")
Back to Journals » Infection and Drug Resistance » Volume 13
Reversal of mcr-1-Mediated Colistin Resistance in Escherichia coli by CRISPR-Cas9 System
Authors Wan P, Cui S, Ma Z, Chen L, Li X, Zhao R, Xiong W, Zeng Z
Received 6 January 2020
Accepted for publication 30 March 2020
Published 22 April 2020 Volume 2020:13 Pages 1171—1178
DOI https://doi.org/10.2147/IDR.S244885
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Joachim Wink
Peng Wan,1– 3,* Shiyun Cui,1– 3,* Zhenbao Ma,1– 3 Lin Chen,1– 3 Xiaoshen Li,1– 3 Ruonan Zhao,1– 3 Wenguang Xiong,1– 3 Zhenling Zeng1– 3
1Guangdong Provincial Key Laboratory of Veterinary Pharmaceutics Development and Safety Evaluation, South China Agricultural University, Guangzhou 510642, People’s Republic of China; 2National Laboratory of Safety Evaluation (Environmental Assessment) of Veterinary Drugs, College of Veterinary Medicine, South China Agricultural University, Guangzhou, 510642, People’s Republic of China; 3National Risk Assessment Laboratory for Antimicrobial Resistance of Animal Original Bacteria, South China Agricultural University, Guangzhou 510642, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Wenguang Xiong Email [email protected]
Zhenling Zeng Tel +86 20 85281204
Fax +86 20 8528 4896
Email [email protected]
Purpose: The plasmid-borne mobilized colistin resistance gene (mcr-1) was discovered in 2015. Subsequently, the rapid horizontal transfer of mcr-1 gene to diverse bacterial species poses a serious threat to public health, which urgently needs the introduction of novel antimicrobial strategies. Therefore, the purpose of this study is to sensitize bacteria to colistin and reduce the propagation of mcr-1 gene by curing mcr-1-harboring plasmid in Escherichia coli (E. coli) using the CRISPR-Cas9 system.
Methods: Two sgRNAs specific to mcr-1 gene were designed and cloned into plasmid pCas9. The recombinant plasmid pCas9-mcr was transformed into E. coli carrying pUC19-mcr-1 or pHNSHP45, separately. The elimination efficiency in strains was evaluated by PCR and quantitative real-time PCR (qPCR). The antimicrobial susceptibility test was performed using the broth microdilution method.
Results: In this study, we constructed the high copy number plasmid pUC19-mcr-1 and recombinant plasmid pCas9-m1 or pCas9-m2, which contain 20 nt or 30 nt sgRNA sequences targeted to mcr-1, respectively. PCR and qPCR results showed that mcr-1-harboring plasmids could be efficiently eliminated, and there was no significant correlation between sgRNA lengths and curing efficiency. However, when comparing restructured high copy number plasmid (pUC19-mcr-1) to natural resistance plasmid (pHNSHP45) in eliminating efficiency, we found that the content of plasmid backbone had an influence on efficiency. Furthermore, the conjugation assays verified that the engineered CRISPR-Cas9 system in bacteria or in bacteria genome can protect the recipient from plasmid-borne mcr-1 transfer via conjugation. Additionally, sequence analysis showed that three different types of defects in CRISPR-Cas9 system lead to escape mutants.
Conclusion: We presented a method that only one plasmid-mediated CRISPR-Cas9 system can be used to efficiently resensitize E. coli to colistin. Moreover, this system provided a great potentiality to counteract the propagation of mcr-1 among bacterial pathogens.
Keywords: sgRNA lengths, curing efficiency, backbone content, quantitative real-time PCR, conjugation assays, escape mutants
Introduction
Antibiotics have significant impacts on health care since their first introduction in the early 20th century. While their irrational use in human medicine and animal husbandry resulted in the emergence and rise of antimicrobial resistance genes (ARGs). Polymyxins serve as the last-line refuge against multidrug-resistant (MDR) Gram-negative bacterial infections. Unfortunately, the discovery of mcr-1 exacerbates the failure in clinical treatment.1 Since then, the dissemination of transferable colistin resistance genes, which involves different ecological niches, bacterial species, replicon types and mcr-like variants,2 has been increasingly reported in over forty countries covering six continents.3 More alarmingly, the detection of coexistence of mcr-1 and blaNDM-5 in clinical isolates heightened the challenge of the treatment in clinical therapy.4 The global distribution of mcr-1 gene has captured the attention of public health communities and efficient strategies are needed to be formulated.
ARGs, often present on mobile genetic elements, can be transferred among bacteria, which led to the global prevalence of antibiotic-resistant bacteria and the demand for novel treatment strategies.5 The search of plasmid curing inspired us to remove plasmid from antimicrobial-resistant (AMR) bacteria. However, most traditional plasmid curing methods, such as high temperature, detergents, biocides, DNA intercalating agents and plant-derived compounds, may favor irrespective chromosome mutation or produce unwanted toxic and side effects.6 In recent years, the CRISPR-Cas9 system has been opened new avenues for plasmid curing. It can be designed to target specific sequences of plasmid rather than cure plasmid without discrimination.7
Clustered regularly interspaced short palindromic repeats (CRISPR) together with CRISPR-associated (Cas) proteins constitute the adaptive immune system of bacteria and archaea, which allows recognition, degradation and memory of foreign DNA sequences.8 Notably, over the past 10 years, the type II CRISPR-Cas system has become the predominant choice for genome engineering applications,9 which requires only the co-expression of Cas9 protein and two RNAs that guide Cas9 to the target site in the invading DNA for subsequent recognition and cleavage.10 Since the two RNAs were further improved into a chimeric, single guide RNA (sgRNA), genome editing with Cas9 has become simpler. After the sgRNA targets specific sequences and recruits Cas9 protein to form the compound, the Cas9 protein acts as a nuclease and generates a blunt end double-strand break (DSB). Owing to the lack of the error-prone non-homologous end-joining (NHEJ) pathway in prokaryotes, Cas9-mediated DSB cannot be repaired spontaneously, so that the DSB occurs in plasmid sequence triggering plasmid curing.10 Based on the CRISPR-Cas9 system, reported studies have been transformed E. coli with plasmids that coded sgRNAs to specifically cleave quinolone resistance gene and Extended-spectrum β-lactamases (ESBLs) resistance gene, respectively.11,12 Together, using the CRISPR-Cas9 system to resensitize colistin-resistant E. coli by curing mcr-1-carrying plasmid is considered as a potentially effective method to resist the dissemination of colistin resistance genes.
It is time to develop unique approaches to combat the dissemination of mcr-1 gene because of possible breach of the last group of antibiotics, polymyxins, by plasmid-mediated resistance.CRISPR-Cas9 technology provides a novel strategy to target any resistance gene and keep the original colony ecologically stable. Given that the threats of mcr-1 gene and the advantages of CRISPR-Cas9 system, this study constructed a recombinant plasmid, which holds all functional parts to recognize and degrade targeting sequences, for eliminating mcr-1 gene and relieving the stress of ARGs. And then we showed the feasibility of the method and explained unexpected escape mutants during the experiment. In conclusion, the study developed a one-plasmid mediated CRISPR-Cas9 system for genome editing in E. coli and offered an appealing option to eliminate mcr-1 gene.
Materials and Methods
Bacterial Strains and Plasmids
All strains and plasmids used in this study are listed in Table 1. E. coli DH5α was used to be made chemically competent cells and transformed via heat shock. Porcine E. coli strain SHP45,1 which is a wild strain containing the mcr-1 gene and E. coli C600 were employed for conjugation experiments. Transconjugants E. coli C600+pHNSHP45 were screened on Luria-Bertani (LB) agar plates containing colistin (2 mg/L) and streptomycin (2000 mg/L). The plasmid pCas9 (Addgene: #42876) was obtained from commercial sources conferring chloramphenicol resistance.13 E. coli strains were grown in LB broth and agar plates at 37°C. If necessary, the following antibiotics were supplemented at the different concentrations: colistin (2 mg/L), chloramphenicol (50 mg/L), ampicillin (50 mg/L) and streptomycin (2000 mg/L).
 |
Table 1 Bacterial Strains and Plasmids Used in This Study |
Plasmid Construction
All oligonucleotides used in this work are shown in Table 2. Plasmids were constructed using standard molecular biology techniques. Firstly, two pairs of sgRNAs (sgRNA1-F/R, sgRNA2-F/R) targeting the mcr-1 gene were designed by the CHOPCHOP tool. Two targeted sgRNA sequences with different lengths, 20 nt or 30 nt, were synthesized in oligonucleotides with the sticky ends AAAC or G adding at the beginning or end of the forward oligos, respectively, while AAAC was added at the beginning of the reverse oligos. The paired oligonucleotides were then annealed and ligated to the BsaI-digested pCas9 using T4 DNA ligase (NEB), resulting in pCas9-m1 and pCas9-m2, respectively. The recombinant plasmids were transformed into competent E. coli DH5α, and the transformants were incubated at 37°C overnight and selected on a plate containing 50 mg/L chloramphenicol. The spacer sequence was confirmed by colony PCR using the primers DR-JD-F/R. And the plasmid pCas9, which does not have the sgRNA cassette, was used as a control plasmid. In order to obtain restructured high copy mcr-1 plasmid, DNA segment containing the promoter and mcr-1 gene was amplified from E. coli SHP45 template DNA using primers SacI-mcr-1-F/R, followed by digestion with SacI and ligated into the corresponding site in pUC19, yielding pUC19-mcr-1. We verified both of recombinant plasmids by PCR and further sequencing.
 |
Table 2 Primers Used in This Study |
Plasmid Transformation Assays
The competent cells including E. coli DH5α containing pUC19-mcr-1 and E. coli C600+pHNSHP45 were prepared and used for transformation assay following the protocol.14 Mixing 100 μL of chemically competent cells with 10 μL of plasmid pCas9-m1 or pCas9-m2 incubated the reaction mixture on ice for 30 min, and subsequently transferred the cells to a constant temperature water bath set at 42°C for 90 s and finally incubated on ice for 2 min. Following transformation, the cells were suspended in 1 mL LB broth and then maintained at 37°C with vigorous shaking (200 rpm) for 1 h. Gradient dilutions of cells were placed on LB agar plates containing 50 mg/L chloramphenicol for selecting the transformants, after standing to dry the plates were inverted cultured at 37°C overnight. The empty plasmid pCas9 was used as negative control.
Editing Efficiency Evaluation
After transformation, 24 colonies were randomly picked for evaluating the editing efficiency in each experiment. Primers mcr-1-JD-F/R were used to screen the colonies with target gene deletion by PCR. The strain with mcr-1 gene was used as a positive control. PCR-confirmed original strains and corresponding deletion strains were subjected to antimicrobial susceptibility tests. The minimum inhibitory concentration (MIC) of colistin was performed by broth microdilution method in accordance with the guidelines of Clinical and Laboratory Standards Institute (2018).15 E. coli ATCC 25922 was routinely used as a quality control strain.
In order to further analyze the efficiency of pCas9-m1/m2 on removing mcr-1 gene, the pUC19-mcr-1 copy numbers at each time point were determined by qPCR. Genomic DNA was extracted using TIANamp Bacteria DNA Kit (Tiangen, Beijing, China). qPCR was performed using SYBR Premix ExTaq II (Takara, Dalian, China) with primers specific for mcr-1 gene and chromosomal 16S ribosomal RNA (16S rRNA) gene. All reactions were run in triplicate. The 2−ΔΔCT method was used to calculate the change of pUC19-mcr-1 copy number in the experimental group compared to the control group.
Conjugation Assays
Conjugation experiments were performed using colistin-resistant E. coli C600+pHNSHP45 as the donor and chloramphenicol-resistant E. coli C600+pCas9-m1 or C600+pCas9 as the recipient strain as mentioned before.16 In brief, donor and recipient strains grown in suspension overnight and then were diluted 1:100 in fresh LB broth and incubated to an OD600 ~0.4 at 37°C. Then, the cultures were mixed at a donor/recipient ratio of 1:4 (vol/vol) and grown for 24 h. Serial dilutions of the mixtures were placed on LB agar plates supplemented with chloramphenicol and colistin to select for transconjugants. After incubation for 24 h, enumerated the number of colony-forming units (CFU).
Sequencing of Escape Mutants
Transformants spread on LB agar plates with chloramphenicol were subcultured on another plate containing colistin and we found that a few survivors grew thereon. Then, plasmid DNA from each escape mutant was extracted using EasyPure HiPure Plasmid MiniPrep kits (TransGen, Beijing, China) for sequencing analysis. To analyze the PAM-flanking sequence, approximately 500bp nearby the protospacer within the mcr-1 gene of each escape mutant was amplified using PCR and subjected to DNA sequencing.
Statistical Analysis
All the experiments described above were repeated at least three times. All statistical analyses were performed using GraphPad Prism version 7.0 (GraphPad Software Inc., San Diego, CA, USA). P values were calculated using the Student’s t-test, and P < 0.05 were considered statistically significant.
Results
Construction of CRISPR-Cas9 Plasmids Targeting mcr-1 Gene and Recombinant High Copy Plasmids
At first, plasmid pCas9 which contains essential elements including the gene cassettes expressing Streptococcus pyogenes Cas9, tracrRNA and crRNA, so that we chose it as the backbone to construct the engineered pCas9 specific to mcr-1 gene. Oligonucleotides with 20 nt or 30 nt sgRNA sequences replaced the crRNA coding sequences of pCas9, yielding pCas9-m1 and pCas9-m2, respectively (Figure 1). Additionally, in order to obtain high copy plasmid containing mcr-1 gene, plasmids pUC19, which has higher copy number compared to its parent plasmid pBR322, was selected as the backbone to construct the high copy numbers plasmid harboring mcr-1 gene.
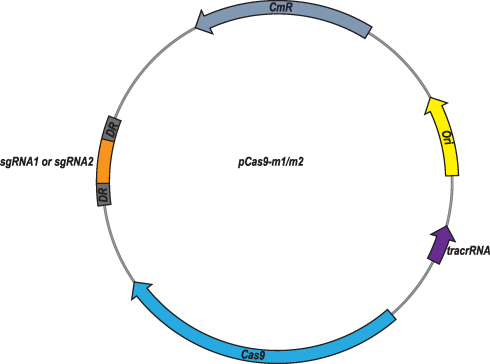 |
Figure 1 Plasmid map of pCas9-m1/m2 targeted to mcr-1 gene. The pCas9-m1/m2 was constructed by inserting spacer targeting the mcr-1 gene along with other essential modules for CRISPR-Cas9 activity. |
Elimination of Plasmid Using the Engineered CRISPR-Cas9 System
To estimate whether CRISPR-Cas9 system can eliminate plasmid from E. coli, the control vector pCas9 and the engineered vector pCas9-m1 were transformed into competent E. coli C600+pHNSHP45. The results showed that the 582bp fragment indicating the presence of mcr-1 gene failed to be amplified from 18/24 colonies transformed with pCas9-m1 (Figure 2A). Meanwhile, 6 colonies still have mcr-1 gene, defined as escape mutants. For antimicrobial susceptibility tests, the colistin MIC of knockout strain was 0.25 mg/L, which was 6 times lower than that of wild-type (2 mg/L).
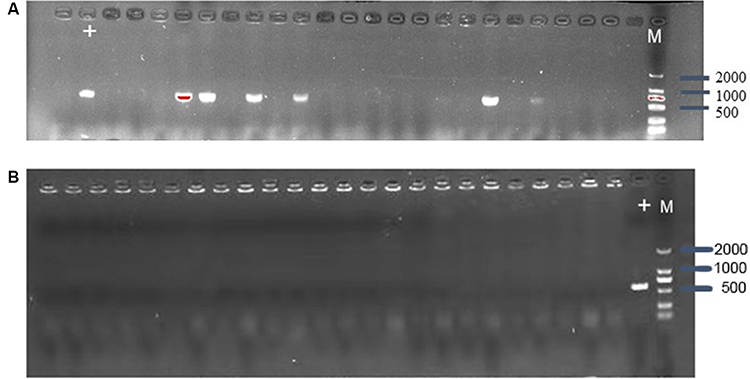 |
Figure 2 (A) Confirmation of mcr-1 gene presence in E. coli C600+pHNSHP45 by PCR amplification with primer mcr-1-JD-F/R. The M means 2000bp marker. The + lane is a C600+pHNSHP45 strain transformed with pCas9 as positive control. The other lanes mean C600+pHNSHP45 strain transformed with pCas9-m1 (B) Confirmation of mcr-1 gene elimination in C600+pUC19-mcr-1 by PCR amplification with primer mcr-1-JD-F/R. |
The inadequate knockout efficiency suggested us that whether different backgrounds of plasmid containing the interested gene would be an influencing factor, so we generated high copy plasmid pUC19-mcr-1 based on pUC19 backbone. The competent cells of E. coli C600+pUC19-mcr-1 were used as host and transformed with pCas9-m1. The colony PCR showed that 24 colonies were mcr-1 negative (Figure 2B).
To demonstrate whether the efficacy of CRISPR-Cas9 is affected by the length of sgRNA sequence, we constructed the pCas9-m2 containing 30 nt sgRNA targeting mcr-1. The pCas9-m2 was transformed into competent cell of E. coli C600+pUC19-mcr-1. The colony PCR results indicated that the efficacy of pCas9-m2 was not significantly different from that of pCas9-m1.
In order to more accurately evaluate editing efficiency, the relative copy number of plasmid at each time point in pCas9-m1 or pCas9-m2 treated group and pCas9 control group was determined by qPCR. Significant differences in plasmid copy numbers can be seen between the experimental and control groups (Figure 3). The elimination efficiency of two experimental groups was greater than 80% at the 8th hour and continued until the 24th hour, which clearly indicates that the engineered CRISPR-Cas9 system can effectively eliminate mcr-1 gene and the length of sgRNA sequence has no significant effect.
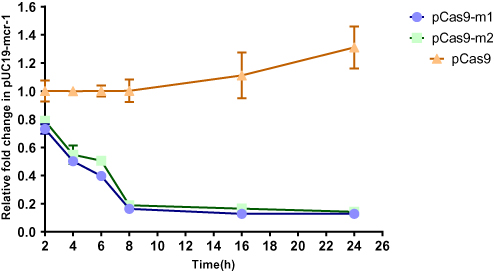 |
Figure 3 The relative copy number of plasmid pUC19-mcr-1 at each time point. pCas9-m1 and pCas9-m2 transformed into competent E. coli C600+pUC19-mcr-1 serve as experimental groups. Plasmid pCas9 transformed into competent E. coli C600+pUC19-mcr-1 is control group. The bars represent mean value of three biological replicates with error-bars showing standard deviation. |
The Engineered CRISPR-Cas9 System in Bacteria Blocks Plasmid Conjugation
Bacterial conjugation is an important way to mediate the horizontal transfer of resistance genes. The CRISPR-Cas system is an adaptive immune system of bacteria and archaea, which can generate immune memory to foreign DNA. We hypothesized that the engineered CRISPR-Cas9 system in bacteria could block plasmid conjugation. The E. coli C600 was transformed with a control plasmid pCas9 lacking the target sgRNA or a recombinant plasmid pCas9-m1 targeting mcr-1 gene, which was used as recipient. The E. coli C600 carrying the conjugation plasmid pHNSHP45 was used as a donor. As shown in Figure 4, the E. coli C600 containing plasmid pCas9-m1 reduced the number of transconjugants by 67-times compared to the control plasmid pCas9 (1.68 + E06 vs 2.5 + E04 CFU/mL; p=0.0022). The results demonstrated that the engineered CRISPR-Cas9 system in the recipient cell could block the horizontal transfer of the conjugative plasmid pHNSHP45.
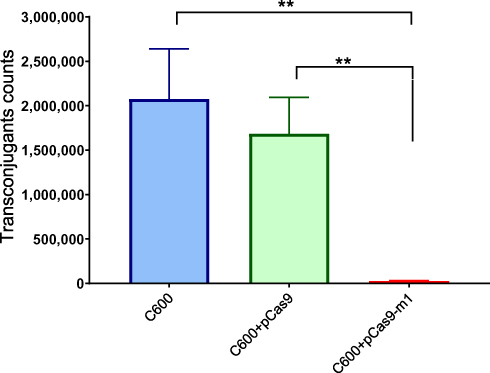 |
Figure 4 E. coli strain C600 harboring pCas9-m-1 limited the conjugation of pHNSHP45. Colistin-resistant E. coli C600+ pHNSHP45 as donor and E. coli C600, chloramphenicol-resistant, E. coli C600+pCas9-m1 or C600+pCas9 as the recipient strain. Double asterisk indicates statistical significance with P< 0.01 in Student’s t-test, compared to the results from E. coli C600+pCas9-m1. The bars represent mean value of three biological replicates with error-bars showing standard deviation. |
Escape Mutants Analysis
As mentioned before, when pCas9-m1 was transformed into E. coli C600+pHNSHP45, there were 6/24 colonies still resistant to colistin. To initially explore the basis of escape, we inoculated 6 colonies for further analysis and extracted plasmids from escape mutants for sequencing. The result showed that mutants escaped through spacer mutations in the CRISPR locus, deletions in tracrRNA and transposon insertions in Cas9 as presented in Figure 5. No mutants were observed in the protospacer within the mcr-1 gene of each escape mutant. In total, an integral active endonuclease, tracrRNA and crRNA are necessary and sufficient to mediate mcr-1 gene elimination in E. coli.
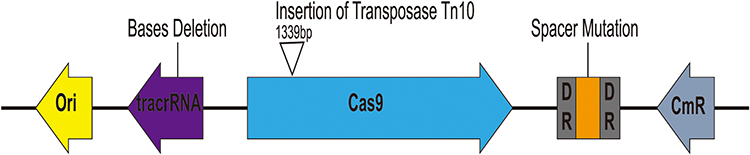 |
Figure 5 Characterization of escape mutants that tolerated transformation of pCas9-m1 construct. Spacer mutations in the CRISPR locus, deletions in tracrRNA and transposon insertions in cas9 led to pCas9-m1 inactivation in successful transformants. |
Discussion
Antimicrobial resistance is an increasingly serious threat to global public health.17 The emergence and dissemination of colistin resistance mcr-1 raise a great challenge to the treatment of clinical infections. Since the first report of mcr-1 in IncI2-type plasmid pHNSHP45, a series of mcr-1-carrying plasmids have been detected, including IncHI2,18 IncX4,19 IncX2,20 IncP,21 IncII,22 and other.3,23 The plasmids can act as reservoirs of horizontal gene transfer. Therefore, plasmid curing is an attractive strategy to slow the spread of colistin resistance in bacteria.
Conventional methods for plasmid curing involve growing at high temperatures or adding chemical agents and based on replicon-incompatibility. These methods would generate unwanted mutations, be time-consuming and require precise knowledge about replication of the targeted plasmid.24 CRISPR-Cas system is a bacterial adaptive immune system protecting itself against invaders and has been used in several bacteria such as E. coli,25 Staphylococcus aureus and Shewanella algae.26,27 This system provided an effective strategy for sequence-specific curing of the plasmid.
In this study, we applied CRISPR-Cas9 system into antibiotic-resistant bacteria to eliminate mcr-1-carrying plasmid. The results demonstrated that mcr-1-positive plasmid in recipient strains can be eliminated, recipient strains are sensitive to colistin and knockout-strain can block the conjugation of initial colistin-resistant plasmid after transformation with CRISPR-Cas9 plasmid targeting mcr-1 gene. For restructured high copy plasmid pUC19-mcr-1, there are no significant differences in curing efficiency with different lengths of sgRNA sequence, presumably caused by identical targeted gene and host. Other study has shown that the knockout efficiency of 17 nt sgRNA in human cells is similar to that of 20 nt sgRNA, but the off-target mutagenic effect is greatly reduced.28 While another study showed that the knockout potency of 17 nt or 20 nt sgRNA was various with different host stem cells and the discrepancy can be explained by differential expression levels of Cas9 in different types of cells.29 These may indicate that the effect of sgRNA sequence length varies from host to host. However, differences in knockout efficiency were observed between pHNSHP45 and pUC19-mcr-1, which could be a result of different backbone content. The pHNSHP45 possesses a typical IncI2-type backbone encoding replication, horizontal transfer, maintenance, and stability functions.1 The pUC19 is a commonly used cloning vector and display a relatively high copy-number based on ColE1-like replication machinery. The previous study showed that the curing rates were comparable between the target plasmids with similar backbone but different replicons, while the curing efficiencies varied among the target plasmids with similar replicons but different backbone content.24 Considering variations in copy-number, two other studies showed that there is no clear correlation between copy-number and curing efficiency.24,30 Together, the backbone content of target plasmids is an important factor affecting the curing efficiency.
Compared to other CRISPR-Cas systems, the type II system is the simplest and present only in bacteria.31 The sgRNAs can be designed with multiple spacers simultaneously target different positions of antibiotic-resistant genes. It can also be used to target several genes or various subtypes of drug resistance genes. Targeting diverse locations within the same gene would reduce the probability of CRISPR-escape mutants by the target. And two sgRNAs on a single construct could be used to target and remove both blaNDM-1 and blaCTX-M-15 genes, this is significant for reducing the dissemination of multidrug-resistance (MDR).32 In this study, we selected mcr-1 gene for target, however, 11 more genetic variants of mcr-1 and additional MCR members have been identified,3 hence further studies are needed to target more genes. The previous study showed that there are many mutations in the sequence of ESBLs, thus they used the homologous regions in TEM and SHV β-lactamases as the target sequence.12 Therefore, the conserved sequence among variants of mcr-1 or additional MCR members can be selected as a universal target.
The off-target property of CRISPR-Cas system has been a bottleneck for this method in biomedical and clinical applications.33 Combined with experimental and analytical approaches to design optimal sgRNA would improve sgRNA activity and specificity.34 And the efficacy of CRISPR-Cas9 mainly depends on the efficiency of CRISPR-Cas9 system delivering to the target organisms.35 As transformation is a non-spontaneous mode, this form is limited in clinical application. Therefore, multiple other delivery approaches including plasmid conjugation, phagemid and bacteriophages have been used in vitro or in vivo models.25,26,32 In general, the CRISPR-Cas9 system has great potential to reduce the dissemination of antibiotic-resistance and cure infections caused by MDR pathogens.
Conclusion
Our study showed that the engineered CRISPR-Cas9 system could be used to efficiently resensitize resistant isolates to colistin and reduce the horizontal transfer of the conjugative plasmid pHNSHP45. Taken together, the CRISPR-Cas9 system is an attractive alternative against antibiotic resistance and further study optimizing delivery methods are required to improve efficiency in clinical application.
Acknowledgment
This work was supported by the National Natural Science Foundation of China (Grant no. 31872524).
Author Contributions
Zhenling Zeng, Wenguang Xiong provided reagents and supplies. Peng Wan, Shiyun Cui designed the experiments, performed the experiments, analyzed the data, and wrote the manuscript. Zhenbao Ma, Lin Chen, Xiaoshen Li and Ruonan Zhao performed the experiments and revised the manuscript. All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; and agreed to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Liu YY, Wang Y, Walsh TR, et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: a microbiological and molecular biological study. Lancet Infect Dis. 2016;16:161–168. doi:10.1016/S1473-3099(15)00424-7
2. Sun J, Zhang HM, Liu YH, et al. Towards understanding MCR-like colistin resistance. Trends Microbiol. 2018;26:794–808. doi:10.1016/j.tim.2018.02.006
3. Nang SC, Li J, Velkov T. The rise and spread of mcr plasmid-mediated polymyxin resistance. Crit Rev Microbiol. 2019;45:131–161. doi:10.1080/1040841X.2018.1492902
4. Sun J, Yang RS, Zhang Q, et al. Co-transfer of blaNDM-5 and mcr-1 by an IncX3-X4 hybrid plasmid in Escherichia coli. Nat Microbiol. 2016;1:16176. doi:10.1038/nmicrobiol.2016.176
5. Buckner MMC, Ciusa ML, Piddock LJV. Strategies to combat antimicrobial resistance: anti-plasmid and plasmid curing. FEMS Microbiol Rev. 2018;42:781–804. doi:10.1093/femsre/fuy031
6. Wang P, Zhu Q, Shang H, et al. Curing of plasmid pBMB28 from Bacillus thuringiensis YBT-020 using an unstable replication region. J Basic Microbiol. 2016;56:206–210. doi:10.1002/jobm.201500256
7. Bikard D, Barrangou R. Using CRISPR-Cas systems as antimicrobials. Curr Opin Microbiol. 2017;37:155–160. doi:10.1016/j.mib.2017.08.005
8. Doudna JA, Charpentier E. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346(6213):1258096. doi:10.1126/science.1258096
9. Luo ML, Leenay RT, Beisel CL. Current and future prospects for CRISPR-based tools in bacteria. Biotechnol Bioeng. 2016;113:930–943. doi:10.1002/bit.25851
10. de la Fuente-nunez C, Lu TK. CRISPR-Cas9 technology: applications in genome engineering, development of sequence-specific antimicrobials, and future prospects. Integr Biol. 2017;9:109–122. doi:10.1039/c6ib00140h
11. Qiu HX, Gong JS, Butaye P, et al. CRISPR/Cas9/sgRNA-mediated targeted gene modification confirms the cause-effect relationship between gyrA mutation and quinolone resistance in Escherichia coli. FEMS Microbiol Lett. 2018;365.
12. Kim JS, D H C, Park M, et al. CRISPR/Cas9-mediated re-sensitization of antibiotic-resistant escherichia coli harboring extended-spectrum beta-lactamases. J Microbiol Biotechnol. 2016;26:394–401. doi:10.4014/jmb.1508.08080
13. Su T, Liu F, Gu P, et al. A CRISPR-Cas9 assisted non-homologous end-joining strategy for one-step engineering of bacterial genome. Sci Rep. 2016;6.
14. Cohen SN, Chang AC, Boyer HW, et al. Construction of biologically functional bacterial plasmids in vitro. Proc Natl Acad. 1973;70:3240–3244. doi:10.1073/pnas.70.11.3240
15. CLSI. Performance standards for antimicrobial susceptibility testing. 28th ed. CLSI supplement M100. Wayne, PA. Clin Lab Stand Ins. 2018.
16. Chen L, Chen ZL, Liu JH, et al. Emergence of RmtB methylase-producing Escherichia coli and Enterobacter cloacae isolates from pigs in China. J Antimicrob Chemother. 2007;59:880–885. doi:10.1093/jac/dkm065
17. World Health Organization (WHO) [homepage on the Internet]. Geneva: antimicrobial resistance; 2018.Available from: http://www.who.int/en/newsroom/factsheets/detail/antimicrobial-resistance.
18. Zhi CP, Lv LC, Yu LF, et al. Dissemination of the mcr-1 colistin resistance gene. Lancet Infect Dis. 2016;16:292–293. doi:10.1016/S1473-3099(16)00063-3
19. Sun J, Fang LX, Wu Z, et al. Genetic analysis of the IncX4 plasmids: implications for a unique pattern in the mcr-1 acquisition. Sci Rep. 2017;7.
20. Guo Q, Su J, McElheny CL, et al. IncX2 and IncX1-X2 hybrid plasmids coexisting in a fosa6-producing Escherichia coli strain. Antimicrob Agents Chemother. 2017;61.
21. Zhao FF, Feng Y, Lü X, et al. IncP plasmid carrying colistin resistance gene mcr-1 in klebsiella pneumoniae from hospital sewage. Antimicrob Agents Chemother. 2017;61.
22. Xavier BB, Lammens C, Butaye P, et al. Complete sequence of an IncFII plasmid harbouring the colistin resistance gene mcr-1 isolated from Belgian pig farms. J Antimicrob Chemother. 2016;71:2342–2344. doi:10.1093/jac/dkw191
23. Li Z, Cao Y, Yi L, et al. Emergent polymyxin resistance: end of an era? Open Forum Infect Dis. 2019;6. doi:10.1093/ofid/ofz368.
24. Lauritsen I, Porse A, Sommer MOA, et al. A versatile one-step CRISPR-Cas9 based approach to plasmid-curing. Microb Cell Fact. 2017;16:135. doi:10.1186/s12934-017-0748-z
25. Citorik RJ, Mimee M, Lu TK. Sequence-specific antimicrobials using efficiently delivered RNA-guided nucleases. Nat Biotechnol. 2014;32:1141–1145. doi:10.1038/nbt.3011
26. Bikard D, Euler CW, Jiang W, et al. Exploiting CRISPR-Cas nucleases to produce sequence-specific antimicrobials. Nat Biotechnol. 2014;32:1146–1150. doi:10.1038/nbt.3043
27. ZY W, Huang Y-T, Chao W-C, et al. Reversal of carbapenem-resistance in Shewanella algae by CRISPR/Cas9 genome editing. J Adv Res. 2019;18:61–69. doi:10.1016/j.jare.2019.01.011
28. Fu YF, Sander JD, Reyon D, et al. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol. 2014;32:279–284. doi:10.1038/nbt.2808
29. Zhang JP, Li XL, Neises A, et al. Different Effects of sgRNA Length on CRISPR-mediated gene knockout efficiency. Sci Rep. 2016;6.
30. Yuen G, Khan FJ, Gao SJ, et al. CRISPR/Cas9-mediated gene knockout is insensitive to target copy number but is dependent on guide RNA potency and Cas9/sgRNA threshold expression level. Nucleic Acids Res. 2017;45:12039–12053. doi:10.1093/nar/gkx843
31. Shabbir MA, Shabbir MZ, Wu Q, et al. CRISPR-Cas system: biological function in microbes and its use to treat antimicrobial-resistant pathogens. Ann Clin Microbiol Antimicrob. 2019;18.
32. Yosef I, Manor M, Kiro R, et al. Temperate and lytic bacteriophages programmed to sensitize and kill antibiotic-resistant bacteria. Proc Natl Acad Sci USA. 2015;112:7267–7272. doi:10.1073/pnas.1500107112
33. Zhang XH, Tee LY, Wang XG, et al. Off-target effects in CRISPR/Cas9-mediated genome engineering. Mol Ther Nucleic Acids. 2015;4.
34. Doench JG, Fusi N, Sullender M, et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol. 2016;34:184–191. doi:10.1038/nbt.3437
35. Park JY, Moon BY, Park JW, et al. Genetic engineering of a temperate phage-based delivery system for CRISPR/Cas9 antimicrobials against Staphylococcus aureus.. Sci Rep. 2017;7.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.