, 101.96, 56.57.
Elemental Analysis: calc C 56.97%, H 3.82%, N 8.86%; found C 56.67%, H 3.71%, N 8.64%.
m.p.: 237°C.
Using Method A the title compound was isolated as a pale-yellow solid. Yield 53%
1H NMR (300 MHz, DMSO-d6) δ 10.11 (s, 1H), 8.41 (d, J = 2.6 Hz, 1H), 8.26 (dd, J = 9.0, 2.6 Hz, 1H), 8.04 (d, J = 9.0 Hz, 1H), 7.63 (dd, J = 8.2, 1.8 Hz, 1H), 7.53 (d, J = 1.8 Hz, 1H), 7.10 (d, J = 8.2 Hz, 1H), 6.16 (s, 2H).
13C NMR (75 MHz, DMSO) δ 164.49, 150.72, 147.55, 144.38, 141.41, 127.99, 127.12, 126.65, 124.85, 123.39, 122.86, 108.12, 107.78, 101.99.
Elemental Analysis: calc C 52.44%, H 2.83%, N 8.74%; found C 52.20%, H 2.85%, N 8.65%.
m.p.: 227°C.
Using Method A the title compound was isolated as a pale-yellow solid. Yield 52%
1H NMR (300 MHz, Chloroform-d) δ 8.86 (d, J = 9.2 Hz, 1H), 8.79 (s, 1H), 8.43–8.37 (m, 1H), 8.31 (d, J = 2.5 Hz, 1H), 8.19 (ddd, J = 9.3, 2.6, 0.5 Hz, 1H), 7.99–7.82 (m, 4H), 7.63–7.49 (m, 2H).
13C NMR (75 MHz, CDCl3) δ 165.44, 143.07, 140.51, 135.31, 132.57, 130.76, 129.30, 129.24, 128.64, 128.32, 127.92, 127.37, 124.76, 123.83, 123.06, 122.59, 120.24.
Elemental Analysis: calc C 62.49%, H 3.39%, N 8.57%; found C 62.72%, H 3.09%, N 8.71%.
m.p.: 225°C.
Using Method A the title compound was isolated as a yellow solid. Yield 10%
1H NMR (300 MHz, DMSO-d6) δ 12.21 (s, 1H), 11.64 (s, 1H), 8.91 (d, J = 9.2 Hz, 1H), 8.75 (s, 1H), 8.46 (d, J = 2.6 Hz, 1H), 8.34 (dd, J = 9.2, 2.7 Hz, 1H), 8.03 (d, J = 8.2 Hz, 1H), 7.81 (dd, J = 8.5, 1.2 Hz, 1H), 7.56 (ddd, J = 8.2, 6.9, 1.3 Hz, 1H), 7.46–7.35 (m, 2H).
13C NMR (75 MHz, DMSO) δ 163.56, 152.25, 142.46, 141.41, 136.28, 133.41, 129.23, 128.86, 127.21, 125.73, 124.81, 124.14, 123.93, 122.35, 120.73, 120.10, 110.95.
Elemental Analysis: calc C 59.58%, H 3.24%, N 8.17%; found C 58.97%, H 3.20%, N 7.67%.
m.p.: 230°C (lit: 233°C).77
Using Method A the title compound was isolated as an off-white solid. Yield 26%
1H NMR (300 MHz, DMSO-d6) δ 11.20 (s, 1H), 10.89 (s, 1H), 8.48 (s, 1H), 8.01 (d, J = 8.4 Hz, 2H), 7.94 (d, J = 8.2 Hz, 1H), 7.76 (dd, J = 8.7, 4.6 Hz, 3H), 7.51 (ddd, J = 8.3, 6.9, 1.3 Hz, 1H), 7.42–7.31 (m, 2H).
13C NMR (75 MHz, DMSO) δ 165.84, 153.25, 142.22, 135.71, 130.63, 128.66, 128.11, 126.83, 126.12, 126.06, 126.01, 125.75, 123.72, 122.51, 120.11, 110.47.
Elemental Analysis: calc C 65.26%, H 3.65%, N 4.23%; found C 64.43%, H 3.69%, N 3.93%.
m.p.: 280°C (lit: 276–278°C).
Using Method A the title compound was isolated as an off-white solid. Yield 29%
1H NMR (300 MHz, DMSO-d6) δ 11.22 (s, 1H), 10.68 (s, 1H), 8.47 (s, 1H), 7.93 (d, J = 8.2 Hz, 1H), 7.83–7.71 (m, 4H), 7.63–7.45 (m, 4H), 7.42–7.30 (m, 2H).
13C NMR (75 MHz, DMSO) δ 165.64, 153.46, 137.91, 135.70, 131.57, 130.44, 128.65, 128.09, 126.82, 125.75, 123.72, 122.26, 122.12, 115.62, 110.48.
Elemental Analysis: calc C 59.67%, H 3.53%, N 4.09%; found C 59.37%, H 3.56%, N 3.98%.
m.p.: 250°C (lit: 248–250°C).
Using Method A the title compound was isolated as a yellow solid. Yield 13%
1H NMR (300 MHz, DMSO-d6) δ 13.06 (s, 1H), 11.92–11.53 (m, 1H), 8.50–8.42 (m, 2H), 8.38 (d, J = 8.2 Hz, 1H), 8.31 (dd, J = 9.1, 2.6 Hz, 1H), 8.07 (d, J = 8.8 Hz, 1H), 7.92 (d, J = 8.1 Hz, 1H), 7.72–7.54 (m, 2H), 7.48 (d, J = 8.8 Hz, 1H).
13C NMR (75 MHz, DMSO) δ 167.42, 143.79, 141.30, 136.40, 129.02, 127.62, 126.20, 125.80, 124.85, 124.75, 124.52, 123.42, 123.35, 109.88.
Elemental Analysis: calc C 59.58%, H 3.24%, N 8.17%; found C 59.36%, H 3.25%, N 8.05%.
m.p.: 238°C.
Using Method A the title compound was isolated as a pale-yellow solid. Yield 47%
1H NMR (300 MHz, Chloroform-d) δ 8.90 (d, J = 9.2 Hz, 1H), 8.46–8.29 (m, 2H), 8.21 (dd, J = 9.3, 2.6 Hz, 1H), 7.57 (ddt, J = 5.3, 3.8, 1.9 Hz, 2H), 7.50–7.39 (m, 2H), 3.80 (q, J = 2.4 Hz, 2H), 2.69 (t, J = 2.4 Hz, 3H).
13C NMR (75 MHz, CDCl3) δ 163.71, 152.04, 145.10, 142.64, 141.96, 140.74, 130.78, 128.36, 127.23, 124.68, 124.04, 123.76, 122.02, 121.40, 119.77, 38.03, 12.76.
Elemental Analysis: calc C 62.11%, H 3.99%, N 8.54%; found C 62.23%, H 3.59%, N 8.29%.
m.p.: 216°C.
Using Method A the title compound was isolated as a yellow solid. Yield 13%
1H NMR (300 MHz, DMSO-d6) δ 11.58 (s, 1H), 8.82 (d, J = 2.6 Hz, 1H), 8.63 (dd, J = 9.1, 2.7 Hz, 1H), 8.37 (d, J = 9.1 Hz, 1H), 7.94–7.84 (m, 2H), 7.78 (dd, J = 8.4, 1.0 Hz, 1H), 7.58 (ddd, J = 8.4, 7.2, 1.3 Hz, 1H), 7.42 (td, J = 7.6, 1.0 Hz, 1H).
13C NMR not performed due to poor solubility.
Elemental Analysis: calc C 55.05%, H 2.77%, N 12.84%; found C 54.77%, H 2.67%, N 12.85%.
m.p.: 237°C.
Using Method A the title compound was isolated as a yellow solid. Yield 54%
1H NMR (300 MHz, Chloroform-d) δ 9.13 (s, 1H), 8.79 (d, J = 9.2 Hz, 1H), 8.29 (d, J = 2.6 Hz, 1H), 8.15 (ddd, J = 9.2, 2.6, 0.6 Hz, 1H), 7.71–7.65 (m, 1H), 7.62 (d, J = 1.0 Hz, 1H), 7.54 (dq, J = 8.5, 1.0 Hz, 1H), 7.44 (ddd, J = 8.4, 7.2, 1.3 Hz, 1H), 7.29 (ddd, J = 8.0, 7.2, 1.1 Hz, 1H).
13C NMR (75 MHz, CDCl3) δ 156.56, 155.05, 147.26, 143.26, 139.86, 128.12, 127.41, 124.84, 124.33, 123.71, 123.13, 122.73, 120.23, 113.21, 112.13.
Elemental Analysis: calc C 56.89%, H 2.86%, N 8.85%; found C 56.99%, H 2.78%, N 8.64%.
m.p.: 203°C.
Using Method A the title compound was isolated as an off-white solid. Yield 21%
1H NMR (300 MHz, DMSO-d6) δ 10.59 (s, 1H), 8.49–8.39 (m, 2H), 8.28 (dd, J = 8.9, 2.6 Hz, 1H), 8.13–7.99 (m, 3H), 7.58–7.45 (m, 2H).
13C NMR (75 MHz, DMSO) δ 160.53, 144.74, 140.72, 140.63, 138.90, 138.05, 128.21, 127.35, 127.03, 126.94, 125.69, 125.20, 124.97, 122.93, 122.90.
Elemental Analysis: calc C 54.14%, H 2.73%, N 8.42%, S 9.63%; found C 54.34%, H 2.46%, N 8.37%, S 9.49%.
m.p.: 228°C.
Chemistry
General Synthesis Procedures
The aniline derivative (6 mmol, 1 eq) and the aromatic acid derivative (6 mmol, 1 eq) were suspended in toluene (10 mL). After heating to reflux and dissolving of the precipitate, phosphor trichloride (2.4 mmol, 0.4 eq) was added in a dropwise manner. The reaction was monitored using TLC (ethyl acetate:n-hexane 1:4). After reaction completion, the hot mixture was filtered and the organic solution was stored at 5 °C. The formed precipitate was filtered off, washed with ice-cold toluene and recrystallized from toluene.17
1,1ʹ-Carbonyldiimidazole (CDI) (9 mmol, 1.5 eq) was dissolved in THF (10 mL). To this solution the respective aromatic acid (9 mmol, 1.5 eq) was added dissolved in THF (10 mL). After 10 min stirring at room temperature, a solution of the aniline derivative (6 mmol, 1 eq) in THF (10 mL) was added. The reaction mixture was stirred for 10 min at room temperature and then refluxed overnight. After cooling to room temperature, the solvent was evaporated under reduced pressure and the residue dissolved in ethylacetate. The organic phase was then washed with saturated aqueous sodium hydrogen carbonate solution and brine. After drying over magnesium sulfate, the solvent was evaporated under reduced pressure and the residue was recrystallized from toluene.18
The respective pyridine derivative (6 mmol, 1 eq) and the benzoyl chloride derivative (6 mmol, 1 eq) were sealed in a glass vial. The reaction mixture was stirred for 4 min at 160°C under microwave irradiation. The resulting mixture was recrystallized from ethanol.19
The respective nitro derivative (6, 9, 26) (6 mmol, 1 eq) was dissolved in dried ethanol (20 mL). After adding Pd/C (0.6 mmol, 0.1 eq) and hydrazine monohydrate (60 mmol, 10 eq), the mixture was refluxed for 2 h and afterwards stirred for another 45 min at room temperature. Filtration via a short Celite column and evaporation of the solvent afforded the desired compounds (5, 8, 25).20
Enzymology
Monoamine oxidase A/B inhibition assays were performed as described previously using the discontinuous fluorimetric method with kynuramine as MAO substrate.21 For the screening (test concentration: 10−6 M) and IC50 determinations (ten suitable concentrations ranging from 10−11 to 10−5 M), the test compounds were pre-mixed with the substrate (2 ⨰ KM final concentration; KM of 20 µm and 30 µm for MAO A and B, respectively). MAO A (1.25 ng mL−1, 900 units mL−1) or MAO B (1.67 ng mL−1, 375 units mL−1), respectively, was added to start the reactions. The reaction mixtures were incubated for 20 min incubation time at 37°C. All enzyme assays were conducted in pre-warmed potassium phosphate buffer (50 mM, pH = 7.4) and a final assay volume of 100 µL. Reactions were stopped by adding 35 µL sodium hydroxide solution (2 N) to the assay mixture. The enzyme activity was determined by measuring the 4-hydroxyquinoline formed during incubation time (expressed as RFU; λEm = 405±20 nm and λEx = 320±20 nm). Compounds showing a MAO B inhibition >90% at 10−6 M were evaluated for their IC50-values. The IC50 curves were fitted (via nonlinear regression) either to the respective bottom plateau (graphically defined by RFUs for the at least two highest concentrations used, see Figure S12 in supplemental material) or set to zero in case when the bottom plateau was not reached with highest concentration tested (leading to “IC50 estimates”). The highest concentrations to be tested had to be identified considering each compound’s solubility and potential interfering fluorescence properties under assay conditions.
Reversibility of inhibition was confirmed via preincubation of inhibitor (10 ⨰ IC50 in preincubation setting) with MAO B (10 ng µL−1 in preincubation setting) for 0, 30, 60 and 90 min (37 °C), followed by 50⨰ dilution in buffer and assayed with an excess of substrate (10 ⨰ KM final concentration) as described above.21 Data were calculated as percentage of vehicle control (DMSO; set to 100% enzyme activity remained) for each time point.
Mode of MAO B inhibition was determined by substrate-dependent (seven concentrations, 5 to 400 µm kynuramine) Michaelis-Menten kinetic analysis without inhibitor and in the presence of five different inhibitor concentrations (0.25⨰, 0.5⨰, 0.75⨰, 1⨰ and 2 ⨰ IC50) and assayed as described above. Data were fitted using nonlinear “Competitive inhibition” fit and transformed into double reciprocal (Lineweaver–Burk) plots. The slopes of respective Lineweaver–Burk linearization were plotted against inhibitor concentration for additional Ki determination.22
All data were analyzed with GraphPad Prism 6.
Molecular Modelling
The crystal structure 2Z5X23 was used as the representative complex structure of MAO A and the crystal structure 6FVZ9 for MAO B. Both crystal structures were chosen due to the lipophilicity of the crystalized ligands (HRM for MAO A and E8Z for MAO B) and the presence of a peptide bond of the MAO B ligand. The enzyme structures were prepared using the Quick Prep tool in MOE 2018.0101 (Chemical Computing Group Inc., Canada) including protonation and energy minimization. Poses were generated using the Dock tool in MOE 2018.0101. A general docking was conducted. In case of MAO A, solvent was ignored. The placement method triangle matcher with the scoring function London dG was chosen. The refinement method rigid receptor with the scoring function Alpha HB was chosen.
Pharmacophore Modeling
The pharmacophore hypotheses of the MAO A and B inhibitors were constructed using the HipHop module of Discovery studio software 2.5.5. A subset of 25 representative compounds was selected and classified as active, moderately active, and inactive (Table S1 in the supplemental material). In total 16 HipHop runs were separately conducted for MAO A and MAO B ligands that ultimately generate 160 models (10 from each run) for each target by varying the types and ranges of pharmacophoric features and the number of features (Table S2 in the supplemental material). The generated models were allowed to compete in Receiver Operating Characteristic (ROC) curve analysis to assess their abilities to selectively capture diverse MAO A or MAO B inhibitors from a large list of decoys. The decoy list was prepared as described by Verdonk et al.24 The testing sets include structurally diverse active inhibitors of 14 MAO A and 18 MAO B retrieved from literature.14,25,26 36 decoys were selected for each active compound in the testing set retrieved from ZINC-database. The ROC testing set was screened by each pharmacophore for ROC analysis employing the “Best rigid search” option, while the conformational spaces of the compounds were generated employing the “CAESAR conformation generation option”. Compounds missing one or more features were discarded from hit lists. The validity of a particular pharmacophore is assessed by the area under the curve (AUC) of the corresponding ROC curve, as well as accuracy, specificity, true positive rate, and false negative rate of the pharmacophore. Table S3 and Figure S10 in the supplemental material show the ROC performance of the best pharmacophore models generated for MAO A and MAO B. The active/inactive classification accuracy of these models is good with ROC-AUC values of 0.779 and 0.750 for MAO A and MAO B models, respectively. Further theoretical and experimental methodology of ROC analyses are described in supplemental material.
Results and Discussion
Chemistry
In 2018 the European Medicines Agency (EMA) recommended 22 biological active small molecules for market authorization.27 All newly approved small molecules exhibit rather complex structures with the need for multistep synthesis, often including stereoselective synthesis steps. Using a one-step synthesis followed by easy purification procedures, we generated a series of promising MAO A/B inhibitors. These small entities might be of interest for the development of selective or multitargeting MAO A/B inhibitors. The substituted aromatic amides were synthesized in low to good yields via activation of the carboxylic acid using either PCl3 or CDI. CDI was used as a coupling agent for compound 47 to prevent condensation to a substituted benzoxazole, which was the major product using PCl3 method. For compounds 2 and 19, solvent free acylation using benzoyl chloride derivatives was performed under microwave irradiation (Figure 1). The aromatic amines (5, 8, 25) were obtained in excellent yields from reduction of the respective nitro derivatives (6, 9, 26). Due to their simple structures, many of our compounds have been described previously, but to our knowledge none of them were determined as potent inhibitors for MAO A or B so far.
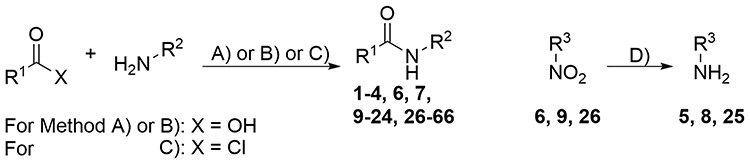 |
Figure 1 Synthetic route to the amide analogs 1–4, 6, 7, 9–24, 26–66. Reagents and conditions: (A) PCl3, toluene, reflux. (B) CDI, THF, reflux. (C) mw, 160 °C. For R1, R2, and R3 see Table 1. |
Biological Activity
MAO A and B Inhibition
An enzyme inhibition screening at a concentration of 1 µm revealed that the synthesized anilides showed the presumed MAO inhibition potency. The majority of derivatives (32 compounds) were found to be MAO B-preferring inhibitors, while 24 compounds were not selective for either of the isoforms and only ten compounds exhibited MAO A preference (Table 1 and see Figure 2 for all compound structures). The most potent MAO inhibitors were chosen for further IC50 evaluation. Six compounds (31, 33, 34, 39, 55, and 65) showed >90% inhibition of MAO B. The most potent MAO A-preferring inhibitor (with MAO B inhibition <50%), compound 7 was also considered for additional investigations. Compounds 31, 33, 34, 55, and 65 showed the expected preference for MAO B with IC50 values in nanomolar concentration ranges (IC50 values between 55 nM and 139 nM; Table 2). A preference for MAO A could be confirmed for compound 7 (ST-2023) (MAO A, IC50 = 126 nM) with an IC50 > 1000 nM for MAO B; selectivity index, SI = 0.1). The potent MAO B inhibition capacity of 39, presumed depending on screening results, could not be verified in IC50 determinations (MAO B, IC50 >1000 nM), where assay limitations did not allow the determination of the bottom plateau due to low solubility and fluorescence crosstalk. Assuming that the real but nondetectable bottom plateau might be » 0, the previous determined percent inhibition value in one-point screening would have been overestimated (as calculated with bottom = 0). The most potent compound 55 (ST-2043) showed high inhibition of MAO B (IC50 = 56 nM), which is in a similar range as described for the marketed drug safinamide (IC50 = 53 nM; Table 1). Compared to safinamide (MAO A/B SI >940), 55 demonstrated only an about five-fold lower inhibition of isoform A, thus being a rather balanced MAO inhibitor. The highest MAO B preference within this series was shown for compound 31 with an SI >10 (MAO B, IC50 = 92 nM).
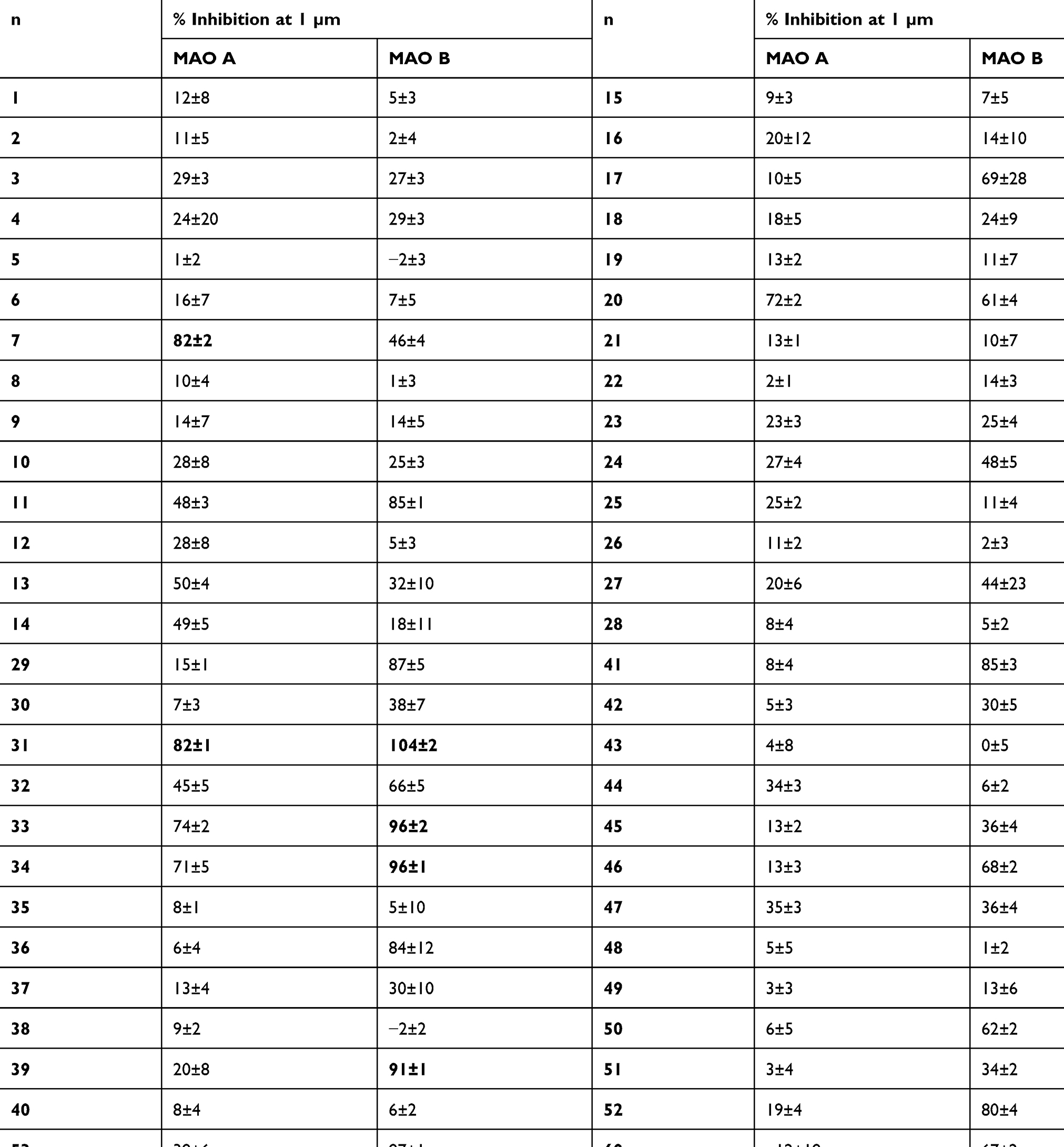 |
Table 1 MAO A and B Inhibition Percentages Measured at One-Point Screening of Anilides (Screening Concentration 1 µm) |
 |
Figure 2 Compounds with anilide motifs taken to MAO A and B screening. |
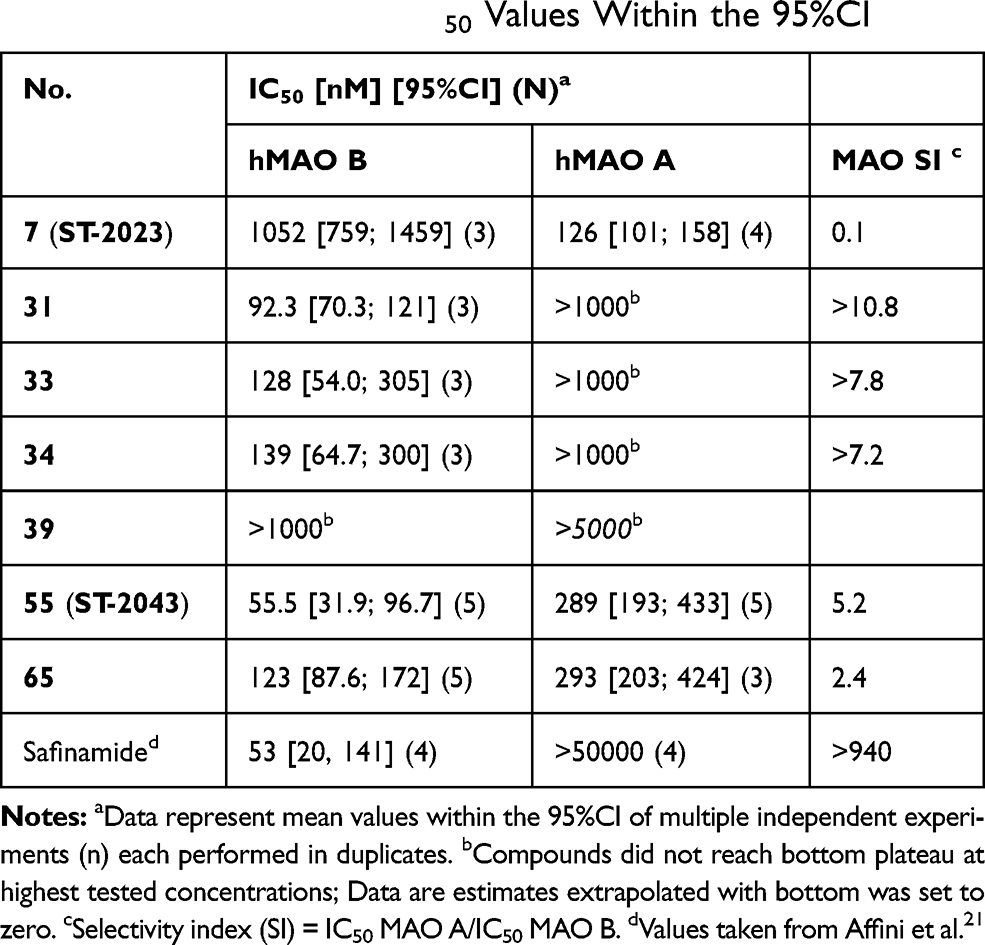 |
Table 2 MAO A and B Inhibition Activities of the Most Potent Compounds Expressed as IC50 Values Within the 95%CI |
The docking experiments revealed a good match of all compounds in the lipophilic binding pockets of the MAO A and B with exception of compound 39, where no suitable docking pose into the MAO A binding pocket could be identified (see Figures S1-S9 in the supplemental material). According to the result of the docking experiments, surface complementarities influence the binding properties of the anilides nearly exclusively, whereas direct interactions are not involved in the binding of the presented ligands. Compounds 31, 33, and 34 showed close structural similarities and comparable inhibition properties. All of these ligands are substituted with a hydroxyl group at the 2-position of the benzylic site and a chloro substituent at the 5-position. The aniline moiety in all cases is para-substituted with a highly lipophilic residue, ie a bromo-substituent (34), a trifluoromethoxy group (33), and a trifluoromethyl group (31) in the para-position. The interaction of these hydrophobic residues with the lipophilic cavity of the MAO B binding pocket, including the nonpolar amino acids Pro102 and Leu164, might be responsible for their potent MAO B inhibition properties. The carbonyl function of the linking amide and the hydroxyl groups in 2-position are directed towards Cys172 and interact via polar aprotic forces (see Figures S5-S7 in the supplemental material). A similar polar system is present in pyrrolo-pyridinyl derivatives synthesized by Tzvetkov et alindicating that a central hydrogen bond donor/acceptor complex is favorable for MAO B binding.28 The docking pose of the MAO A preferring inhibitor 7 (ST-2023) is inverted by 180° in the binding pocket of the MAO B model, where the polar nitro-group is interacted with Cys172 (Figure 3). This would enlarge the distance to the FAD and could be responsible for the low MAO B activity compared with MAO A. This reversed pose was also observed in compounds 39 and 65. In case of inhibitor 39, the salicylic moiety is shifted towards the lipophilic cavity at the entrance of the binding pocket, which might be responsible for its rather poor inhibition properties (see Figures S8 in the supplemental material). In contrast, inhibitor 65 maintained the lipophilic interaction with its benzofuran residue resulting in an orientation of the polar nitro group towards the FAD (see Figures S9 in the supplemental material). Carboxamides synthesized by Tzvetkov et al showed the same orientation resulting from a polar interaction with a bridging water molecule and a tyrosine close to the FAD.29 The most potent inhibitor 55 (ST-2043) in this series is oriented with its piperonylic acid moiety towards the FAD (Figure 3). Using the same moiety, Vishnu et al designed MTLs where the piperonylic acid aligned within the hydrophobic region opposite to the FAD, and the high inhibition capacities resulted from irreversible binding via a propargyl amine residue.30
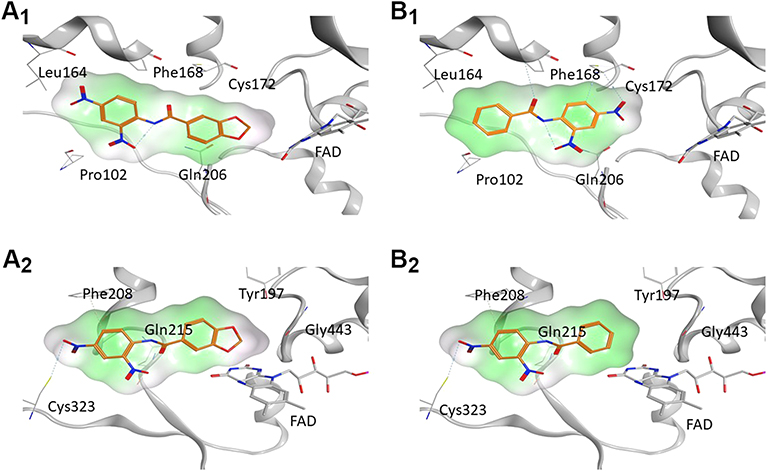 |
Figure 3 Visualization of 55 (ST-2043, A1, A2) and 7 (ST-2023, B1, B2) in the binding pockets of MAO A and B. Compound 55 (ST-2043) binding to the crystal structure of A1) hMAO B (PDB: 6FVZ) and A2) hMAO A (PDB: 2Z5X). Compound 7 (ST-2023) binding to the crystal structure of B1) hMAO B (PDB: 6FVZ) and B2) hMAO A (PDB: 2Z5X). Surface coloring: white: neutral, green: lipophilic; magenta: hydrophilic. |
Due to their small size and molecular weight, the presented structures are useful starting skeletons to design MTLs with MAO B activity. The docking experiments suggest using the aniline residue of 55 (ST-2043) as an attachment point to implement pharmacophores of additional targets. Zhang et al showed that larger substituents at this position are tolerated, where a bulky fluorobenzyl group is located further outside of the enzymes binding pocket than the here presented aniline groups.31 The hypothesis that substitution at this position might be suitable for designing MTLs is coherent with the linear structures synthesized by Pisani et al that combine MAO B inhibition with nitric oxide releasing precursors and acetylcholine esterase (AChE) inhibition moieties.32
In the MAO A binding pocket, the polar salicylic group of compounds 31, 33, 34, and 39 did not match the surrounding hydrophobic cavity, which might explain the loss of inhibition properties for MAO A (see Figures S1-S3 in the supplemental material). The three compounds, missing the salicylic moiety (7, 55, 65), are aligned well into the MAO A active site with respect to surface complementarity, showing that small alteration in the molecular structure can have a major impact on the modulation of the MAO isoform selectivity. The modeling results are further supported by ROC-selected pharmacophore models based on inhibitors presented here (see Figure 4 and supplemental material). The essential predicted characteristics for MAO A and MAO B binding are a central hydrogen bond acceptor flanked by aromatic or hydrophobic features. This is in good accordance with the docking poses, with the limitation that the molecular docking showed no direct hydrogen bond interactions between the ligands and the amino acids of the binding pocket.
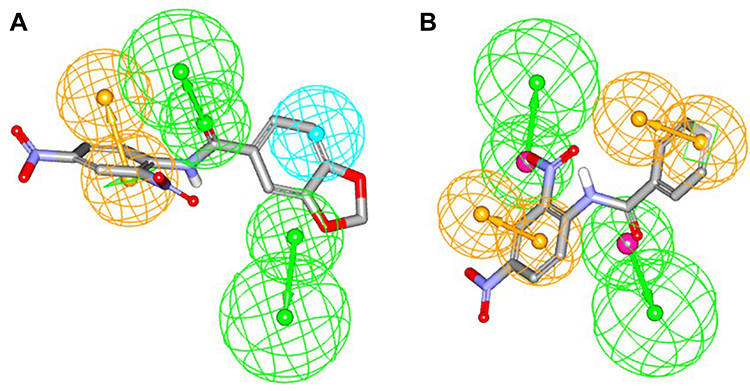 |
Figure 4 ROC-selected pharmacophores of MAO A and MAO B. (A) Overlay of compound 55 (ST-2043) and MAO B pharmacophore model. (B) Overlay of compound 7 (ST-2013) and MAO A pharmacophore model. Color code: green vectored sphere: hydrogen bond acceptor; orange vectored sphere: aromatic feature; blue sphere: hydrophobic feature. |
The most potent MAO B inhibitor 55 (ST-2043) was further investigated to determine mode and type of inhibition. Reversibility studies with excess of substrate verified the expected reversible mode of inhibition. Compared to the suicide inhibitor L-deprenyl, inhibition by compound 55 (ST-2043) (10-fold IC50 concentrations) preincubated with MAO B for 30, 60 or even 90 min was completely reversed after 50-fold dilution in assays mixture (Figure 5). It could be assumed, that compound 55 was readily displaced from the MAO B active side, while a more tight-binding interaction, e.g. as shown for safinamide, is considered to be more favorable in terms of pharmacological activity.9
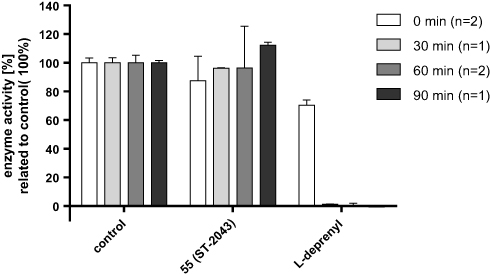 |
Figure 5 Reversibility of inhibition after preincubation of 55 (ST-2043) and L-deprenyl with MAO B. Inhibitors (10 ⨰ IC50) were preincubated with enzyme for 0, 30, 60 or 90 min prior to 50-fold dilution in assays mixture containing kynuramine as substrate (10 ⨰ KM). Data represent mean values ± SD of n independent experiments, each performed in duplicates. |
Inhibition studies using different concentrations of 55 (ST-2043) with seven appropriate substrate concentrations suggest a competitive inhibition type as demonstrated by Lineweaver–Burk plots (Figure 6). Compound 55 (ST-2043) showed promising Ki values of 6.3 nM with 95%CI = [5.0; 7.5] (Michaelis Menten fit “competitive inhibition”) and 9.5 nM (slopes from Lineweaver–Burk plots vs inhibitor concentration) for MAO B, respectively.
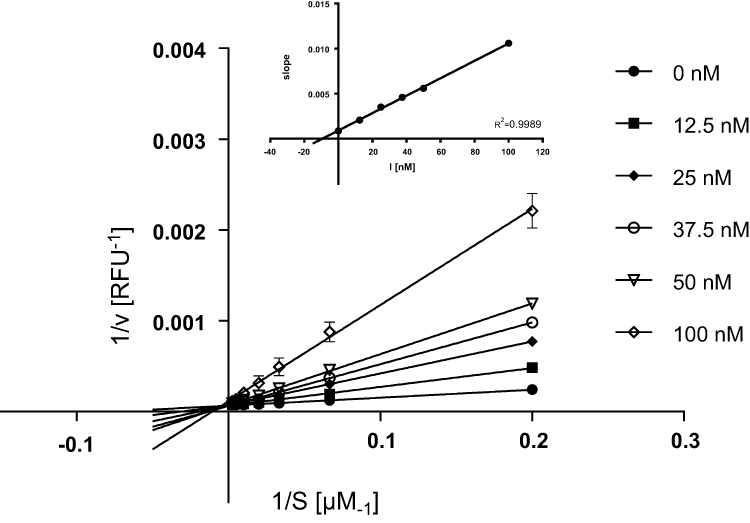 |
Figure 6 Lineweaver–Burk plots of compound 55 (ST-2043) (12.5 to 100 nM) with seven suitable concentrations of kynuramine. Data represent mean values ± SD of one representative experiment performed in duplicates. |
Cholinesterase Inhibition
Cholinesterase enzymes are of potential interest in MTL drug design for the treatment of neurodegenerative diseases.33 Therefore, compound 55 (ST-2043) was screened for inhibition of acetyl- and butyrylcholine esterases (AChE/BuChE). At a concentration of 1 µm, 55 (ST-2043) showed only moderate inhibition capacity with 24±1.8% and 57±4.5% for AChE and BuChE, respectively (see Table S4 and Figure S11 in supplemental material). Thus, no further characterization in this direction has been performed.
Conclusion
This study presented small anilides as MAO A and B inhibitors. Out of 66 screened compounds seven showed promising inhibition properties at a concentration of 1 µm (>90% or >80% inhibition for MAO B and A, respectively). Evaluation of their IC50 values revealed six MAO B preferring (31, 33, 34, 55 (ST-2043), 65) inhibitors and one MAO A preferring (7 (ST-2023)) inhibitor with activities in low nanomolar concentration ranges. The MAO B preferring inhibitors showed IC50 values ranging from 56 nM to 128 nM with selectivity indices between 2.4 and >10. Computational analysis confirmed in vitro binding properties by demonstrating good surface complementarity of the inhibitors with the binding-pockets of the two MAO isoforms. The highest affinity for MAO A was found for ST-2023 (7, IC50 = 126 nM) with 8.3-fold lower affinity towards MAO B. ST-2043 (55) was identified as most potent MAO B inhibitor (IC50 = 56 nM, Ki = 6.3 nM) within this series, which demonstrated a similar inhibition capacity as the reference reversible MAO B inhibitor safinamide, but with rather low MAO selectivity (SI = 5.2). Further characterization suggested a competitive mode of MAO B inhibition for 55, however, with a readily reversible binding rather than tight binding behavior as anticipated for pharmacological efficacy. Nevertheless, these small-sized ligands might be a promising starting point for the design of new selective or multitargeting MAO inhibitors.
Acknowledgments
This work was kindly supported by COST action CM1103, CA15135 and CA18133 as well as DFG INST 208/664e1 FUGG (Germany). Mohammad Khanfar and Holger Stark have been supported by the George Forster Research Fellowship granted by the Alexander von Humboldt-Foundation.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Tong J, Meyer JH, Furukawa Y, et al. Distribution of monoamine oxidase proteins in human brain: Implications for brain imaging studies. J Cereb Blood Flow Metab. 2013;33(6):863–871. doi:10.1038/jcbfm.2013.19
2. Ramsay RR, Albreht A. Kinetics, mechanism, and inhibition of monoamine oxidase. J Neural Transm. 2018;125(11):1659–1683. doi:10.1007/s00702-018-1861-9
3. Kim J, Kang S, Hong S, Yum S, Kim YM, Jung Y. Structure-activity relationship of salicylic acid derivatives on inhibition of TNF-α dependent NFκB activity: Implication on anti-inflammatory effect of N-(5-chlorosalicyloyl)phenethylamine against experimental colitis. Eur J Med Chem. 2012;48:36–44. doi:10.1016/j.ejmech.2011.11.030
4. Hilton S, Jaber B, Ruch R. Moclobemide safety: Monitoring a newly developed product in the 1990s. J Clin Psychopharmacol. 1995;15(4):76S–83S. doi:10.1097/00004714-199508001-00013
5. Ulrich S, Ricken R, Adli M. Tranylcypromine in mind (Part I): Review of pharmacology. Eur Neuropsychopharmacol. 2017;27(8):697–713. doi:10.1016/j.euroneuro.2017.05.007
6. Borštnar R, Repič M, Kržan M, Mavri J, Vianello R. Irreversible inhibition of monoamine oxidase B by the antiparkinsonian medicines rasagiline and selegiline: A computational study. Eur J Org Chem. 2011;2011(32):6419–6433. doi:10.1002/ejoc.v2011.32
7. Bette S, Shpiner DS, Singer C, Moore H. Safinamide in the management of patients with Parkinson’s disease not stabilized on levodopa: A review of the current clinical evidence. Ther Clin Risk Manag. 2018;14:1737–1745. doi:10.2147/TCRM.S139545
8. Kumar B, Sheetal MAK, Kumar V. Synthesis, biological evaluation and molecular modeling studies of phenyl-/benzhydrylpiperazine derivatives as potential MAO inhibitors. Bioorg Chem. 2018;77:252–262. doi:10.1016/j.bioorg.2018.01.020
9. Reis J, Manzella N, Cagide F, et al. Tight-binding inhibition of human monoamine oxidase B by chromone analogs: A kinetic, crystallographic, and biological analysis. J Med Chem. 2018;61(9):4203–4212. doi:10.1021/acs.jmedchem.8b00357
10. Ramsay RR, Popovic-Nikolic MR, Nikolic K, Uliassi E, Bolognesi ML. A perspective on multi-target drug discovery and design for complex diseases. Clin Transl Med. 2018;7(1):3–17. doi:10.1186/s40169-017-0181-2
11. Mathew B, Parambi DGT, Mathew GE, et al. Emerging therapeutic potentials of dual-acting MAO and AChE inhibitors in Alzheimer’s and Parkinson’s diseases. Arch Pharm. 2019;352(11):1–13. doi:10.1002/ardp.201900177
12. Takao K, Yahagi H, Uesawa Y, Sugita Y. 3-(E)-Styryl-2H-chromene derivatives as potent and selective monoamine oxidase B inhibitors. Bioorg Chem. 2018;77:436–442. doi:10.1016/j.bioorg.2018.01.036
13. Van Dyk AS, Petzer JP, Petzer A, Legoabe LJ. 3-Coumaranone derivatives as inhibitors of monoamine oxidase. Drug Des Devel Ther. 2015;9:5479–5489. doi:10.2147/DDDT.S89961
14. Reis J, Cagide F, Chavarria D, et al. Discovery of new chemical entities for old targets: Insights on the lead optimization of chromone-based monoamine oxidase B (MAO-B) inhibitors. J Med Chem. 2016;59(12):5879–5893. doi:10.1021/acs.jmedchem.6b00527
15. Legoabe LJ, Petzer A, Petzer JP. 2-Acetylphenol analogs as potent reversible monoamine oxidase inhibitors. Drug Des Devel Ther. 2015;9:3635–3644. doi:10.2147/DDDT.S86225
16. Ahmad S, Zaib S, Jalil S, et al. Synthesis, characterization, monoamine oxidase inhibition, molecular docking and dynamic simulations of novel 2,1-benzothiazine-2,2-dioxide derivatives. Bioorg Chem. 2018;80:498–510. doi:10.1016/j.bioorg.2018.04.012
17. Ai N, Wood RD, Yang E, Welsh WJ. Niclosamide is a negative allosteric modulator of Group I metabotropic glutamate receptors: implications for neuropathic pain. Pharm Res. 2016;33(12):3044–3056. doi:10.1007/s11095-016-2027-9
18. Kumar A, Narasimhan B, Kumar D. Synthesis, antimicrobial, and QSAR studies of substituted benzamides. Bioorg Med Chem. 2007;15:4113–4124. doi:10.1016/j.bmc.2007.03.074
19. Li Y, Wang Y, Wang J. Microwave-promoted conversion of heterocyclic amines to corresponding amides under solvent-free conditions. Heterocycl Commun. 2007;13(4):251–256. doi:10.1515/HC.2007.13.4.251
20. Li L, Han J, Nguyen B, Burgess K. Syntheses and spectral properties of functionalized, water-soluble BODIPY derivatives. J Org Chem. 2008;73(5):1963–1970. doi:10.1021/jo702463f
21. Affini A, Hagenow S, Zivkovic A, Marco-Contelles J, Stark H. Novel indanone derivatives as MAO B/H3R dual-targeting ligands for treatment of Parkinson’s disease. Eur J Med Chem. 2018;148:487–497. doi:10.1016/j.ejmech.2018.02.015
22. Mostert S, Petzer A, Petzer JP. Inhibition of monoamine oxidase by benzoxathiolone analogues. Bioorg Med Chem Lett. 2016;26(4):1200–1204. doi:10.1016/j.bmcl.2016.01.034
23. Son S-Y, Ma J, Kondou Y, Yoshimura M, Yamashita E, Tsukihara T. Human monoamine oxidase A: Structure and control of opening the entry for substrates/inhibitors. Proc Natl Acad Sci USA. 2008;105(15):5739–5744. doi:10.1073/pnas.0710626105
24. Verdonk ML, Berdini V, Hartshorn MJ, et al. Virtual screening using protein-ligand docking: Avoiding artificial enrichment. J Chem Inf Comput Sci. 2004;44(3):793–806. doi:10.1021/ci034289q
25. Frédérick R, Dumont W, Ooms F, et al. Synthesis, structural reassignment, and biological activity of type B MAO inhibitors based on the 5H-indeno[1,2-c]pyridazin-5-one core. J Med Chem. 2006;49(12):3743–3747. doi:10.1021/jm051091j
26. La Regina G, Silvestri R, Artico M, et al. New pyrrole inhibitors of monoamine oxidase: Synthesis, biological evaluation, and structural determinants of MAO-A and MAO-B selectivity. J Med Chem. 2007;50(5):922–931. doi:10.1021/jm060882y
27. European Medicines Agency [homepage on the Internet]. Human medicines highlights 2018. Authorisation of new medicines. Available from: https://www.ema.europa.eu/en/news/human-medicines-highlights-2018. Accessed September 5, 2019.
28. Tzvetkov NT, Stammler HG, Hristova S, Atanasov AG, Antonov L. (Pyrrolo-pyridin-5-yl)benzamides: BBB permeable monoamine oxidase B inhibitors with neuroprotective effect on cortical neurons. Eur J Med Chem. 2019;162:793–809. doi:10.1016/j.ejmech.2018.11.009
29. Tzvetkov NT, Stammler HG, Georgieva MG, et al. Carboxamides vs. methanimines: crystal structures, binding interactions, photophysical studies, and biological evaluation of (indazole-5-yl)methanimines as monoamine oxidase B and acetylcholinesterase inhibitors. Eur J Med Chem. 2019;179:404–422. doi:10.1016/j.ejmech.2019.06.041
30. Vishnu MS, Pavankumar V, Kumar S, Raja AS. Experimental and computational evaluation of piperonylic acid derived hydrazones bearing isatin moieties as dual inhibitors of cholinesterases and monoamine oxidases. Chem Med Chem. 2019;14:1359–1376. doi:10.1002/cmdc.v14.14
31. Zhang C, Yang K, Yu S, et al. Design, synthesis and biological evaluation of hydroxypyridinone-coumarin hybrids as multimodal monoamine oxidase B inhibitors and iron chelates against Alzheimer’s disease. Eur J Med Chem. 2019;180:367–382. doi:10.1016/j.ejmech.2019.07.031
32. Pisani L, Iacobazzi RM, Catto M, et al. Investigating alkyl nitrates as nitric oxide releasing precursors of multitarget acetylcholinesterase-monoamine oxidase B inhibitors. Eur J Med Chem. 2019;161:292–309. doi:10.1016/j.ejmech.2018.10.016
33. Ramsay RR, Majekova M, Medina M, Valoti M. Key targets for multi-target ligands designed to combat neurodegeneration. Front Neurosci. 2016;10. doi:10.3389/fnins.2016.00375
34. Desai KR, Shaikh MS, Coutinho EC. Molecular modeling studies, synthesis and biological evaluation of derivatives of N-phenylbenzamide as Plasmodium falciparum dihydroorotate dehydrogenase (PfDHODH) inhibitors. Med Chem Res. 2011;20(3):321–332. doi:10.1007/s00044-010-9323-4
35. Xu X, Li P, Huang Y, Tong C, Yan YY, Xie Y. Atmospheric oxidative catalyst-free cross-dehydrogenative coupling of aldehydes with N-hydroxyimides. Tetrahedron Lett. 2017;58(18):1742–1746. doi:10.1016/j.tetlet.2017.03.064
36. Ferrins L, Gazdik M, Rahmani R, et al. Pyridyl benzamides as a novel class of potent inhibitors for the kinetoplastid Trypanosoma brucei. J Med Chem. 2014;57(15):6393–6402. doi:10.1021/jm500191u
37. Ockey DA, Dotson JL, Struble ME, et al. Structure-activity relationships by mass spectrometry: Identification of novel MMP-3 inhibitors. Bioorg Med Chem. 2004;12(1):37–44. doi:10.1016/j.bmc.2003.10.053
38. Wen Q, Jin J, Mei Y, Lu P, Wang Y. Copper-mediated cyanation of aryl halides by activation of benzyl cyanide as the cyanide source. Eur J Org Chem. 2013;2013(19):4032–4036. doi:10.1002/ejoc.201300052
39. Deshidi R, Rizvi MA, Shah BA. Highly efficient dehydrogenative cross-coupling of aldehydes with amines and alcohols. RSC Adv. 2015;5(110):90521–90524. doi:10.1039/C5RA17425B
40. Hong G, Wu S, Zhu X, Mao D, Wang L. Peroxide-mediated direct synthesis of amides from aroyl surrogates. Tetrahedron. 2016;72(3):436–441. doi:10.1016/j.tet.2015.11.063
41. Scarborough R, Jantzen H-M, Huang W, et al. Platelet ADP receptor inhibitors. European Patent EP 2314593A1. 2011.
42. Srinivasachari R, Ramaswami S, Asha K. Quinone imine route to benzimidazol-2-ylcarbamates. Part 1. Synthesis of open-chain and cyclic 5-acylamino derivatives. J Chem Res Miniprint. 1986;1657–1675.
43. Sun Y, Wang G, Guo W. Colorimetric detection of cyanide with N-nitrophenyl benzamide derivatives. Tetrahedron. 2009;65(17):3480–3485. doi:10.1016/j.tet.2009.02.023
44. Sheng WJ, Ye Q, Bin YW, et al. CuSO4 -mediated decarboxylative C-N cross-coupling of aromatic carboxylic acids with amides and anilines. Tetrahedron Lett. 2015;56(4):599–601. doi:10.1016/j.tetlet.2014.12.085
45. Downer NK, Jackson YA, Indies W. Synthesis of benzothiazoles via ipso substitution of ortho-methoxythiobenzamides. Org Biomol Chem. 2004;2:3039–3043. doi:10.1039/b410373d
46. Carbone G, Burnley J, Moses JE. A catalytic and tert-butoxide ion-mediated amidation of aldehydes with para-nitro azides. Chem Commun. 2013;49(27):2759–2761. doi:10.1039/c3cc40452h
47. Basri AM, Lord RM, Allison SJ, et al. Bis-Picolinamide ruthenium (III) dihalide complexes: dichloride to diiodide exchange generates single trans isomers with high potency and cancer cell selectivity. Chem - A Eur J. 2017;23(26):6341–6356. doi:10.1002/chem.201605960
48. Martínez ÁM, Rodríguez N, Arrayás RG, Carretero JC. Copper-catalyzed ortho-C-H amination of protected anilines with secondary amines. Chem Commun. 2014;50(21):2801–2803. doi:10.1039/c3cc49633c
49. Rastelli G, Ferrari AM, Costantino L, Gamberini MC. Discovery of new inhibitors of aldose reductase from molecular docking and database screening. Bioorg Med Chem. 2002;10(5):1437–1450. doi:10.1016/S0968-0896(01)00410-2
50. Zhu ZW, Shi L, Ruan XM, et al. Synthesis and antiproliferative activities against Hep-G2 of salicylanide derivatives: potent inhibitors of the epidermal growth factor receptor (EGFR) tyrosine kinase. J Enzyme Inhib Med Chem. 2011;26(1):37–45. doi:10.3109/14756361003671060
51. Guo L, Wang QL, Jiang QQ, Jiang QJ, Jiang YB. Anion-triggered substituent-dependent conformational switching of salicylanilides. New hints for understanding the inhibitory mechanism of salicylanilides. J Org Chem. 2007;72(26):9947–9953. doi:10.1021/jo701823d
52. Hsi RSP, Kalamazoo M. Alkylcarbamates of cyanosalicyclanilides. United States Patent US 3317583. 1967.
53. Feng Z, Shen Z, Yuan C, et al. p-Nitroaniline compound, preparation method thereof, pharmaceutical composition and application. Chinese Patent CN107434770. 2017.
54. Nair PM, Asas RS, Venkataraman K. Condensation of 2,4-Dinitrochlorobenzene with o-Hydroxyarylamides. Tetrahedron. 1969;11:140–147. doi:10.1016/0040-4020(60)80063-4
55. Biagi G, Giorgi I, Livi O, et al. Synthesis and biological activity of novel substituted benzanilides as potassium channel activators. V. Eur J Med Chem. 2004;39(6):491–498. doi:10.1016/j.ejmech.2004.02.006
56. Wood RD, Welsh WJ, Ekins S, Ni A. Glutamate Receptor Modulators and Therapeutic Agents. United States Patent US2009239919A1. 2009.
57. Zhang J, Ma Y, Ma Y. Synthesis of secondary amides through the Palladium(II)-Catalyzed aminocarbonylation of arylboronic acids with amines or hydrazines and carbon dioxide. Eur J Org Chem. 2018;2018(14):1720–1725. doi:10.1002/ejoc.201701802
58. Devi ES, Alanthadka A, Tamilselvi A, Nagarajan S, Sridharan V, Maheswari CU. Metal-free oxidative amidation of aldehydes with aminopyridines employing aqueous hydrogen peroxide. Org Biomol Chem. 2016;14:8228–8231. doi:10.1039/C6OB01454B
59. Ronson TO, Renders E, Van Steijvoort BF, et al. Ruthenium-catalyzed reductive arylation of N-(2-Pyridinyl)amides with isopropanol and arylboronate esters. Angew Chemie - Int Ed. 2019;58(2):482–487. doi:10.1002/anie.v58.2
60. Lo WS, Hu WP, Lo HP, et al. Synthesis of sulfur-sulfur bond formation from thioamides promoted by 2,3-dichloro-5,6-dicyanobenzoquinone. Org Lett. 2010;12(23):5570–5572. doi:10.1021/ol102455x
61. Zuo M, Zheng YW, Lu SM, Li Y, Zhang SQ. Synthesis and biological evaluation of N-aryl salicylamides with a hydroxamic acid moiety at 5-position as novel HDAC-EGFR dual inhibitors. Bioorg Med Chem. 2012;20(14):4405–4412. doi:10.1016/j.bmc.2012.05.034
62. Islam AM, Hannout IB, Hassan EA, Ihsan AE. Synthesis of some substituted salicylanilides of expected biological activity. J Für Prakt Chemie. 1972;314:727–734. doi:10.1002/prac.19723140505
63. Lee IY, Gruber TD, Samuels A, et al. Structure-activity relationships of antitubercular salicylanilides consistent with disruption of the proton gradient via proton shuttling. Bioorg Med Chem. 2013;21(1):114–126. doi:10.1016/j.bmc.2012.10.056
64. Muto S, Itai A. PAI-1 production inhibitor. European Patent EP2201946A1. 2008.
65. Waisser K, Bureš O, Holý P, et al. Relationship between the structure and antimycobacterial activity of substituted salicylanilides. Arch Pharm Pharm Med Chem. 2003;336(1):53–71. doi:10.1002/ardp.200390004
66. Kang S, Min HJ, Kang MS, Jung MG, Kim S. Discovery of novel 2-hydroxydiarylamide derivatives as TMPRSS4 inhibitors. Bioorg Med Chem Lett. 2013;23(6):1748–1751. doi:10.1016/j.bmcl.2013.01.055
67. Kozic J, Novotná E, Volková M, Stolaříková J, Trejtnar F, Vinšová J. Synthesis and in vitro antimycobacterial activity of 2-methoxybenzanilides and their thioxo analogues. Eur J Med Chem. 2012;56:387–395. doi:10.1016/j.ejmech.2012.07.044
68. Paraskevopoulos G, Monteiro S, Vosátka R, et al. Novel salicylanilides from 4,5-dihalogenated salicylic acids: synthesis, antimicrobial activity and cytotoxicity. Bioorg Med Chem. 2017;25(4):1524–1532. doi:10.1016/j.bmc.2017.01.016
69. Ozawa I, Takeuchi I, Yamamoto K, et al. Synthesis and antimicrobial activity of salicylanilide derivatives. II. Chem Pharm Bull. 1984;32(1):305–312. doi:10.1248/cpb.32.305
70. Muto S, Itai A. NF-KB activation inhibitors. European Patent EP1535609A1. 2005.
71. Mook RA, Chen M, Lu J, Barak LS, Lyerly HK, Chen W. Small molecule modulators of Wnt/β-catenin signaling. Bioorg Med Chem Lett. 2013;23(7):2187–2191. doi:10.1016/j.bmcl.2013.01.101
72. Calderone V, Coi A, Fiamingo FL, et al. Structural modifications of benzanilide derivatives, effective potassium channel openers. X. Eur J Med Chem. 2006;41(12):1421–1429. doi:10.1016/j.ejmech.2006.07.016
73. Dimitrova N, Zamudio JR, Jong RM, et al. Salicylanilide Inhibitors of Toxoplasma gondii. PLoS ONE. 2017;32(7):736–740.
74. Ríos Martínez CH, Nué Martínez JJ, Ebiloma GU, De Koning HP, Alkorta I, Dardonville C. Lowering the pKa of a bisimidazoline lead with halogen atoms results in improved activity and selectivity against Trypanosoma brucei in vitro. Eur J Med Chem. 2015;101:806–817. doi:10.1016/j.ejmech.2015.07.013
75. Saladiioh R, Gent G, Summac V, Nicolettic R, Fochetd F, Spadarid S. Researches on antiviral agents. 41. studies on the chemistry of acyloxypyrimidine derivatives as new acylation reagents and inhibitors of Uracil DNA glycosylases. Tetrahedron. 1994;50(11):3603–3618. doi:10.1016/S0040-4020(01)87037-X
76. Botta M, De Angelis F, Nicoletti R, Pani A, Marongiu ME, La Colla P. 6-Alkyl-2-hethoxy-4-(3h)-pyrimidinones in the transformation of pyrimidines: regiospecific preparation, antitumor and antimicrobial activity of 4-O-acylated pyrimidine derivatives. New agents for selective acylation of amines. Tetrahedron Lett. 1988;29(22):2741–2744. doi:10.1016/0040-4039(88)85275-4
77. Dubey SK, Singh AK, Singh H, et al. Synthesis of substituted l-Hydroxy-2-naphthanilides as potential cestodicidal agents. J Med Chem. 1978;21(11):1178–1181. doi:10.1021/jm00209a020
78. Li BX, Yamanaka K, Xiao X. Structure – activity relationship studies of naphthol AS-E and its derivatives as anticancer agents by inhibiting CREB-mediated gene transcription. Bioorg Med Chem. 2012;20(23):6811–6820. doi:10.1016/j.bmc.2012.09.056
79. Saikatsu H, Fukuda T, Okamoto H, et al. Production method of asymmetrical polyazo dyes with good heat and light resistance for color filters and liquid crystal displays. International Patent WO 2011/078163 A1. 2011.
80. Han SB, Achari R, Kim MH, Jong YS, Kim PH. Novel compound or pharmaceutically acceptable salt thereof and pharmaceutical composition for prevention or treatment of disease caused by influenza virus infection containing the same as an active ingredient. Korean Patent KR2016/21163. 2016.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.