")
Back to Journals » Clinical Ophthalmology » Volume 16
Retinitis Pigmentosa in the Puerto Rican Population: A Geographic Distribution
Authors Santos DF, Molina Thurin LJ, Vargas JG, Izquierdo NJ, Oliver A
Received 8 August 2022
Accepted for publication 12 September 2022
Published 28 September 2022 Volume 2022:16 Pages 3175—3179
DOI https://doi.org/10.2147/OPTH.S375365
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
David F Santos,1 Leonardo J Molina Thurin,2 José Gustavo Vargas,3 Natalio J Izquierdo,4 Armando Oliver5
1School of Medicine, Medical Sciences Campus, University of Puerto Rico, San Juan, PR, USA; 2San Juan Bautista School of Medicine, Caguas, PR, USA; 3The Johns Hopkins University School of Medicine, Baltimore, MD, USA; 4Department of Surgery, School of Medicine, Medical Sciences Campus, University of Puerto Rico, San Juan, PR, USA; 5Department of Ophthalmology, School of Medicine, Medical Sciences Campus, University of Puerto Rico, San Juan, PR, USA
Correspondence: Natalio J Izquierdo, Email [email protected]
Background: Previous studies have reported on retinitis pigmentosa (RP) in Puerto Rico. Information on the geographic distribution of RP mutations in Puerto Rico may lead to higher rates of diagnosis and co-management. We aimed to evaluate whether there are areas with increased incidence of genes leading to RP in the Island.
Materials and Methods: We conducted a non-concurrent prospective study on the genotype of 241 patients with RP in Puerto Rico. We evaluated their townships to determine whether there are clusters of genes leading to RP. Genetic studies were done using the Invitae inherited retinal diseases panel analyzing 330 genes.
Results: A total of 100 patients were evaluated. Clusters of patients with mutations were found in certain townships. As depicted in the map, a cluster of patients with a mutation in the PDE6B gene was found in San Juan (9), those with the BBS1 gene occurred in San Juan (6) and Bayamón (4), mutations on the USH2A gene were found in Toa Baja (5), mutations in the CRB1 gene appeared in Ciales (4), and mutations in the BBS7 were found in Aibonito (2). Other mutations are scattered throughout the Island.
Conclusion: Clusters of mutations were identified in several townships including San Juan, Bayamón, Toa Baja, Ciales, and Aibonito. Some of these are isolated geographically. Additional mutations were identified but only the most pertinent were reported. Genetic studies are warranted in all patients with RP in Puerto Rico.
Keywords: retinitis pigmentosa, inherited retinal eye diseases, Usher syndrome, Bardet–Biedl syndrome
Introduction
Retinitis pigmentosa (RP) is a heterogeneous group of inherited disorders affecting 1 in 3000–8000 people caused by abnormalities of photoreceptors or retinal pigment epithelium of the retina which leads to progressive visual loss.1 RP can be inherited in an autosomal dominant, autosomal recessive, or X-linked manner.1,2
RP is considered a rod-cone dystrophy because it is characterized by primary degeneration of photoreceptor rods which is followed by secondary degeneration of cones.1 Therefore, patients typically experience night blindness early in the disease course which later progresses to visual loss in diurnal conditions.3 Degeneration of the retina begins in the mid-periphery and advances towards the macula lutea and fovea.1 This explains why night blindness is followed by visual field loss in a concentric pattern.4 RP then progresses to include degeneration of central cone photoreceptors resulting in central vision loss.4 Typically, patients will begin to experience vision loss in their teenage years which progresses to severe vision loss by 40–50 years of age.1 Patients may also experience photophobia usually in more advanced stages of the disease.3
Genetic mutations causative of RP lead to alterations in proteins which are essential for function of the neuroretina and retinal pigment epithelium.4 Altered protein function damages photoreceptor and retinal pigment epithelial cells possibly due to excessive reactive oxygen species.5 This leads to apoptosis of photoreceptors, a reduction in the thickness of the outer nuclear layer in the retina and deposition of retinal pigment in the fundus.1
The diagnostic criteria for RP are multifaceted and findings can vary significantly between individuals. RP is considered to have a classic triad of fundus abnormalities which include bone spicule pigmentation in the periphery and/or mid periphery, attenuation of retinal vessels, and waxy pallor of the optic nerve.4 Electroretinogram findings demonstrate decreased amplitude of a- and b-waves and can be used for diagnosing RP as well as monitoring disease progression.3 Optical coherence tomography (OCT) and fundus autofluorescence (FAF) imaging show loss of outer retinal layers and altered lipofuscin distribution.4
RP is usually limited to the eye but can also occur in a syndromic manner as part of the Usher or Bardet-Biedl syndrome (BBS).1,6 Usher syndrome is the most frequent syndromic form of RP.3 It is characterized by RP and neurosensory deafness. It is classified into three types based on characteristics of hearing loss. Type 1 is associated with profound hearing loss, type 2 with moderate hearing loss, and type 3 with deafness which occurs in the first decade of life and progressively worsens. BBS is less frequent and characterized by RP with associated obesity, intellectual impairment, polydactyly, hypogenitalism, and renal abnormalities.3
Over 80 different genes have been associated with non-syndromic RP and at least 11 genes have been associated with Usher syndrome and BBS.3,4 The incidence of genetic mutation which cause syndromic and non-syndromic RP are different depending on regions and ethnicities.7 Studies have identified clusters of gene mutations leading to RP in Spain, Mexico, Brazil, China, South Africa, France, South Korea, and other countries.2,7–10 Puerto Rico’s geographic isolation leads to inbreeding which could lead to a higher incidence of particular genetic mutations. However, no studies have determined the incidence of specific gene mutations in this population.
We sought to study the distribution of the various gene mutations leading to RP according to the Puerto Rican townships. This led us to evaluate if there are clusters of genes in certain townships in PR. Identification of these clusters can lead to improvements in the diagnosis and treatment of patients.
Materials and Methods
This a non-concurrent prospective study on the genotype of 241 patients with a clinical diagnosis of RP in Puerto Rico as delineated in a previous study.11 Institutional Review Board approval was obtained from the Medical Sciences Campus of the University of Puerto Rico (B1960120). Our study complies with the Declaration of Helsinki.
We obtained saliva samples from patients with an RP diagnosis recruited at an RP symposium on November 8th, 2019. All patients were Puerto Rican and required to sign a consent form. All patients were evaluated by at least one of the authors. Patients under the age of 18 were required to have a legal custodian provide written consent.
Saliva samples were obtained from patients and submitted for DNA analysis. Samples were analyzed using an Inherited Retinal Disorders (IRD) panel from the Invitae corporation. This laboratory does full gene sequencing and deletion/duplication analysis with next-generation sequencing (NGS) technology. The Invitae corporation panel analyzes a total of 330 genes.
A genotyping microarray from the Invitae corporation was used to screen for mutations. DNA was enriched for targeted regions and sequenced with Illumina technology to identify significant variants. Patient demographic data included township, age, and gender. We only included previously described pathogenic variants leading to RP as being associated with RP.
Inclusion criteria were based on genetic results, clinical diagnosis, and demographic data. For patients to meet inclusion criteria their genetic results must be classified as one of the following categories: patients with homozygous pathogenic variants in autosomal recessive (AR) genes; compound heterozygous pathogenic variants in AR genes; autosomal dominant (AD) pathogenic variants; and pathogenic variants in X-linked genes. Patients were also required to have a clinical diagnosis of RP. This diagnosis was based on symptoms of nyctalopia and decreased visual acuity as well as retinal findings, a visual field test, and electroretinography results compatible with RP. Finally, patients were required to provide demographic data which identified their township.
The exclusion criteria were patients whose genetic results did not comply with one of the previously mentioned categories. These were patients who had a single heterozygous pathogenic variant in an AR gene or whose identified variants were of unknown significance. Other exclusion criteria were not being able to provide a saliva sample for genetic testing, presence of congenital nystagmus, and patients which lacked township data.
Frequency counts were calculated to determine the incidence of each mutation by township in Puerto Rico and plotted on a map of the island.
Results
Demographics
A total of 100 patients met inclusion criteria. Of these, there were 51 (51%) male and 49 (49%) female patients. Their ages ranged from 10 to 81 years (average age = 42 years).
Mutational Spectrum
64 cases (64%) carried homozygous pathogenic variants in an AR gene. Twenty-nine (29%) carried compound heterozygous variants in an AR gene. Only seven cases (7%) had AD gene variants; and none had XL gene pathogenic variants.
25 patients (25%) had mutations in the BBS1 gene; 13 (13%) had pathogenic mutations in the PDE6B gene; 12 (12%) had pathogenic mutations in the CNGB1 gene; 11 patients (11%) had pathogenic mutations in the CRB1 gene; and 9 patients (9%) had pathogenic mutations in the USH2A gene. These mutations represent 70% of cases. Refer to Supplementary Table 1 for a comprehensive list of all mutations identified.
Geographic Distribution
As depicted in Figure 1, a cluster of 6 patients (6/25; 24%) with a mutation in the BBS1 gene was identified in the township of San Juan and a cluster of 4 patients (4/25; 16%) was identified in Bayamon. The p.Met390Arg variant was the most common variant in the BBS1 gene in our patient population. All 6 patients in the San Juan cluster had the p.Met390Arg variant. Of these, 3 were compound heterozygotes having both the p.Met390Arg and the p.Glu549* variants. In the Bayamon cluster, 2 patients were homozygous for the p.Met390Arg variant, 1 was homozygous for the p.Glu549* variant and 1 was a compound heterozygote for both the p.Met390Arg and the p.Glu549* variants.
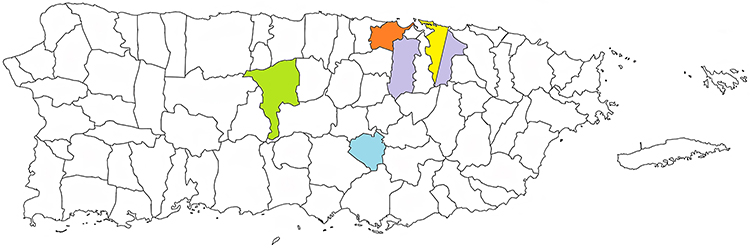 |
Figure 1 Map of the US Commonwealth of Puerto Rico. The map shows the location of the townships with a cluster of mutations in the PDE6B gene (yellow), BBS1 gene (purple), CRB1 gene (green), USH2A gene (Orange), and BBS7 gene (blue). |
A cluster of 9 patients (9/13; 69%) with a mutation in the PDE6B gene was identified in San Juan. All 9 patients had the p.Leu514Trpfs*61 variant with one being compound heterozygote for the p.Leu514Trpfs*61 variant and the p.His620Glnfs*23 variant.
A cluster of 5 patients (5/9; 56%) with a mutation in the USH2A gene was identified in Toa Baja. Of these, one patient was homozygous for the p.Glu767Serfs*21 variant and 4 patients were compound heterozygotes with the p.Glu767Serfs*21 and the p.Cys575Tyr variants.
A cluster of 4 patients (4/11; 36.4%) with a mutation in the CRB1 gene was identified in Ciales. Of these, 3 are homozygous for the p.Ile167_Gly169del variant and one is homozygous for the p.Ile205Aspfs*13 variant.
A cluster of 2 patients (2/4; 50%) with a mutation in the BBS7 gene was identified in Aibonito. Both patients were homozygous for the p.Thr211Ile variant.
Discussion
We conducted a non-concurrent prospective study on the genotype of a cohort of Puerto Rican patients with a clinical diagnosis of RP. We evaluated their townships to determine the presence of clusters of genetic mutations which lead to RP. In our study, we found 5 main clusters due to mutations in the BBS1, PDE6B, CRB1, and USH2A genes. All clusters are from townships on the northern part of the island except for the CRB1 cluster which is in the middle of the island.
Studies in a Spanish cohort showed that mutations in CRB1 contributed to 8% of autosomal recessive cases of RP.12 This value is close to our frequency of 11% and may be due to the Spanish colonization of the island. The presence of a cluster in the township of Ciales suggests this Spanish influence may be contributing to RP cases in that area of the island.
Previous studies have identified that mutations in the CRB1 gene are one of the major contributors to RP in the Mexican population.10 Mutations in CRB1 contributed to 7% of pathogenic mutations associated with inherited retinal dystrophies.10 In our cohort, mutations in CRB1 were associated with 11% of cases. The relatively higher frequencies in our cohort could be explained by consanguinity. The fact that CRB1 mutations are major contributors in both the Mexican and Puerto Rican populations could be explained by Maya population migrations in the Caribbean Sea.
Both the p.Ile205Aspfs*13 and p.Ile167_Gly169del variants identified in the township of Ciales have previously been identified in France.13 Meanwhile, the p.Ile167_Gly169del variant has been identified in Brazilian and Spanish cohorts.14,15 A study based on a small European cohort of patients identified this variant in all their patients indicating further European influence.16
Studies of Spanish cohorts identified that mutations in the BBS1 gene accounted for a large proportion of patients with syndromic RP.12,17 A study also identified the p.Met390Arg as the most frequent variant and reported compound heterozygotes with the same combination of p.Met390Arg and p.Glu549*.17 Likewise, in our cohort the p.Met390Arg variant was present in all but one patient in the San Juan and Bayamon clusters. This is further evidence of the Spanish influence on clusters of RP in the island.
Mutations in the BBS7 gene were associated with 4% of cases. This is a higher incidence than has been identified in other populations.10,12 Studies have also identified BBS7 mutations in Spanish and Mexican cohorts.12 However, the p.Thr211Ile variant has not been previously identified in other populations. This provides an interesting opportunity for further study to trace this variant to other populations.
Mutations in PDE6B accounted for 13% of patients with RP. Studies of cohorts in South Korea have demonstrated that PDE6B mutations accounted for 6.3% of patients with RP and 17% of patients with AR RP.7 Studies in the Spanish population showed that mutations in the PDE6B gene caused fewer than 1% of AR RP cases.12 In our cohort, PDE6B gene mutations account for 12.7% of cases. This is closer to the frequencies found in the South Korean population than in the Spanish population which is contrary to what would be expected. However, the cluster of patients with PDE6B mutations in San Juan could be due to Spanish ancestors with a higher frequency due to consanguinity. The p.His620Glnfs*23 found in one patient in the cluster has been identified in an African cohort. This could be explained by the presence of African ancestry on the island.
Mutations in the USH2A gene were associated with 9% of cases. Mutations in USH2A contributed to 19% of AR RP cases in the Spanish population.12 All patients in the Toa Baja cluster were compound heterozygotes for the p.Glu767Serfs*21 and the p.Cys575Tyr variants. The former has been identified as an ancestral European pathogenic variant and has been identified in the Spanish population.12
Limitations of our study include that demographic data was not available for all patients tested. A larger number of patients could help elucidate other clusters or add confidence to the ones already identified. Moreover, due to convenience sampling our patient population did not include representation from all townships. However, our sample included patients from a large variety of areas. Strengths include many mutations identified, a comprehensive inclusion and exclusion criteria, and identification of rare mutations.
Conclusions
This study is the first report on the geographic distribution of patients with RP in the Puerto Rican population. Clusters of mutations in the BBS1, PDE6B, CRB1, USH2A, and BBS7 genes were identified in certain townships. Mutations in these genes have been identified in Mexican and Spanish populations which suggests there may be an effect from European colonization and Mexican migration in these townships. These results can assist in determining any founder effects on the Island.
Funding
Supported in part by Invitae Corp. (mutational screening tests) and the Fundación de retinitis pigmentosa de PR.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Ali MU, Rahman MSU, Cao J, Yuan PX. Genetic characterization and disease mechanism of retinitis pigmentosa; current scenario. Biotech. 2017;7(4). doi:10.1007/S13205-017-0878-3
2. Bravo-Gil N, González-Del Pozo M, Martín-Sánchez M, et al. Unravelling the genetic basis of simplex Retinitis Pigmentosa cases OPEN. Sci Rep. 2017;7(1). doi:10.1038/srep41937
3. Hamel C. Retinitis pigmentosa. Orphanet J Rare Dis. 2006;1(1):1–12. doi:10.1186/1750-1172-1-40
4. Verbakel SK, van Huet RAC, Boon CJF, et al. Non-syndromic retinitis pigmentosa. Prog Retin Eye Res. 2018;66:157–186. doi:10.1016/j.preteyeres.2018.03.005
5. Scimone C, Donato L, Alibrandi S, et al. N-retinylidene-N-retinylethanolamine adduct induces expression of chronic inflammation cytokines in retinal pigment epithelium cells. Exp Eye Res. 2021;209:108641. doi:10.1016/j.exer.2021.108641
6. Daiger SP, Sullivan LS, Bowne SJ. Genes and mutations causing retinitis pigmentosa. Clin Genet. 2013;84(2):132–141. doi:10.1111/cge.12203
7. Kim YN, Kim YJ, Seol CA, Seo EJ, Lee JY, Yoon YH. Genetic profile and associated characteristics of 150 Korean patients with retinitis pigmentosa. J Ophthalmol. 2021;2021. doi:10.1155/2021/5067271
8. Jimenez Diaz F, Autonoma madrid U, Najera C, Valverde D, Carballo M, Antiñolo G. Retinitis pigmentosa in Spain. Clin Genet. 1995;48:120–122.
9. Roberts L, Ratnapriya R, du Plessis M, Chaitankar V, Ramesar RS, Swaroop A. Molecular diagnosis of inherited retinal diseases in indigenous African populations by whole-exome sequencing. Investig Ophthalmol Vis Sci. 2016;57(14):6374–6381. doi:10.1167/iovs.16-19785
10. Villanueva-Mendoza C, Tuson M, Apam-Garduño D, et al. The genetic landscape of inherited retinal diseases in a Mexican cohort: genes, mutations and phenotypes. Genes. 2021;12(11):1824. doi:10.3390/genes12111824
11. Vargas JG, Izquierdo NJ, Oliver A, et al. Genetic analysis of patients with nonsyndromic and syndromic retinitis pigmentosa in Puerto Rico: a genetic legacy. Ophthalmic Genet. 2022;43(4):454–461. doi:10.1080/13816810.2022.2050764
12. Perea-Romero I, Gordo G, Iancu IF, et al. Genetic landscape of 6089 inherited retinal dystrophies affected cases in Spain and their therapeutic and extended epidemiological implications. Sci Rep. 2021;11(1). doi:10.1038/s41598-021-81093-y
13. Mairot K, Smirnov V, Bocquet B, et al. Crb1‐related retinal dystrophies in a cohort of 50 patients: a reappraisal in the light of specific müller cell and photoreceptor crb1 isoforms. Int J Mol Sci. 2021;22(23):12642. doi:10.3390/ijms222312642
14. Louise Motta F, Vallim Salles M, Antunes Costa K, Filippelli-Silva R, Paulo Martin R, Maria Ferraz Sallum J. The correlation between CRB1 variants and the clinical severity of Brazilian patients with different inherited retinal dystrophy phenotypes. Sci Rep. 2017;7(1):1–9. doi:10.1038/s41598-017-09035-1
15. Corton M, Tatu SD, Avila-Fernandez A, et al. High frequency of CRB1 mutations as cause of early-onset retinal dystrophies in the Spanish population; 2013. Available from: http://www.ojrd.com/content/8/1/20.
16. Khan KN, Robson A, Mahroo OAR, et al. A clinical and molecular characterisation of CRB1-associated maculopathy. Eur J Hum Genet. 2018;26:687–694. doi:10.1038/s41431-017-0082-2
17. Castro-Sánchez S, Álvarez-Satta M, Cortón M, Guillén E, Ayuso C, Valverde D. Exploring genotype-phenotype relationships in Bardet-Biedl syndrome families. J Med Genet. 2015;52(8):503–513. doi:10.1136/jmedgenet
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.