")
Back to Journals » Eye and Brain » Volume 8
Retinopathy of prematurity: the need for prevention
Authors Liegl R, Hellström A, Smith L
Received 24 October 2015
Accepted for publication 16 January 2016
Published 20 May 2016 Volume 2016:8 Pages 91—102
DOI https://doi.org/10.2147/EB.S99038
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Margaret Wong-Riley
Raffael Liegl,1 Ann Hellström,2 Lois EH Smith1
1Department of Ophthalmology, Boston Children's Hospital, Harvard Medical School, Boston, MA, USA; 2Department of Ophthalmology, Institute of Neuroscience and Physiology, Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden
Abstract: More than 450,000 babies are born prematurely in the USA every year. The improved survival of even the most vulnerable low body weight preterm infants has, despite improving health outcomes, led to the resurgence in preterm complications including one of the major causes for blindness in children, retinopathy of prematurity (ROP). The current mainstay in ROP therapy is laser photocoagulation and the injection of vascular endothelial growth factor (VEGF) antibodies in the late stages of the disease after the onset of neovascularization. Both are proven options for ophthalmologists to treat the severe forms of late ROP. However, laser photocoagulation destroys major parts of the retina, and the injection of VEGF antibodies, although rather simple to administer, may cause a systemic suppression of normal vascularization, which has not been studied in sufficient depth. However, the use of neither VEGF antibody nor laser treatment prevents ROP, which should be the long-term goal. It should be possible to prevent ROP by more closely mimicking the intrauterine environment after preterm birth. Such preventive measures include preventing the toxic postbirth influences (eg, oxygen excess) as well as providing the missing intrauterine factors (eg, insulin growth factor 1) and are likely to also reduce other complications of premature birth as well as ROP. This review is meant to summarize the current knowledge on the prevention of ROP with a particular emphasize on the use of insulin growth factor 1 supplementation.
Keywords: ROP, IGF-1, insulin growth factor 1
Introduction
Among the ~4 million babies who are yearly born in the USA,1 >450,000 are born prematurely, defined as birth before 37 weeks of gestation.2,3 Preterm birth has been associated with many complications, occurring during delivery, within the first few weeks after delivery and even many years later.4 Preterm birth is the most frequent reason for neonatal death5,6 and among the most common causes for death in children younger than 5 years.7 A significant improvement of the standards in neonatal intensive care units (NICUs) and perinatal care over the last decades has increased the survival rate of even the youngest preterm babies. Delivery after gestational week 26 has survival rates of >80% today.8–10
Despite improving the health outcomes and survival rates in even the youngest infants, preterm birth still carries a high risk of negatively affecting most organ systems, such as the heart,11,12 lungs,13,14 brain,15–17 and the eye.18,19 The improved survival of very small and particularly vulnerable preterm infants has led to the resurgence in preterm complications including one of the major causes for blindness in children, retinopathy of prematurity (ROP).20
Laser photocoagulation and the intravitreal injection of vascular endothelial growth factor (VEGF) antibodies have proven to be useful treatment options for severe late ROP. However, laser photocoagulation destroys major parts of the retina, and the injection of VEGF antibodies, though simple to apply, may cause a systemic suppression of vascular growth affecting the other organs, a process which has not been studied in sufficient depth. However, the simple mode of application of VEGF antibodies, and the so far promising clinical data should, not obscure the fact that the prevention of ROP should be the first priority.
There is a great interest in preventing ROP by more closely mimicking the intrauterine environment after preterm birth. Such preventative measures are also likely to reduce other complications besides ROP. These measures include preventing the toxic postbirth influences (eg, oxygen excess) as well as providing missing intrauterine factors (eg, insulin growth factor 1 [IGF-1]). This review is, therefore, meant to summarize the current knowledge on the prevention of ROP with a particular emphasize on the use of IGF-1 supplementation.
History of ROP
In 1984, the first part of the International Classification of Retinopathy of Prematurity was published by an international expert group, which was expanded in 1987.21,22 This classification permits the examiner to specify the location of the late-stage neovascular disease and the extent of the developing vasculature involved. In order to assess the location, the retina is divided into three zones, with the optic disk being the center for each zone, and the extent is specified as clock hours (Figure 1). In addition to these two parameters, four stages are used to describe the severity of vascular abnormality ranging from stage 1, which refers to a demarcation line, separating the avascular anterior retina from the vascularized posterior part of the retina, to stage 4, which describes retinal detachment. The term plus disease was introduced to account for dilated veins and/or tortuous arterioles and was added to any stage that had these vessel characteristics associated with the poorer outcomes. The expansion of this initial classification in 1987 added a fifth stage that applied when a total retinal detachment was present. Stage 4 was, therefore, defined as a retinal detachment that would not affect the whole retina. In order to be able to distinguish between a retinal detachment with and without foveal involvement, stages 4a (extrafoveal) and 4b (including fovea) were introduced.
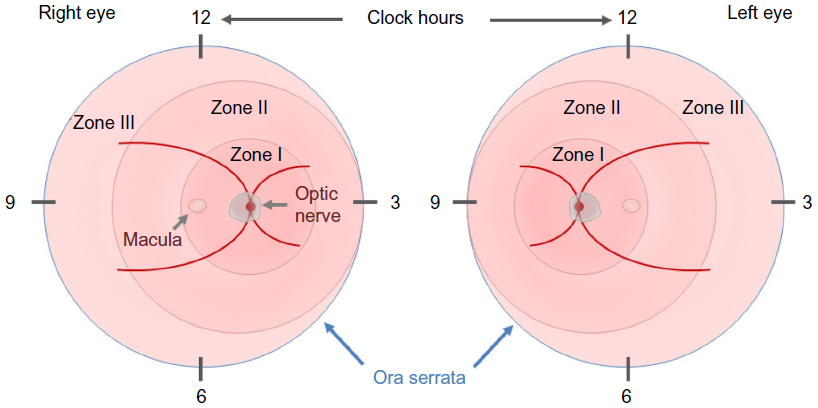 | Figure 1 The ICROP committee published three parts on the classification of retinopathy of prematurity. |
The latest revision of this classification was published in 2005 and it introduced the terms aggressive posterior ROP and preplus disease and clarified the definition of zone I.23
ROP was initially understood and described in terms of oxygen toxicity, without understanding, at that time, the contribution of other risk factors. In the 1940s, Terry described a disease in preterm infants that was characterized by an opaque tissue behind the lens that he referred to as retrolental fibroplasia.24,25
The term ROP was later coined secondary to the observation that premature birth is a key aspect in the development of this disease. At that time, the survival of preterm infants with respiratory problems was improved through the use of (unmonitored) oxygen supplementation.26 In the 1940s, ROP was the single most common reason for childhood blindness in the developed world.27 The animal studies by Ashton et al and a clinical study in 1952 and 1953 by Patz et al identified unmonitored use of high oxygen supplementation as a main driver of ROP.28–31 These first observations were affirmed by other groups30,32 and supported in animal models.28,33 A restriction of the oxygen supply in neonates resulted in a decrease in the incidence of ROP but an increase in perinatal death.34 The use of oxygen supplementation in the following years was more liberal in the nurseries, resulting in another epidemic of ROP that was, however, more treatable owing to improvements in neonatal intensive care35,36 and the introduction of retinal ablative therapies for ROP.37,38 The difficulty in balancing the oxygen delivery to minimize ROP (relatively low oxygen tension) versus minimizing the risk of developing brain hypoxia or death (relatively high oxygen tension) has not been fully resolved. With a better, if not perfect, control of supplemental oxygen, the other risk factors of ROP are more prominent. Over the last years, gestational age has remained as a prominent risk factor. With the lower gestational age, the degree of retinal vascular maturity will decrease, making the retina more susceptible to damage due to a lack of (postnatal) growth factors (GFs) and eventually decelerated vascularization.
Pathogenesis of ROP
The retinal vasculature in human is fully developed by the end of a full-term pregnancy.39 After preterm birth, compared to the third trimester in utero, there are several insults that prevent the immature partially vascularized retina from vascularizing normally. The partial pressure of oxygen (PaO2) in utero is ~50 mmHg by the end of pregnancy when compared to ~160 mmHg PaO2 found in ambient room air.40 Some infants are given as high as 100% oxygen as supplementation. When a preterm infant is born, he or she is immediately exposed to this dramatic increase in oxygen pressure. The high oxygen levels of even ambient room air and certainly with oxygen supplementation pose a risk for developing ROP. The reason behind this phenomenon is found in the development of the retinal vasculature: by the beginning of the fourth month of gestational age, vessels start to sprout radially from the optic nerve toward the ora serrata.41 This process is usually completed shortly before full-term birth.42 However, in preterm infants, this process is incomplete at birth. Studies suggest that a PaO2 of ≥80 mmHg besides preventing the normal vascularization will also damage the existing newly developed retinal capillaries. Increased oxygen pressure as found in ambient room air, which is around double that found to be harmful to the retinal vasculature, causes attenuation in vessel growth and a constriction of already developed vessels.43,44 As a result, avascular areas form in the retina.45 This phenomenon is further amplified when preterm babies are ventilated with high oxygen concentrations. The size of the initial avascular zone is dependent on the gestational age at birth of the preterm infant. If Phase I of ROP with an impaired vessel growth could be prevented with the control of oxygen but also with a restoration of factors missing in the preterm infant, such as IGF-1, then the second neovascular phase that depends on the extent of avascular retina would not occur. Restoring the normal vascularization that would occur in utero would prevent end-stage ROP.
Following this first phase of impaired vascular growth and vasoattenuation, the increasing metabolism of the growing infant’s retina leads to a rising demand for oxygen and nutrients and a subsequent upregulation of angiogenic GFs, such as VEGF and erythropoietin.46,47 This second phase of the disease, driven prominently by VEGF, is clinically characterized by the outgrowth of new but pathological vessels into the vitreous. These neovascular tufts are highly perfused, yet they do not contribute to the physiological needs of the infant’s retina and may be leaky due to an impaired blood–retina barrier. As the retina develops further, these pathological vessels may regress. However, fibrous scar tissue often remains, which tends to cause traction on the retina, and can, in the worst case, lead to retinal detachment and possibly blindness (Figure 2).48
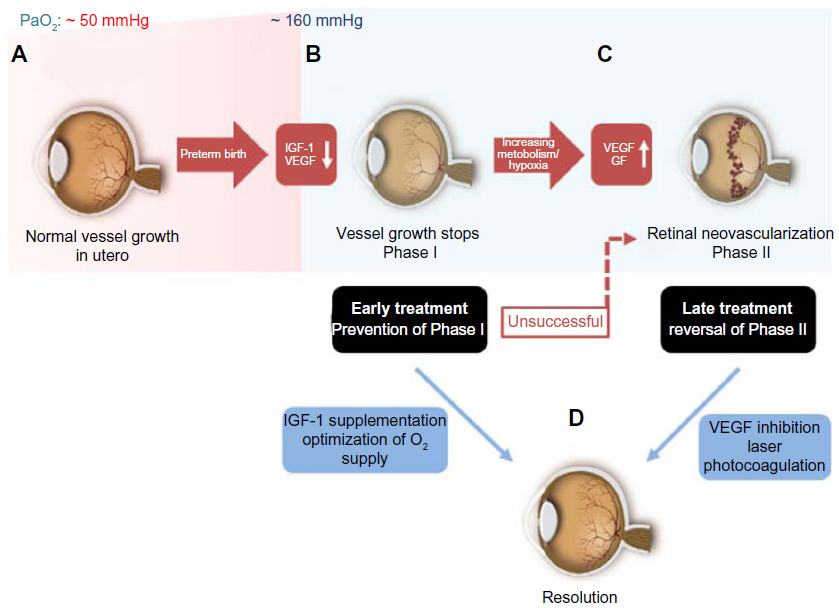 | Figure 2 Timecourse of ROP. |
The transition of Phase I to Phase II usually occurs around the 32nd postmenstrual week, but the onset as well as the severity of the neovascular phase can be variable.49,50 Interestingly, a multicenter trial found that the onset of retinal vascular changes in preterm infants seems to correlate more with gestational age at birth (time elapsed between the first day of last menstrual period and the day of delivery) rather than the chronological age (time elapsed from birth).51,52 For infants born before gestational week 27, the onset of ROP may correlate a little more with the chronological age than the gestational age. However, even in these severely preterm babies, stage 3 ROP was never seen before a postmenstrual age (PMA; gestational age + chronological age) of 31 weeks.49,53
Even mild ROP in more mature infants can disturb proper central retinal development.54 ROP-related visual impairment both in the central part of the retina55,56 and in the periphery55,57 can often be detected many years after the initial onset of the disease. Thus, the area of undeveloped retina at birth and the lack of postnatal growth are strong determinants of proliferative ROP.
Risk factors for ROP
Oxygen
Prevention of ROP must start with optimizing the oxygen delivery to minimizing the vessel loss and vessel growth cessation that set the stage for proliferative ROP. However, oxygen must also be optimized to prevent the brain damage and death. As discussed earlier, ROP is associated with an excessive oxygen use in the perinatal period in preterm infants. Today, the controlled supplemental oxygen delivery to these infants is intended to balance the need for adequate blood levels of oxygen and the risk of developing ROP.31 It is surprisingly difficult to address this issue as history shows. In the 1940s, excessive oxygen supplementation (100%) significantly reduced the number of infant deaths but precipitated the first epidemic of ROP. Against this background, oxygen supply was limited, even in respiratory distressed preterm infants, resulting in lower incidents of ROP and other oxygen- toxicity-associated diseases, such as hyaline membrane disease (HMD) now termed as bronchopulmonary dysplasia (BPD).58 The downside of this success was an increase in the mortality rate of these babies.34 Even 60 years later, no definitive answer can be given on the right amount of oxygen at the different developmental time points that gives sufficient attention to ROP pathogenesis and consideration of the overall survival of these infants. In the 1990s, several studies showed that a PaO2 of >80 mmHg is associated with a higher incidence of ROP59 and that NICUs that had lower SpO2 alarms (70%–90%) had a significantly reduced occurrence of severe ROP outcome (6% vs 27%) compared to higher SpO2 alarms (88%–98%).60
Larger studies addressed this issue more recently, including the SUPPORT trial,61–63 three BOOST II trials from the UK, Australia, and New Zealand,64,65 and the Canadian Oxygen Trial.66 All of these five trials included premature infants <28 weeks of gestational age and were randomized to either a low (85%–89%) or a high (90%–95%) SpO2 oxygen supplementation group. All these studies were later combined and analyzed by the neonatal oxygenation prospective meta-analysis study.67 Unfortunately, the results taken together are still unsatisfactory. The lower oxygen group showed a significantly smaller degree of development of severe ROP but a significantly higher incidence of infant death in the study population (preterm infants <28 weeks with oxygen supply until postmenstrual age 36 weeks). Conversely, higher oxygen supplementation lowered the infant death but was associated with more ROP.68 Conclusions from these studies, which have also been integrated into the European guidelines on functional SpO2, now recommend targeting the higher SpO2 levels (90%–95%) in these babies, despite the higher chance of provoking ROP.68,69 Furthermore, none of these studies can answer the question of whether SpO2 levels should be constant throughout the supplementation phase or adjusted with postnatal age.70 Overall, the new recommendations for higher oxygen levels to prevent death will result in more cases of ROP.
Gestational age, birth weight, and growth
Postnatal growth is a major risk factor that can be addressed to prevent ROP. Major risk factors for developing ROP include low gestational age and low birth weight in relation to age.71 However, gestational age and birth weight might not be independent risk factors.72 A lower gestational age goes along with a shorter duration of maternal protective factors that the infant might not be able to produce by himself and also a longer duration of exposure to extrauterine factors that might be harmful.
A strong independent risk factor that has been recognized over the past few years has been poor prenatal growth. Unlike poor postnatal growth where the data are very strong, the data for poor prenatal growth as a risk factor are somewhat inconsistent but imply that prenatal growth restriction may increase the risk for ROP infants born at older gestational ages, but not younger gestational ages.73
Poor postnatal growth on the other hand has been clearly linked with an increased risk for ROP, both in clinical and in animal studies.74–76 There seems to be a pivotal role for IGF-1, as lower serum levels after preterm birth are associated with an impaired postnatal growth and later development of proliferative ROP. This finding raises the important issue of potentially replacing IGF-1 to in-utero levels to prevent ROP.
Hyperglycemia and insulin
Arising from the observation that poor weight gain is associated with the development of late severe ROP, several approaches to optimize nutrition in preterm infants have been proposed. It was generally shown that very low birth weight infants can tolerate increased rates of infusion of intravenous fat emulsion solutions from the first week without severe side effects.77 The goal of reducing ROP through this measure has only been partially reached perhaps because of the difficulty in maintaining this regimen.78,79 Another study recommended a more aggressive nutrition practice in the NICUs, which was adopted by some neonatologists.80 Interestingly, this practice was associated with hyperglycemia and an increased demand for insulin, which is also seen in diabetes mellitus.81 In this study, the number of infants with stages 3–4 ROP born at <30 weeks PMA increased from 4% in 2001–2002 to 9% in 2006–2007. Hyperglycemia in the context of ROP has been mainly linked to both relative insulin resistance and defective proinsulin processing.82 In addition, a recent study proposed that hyperglycemia may not be linked to a higher risk of developing severe ROP in very small preterm babies, but the use of insulin significantly increases the risk.83 It may be that treating preterm infants who may have both an increased insulin sensitivity and hyperglycemia is counter productive, even though the body of evidence is not very strong yet. The use of insulin in preterm babies in general seems to offer little clinical benefit but increases the risk of hypoglycemia.84
Genetic factors in ROP
ROP is a complex disease that is influenced by both genetic and environmental factors. Several very small studies have found genetic variants in EPAS1, VEGF, SOD, and members of the WNT family in association with ROP. However, a larger study (817 infants and 543 infants in a replication cohort) to determine genetic variants associated with the severe ROP in a candidate gene cohort study of US preterm infants using the whole genome amplified DNA from the stored blood spot samples and the analyses such as, controlled for multiple comparisons, ancestral eigenvalues, family relatedness, and significant epidemiologic variables in both cohorts, showed that only two intronic single-nucleotide polymorphism in the gene BDNF were associated with the severe ROP.85,86
Treatment of ROP
Current treatment is not preventative but deals with the second proliferative phase and not the first phase of ROP, which sets the stage for neovascularization. The first treatment option in Phase II ROP was cryocoagulation of the avascular retina. The results and recommendations following this treatment approach were published in 1990 as the CRYO-ROP study.38,87 However, today, laser photocoagulation has replaced cryotherapy in most countries.88 Yet, the therapeutic principle stays the same: by destroying the avascular retina, the driver of an increased production of vascular GFs is reduced. When applied properly, this procedure is fairly safe and prevents most infants from developing retinal detachment (ROP stages 4 and 5). This intervention at early Phase II is important, as repairing late stage (4 and 5) ROP is unfortunately often associated with poor clinical outcomes.89
Anti-VEGF therapy has been a clinical treatment option in most proliferative eye disease in adults and is today the most important therapy for neovascular age-related macular degeneration. Not surprisingly, anti-VEGF therapy has also found its way into (off-label) Phase II ROP treatment with neovascularization. However, treating preterm infants is challenging, and the right dose or right drug has yet to be determined. The CARE-ROP trial (clinical trial identifier: NCT02134457) investigates, among other questions, if two different lower doses (compared to the BEAT-ROP study) of an even smaller molecule, the anti-VEGF fragment ranibizumab, are similarly effective in the treatment of neovascular Phase II ROP. Phase I trial of de-escalating doses of bevacizumab for Phase II ROP is also underway (clinical trial identifier: NCT02390531). The dose issue is critical as anti-VEGF treatment leaks into the systemic circulation to suppress systemic VEGF levels for many weeks with the potential of suppressing normal brain and other organs’ growth in fragile preterm neonates.
IGF-1 and its deficiency in the preterm infant
IGF-1 has a dominant role in somatic growth,90 especially in the third trimester of pregnancy, therefore, in the growth of preterm infants. IGF-1 levels drop to very low levels after preterm birth and remain low for many weeks postnatally. To understand the effects of lack of IGF-1 after preterm birth, genetic deletion of IGF-1 in mice is illustrative. Mice that do not produce IGF-1 are 40% smaller in body weight at birth compared to the wild-type controls.91 Additionally, body weight and bone length in human fetuses with a gestational age of 15–37 weeks correlate positively with serum IGF-1 concentrations.92,93 While some data suggest a role for IGF-1 in the development of many tissues even from the start of the first trimester,94,95 the increase in circulating fetal IGF-1 during the third trimester suggests that IGF-1 is particularly important at the later stages of pregnancy.
IGF-1 in the fetus during pregnancy is supplied by different sources, including fetal tissue. It is also received through swallowed amniotic fluid that contains IGF-1.96,97 Nutritional intake by the mother also affects IGF-1 production, especially in response to increasing the levels of (placental) growth hormone, which in turn may regulate fetal growth.98 The IGF-1 supply through the mother is particularly important in preterm infants, as the fetus at this early stage does not have an adult pattern of growth hormone (GH) regulation and, thus, is lacking this source of IGF-1 production at birth. Hence, it does not come as a surprise that preterm born infants have a very low serum level of IGF-1 compared to full-term babies. Without external supplementation of IGF-1, these preterm infants do not reach in-utero IGF-1 levels of infants of correspondent gestational age.93,99–101 In these preterm infants, the IGF-1 levels correlate with postnatal weight. The loss of this important GF after preterm birth and the association of low IGF-1 levels with the later development of ROP suggest that the replacement of IGF-1 may prevent ROP by stimulating normal postnatal growth in the retina.
Being a single chain polypeptide, IGF-1 is similar in structure to (pro)insulin. However, both IGF-1 and insulin have their own distinctive receptors that share a homology of ~60%.102 The IGF-1 receptor has a 1,000× higher affinity for IGF-1 than for insulin, while the insulin receptor has a 100× higher affinity for insulin than for IGF-1.103 Aside from IGF-1 and growth hormone, the IGF system also includes IGF-2, two receptors, types 1 and 2, IGF-binding proteins (IGFBPs), and IGFBP proteases.103 There are six IGFBPs, and ~98% of all IGF-1 is bound to IGFBP-3,104 which is also the most abundant form of all IGFBPs.105,106 IGFBP-3 is important in regulating the action of IGF-1 as it can prolong the half-life of IGF-1107 due to a stronger affinity of IGFBP-3 to IGF-1 than IGF-1 has to its receptor. The effect of IGF-1 will be augmented when released in proximity to the IGF-1 receptor and attenuated when IGF-1 stays bound to IGFBP-3. IGF-1’s mechanism of action is believed to be at least partly through MAPK and AKT signaling pathways, which stimulate cell growth and proliferation. IGF-1 receptor signals through multiple pathways. One key pathway is regulated by phosphatidylinositol-3 kinase and its downstream partner, the mammalian target of rapamycin, responsible for upregulating the AKT and thereby driving the growth. In addition, IGF-1 is a potent inhibitor of programmed cell death.108,109
IGF-1 also influences glucose metabolism. Insulin is the major regulator of metabolic processes under normal physiologic conditions and exerts its effect, especially in fat, muscle, and liver cells. However, even though IGFs are better known to induce growth and anabolism, they can also influence glucose homeostasis.110 Laboratory studies confirm that IGF-1 promotes glucose uptake in peripheral tissues.111,112 The metabolic consequences of IGF-1 deficiency have been studied in a liver-specific IGF-1-deficient mouse model, which show a 75% reduction in circulating IGF-1 and insulin insensitivity in muscle. Treatment with IGF-1 reduces insulin concentrations and improves insulin sensitivity, providing evidence for IGF-1 as an important component of overall insulin action in peripheral tissues.113 These data suggest that IGF-1 has an important role in metabolism; therefore, it is not surprising that a clear association between preterm babies with ROP and low early postnatal IGF-1 levels is found.114 While full-term babies physiologically show a rapid increase in IGF-1 levels postnatally, preterm infants born before gestational week 33 have a very slow increase in IGF-1.115 After preterm birth, energy requirements will strongly increase, while the maternal supply of nutrition and GFs, such as IGF-1, abruptly stop and often are not replaced, resulting in disturbed glucose metabolism. A recent study shows that plasma insulin levels are inversely correlated with gestational age,116 and increased glucose levels during the first week of life are strongly associated with ROP. Others have shown that premature babies versus full-term infants have some clinical signs of metabolic syndrome, including decreased insulin sensitivity117–119 and altered adiposity120,121 that share similarities with type 2 diabetes mellitus. ROP on the other hand shares some similarities with proliferative diabetic retinopathy, such as ischemic and vasoattenuated retinal areas, that may possibly lead to neovascularization. Interestingly, even though early data on insulin treatment within the first week of life show that it seems to increase IGF-I concentrations and improve longitudinal growth, respectively122 as mentioned earlier, it may also pose an increased risk to develop ROP.83
A direct association between low IGF-1 and poor retinal vascular growth in IGF-1-deficient mice suggests that the size of the avascular zone (Phase I) may also be dependent on the level of IGF-1, as low IGF-1 levels may add to a decrease in vascular growth in ROP.123
The retina of an infant is incompletely vascularized after preterm birth and has low IGF-I concentrations and low VEGF concentrations compared to those in utero. Thus, vascularization is delayed, and the retina becomes hypoxic. Neovascularization and blindness can occur.48,114,123,124 The concentrations of serum IGF-I and the duration of low IGF-I strongly correlate with the severity of ROP.48,123,125,126 Clinical studies mentioned earlier, correlate low postnatal IGF-I with the later development of ROP. Hence, IGF-1 supplementation in infants who are at risk for ROP may have the potential to prevent or at least attenuate ROP severity.
IGF-1 in Phases I and II of ROP
The severity of the second or neovascular phase of ROP is predominantly dictated by the inadequate vascularization in Phase I and eventually by the extent of the avascular zone of the retina. IGF-1 knockout mice have a slower growth of normal retinal blood vessels than their wild-type controls.123 This paucity in IGF-1 is also found in human subjects, where low IGF-1 serum levels directly correlate with the severity of ROP, and interestingly may also account for abnormal brain development and possibly aberrant neural retinal function.127 Other data derived from studies carried out in humans, link genetic defects of the GH/IGF-1 axis to very low levels of IGF-1, accounting for a decreased retinal vascular density in these patients.128 Additionally, IGF-1 controls the maximal VEGF-induced activation of MAPK and AKT in endothelial cells and, therefore, modulates vessel proliferation and survival,123 essential events in preventing Phase I of ROP.
The second phase of ROP is characterized by vessel proliferation driven by vascular GFs that are produced as a result of retinal hypoxia due to insufficient vascularization. Many evidence exists correlating low IGF-1 with the occurrence of Phase I ROP. Interestingly, the first data that indirectly linked IGF-1 with ROP obtained from work showing that transgenic mice expressing a GH receptor antagonist display significantly less retinal neovascularization at the second phase of ROP-like disease.129 These findings were supported by later studies showing that an IGF-1 receptor antagonist reduces retinal neovascularization in vivo. This study also proved that IGF-1 is required for maximum VEGF-induced activation of p44/42 MAPK – an essential pathway for endothelial cell proliferation – and, thus, the neovascularization observed in Phase II of ROP. VEGF alone would be insufficient to induce maximal angiogenesis associated with ROP, and IGF-1 acts as a permissive factor in ROP and possibly in other proliferative retinopathies.130
IGF-1 supplementation: a preventative treatment for ROP in preterm infants
Current options in ROP treatment aim at limiting the disastrous effects arising from severe development of Phase II ROP. Laser ablative therapy destroys avascular retina in order to reduce the production of hypoxia driven GFs. Later, these (although peripheral) parts of the retina will never be able to contribute to normal visual perception.
Low systemic IGF-1 levels are associated with the later development of ROP.114 Hence, the importance of sufficient systemic concentrations of IGF-1 reaching the retina through circulation raises the possibility of pharmacological restoration of IGF-1 to in-utero levels as a strategy for countering the vascular degeneration associated with the first phase of ROP. An interventional preclinical study of oxygen-induced retinopathy found that mice that received recombinant human insulin-like growth factor (rhIGF)-1 treatment developed less retinopathy, which supports the possible role of supplemental IGF-1 in preventing ROP.131
These observations led to the initiation of clinical trials to evaluate the effect of a complex of IGF-1 with its most important binding protein, IGFBP-3, on ROP development. While the half-life of IGF-1 in adults was calculated to be ~17 hours,132 the half-life of IGF-1 in preterm infants is significantly shorter, suggesting that a continuous intravenous infusion would be beneficial. For a better understanding of the pharmacokinetics of IGF-1 and IGFBP-3 in preterm infants, a first clinical study was performed using fresh-frozen adult plasma (FFP).133 FFP from healthy adults has a considerably higher concentration of IGF-1 and IGFBP-3 than those reported for very preterm infants.134 In this study, a total of 20 extremely preterm infants were included with a mean gestational age of 20 weeks. FFP was then administered between days 1 and 7 (median day 2) at a mean volume of 11 mL/kg over a time period of ~120 minutes (range: 90–240 minutes). This one time continuous infusion led to an increase of 133% in IGF-1 levels and 61% in IGFBP-3 levels. The first pharmacokinetic and dosing study of an intravenous IGF-1 and IGFBP-3 complex to preterm infants was carried out in Sweden. This study included preterm infants with a gestational age of 26–29 weeks and a mean birth weight of 1,022 g. This study concluded that an intravenous infusion of an equimolar preparation of rhIGF-1/rhIGFBP-3 to these very preterm infants normalized systemic IGF-1 levels to that found in utero at the corresponding gestational age. The administration of rhIGF-1/rhIGFBP-3 in this study was safe without any incidents of hypoglycemia and was well tolerated by all infants.135
A Phase II trial in preterm infants with a median age of 27 weeks confirmed these results and also showed that a continuous infusion of an IGF-1/IGFBP-3 complex can bring IGF-1 levels back to the lower end of normal values. However, per the protocol, continuous infusion was discontinued after 7 days, and it was observed that thereafter serum IGF-1 concentrations fell below intrauterine levels for the corresponding gestational age. Therefore, a continuous infusion for >7 days was proposed.136 The clinical trial (clinical trial identifier: NCT01096784) is recruiting patients, and further results are expected.
Outlook and conclusion
Destroying the avascular retina with laser photocoagulation and injecting a drug in the vitreous that may leak into systemic circulation are problematic in the context of very premature infants, yet both approaches are the only therapeutic means currently available to treat severe ROP.
The results of preclinical and early clinical studies support conducting the trials to examine ROP prevention by supplementing IGF-1 and IGFBP-3 to normal in-utero levels. Clinical studies are currently ongoing to evaluate the effect of this replacement treatment on preventing ROP. If prevention is effective, anti-VEGF injections and particularly laser photocoagulation could be used only in cases where preventative interventions were missed or an insufficient response to initial treatment was observed.
Disclosure
Ann Hellström and Lois EH Smith are consultants for Shire Pharmaceuticals. Raffael Liegl reports no conflicts of interest in this work.
References
Hamilton BE, Martin JA, Osterman MJK. National Vital Statistics Reports. Vol. 63. Hyattsville, MD: National Center for Health Statistics; 2014. | |
CDC [webpage on the Internet]. Preterm Birth. Available from: http://www.cdc.gov/reproductivehealth/maternalinfanthealth/pretermbirth.htm. Accessed October 2015. | |
Goldenberg RL, Culhane JF, Iams JD, Romero R. Epidemiology and causes of preterm birth. Lancet. 2008;371(9606):75–84. | |
McCormick MC. The contribution of low birth weight to infant mortality and childhood morbidity. N Engl J Med. 1985;312(2):82–90. | |
Lawn JE, Gravett MG, Nunes TM, Rubens CE, Stanton C; GAPPS Review Group. Global report on preterm birth and stillbirth (1 of 7): definitions, description of the burden and opportunities to improve data. BMC Pregnancy Childbirth. 2010;10(suppl 1):S1. | |
Callaghan WM, MacDorman MF, Rasmussen SA, Qin C, Lackritz EM. The contribution of preterm birth to infant mortality rates in the United States. Pediatrics. 2006;118(4):1566–1573. | |
Liu L, Johnson HL, Cousens S, et al; Child Health Epidemiology Reference Group of WHO and UNICEF. Global, regional, and national causes of child mortality: an updated systematic analysis for 2010 with time trends since 2000. Lancet. 2012;379(9832):2151–2161. | |
Stoll BJ, Hansen NI, Bell EF, et al; Eunice Kennedy Shriver National Institute of Child Health and Human Development Neonatal Research Network. Trends in care practices, morbidity, and mortality of extremely preterm neonates, 1993–2012. JAMA. 2015;314(10):1039–1051. | |
Guillen U, Weiss EM, Munson D, et al. Guidelines for the management of extremely premature deliveries: a systematic review. Pediatrics. 2015;136(2):343–350. | |
Iams JD, Romero R, Culhane JF, Goldenberg RL. Primary, secondary, and tertiary interventions to reduce the morbidity and mortality of preterm birth. Lancet. 2008;371(9607):164–175. | |
Laas E, Lelong N, Thieulin AC, et al; EPICARD Study Group. Preterm birth and congenital heart defects: a population-based study. Pediatrics. 2012;130(4):e829–e837. | |
Weinberg JG, Evans FJ, Burns KM, Pearson GD, Kaltman JR. Surgical ligation of patent ductus arteriosus in premature infants: trends and practice variation. Cardiol Young. Epub 2015 Sep 23. | |
Jobe AH, Ikegami M. Prevention of bronchopulmonary dysplasia. Curr Opin Pediatr. 2001;13(2):124–129. | |
Ho JJ, Subramaniam P, Davis PG. Continuous distending pressure for respiratory distress in preterm infants. Cochrane Database Syst Rev. 2015;7:CD002271. | |
Back SA. Brain injury in the preterm infant: new horizons for pathogenesis and prevention. Pediatr Neurol. 2015;53(3):185–192. | |
Glass HC, Costarino AT, Stayer SA, Brett CM, Cladis F, Davis PJ. Outcomes for extremely premature infants. Anesth Analg. 2015; 120(6):1337–1351. | |
Volpe JJ. Brain injury in premature infants: a complex amalgam of destructive and developmental disturbances. Lancet Neurol. 2009;8(1):110–124. | |
Smith LE. Pathogenesis of retinopathy of prematurity. Acta Paediatr Suppl. 2002;91(437):26–28. | |
Smith LE. Pathogenesis of retinopathy of prematurity. Semin Neonatol. 2003;8(6):469–473. | |
Kong L, Fry M, Al-Samarraie M, Gilbert C, Steinkuller PG. An update on progress and the changing epidemiology of causes of childhood blindness worldwide. J AAPOS. 2012;16(6):501–507. | |
An international classification of retinopathy of prematurity. The Committee for the Classification of Retinopathy of Prematurity. Arch Ophthalmol. 1984;102(8):1130–1134. | |
An international classification of retinopathy of prematurity. II. The classification of retinal detachment. The International Committee for the Classification of the Late Stages of Retinopathy of Prematurity. Arch Ophthalmol. 1987;105(7):906–912. | |
International Committee for the Classification of Retinopathy of Prematurity. The international classification of retinopathy of prematurity revisited. Arch Ophthalmol. 2005;123(7):991–999. | |
Terry TL. Retrolental fibroplasia in the premature infant: V. Further studies on fibroplastic overgrowth of the persistent tunica vasculosa lentis. Trans Am Ophthalmol Soc. 1944;42:383–396. | |
Terry TL. Fibroblastic overgrowth of persistent tunica vasculosa lentis in infants born prematurely: II. Report of cases-clinical aspects. Trans Am Ophthalmol Soc. 1942;40:262–284. | |
Zin A, Gole GA. Retinopathy of prematurity-incidence today. Clin Perinatol. 2013;40(2):185–200. | |
Silverman W. Retrolental Fibroplasia: A Modern Parable. New York, NY: Grune and Stratton;1980. | |
Ashton N, Ward B, Serpell G. Effect of oxygen on developing retinal vessels with particular reference to the problem of retrolental fibroplasia. Br J Ophthalmol. 1954;38(7):397–432. | |
Patz A, Eastham A, Higginbotham DH, Kleh T. Oxygen studies in retrolental fibroplasia. II. The production of the microscopic changes of retrolental fibroplasia in experimental animals. Am J Ophthalmol. 1953;36(11):1511–1522. | |
Patz A, Hoeck LE, De La Cruz E. Studies on the effect of high oxygen administration in retrolental fibroplasia. I. Nursery observations. Am J Ophthalmol. 1952;35(9):1248–1253. | |
Kinsey VE, Hemphill FM. Etiology of retrolental fibroplasia and preliminary report of cooperative study of retrolental fibroplasia. Trans Am Acad Ophthalmol Otolaryngol. 1955;59(1):15–24. [discussion, 11–40]. | |
Campbell K. Intensive oxygen therapy as a possible cause of retrolental fibroplasia; a clinical approach. Med J Aust. 1951;2(2):48–50. | |
Ashton N, Ward B, Serpell G. Role of oxygen in the genesis of retrolental fibroplasia; a preliminary report. Br J Ophthalmol. 1953;37(9):513–520. | |
Bolton DP, Cross KW. Further observations on cost of preventing retrolental fibroplasia. Lancet. 1974;1(7855):445–448. | |
Shohat M, Reisner SH, Krikler R, Nissenkorn I, Yassur Y, Ben-Sira I. Retinopathy of prematurity: incidence and risk factors. Pediatrics. 1983;72(2):159–163. | |
Gunn TR, Easdown J, Outerbridge EW, Aranda JV. Risk factors in retrolental fibroplasia. Pediatrics. 1980;65(6):1096–1100. | |
Multicenter trial of cryotherapy for retinopathy of prematurity. Preliminary results. Cryotherapy for Retinopathy of Prematurity Cooperative Group. Arch Ophthalmol. 1988;106(4):471–479. | |
Multicenter trial of cryotherapy for retinopathy of prematurity. Three-month outcome. Cryotherapy for Retinopathy of Prematurity Cooperative Group. Arch Ophthalmol. 1990;108(2):195–204. | |
Provis JM. Development of the primate retinal vasculature. Prog Retin Eye Res. 2001;20(6):799–821. | |
Nicolaides KH, Economides DL, Soothill PW. Blood gases, pH, and lactate in appropriate- and small-for-gestational-age fetuses. Am J Obstet Gynecol. 1989;161(4):996–1001. | |
Fledelius HC, Dahl H. Retinopathy of prematurity, a decrease in frequency and severity. Trends over 16 years in a Danish county. Acta Ophthalmol Scand. 2000;78(3):359–361. | |
Roth AM. Retinal vascular development in premature infants. Am J Ophthalmol. 1977;84(5):636–640. | |
Reynolds JD. The management of retinopathy of prematurity. Paediatr Drugs. 2001;3(4):263–272. | |
Hartnett ME, Lane RH. Effects of oxygen on the development and severity of retinopathy of prematurity. J AAPOS. 2013;17(3):229–234. | |
Pierce EA, Foley ED, Smith LE. Regulation of vascular endothelial growth factor by oxygen in a model of retinopathy of prematurity. Arch Ophthalmol. 1996;114(10):1219–1228. | |
Aiello LP, Pierce EA, Foley ED, et al. Suppression of retinal neovascularization in vivo by inhibition of vascular endothelial growth factor (VEGF) using soluble VEGF-receptor chimeric proteins. Proc Natl Acad Sci USA. 1995;92(23):10457–10461. | |
Watanabe D, Suzuma K, Matsui S, et al. Erythropoietin as a retinal angiogenic factor in proliferative diabetic retinopathy. N Engl J Med. 2005;353(8):782–792. | |
Smith LE, Hard AL, Hellstrom A. The biology of retinopathy of prematurity: how knowledge of pathogenesis guides treatment. Clin Perinatol. 2013;40(2):201–214. | |
Austeng D, Kallen KB, Hellstrom A, Tornqvist K, Holmström GE. Natural history of retinopathy of prematurity in infants born before 27 weeks’ gestation in Sweden. Arch Ophthalmol. 2010;128(10):1289–1294. | |
Good WV, Hardy RJ, Dobson V, et al; Early Treatment for Retinopathy of Prematurity Cooperative Group. The incidence and course of retinopathy of prematurity: findings from the early treatment for retinopathy of prematurity study. Pediatrics. 2005;116(1):15–23. | |
Engle WA. Age terminology during the perinatal period. Pediatrics. 2004;114(5):1362–1364. | |
Palmer EA, Flynn JT, Hardy RJ, et al. Incidence and early course of retinopathy of prematurity. The Cryotherapy for Retinopathy of Prematurity Cooperative Group. Ophthalmology. 1991;98(11):1628–1640. | |
Austeng D, Kallen KB, Hellstrom A, et al. Screening for retinopathy of prematurity in infants born before 27 weeks’ gestation in Sweden. Arch Ophthalmol. 2011;129(2):167–172. | |
Barnaby AM, Hansen RM, Moskowitz A, Fulton AB. Development of scotopic visual thresholds in retinopathy of prematurity. Invest Ophthalmol Vis Sci. 2007;48(10):4854–4860. | |
Reisner DS, Hansen RM, Findl O, Petersen RA, Fulton AB. Dark-adapted thresholds in children with histories of mild retinopathy of prematurity. Invest Ophthalmol Vis Sci. 1997;38(6):1175–1183. | |
Hammer DX, Iftimia NV, Ferguson RD, et al. Foveal fine structure in retinopathy of prematurity: an adaptive optics Fourier domain optical coherence tomography study. Invest Ophthalmol Vis Sci. 2008;49(5):2061–2070. | |
Moskowitz A, Hansen R, Fulton A. Early ametropia and rod photoreceptor function in retinopathy of prematurity. Optom Vis Sci. 2005;82(4):307–317. | |
Avery ME. Recent increase in mortality from hyaline membrane disease. J Pediatr. 1960;57:553–559. | |
Flynn JT. Acute proliferative retrolental fibroplasia: multivariate risk analysis. Trans Am Ophthalmol Soc. 1983;81:549–591. | |
Tin W, Milligan DW, Pennefather P, Hey E. Pulse oximetry, severe retinopathy, and outcome at one year in babies of less than 28 weeks gestation. Arch Dis Child Fetal Neonatal Ed. 2001;84(2):F106–F110. | |
Carlo WA, Bell EF, Walsh MC. Oxygen-saturation targets in extremely preterm infants. N Engl J Med. 2013;368(20):1949–1950. | |
Finer NN, Carlo WA, Walsh MC, et al; SUPPORT Study Group of the Eunice Kennedy Shriver NICHD Neonatal Research Network. Early CPAP versus surfactant in extremely preterm infants. N Engl J Med. 2010;362(21):1970–1979. | |
Carlo WA, Finer NN, Walsh MC, et al; SUPPORT Study Group of the Eunice Kennedy Shriver NICHD Neonatal Research Network. Target ranges of oxygen saturation in extremely preterm infants. N Engl J Med. 2010;362(21):1959–1969. | |
Stenson BJ, Tarnow-Mordi WO, Darlow BA, et al; BOOST II United Kingdom Collaborative Group; BOOST II Australia Collaborative Group; BOOST II New Zealand Collaborative Group. Oxygen saturation and outcomes in preterm infants. N Engl J Med. 2013;368(22):2094–2104. | |
Stenson B, Brocklehurst P, Tarnow-Mordi W. Increased 36-week survival with high oxygen saturation target in extremely preterm infants. N Engl J Med. 2011;364(17):1680–1682. | |
Schmidt B, Whyte RK, Asztalos EV, et al; Canadian Oxygen Trial (COT) Group. Effects of targeting higher vs lower arterial oxygen saturations on death or disability in extremely preterm infants: a randomized clinical trial. JAMA. 2013;309(20):2111–2120. | |
Askie LM, Brocklehurst P, Darlow BA, et al; NeOProM Collaborative Group. NeOProM: neonatal oxygenation prospective meta-analysis collaboration study protocol. BMC Pediatr. 2011;11:6. | |
Saugstad OD, Aune D. Optimal oxygenation of extremely low birth weight infants: a meta-analysis and systematic review of the oxygen saturation target studies. Neonatology. 2014;105(1):55–63. | |
Sweet DG, Carnielli V, Greisen G, et al; European Association of Perinatal Medicine. European consensus guidelines on the management of neonatal respiratory distress syndrome in preterm infants – 2013 update. Neonatology. 2013;103(4):353–368. | |
Chen ML, Guo L, Smith LE, Dammann CE, Dammann O. High or low oxygen saturation and severe retinopathy of prematurity: a meta-analysis. Pediatrics. 2010;125(6):e1483–e1492. | |
Darlow BA, Hutchinson JL, Henderson-Smart DJ, et al; Australian and New Zealand Neonatal Network. Prenatal risk factors for severe retinopathy of prematurity among very preterm infants of the Australian and New Zealand Neonatal Network. Pediatrics. 2005;115(4):990–996. | |
EXPRESS Group. Incidence of and risk factors for neonatal morbidity after active perinatal care: extremely preterm infants study in Sweden (EXPRESS). Acta Paediatr. 2010;99(7):978–992. | |
Hellström A, Smith LE, Hard AL. Effects of body weight gain on retinopathy of prematurity. In: Hartnett ME, editor. Pediatric Retina. Netherlands: Wolters Kluwer; 2013. | |
Cooke RJ, Ainsworth SB, Fenton AC. Postnatal growth retardation: a universal problem in preterm infants. Arch Dis Child Fetal Neonatal Ed. 2004;89(5):F428–F430. | |
Hellstrom BE. Experimental approach to the pathogenesis of retrolental fibroplasia. V. The influence of the state of nutrition on oxygen-induced changes in the mouse eye. Acta Paediatr. 1956;45(1):43–57. | |
Stahl A, Chen J, Sapieha P, et al. Postnatal weight gain modifies severity and functional outcome of oxygen-induced proliferative retinopathy. Am J Pathol. 2010;177(6):2715–2723. | |
Drenckpohl D, McConnell C, Gaffney S, Niehaus M, Macwan KS. Randomized trial of very low birth weight infants receiving higher rates of infusion of intravenous fat emulsions during the first week of life. Pediatrics. 2008;122(4):743–751. | |
Hellstrom A, Hard AL. Editorial on ‘Hyperglycemia, insulin and slower growth velocity may increase the risk of retinopathy of prematurity’ Kaempf JW et al. J Perinatol. 2011;31(4):228–229. | |
Embleton ND. Optimal protein and energy intakes in preterm infants. Early Hum Dev. 2007;83(12):831–837. | |
Ziegler EE, Thureen PJ, Carlson SJ. Aggressive nutrition of the very low birth weight infant. Clin Perinatol. 2002;29(2):225–244. | |
Kaempf JW, Kaempf AJ, Wu Y, Stawarz M, Niemeyer J, Grunkemeier G. Hyperglycemia, insulin and slower growth velocity may increase the risk of retinopathy of prematurity. J Perinatol. 2011;31(4):251–257. | |
Mitanchez-Mokhtari D, Lahlou N, Kieffer F, Magny JF, Roger M, Voyer M. Both relative insulin resistance and defective islet beta-cell processing of proinsulin are responsible for transient hyperglycemia in extremely preterm infants. Pediatrics. 2004;113(3 pt 1):537–541. | |
Lee JH, Hornik CP, Testoni D, et al. Insulin, hyperglycemia, and severe retinopathy of prematurity in extremely low-birth-weight infants. Am J Perinatol. Epub 2015 Oct 20. | |
Beardsall K, Vanhaesebrouck S, Ogilvy-Stuart AL, et al. Early insulin therapy in very-low-birth-weight infants. N Engl J Med. 2008;359(18):1873–1884. | |
Hartnett ME, Cotten CM. Genomics in the neonatal nursery: focus on ROP. Semin Perinatol. 2015;39(8):604–610. | |
Hartnett ME, Morrison MA, Smith S, et al; Genomics Subcommittee. Genetic variants associated with severe retinopathy of prematurity in extremely low birth weight infants. Invest Ophthalmol Vis Sci. 2014;55(10):6194–6203. | |
Multicenter trial of cryotherapy for retinopathy of prematurity. One-year outcome – structure and function. Cryotherapy for Retinopathy of Prematurity Cooperative Group. Arch Ophthalmol. 1990;108(10):1408–1416. | |
Simpson JL, Melia M, Yang MB, Buffenn AN, Chiang MF, Lambert SR. Current role of cryotherapy in retinopathy of prematurity: a report by the American Academy of Ophthalmology. Ophthalmology. 2012; 119(4):873–877. | |
Repka MX, Tung B, Good WV, Capone A Jr, Shapiro MJ. Outcome of eyes developing retinal detachment during the early treatment for retinopathy of prematurity study. Arch Ophthalmol. 2011;129(9):1175–1179. | |
Daughaday WH. The possible autocrine/paracrine and endocrine roles of insulin-like growth factors of human tumors. Endocrinology. 1990;127(1):1–4. | |
Powell-Braxton L, Hollingshead P, Warburton C, et al. IGF-I is required for normal embryonic growth in mice. Genes Dev. 1993; 7(12B):2609–2617. | |
Ashton IK, Zapf J, Einschenk I, MacKenzie IZ. Insulin-like growth factors (IGF) 1 and 2 in human foetal plasma and relationship to gestational age and foetal size during midpregnancy. Acta Endocrinol. 1985;110(4):558–563. | |
Lassarre C, Hardouin S, Daffos F, Forestier F, Frankenne F, Binoux M. Serum insulin-like growth factors and insulin-like growth factor binding proteins in the human fetus. Relationships with growth in normal subjects and in subjects with intrauterine growth retardation. Pediatr Res. 1991;29(3):219–225. | |
Han VK, Lund PK, Lee DC, D’Ercole AJ. Expression of somatomedin/insulin-like growth factor messenger ribonucleic acids in the human fetus: identification, characterization, and tissue distribution. J Clin Endocrinol Metab. 1988;66(2):422–429. | |
Roberts CT Jr, Lasky SR, Lowe WL Jr, Seaman WT, LeRoith D. Molecular cloning of rat insulin-like growth factor I complementary deoxyribonucleic acids: differential messenger ribonucleic acid processing and regulation by growth hormone in extrahepatic tissues. Mol Endocrinol. 1987;1(3):243–248. | |
Gluckman PD, Harding JE. The physiology and pathophysiology of intrauterine growth retardation. Horm Res. 1997;48(suppl 1):11–16. | |
Bona G, Aquili C, Ravanini P, et al. Growth hormone, insulin-like growth factor-I and somatostatin in human fetus, newborn, mother plasma and amniotic fluid. Panminerva Med. 1994;36(1):5–12. | |
Mirlesse V, Frankenne F, Alsat E, Poncelet M, Hennen G, Evain-Brion D. Placental growth hormone levels in normal pregnancy and in pregnancies with intrauterine growth retardation. Pediatr Res. 1993;34(4):439–442. | |
Bang P, Westgren M, Schwander J, Blum WF, Rosenfeld RG, Stangenberg M. Ontogeny of insulin-like growth factor-binding protein-1, -2, and -3: quantitative measurements by radioimmunoassay in human fetal serum. Pediatr Res. 1994;36(4):528–536. | |
Lofqvist C, Hansen-Pupp I, Andersson E, et al. Validation of a new retinopathy of prematurity screening method monitoring longitudinal postnatal weight and insulinlike growth factor I. Arch Ophthalmol. 2009;127(5):622–627. | |
Hansen-Pupp I, Hellstrom-Westas L, Cilio CM, Andersson S, Fellman V, Ley D. Inflammation at birth and the insulin-like growth factor system in very preterm infants. Acta Paediatr. 2007;96(6):830–836. | |
Ullrich A, Gray A, Tam AW, et al. Insulin-like growth factor I receptor primary structure: comparison with insulin receptor suggests structural determinants that define functional specificity. EMBO J. 1986;5(10):2503–2512. | |
Kim MS, Lee DY. Insulin-like growth factor (IGF)-I and IGF binding proteins axis in diabetes mellitus. Ann Pediatr Endocrinol Metab. 2015;20(2):69–73. | |
Frystyk J, Bek T, Flyvbjerg A, Skjaerbaek C, Ørskov H. The relationship between the circulating IGF system and the presence of retinopathy in type 1 diabetic patients. Diabet Med. 2003;20(4):269–276. | |
Jogie-Brahim S, Feldman D, Oh Y. Unraveling insulin-like growth factor binding protein-3 actions in human disease. Endocr Rev. 2009;30(5):417–437. | |
Muzumdar RH, Ma X, Fishman S, et al. Central and opposing effects of IGF-I and IGF-binding protein-3 on systemic insulin action. Diabetes. 2006;55(10):2788–2796. | |
Firth SM, Baxter RC. Cellular actions of the insulin-like growth factor binding proteins. Endocr Rev. 2002;23(6):824–854. | |
LeRoith D, Werner H, Beitner-Johnson D, Roberts CT Jr. Molecular and cellular aspects of the insulin-like growth factor I receptor. Endocr Rev. 1995;16(2):143–163. | |
Fernandez M, Sanchez-Franco F, Palacios N, Sánchez I, Fernández C, Cacicedo L. IGF-I inhibits apoptosis through the activation of the phosphatidylinositol 3-kinase/Akt pathway in pituitary cells. J Mol Endocrinol. 2004;33(1):155–163. | |
Bassett NS, Breier BH, Hodgkinson SC, Davis SR, Henderson HV, Gluckman PD. Plasma clearance of radiolabelled IGF-1 in the late gestation ovine fetus. J Dev Physiol. 1990;14(2):73–79. | |
Moses AC, Young SC, Morrow LA, O’Brien M, Clemmons DR. Recombinant human insulin-like growth factor I increases insulin sensitivity and improves glycemic control in type II diabetes. Diabetes. 1996;45(1):91–100. | |
Scavo LM, Karas M, Murray M, Leroith D. Insulin-like growth factor-I stimulates both cell growth and lipogenesis during differentiation of human mesenchymal stem cells into adipocytes. J Clin Endocrinol Metab. 2004;89(7):3543–3553. | |
Yakar S, Liu JL, Fernandez AM, et al. Liver-specific IGF-1 gene deletion leads to muscle insulin insensitivity. Diabetes. 2001;50(5):1110–1118. | |
Hellstrom A, Engstrom E, Hard AL, et al. Postnatal serum insulin-like growth factor I deficiency is associated with retinopathy of prematurity and other complications of premature birth. Pediatrics. 2003;112(5):1016–1020. | |
Lineham JD, Smith RM, Dahlenburg GW, et al. Circulating insulin-like growth factor I levels in newborn premature and full-term infants followed longitudinally. Early Hum Dev. 1986;13(1):37–46. | |
Wang G, Divall S, Radovick S, et al. Preterm birth and random plasma insulin levels at birth and in early childhood. JAMA. 2014;311(6):587–596. | |
Hofman PL, Regan F, Jackson WE, et al. Premature birth and later insulin resistance. N Engl J Med. 2004;351(21):2179–2186. | |
Mathai S, Cutfield WS, Derraik JG, et al. Insulin sensitivity and beta-cell function in adults born preterm and their children. Diabetes. 2012;61(10):2479–2483. | |
Hovi P, Andersson S, Eriksson JG, et al. Glucose regulation in young adults with very low birth weight. N Engl J Med. 2007; 356(20):2053–2063. | |
Thomas EL, Parkinson JR, Hyde MJ, et al. Aberrant adiposity and ectopic lipid deposition characterize the adult phenotype of the preterm infant. Pediatr Res. 2011;70(5):507–512. | |
Vohr BR, Allan W, Katz KH, Schneider KC, Ment LR. Early predictors of hypertension in prematurely born adolescents. Acta Paediatr. 2010;99(12):1812–1818. | |
Beardsall K, Ogilvy-Stuart AL, Frystyk J, et al. Early elective insulin therapy can reduce hyperglycemia and increase insulin-like growth factor-I levels in very low birth weight infants. J Pediatr. 2007;151(6):611–617,617e611. | |
Hellstrom A, Perruzzi C, Ju M, et al. Low IGF-I suppresses VEGF-survival signaling in retinal endothelial cells: direct correlation with clinical retinopathy of prematurity. Proc Natl Acad Sci USA. 2001;98(10):5804–5808. | |
Can E, Bulbul A, Uslu S, Cömert S, Bolat F, Nuholu A. Effects of aggressive parenteral nutrition on growth and clinical outcome in preterm infants. Pediatr Int. 2012;54(6):869–874. | |
Lofqvist C, Andersson E, Sigurdsson J, et al. Longitudinal postnatal weight and insulin-like growth factor I measurements in the prediction of retinopathy of prematurity. Arch Ophthalmol. 2006;124(12):1711–1718. | |
Perez-Munuzuri A, Fernandez-Lorenzo JR, Couce-Pico ML, Blanco-Teijeiro MJ, Fraga-Bermúdez JM. Serum levels of IGF1 are a useful predictor of retinopathy of prematurity. Acta Paediatr. 2010;99(4):519–525. | |
Lofqvist C, Engstrom E, Sigurdsson J, et al. Postnatal head growth deficit among premature infants parallels retinopathy of prematurity and insulin-like growth factor-1 deficit. Pediatrics. 2006;117(6):1930–1938. | |
Hellstrom A, Carlsson B, Niklasson A, et al. IGF-I is critical for normal vascularization of the human retina. J Clin Endocrinol Metab. 2002;87(7):3413–3416. | |
Smith LE, Kopchick JJ, Chen W, et al. Essential role of growth hormone in ischemia-induced retinal neovascularization. Science. 1997;276(5319):1706–1709. | |
Smith LE, Shen W, Perruzzi C, et al. Regulation of vascular endothelial growth factor-dependent retinal neovascularization by insulin-like growth factor-1 receptor. Nat Med. 1999;5(12):1390–1395. | |
Vanhaesebrouck S, Daniels H, Moons L, Vanhole C, Carmeliet P, De Zegher F. Oxygen-induced retinopathy in mice: amplification by neonatal IGF-I deficit and attenuation by IGF-I administration. Pediatr Res. 2009;65(3):307–310. | |
Mizuno N, Kato Y, Iwamoto M, et al. Kinetic analysis of the disposition of insulin-like growth factor 1 in healthy volunteers. Pharm Res. 2001;18(8):1203–1209. | |
Hansen-Pupp I, Engstrom E, Niklasson A, et al. Fresh-frozen plasma as a source of exogenous insulin-like growth factor-I in the extremely preterm infant. J Clin Endocrinol Metab. 2009;94(2):477–482. | |
Yu H, Mistry J, Nicar MJ, et al. Insulin-like growth factors (IGF-I, free IGF-I and IGF-II) and insulin-like growth factor binding proteins (IGFBP-2, IGFBP-3, IGFBP-6, and ALS) in blood circulation. J Clin Lab Anal. 1999;13(4):166–172. | |
Lofqvist C, Niklasson A, Engstrom E, et al. A pharmacokinetic and dosing study of intravenous insulin-like growth factor-I and IGF-binding protein-3 complex to preterm infants. Pediatr Res. 2009;65(5):574–579. | |
Ley D, Hansen-Pupp I, Niklasson A, et al. Longitudinal infusion of a complex of insulin-like growth factor-I and IGF-binding protein-3 in five preterm infants: pharmacokinetics and short-term safety. Pediatr Res. 2013;73(1):68–74. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.