")
Back to Journals » Journal of Inflammation Research » Volume 14
Resveratrol Relieves Gouty Arthritis by Promoting Mitophagy to Inhibit Activation of NLRP3 Inflammasomes
Authors Fan W, Chen S, Wu X, Zhu J, Li J
Received 22 May 2021
Accepted for publication 29 June 2021
Published 24 July 2021 Volume 2021:14 Pages 3523—3536
DOI https://doi.org/10.2147/JIR.S320912
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Ning Quan
Weimin Fan,1,2 Shixian Chen,1 Xianghui Wu,3 Junqing Zhu,1 Juan Li1,2
1Department of Rheumatic & TCM Medical Center, Nanfang Hospital, Southern Medical University, Guangzhou, People’s Republic of China; 2School of Traditional Chinese Medicine, Southern Medical University, Guangzhou, People’s Republic of China; 3Laboratory Animal Research Center, Nanfang Hospital, Southern Medical University, Guangzhou, People’s Republic of China
Correspondence: Juan Li
Department of Rheumatic & TCM Medical Center, Nanfang Hospital, Southern Medical University, No. 1838, Guangzhou North Road, Baiyun District, Guangzhou, People’s Republic of China
Tel + 0086-020-61641675
Fax +86 20 61648308
Email [email protected]
Background: Gouty arthritis (GA) is a common inflammatory disease with pain caused by the deposition of monosodium urate (MSU) crystals into joints and surrounding tissues. Resveratrol (Res), derived from grapes and peanuts and the traditional Chinese medicine (TCM) Reynoutria japonica for GA, acts against oxidation and inflammation. The present study aimed to investigate the therapeutic effect and mechanism of Res on GA.
Methods: Arthritis rat models, MSU-induced peritonitis mouse models, and inflammatory models of mouse bone marrow-derived macrophage (BMDM) were used in this study. Enzyme-linked immunosorbent assay (ELISA), JC-1, histopathological, immunofluorescence, flow cytometry, Western blot methods were applied to observe the effects of resveratrol on NLRP3 inflammasomes and mitophagy.
Results: Res significantly improves the gait score and synovitis of rats with GA and inhibits the peritoneal inflammation induced by MSU. Res inhibits the MSU-induced activation of NLRP3 inflammasomes by reducing the levels of IL-1β, IL-18, and Caspase-1 and the pyroptosis of macrophages. In addition, Res raises the level of mitochondrial membrane potential, inhibits the expression of P62 and Pink1, enhances the expressions of LC3B-II, Parkin, and TOMM20, and promotes mitophagy, while mitophagy inhibitors reverse the inhibitory effect of Res on the activation of NLRP3 inflammasomes.
Conclusion: Res significantly improves GA, and the underlying mechanism might be inhibiting the activation of NLRP3 inflammasomes by triggering the Pink1/Parkin pathway to promote mitophagy.
Keywords: gouty arthritis, inflammation, macrophages, Pink1/Parkin
Introduction
Gouty arthritis (GA) constitutes a group of clinical syndromes caused by monosodium urate (MSU) crystals1 and characterized by the swelling and pain of joints and recurrence.2−3 In recent years, the number of GA patients has increasing greatly in China and worldwide.4 It has been increasing every year in mainland China5 and is affecting at least 1% of the population in developed areas in Western countries.6 It might also lead to a series of complications, including hypertension, type 2 diabetes, and kidney disease.7,8 Therefore, studying GA is rather significant. Previous studies have proved that NLRP3 (NOD-, LRR-, and pyrano-domain-containing protein 3) inflammasomes are closely associated with GA.9 NLRP3 inflammasomes are protein complexes composed of NLRP3, apoptosis-associated speck-like protein (ASC), and caspase-1.10,11 After the NLRP3 effector domain, PYD recruits ASC and caspase-1 to assemble NLRP3 inflammasomes, caspase-1 precursors self-cleave into the mature form, cleave pro-Interleukin-1β (pro-IL-1β) into IL-1β, and are secreted outside the cells to effectuate various immune responses.12 Currently, Dapansutrile, a selective inhibitor of NLRP3, has entered Phase 2A clinical trials.13 MSU activates NLRP3 inflammasomes through multiple pathways and promotes the release of IL-1β. This process mainly includes lysosome destruction,14 potassium ion outflow,15 mitochondrial damage, and reactive oxygen species.16,17 Currently, the factors promoting the activation of NLRP3 inflammasomes by mitochondrial dysfunction are under intensive focus.18
Resveratrol (Res) is a stilbene compound widely existing in natural plants, such as grapes, peanuts, mulberries, and Reynoutria japonica. Pharmacologically, it acts against oxidation,19 aging,20 inflammation,21 and tumors.22 Previous studies have shown that Res reduces the content of uric acids in mice with hyperuricemia,23 improves the joint condition of GA mice, prevents the recurrence of GA,24 and also improves the inflammatory response in GA patients by upregulating SIRT1 to promote autophagy.25
Thus, arthritis rat model, MSU-induced peritonitis mouse model, and inflammatory model of mouse bone marrow-derived macrophage (BMDM) were used in this study to investigate the therapeutic effect and mechanism of Res on GA.
Materials and Methods
Reagents and Antibodies
MSU crystal, lipopolysaccharide (LPS): Sigma–Aldrich, USA; Res: Chengdu Herbpurify, China; colchicine tablets: Xishuangbanna Pharmaceutical, China; M-CSF: PEPROTECH USA; 3-methyladenine (3-MA): MedChemExpress, USA; thioglycolate medium (the Brewer): Qingdao Rishui Biotechnology, China; CCK-8 reagent: Glpbio, USA; IL-1β enzyme-linked immunosorbent assay (ELISA) kit, Interleukin-18 (IL-18) ELISA kit: Hang-zhou Multi Sciences, China; NLRP3 antibody, ASC antibody, IL-1β antibody, LC3 anti-body, P62 antibody, Pink-1 antibody, TOMM20 antibody: Abcam, UK; Caspase-1 anti-body, rabbit Parkin antibody, rat Parkin antibody: Wuhan Proteintech, China; Cleaved-Caspase-1 antibody, Cleaved-IL-1β antibody: Cell Signaling Technology, USA; CD11b-PE, Ly6G-FITC, F4/80-APC: BioLegend, USA; Mitochondrial membrane potential (JC-1) assay kit, Hoechst/Propidium Iodide (PI) assay kit: Beijing Beyotime Biotech-nology, China; Acridine Orange (AO)/Ethidium Bromide (EB) dual fluorescence staining kit: Beijing Leagene Biotechnology, China.
Building of MSU-Induced Arthritis Rat Model
After 1 week of adaptive feeding, 36 specific pathogen-free (SPF) male Sprague–Dawley (SD) rats, 7-weeks-old, were randomly assigned to the blank control (Con) group, model (Mod) group, colchicine (Col) group, low-dose Res (Res L) group, medium-dose Res (Res M) group, and high-dose Res (Res H) group (n = 6/group). The Con and Mod groups were given an equivalent volume of normal saline, the Col group was given 0.3 mg/kg colchicine-water solution, and the Res L, Res M, and Res H groups were administered 10 mg/kg, 25 mg/kg, and 50 mg/kg Res-water solution,26,27 respectively intragastrically for 7 consecutive days. After administration on day 5, all groups, except the Con group, were given an articular luminal injection of 0.1 mL containing 25 mg/mL MSU solution to the right knee to establish the model.28 Subsequently, the gait scores of rats were evaluated at 12 and 24 h, according to Coderre et al.29 After administration on day 7, synovial tissues from the knee joints of the rats were exfoliated for pathological sample preparation.
Building of MSU-Induced Peritonitis Mouse Model
SPF male C57BL/6J mice, 6–8-weeks-old, were assigned to the Con, Mod, and Res groups (n = 3/group). The Mod and Res groups were injected 2 mL of 40.5 mg/mL Brewer intraperitoneally, and the Res group was intraperitoneally injected with 15 mg/kg Res on day 2 for pretreatment.30 On day 3, the Con group was injected 0.5 mL of phosphate-buffered saline (PBS), and the other groups were injected 0.5 mL of 6 mg/mL MSU, respectively, to induce peritonitis model.31 After 6 h, the mice were given CO2 for euthanasia and injected with 9 mL of sterile and precooled PBS intraperitoneally. The lavage fluids were collected, and the supernatant was clarified by centrifugation at 800 rpm for 5 min; IL-1β and IL-18 expression was estimated by ELISA, and cells were collected for flow cytometry.
Cell Culture and Treatment
The bone marrow of male C57BL/6J mice was collected, mixed with 20 ng/mL M-CSF, and cultured in a 5% CO2 incubator for BMDM induction. The cell media were replaced with new M-CSF-containing DMEM on days 3 and 5,32 and cell samples were treated with 500 ng/mL LPS for 3 h on day 7; then, the cells were washed twice with PBS, and was replaced with media containing or not containing FBS. Subsequently, the cell samples were treated with 30, 15, and 7.5 μM Res for 6 h (or with 5 mM 3-MA for 6 h together) and 300 μg/mL MSU for 6 h.33
CCK-8 Cell Survival Test
BMDM was added to a 96-well plate and induced as described above. On days 7, 30, 15, and 7.5 μM Res was added for 6 h of treatment to set blank wells, control wells, and test wells, with three wells in each group. Subsequently, 10 μL CCK-8 reagent was added, and the reaction incubated at 37°C for 4 hours. The absorbance was measured at 450 nm on a microplate reader. The cell survival was calculated by the formula as follows: Cell survival = [(test well - blank well)/(control well - blank well)] × 100%.
IL-1β and IL-18 Detection by ELISA
The cell supernatant or peritoneal lavage fluid of each group was collected and added to the 96-well ELISA plate, according to the instructions of the kit. The absorbance was measured at 450 nm on a microplate reader to construct the standard curve, and the ELISA value of each group was estimated.
Detection of Cell Membrane Potential by JC-1
BMDM was added to a 6-well plate, and cell induction and drug intervention were performed as above. The JC-1 fluorescent probe was assembled according to the instructions of the kit and images acquired by a fluorescence microscope. In the event of high mitochondrial membrane potential, JC-1 aggregates in the matrix of mitochondria and forms polymers, showing red fluorescence. In the event of low mitochondrial membrane potential, JC-1 cannot aggregate in the matrix of mitochondria, as it is a monomer; consequently, green fluorescence is observed. The change in the mitochondrial membrane potential was detected according to the change in the fluorescence color.
Immunofluorescence Detection
Slides with crawled cells were collected, fixed with paraformaldehyde, and permeabilized with 0.1% Triton X-100 at room temperature. After Parkin and LC3 anti-bodies were incubated on the slides, secondary antibodies, goat anti-rabbit IgG H&L (Alexa Fluor 568), and goat anti-mouse IgG H&L (Alexa Fluor 488) were added. Subsequently, the slides were sealed, and images were captured under a fluorescence microscope. Hoechst/PI and AO/EB are completed according to the kit instructions.
Flow Cytometry
Peritoneal cells were collected. CD11b-PE, F4/80-APC, and Ly6G-FITC antibodies were added to the cells and incubated for 30 min at 4°C in the dark. Then, the cells were rinsed, fixed with paraformaldehyde, and detected by flow cytometry.
Histopathological Analysis
Hematoxylin-eosin (HE) staining was used to detect the degree of inflammatory infiltration in the synovial tissues of rats. The synovial tissues of the knee joint of rats were excised, fixed, embedded, sectioned, stained, and observed under a light micro-scope at 400× to assess the histopathological status, and the synovitis score was performed.34
Protein Detection by Western Blot
Adherent cells were scraped and lysates prepared by ultrasonication, and the supernatant is collected by centrifugation at 1200 rpm for 20 min at 4°C. The cell super-natant was obtained by centrifugation at 4000 rpm for 40 min at 4°C, and the dry protein pellet was collected in 1mL acetone. The protein was quantitated by the bicinchoninic acid (BCA) method. An equivalent of proteins was loaded onto 10% SDS-PAGE gels and transferred to PVDF membranes. Then, the membranes were sealed, probed with NLRP3, ASC, Caspase-1, IL-1β, LC3, P62, Pink1, Parkin, Cleaved-Caspase-1 and Cleaved-IL-1β antibodies at 4°C overnight, followed by incubation with goat anti-rabbit IgG-HRP secondary antibody for 2 h. Finally, exposure was performed to generate images.
Statistical Analysis
All data were collected after three or more independent and repeated tests. SPSS22.0 software was used for statistical analysis. The measurement data were expressed as mean ± standard deviation. Normal distribution data were compared using one-way analysis of variance (ANOVA) among multiple groups, and LSD was used for comparison between the two groups, and for the measurement data not conforming to the normal distribution, the rank-sum test was used for comparison among multiple groups. P < 0.05 indicated statistical significance.
Results
Res Alleviated GA in Rats
MSU-induced arthritis rat model was replicated to observe the effects of Res on the gait score and synovial pathology of the joints of rats (Figure 1A–C). At 12 and 24 h after MSU induction in rats, both Res and Col improved the gait score of rats (Figure 1A), while Res reduced the gait score and improved the joint functions of rats; however, no significant difference was detected between Res and Col. Moreover, after pathological section and HE staining of the synovium of the joint, the Mod group showed pathological changes in arthritis, with a multilayered arrangement of the proliferated synovial epithelia, a large number of inflammatory cells infiltrating between tissues, and fibroblast proliferation accompanied with angiogenesis. Nonetheless, after Res and Col treatment, only mild inflammatory infiltration and fibroblast proliferation were observed (Figure 1B and C).
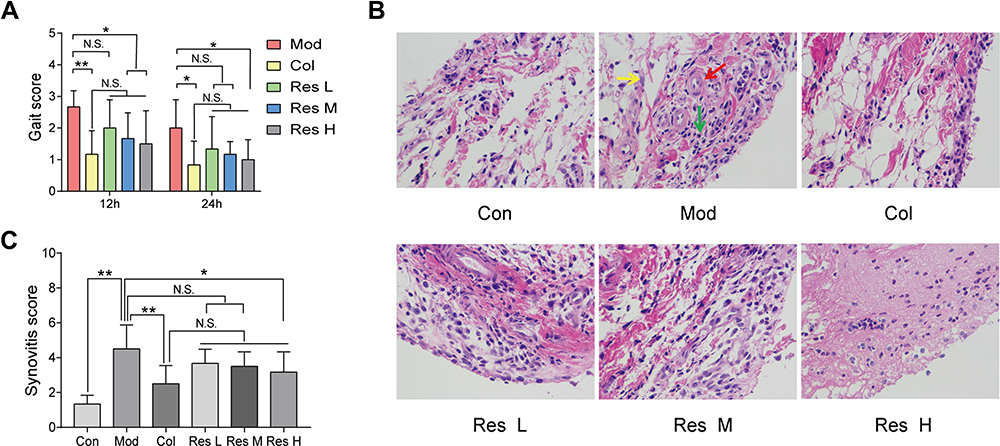 |
Figure 1 Res improved GA in rats. Intragastric administration of 10, 25, or 50 mg/kg Res-water solution or 0.3 mg/kg Col-water solution for 7 days; articular luminal injection of 0.1 mL of 25 mg/mL MSU solution on day 5; subsequently, gait scoring was performed at 12 and 24 h (A). Articular luminal synovial tissues were exfoliated on day 7 to observe the pathological status (B) 400× magnification (yellow arrow: fibroblast proliferation; red arrow: angiogenesis; green arrow: inflammatory cell infiltration), and synovial histopathology is measured by semi-quantitative score (C). Data are expressed as mean ± standard deviation. N.S. indicates not significant, * indicates P < 0.05, and ** indicates P < 0.01. n = 6/group. |
Res Alleviated MSU-Induced Peritonitis in Mice
MSU-induced peritonitis mice model was used for further investigation of the anti-inflammatory activity of Res. ELISA was used to detect IL-1β and IL-18 in peritoneal lavage fluids (Figure 2A and B). After 6 h of MSU treatment, the secretion of IL-1β and IL-18 in the supernatant of the lavage fluid increased significantly but decreased after Res pretreatment. In addition, cells were collected peritoneal lavage fluids by centrifugation and assessed by flow cytometry. After MSU induction, neutrophils increased, while the proportion of macrophages decreased significantly. After Res treatment, neutrophils decreased (Figure 2C and D), and the proportion of macrophages increased significantly (Figure 2E and F).
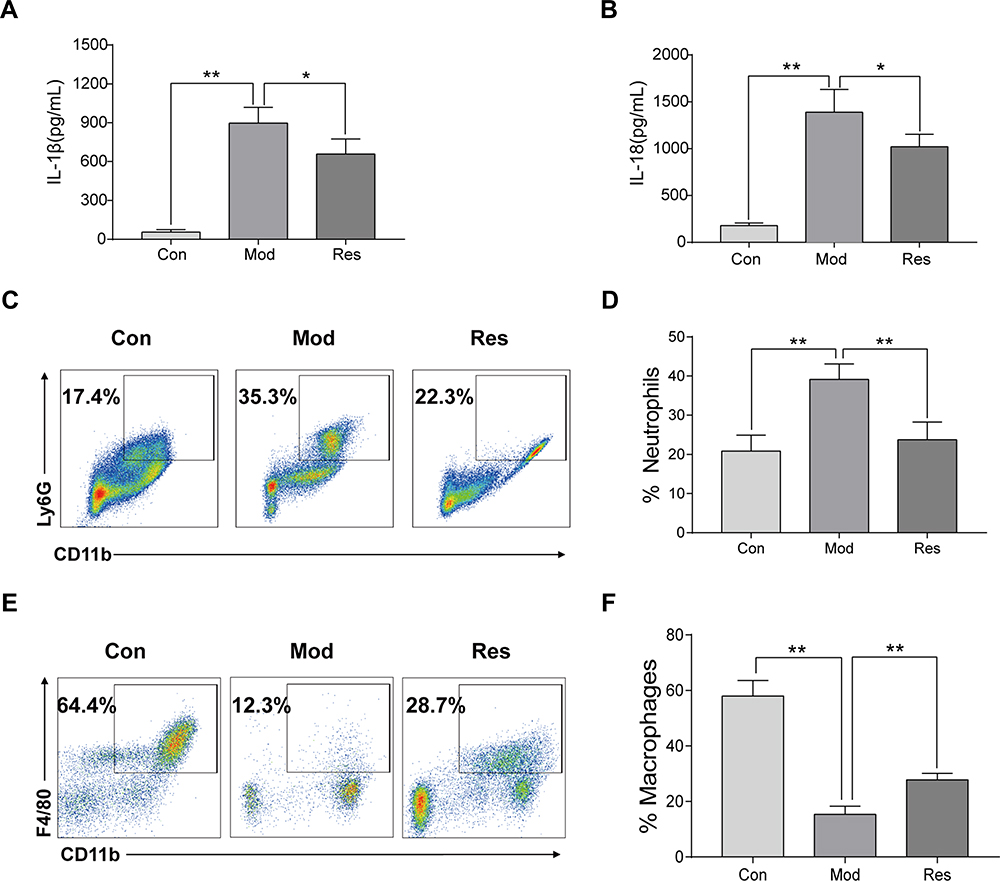 |
Figure 2 Res improved MSU-induced peritonitis in mice. Intraperitoneal injection of 2 mL of 40.5 mg/mL Brewer, intraperitoneal injection of 15 mg/kg Res on day 2 for pretreatment, injected 0.5 mL of 6 mg/mL MSU on day 3 to induce peritonitis model. Lavage fluid was collected at 6 h after induction, and ELISA was used to detect the expression of IL-1β (A) and IL-18 (B), while flow cytometry was used to detect the proportion of neutrophils (C) and macrophages (E). Statistical analysis of neutrophils (D) and macrophages (F). Data are expressed as mean ± standard deviation. * indicates P < 0.05, and ** indicates P < 0.01. n = 3/group. |
Res Inhibited Activation of NLRP3 Inflammasomes in BMDM Inflammatory Cells
BMDM was induced by LPS/MSU (L+M) to observe the inhibitory effect of Res on the activation of NLRP3 inflammasomes. To avoid the cytotoxicity caused by Res, the CCK-8 method was used to measure the BMDM survival rate after the intervention of Res at various different concentrations; the results suggested that 0–30 μM is the safe concentration of Res for cells (Figure 3A). To avoid the possible effect of Res on the LPS-induced signaling pathway, Res treatment was performed after LPS pretreatment. The production of NLRP3 inflammasomes and secretion of IL-1β in BMDM were activated by LPS induction and MSU. As detected by ELISA, after 3 h of LPS induction and 6 h of MSU activation, the secretion of IL-1β in BMDM was significantly increased but decreased after Res pretreatment in a dose-dependent manner (Figure 3B). As expected, similar results were observed for IL-18 (Figure 3C). Western Blot was employed to detect the expression of NLRP3, ASC, Pro-Caspase-1, and Pro-IL-1β in cell lysates and Cleaved-Caspase-1 and Cleaved-IL-1β in the cell supernatant (Figure 3D–J). Res inhibited the activated or Cleaved Caspase-1 and mature Cleaved IL-1β in the supernatant and reduced their expression and secretion (Figure 3D–F). However, Res did not affect the assembly or synthesis of NLRP3 and ASC (Figure 3D, G, and H) and did not inhibit the Pro- form of Caspase-1 or IL-1β (Figure 3D, I, and J).
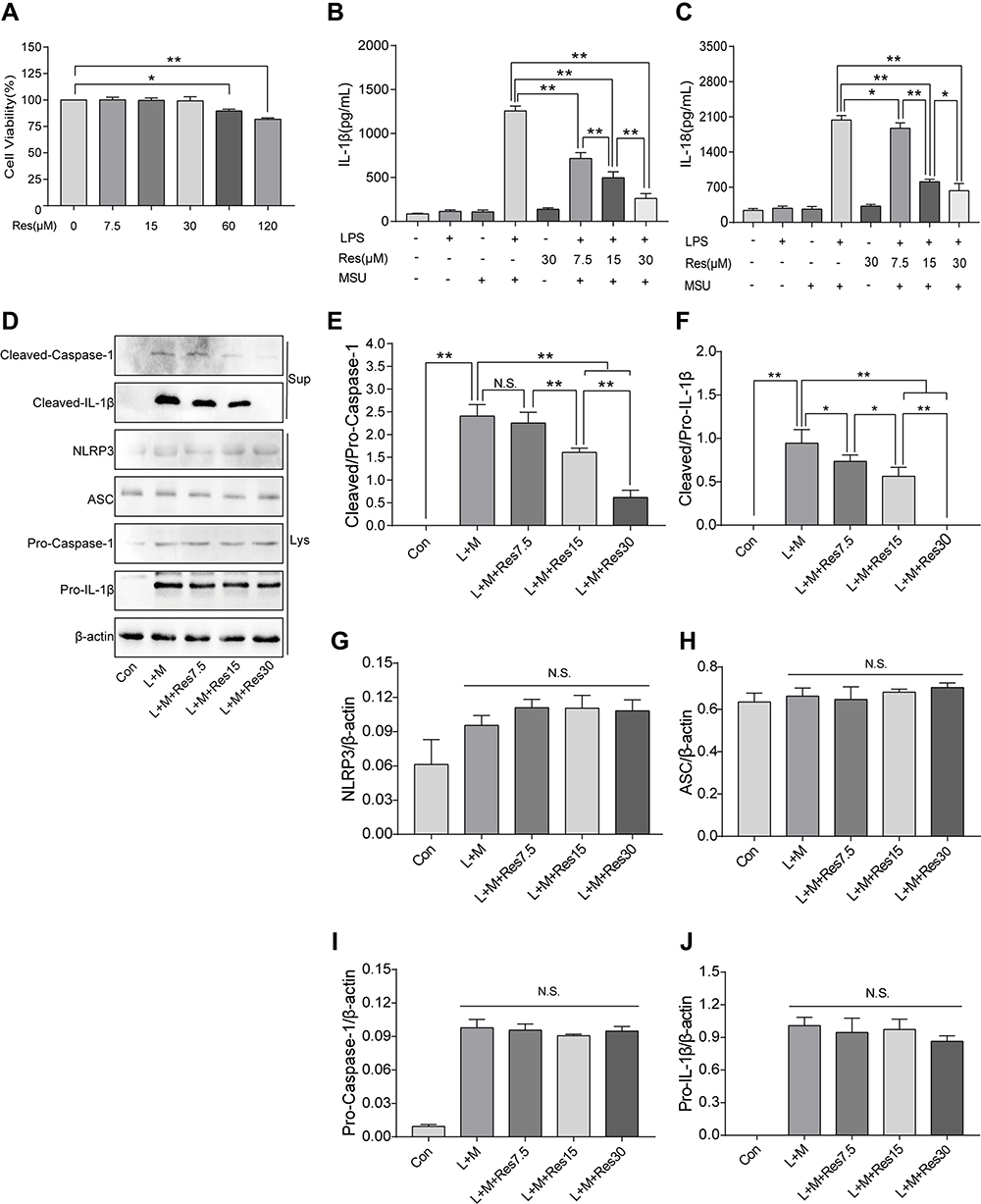 |
Figure 3 Res inhibited the activation of NLRP3 inflammasomes in BMDM inflammatory cells. BMDM treated with 500 ng/mL LPS for 3 h or 7.5, 15, and 30 μM Res for 6 h and 300 μg/mL MSU for 6 h. (A) detection of the effect of Res on BMDM survival rate by CCK-8; detection of IL-1β (B) and IL-18 (C) in cell supernatant by ELISA; detection of protein expressions in the supernatant (Sup) and cell lysates (Lys) by Western blot (D). Statistical analysis of Cleaved Caspase-1 (E) and Cleaved IL-1β (F) in cell supernatant and NLRP3 (G), ASC (H), Pro-Caspase-1 (I), and Pro-IL-1β (J) in cell lysates. Data are expressed as mean ± standard deviation. N.S. indicates not significant, * indicates P < 0.05, and ** indicates P < 0.01. |
The effects of Res on pyroptosis of BMDM inflammatory model cells were also observed under a light microscope, and the results showed that the cells swelled, ruptured, and had a degenerated membrane after LPS/MSU treatment. After Res treatment, the pathological status of the cells improved significantly (Figure 4A). Subsequently, immunofluorescence was used to observe the effect of Res on pyroptosis. After Hoechst/propidium iodide (PI) staining, LPS-activated, and MSU-induced cells showed a strong red and blue fluorescence, which was weakened after Res treatment (Figure 4B). Also, AO/EB staining showed an enhanced red fluorescence and weakened green fluorescence, which improved after Res treatment (Figure 4C). These findings suggested that Res inhibits pyroptosis.
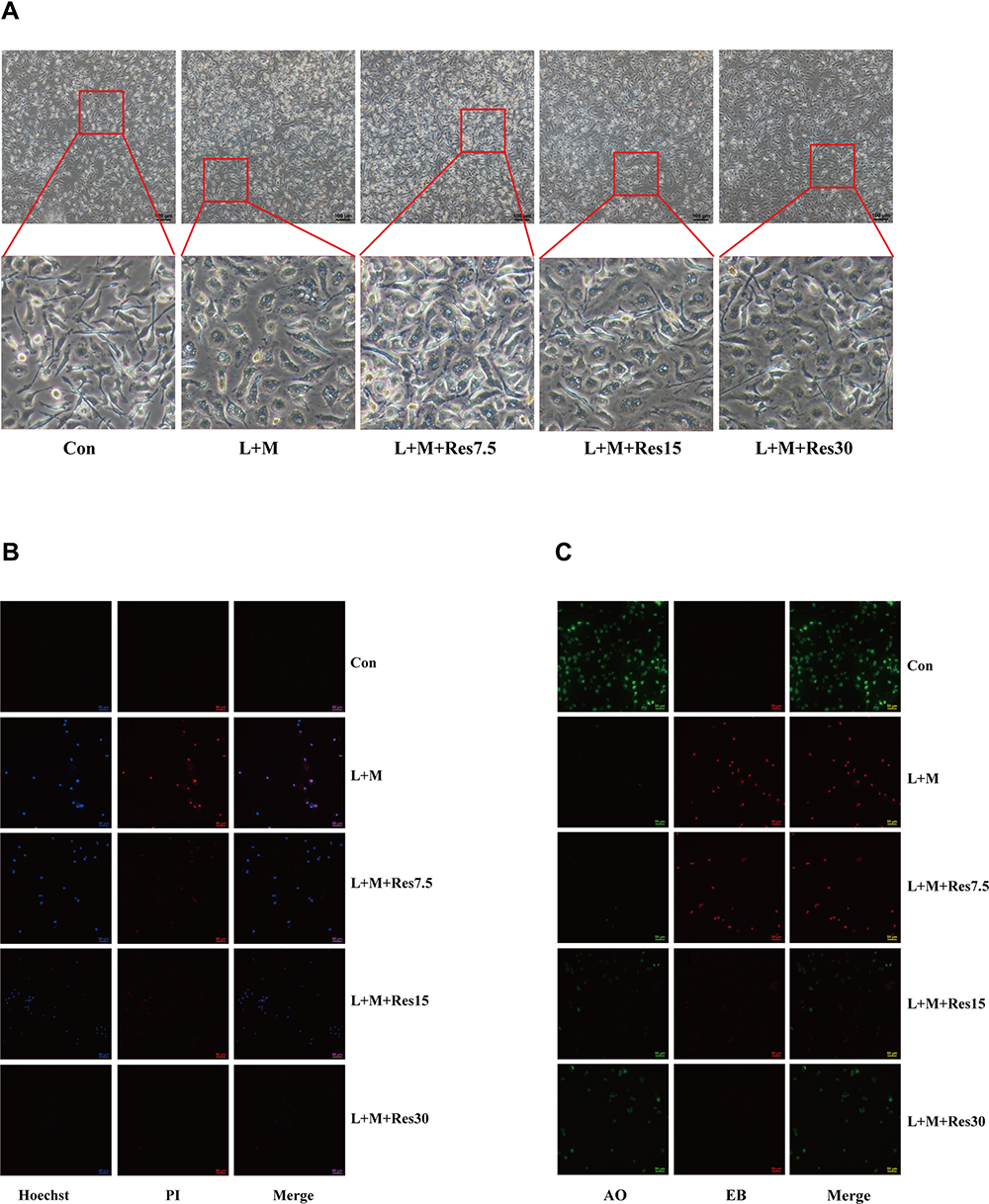 |
Figure 4 Effects of Res on pyroptosis of BMDM. Effects of Res on cell status (at 100×, 100 μM per scale) (A). Detection of py-roptosis by Hoechst/PI staining (dead cells show strong red fluorescence and strong blue fluorescence) (B) and AO/EB staining (dead cells show strong red fluorescence and weak green fluorescence) (C) (at 200×, 50 μM per scale). |
Res Promoted Mitophagy
JC-1 was used to observe the effects of Res on the mitochondrial membrane potential of BMDM (Figure 5A). In normal cells, JC-1 aggregated in the matrix of mitochondria and formed polymers, producing red fluorescence, which suggested a normal mitochondrial membrane potential. In cells induced by LPS/MSU, the mitochondrial membrane potential was low, and JC-1 could not aggregate in the matrix of mitochondria but existed in the form of a monomer, producing green fluorescence, thereby suggesting a reduced membrane potential and damaged mitochondria. After Res intervention, the fluorescence gradually changed the color to red, suggesting that Res raises the mitochondrial membrane potential.
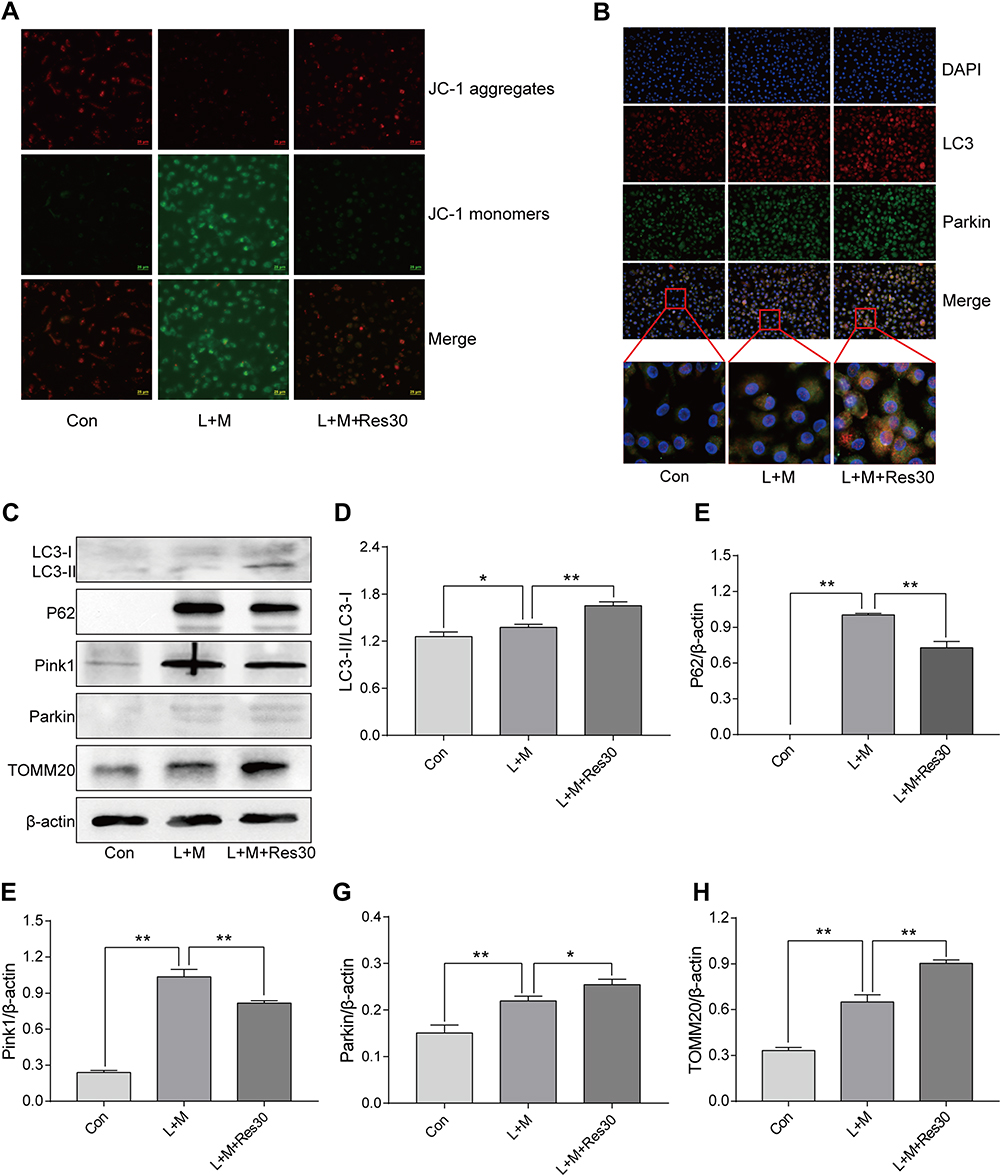 |
Figure 5 Res promotes mitophagy. Detection of changes in the cell membrane potential by JC-1 (JC-1 show red fluorescence in normal cells and green fluorescence when the mitochondrial membrane potential declines (400×, 25 μM per scale)) (A); LC3 and Parkin co-localization by immunofluorescence, showing fluorescence aggregates and mitophagy after Res treatment (400×, 20 μM per scale) (B); Detection of protein expressions in cells by Western Blot (C). Statistical analysis of LC3 (D), P62 (E), Pink1 (F), Parkin (G), and TOMM20 (H). Data are expressed as mean ± standard deviation. N.S. indicates not significant, * indicates P < 0.05, and ** indicates P < 0.01. |
In order to elucidate the mechanism underlying mitophagy and the interventional effect of Res in BMDM inflammation model, immunofluorescence for cell staining was used to observe LC3 and Parkin co-localization (Figure 5B). Only a few Parkin spots were detected in normal cells with no aggregation and almost no damage to the mitochondria and mitophagy, while in LPS/MSU-treated cells, only a few aggregations of Parkin spots were observed. After Res treatment, the number of Parkin spots increased significantly, with many aggregates, suggesting that Res promotes mitophagy in damaged BMDM.
Western Blot was used to detect the level of LC3, P62, Pink1, Parkin, and TOMM20 proteins in BMDM inflammatory model. In LPS/MSU-induced BMDM, a small amount of LC3-I converted into LC3-II, and the ratio of LC3B-II to LC3B-I increase gradually, which increased significantly after Res intervention, along with activated autophagy flow (Figure 5C and D). P62, a selective autophagy receptor, links LC3 and the ubiquitylation substrate for degradation. In LPS/MSU-induced cells, the content of P62 increased, suggesting that the autophagy flow was inhibited partially, while in Res-treated cells, the same decreased, suggesting significantly activated autophagy flow (Figure 5C and E). Then, the expression of Pink1 and Parkin was detected. In normal cells, Pink1 was lowly expressed, while in cells with damaged mitochondria, the pathway of Pink1 was blocked in the inner membrane of mitochondria, causing aggregation of Pink1 in the outer membrane of the mitochondria and recruiting Parkin to the damaged mitochondria. After Res pretreatment, a decreased number of damaged mitochondria caused a decline in the expression of Pink1. Moreover, Res promoted Pink1 recruitment and Parkin activation and activated Parkin clarified the damaged mitochondria and completed the mitophagy process (Figure 5C, F, and G). TOMM20 is a protein in the outer membrane of mitochondria that reflects the changes in the mitochondrial content. In LPS/MSU-induced cells, the mitochondria in the cells were damaged significantly, and the TOMM20 expression increased; both phenomena were improved after Res pretreatment that promoted mitophagy (Figure 5C and H), suggesting that Res could promote mitophagy through the Pink1/Parkin pathway.
Res Inhibited the Activation of NLRP3 Inflammasomes by Promoting Mitophagy
In order to determine the role of mitophagy in NLRP3 inflammasomes, cells were treated with mitophagy inhibitor 3-MA for 6 h after LPS induction, and ELISA was used to detect the changes in IL-1β and IL-18 levels in cell supernatant (Figure 6A and B). After LPS and MSU induction, the secretion of IL-1β and IL-18 increased significantly but decreased after Res intervention. However, after 3-MA pretreatment, the secretions increased gradually, suggesting that 3-MA partially blocked the inhibition of activation of NLRP3 inflammasomes by Res.
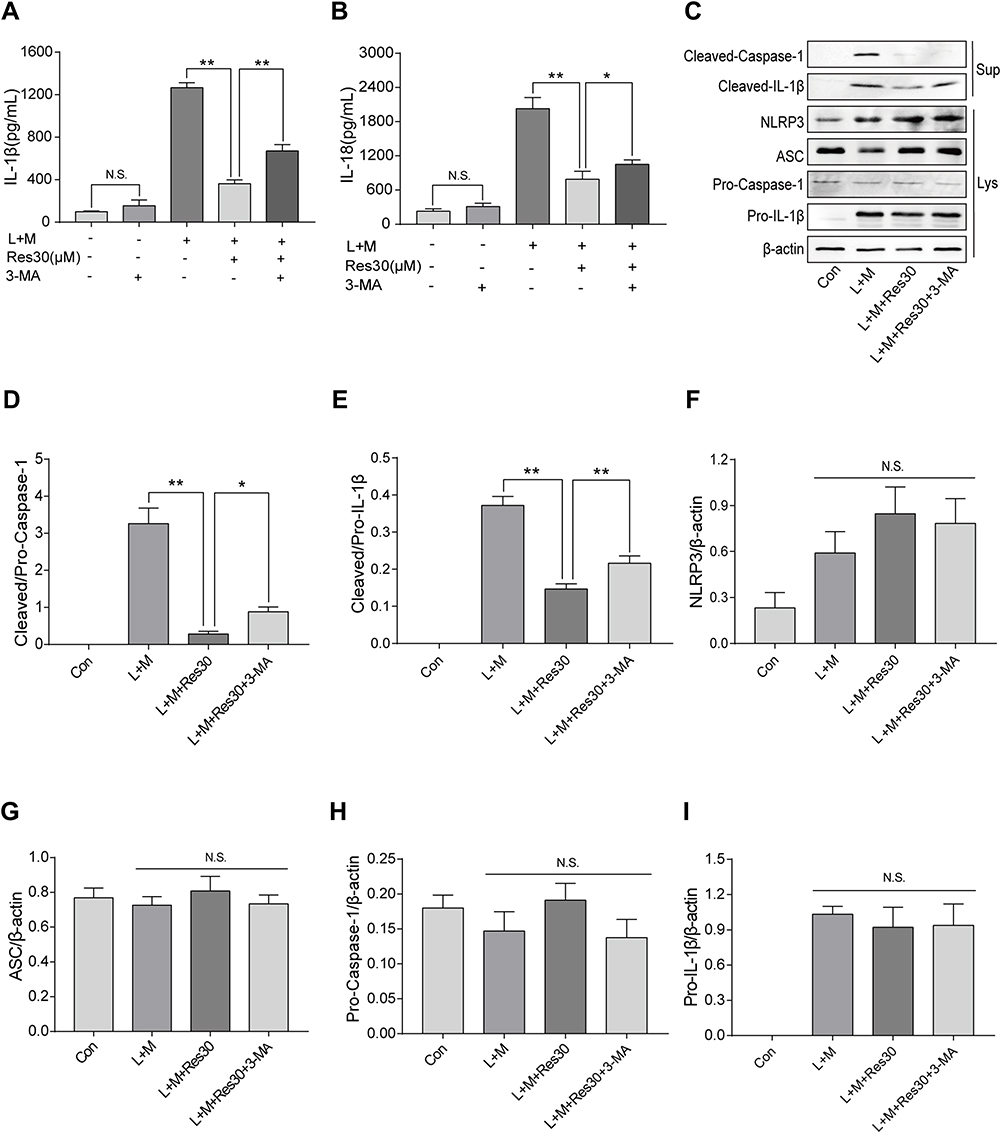 |
Figure 6 Res inhibited the activation of NLRP3 inflammasomes by promoting mitophagy. BMDM treated with 500 ng/mL LPS for 3 h or 30 μM Res for 6 h, or together with 5 mM 3-MA for 6 h and 300 μg/mL MSU for 6 h. The expression of IL-1β (A) and IL-18 (B) in the supernatant by ELISA; detection of protein expressions in the supernatant (Sup) and cell lysate (Lys) by Western blot (C). Statistical analysis of Cleaved Caspase-1 (D) and Cleaved IL-1β (E) in cell superna-tant and NLRP3 (F), ASC (G), Pro-Caspase-1 (H), and Pro-IL-1β (I) in cell lysates. Data are expressed as mean ± standard deviation. N.S. indicates not significant, * indicates P < 0.05, and ** indicates P < 0.01. |
Western blot was utilized to detect the expression of NLRP3, ASC, Pro-Caspase-1, and Pro-IL-1β in cell lysates and Cleaved Caspase-1 and Cleaved IL-1β in the cell supernatant (Figure 6C). After LPS was used to stimulate the first signal and MSU was used to induce the second signal, the expression of Cleaved Caspase-1 and Cleaved IL-1β in the cell supernatant increased but decreased significantly after Res treatment. After 3-MA treatment, the inflammatory effect of Res on the activation or secretion of NLRP3 inflammasomes and IL-1β was partially reversed (Figure 6D and E). Also, no significant changes were detected in NLRP3, ASC, Pro-Caspase-1, or Pro-IL-1β in cell lysates between different groups (Figure 6F–I), suggesting that Res, at least, partially inhibits the activation of NLRP3 inflammasomes by promoting mitophagy.
Discussion
GA is an inflammatory disease with pain caused by the deposition of MSU crystals into joints and surrounding tissues.35 Activated NLRP3 inflammasomes lead to a series of inflammatory diseases, including GA.36 MSU increases the expression of proteins in NLRP3 inflammasomes,37,38 activates NLRP3 inflammasomes, induces macrophages to release IL-1β, and promotes inflammatory response.36 In GA induced by MSU, IL-1β serves as an effector cytokine and the main mediator.39 IL-18 is a member of the interleukin-1 cytokine superfamily and is recognized as an important regulator of both innate and acquired immunity. It is produced in an inactive 24 kDa precursor form, which is cleaved by IL-1b converting enzyme (caspase-1) and proteinase 3 to generate a biologically active 18 kDa mature form.40 The activation of NLRP3 inflammasomes and the activation and release of IL-1β is stimulated by two signals:41 the pathogen-associated molecular pattern (PAMP), such as LPS, viral DNA, and bacterial RNA, which induces the expression of NLRP3 and Pro-IL-1β, and the damage-associated molecular pattern (DAMP), such as adenosine triphosphate (ATP), nigericin, unsaturated fatty acids, and MSU crystals, which completes the assembly of NLRP3 inflammasomes and cleaves Pro-IL-1β into mature IL-1β. MSU crystals alone cannot induce the production of IL-1β but work together with LPS to induce the production of mature IL-1β.42 Therefore, LPS-induced and MSU-activated BMDM were used as the model for in vitro cellular inflammation. These findings showed that LPS-induced and MSU-activated BMDM in the supernatant, the expression of IL-1β and IL-18 increased significantly. Previous studies have proved that Res improves LPS-induced acute lung injury by inhibiting NLRP3 inflammasomes43 and alleviates cerebral ischemia/reperfusion injury.44 On the other hand, the current study showed that Res improves the gait score and pathological status of the synovium in rats, Its effect is comparable with colchicine, lowers the levels of IL-1β and IL-18 in cell supernatant and lavage fluids, reduces the neutrophils chemotaxis, improves the damage of cells, and alleviates pyroptosis of macrophages, and the preventive effect is more prominent than the therapeutic effect, suggesting that Res inhibits the activation of NLRP3 inflammasomes and the secretion of IL-1β in LPS/MSU-induced BMDM.
The molecular mechanisms underlying NLRP3 inflammasome activation mainly include potassium ion outflow, calcium ion loss, lysosome destruction, and mitochondrial dysfunction. The damaged mitochondria can activate NLRP3 inflammasomes45 to damage the cells and cause pathological changes. The JC-1 method was used in this study to detect the mitochondrial membrane potential. LPS/MSU-induced BMDM had a low mitochondrial membrane potential, which increased markedly after Res intervention, suggesting that Res improves the impaired mitochondrial dysfunction.
Mitophagy, as selective autophagy, is realized through the macroautophagy mechanism. In the process of mitophagy, all damaged mitochondria are wrapped in the double-membrane structure, form autophagosomes, and fuse with lysosomes for degradation.46 Mitophagy inhibits the activation of NLRP3 inflammasomes by clearing damaged mitochondria, regulating the quality of mitochondria, and maintaining mitochondrial homeostasis.47–49 As a natural antioxidant, Res activates mitophagy and inhibits NLRP3 inflammasomes as an effective anti-inflammation strategy.50 In the present study, immunofluorescence and Western blot showed that Res improves the level of the autophagy marker LC3 and reduces the content of P62 to further activate mitophagy. Pink1/Parkin is the main signaling pathway in the studies on mitophagy.51 When the mitochondrial membrane potential decreases, Pink1 is translocated out of the mitochondria through the membrane, and Parkin is recruited from the cytoplasm to depolarized mitochondria activating mitophagy.52,53 Furthermore, immunofluorescence evaluated the co-localization of Parkin and LC3, and Western blot confirmed that Res promotes mitophagy through the Pink1/Parkin pathway. To further investigate whether Res inhibits the activation of NLRP3 inflammasomes through mitophagy, a rescue experiment was carried out with the mitophagy inhibitor 3-MA, indicating that 3-MA could partially reverse the inhibitory effect of Res on the activation of NLRP3 inflammasomes. Together, these results showed that Res alleviated GA by promoting mitophagy to inhibit the activation of NLRP3 inflammasomes.
In clinical practice, Colchicine, and non-steroid anti-inflammatory drugs are the common drugs for GA but the long-term usage causes side effects, including liver or kidney damage and gastrointestinal reactions.54 In terms of drug development, traditional Chinese medicine (TCM) is a major resource for the treatment of inflammatory diseases.55 The development of anti-GA drugs with adequate tolerance and safety from TCM is gaining increasing attention.56 Res exists abundantly in grapes, mulberries, peanuts, and Reynoutria japonica and is a drug with great potential against GA.
Conclusion
In conclusion, the present study proved that Res has a significant preventive and therapeutic effect on arthritis rat model, MSU-induced peritonitis mouse model, and inflammatory model of BMDM. The putative mechanism might be promoting mitophagy through the Pink1/Parkin pathway to inhibit the activation of NLRP3 inflammasomes and improve GA (Figure 7).
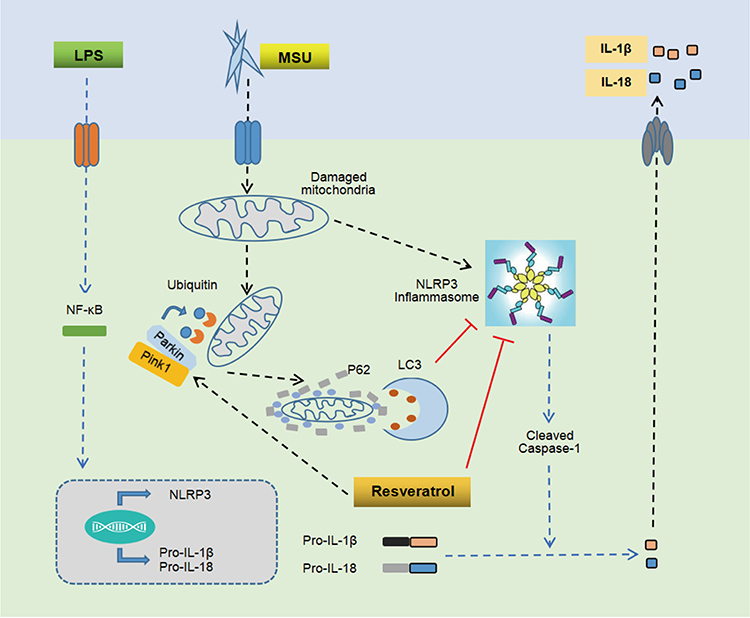 |
Figure 7 Mechanism of Res-inhibited activation of NLRP3 inflammasomes via mitophagy. MSU induces joint inflammation by causing mitochondrial damage, activating NLRP3 inflammasomes, and promoting the secretion of IL-1β and IL-18. Res alleviates GA by promoting mitophagy to clear damaged mitochondria and inhibit the activation of NLRP3 inflammasomes. |
Ethical Statement
The study was approved by the Animal Ethics Committee of Nanfang Hospital, Southern Medical University (NFYY-2019-106, 11/10/2019). All the applied procedures followed the Chinese guidelines for the welfare of the laboratory animals (GB/T 35823-2018).
Acknowledgments
This study was supported by the General projects of China Postdoctoral Science Foundation (2019M652984) and Scientific research projects of Guangdong Bureau of traditional Chinese Medicine (20201222).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Cleophas MCP, Crişan TO, Klück V, et al. Romidepsin suppresses monosodium urate crystal-induced cytokine production through upregulation of suppressor of cytokine signaling 1 expression. Arthritis Res Ther. 2019;21(1):50. doi:10.1186/s13075-019-1834-x
2. Desai J, Steiger S, Anders HJ. Molecular Pathophysiology of Gout. Trends Mol Med. 2017;23(8):756–768.
3. Dalbeth N, Choi HK, Terkeltaub R. Review gout: a roadmap to approaches for improving global outcomes. Arthritis Rheumatol. 2017;69(1):22–34. doi:10.1002/art.39799
4. Gupta MK, Singh JA. Cardiovascular disease in gout and the protective effect of treatments including urate-lowering therapy. Drugs. 2019;79(5):531–541. doi:10.1007/s40265-019-01081-5
5. Liu R, Han C, Wu D, et al. Prevalence of hyperuricemia and gout in Mainland China from 2000 to 2014: a systematic review and meta-analysis. Biomed Res Int. 2015;2015:762820. doi:10.1155/2015/762820
6. Lu X, Zeng R, Lin J, et al. Pharmacological basis for use of madecassoside in gouty arthritis: anti-inflammatory, anti-hyperuricemic, and NLRP3 inhibition. Immunopharmacol Immunotoxicol. 2019;41(2):277–284. doi:10.1080/08923973.2019.1590721
7. Huang HC, Chiang HP, Hsu NW, Huang CF, Chang SH, Lin KC. Differential risk group of developing stroke among older women with gouty arthritis: a latent transition analysis. Eur J Clin Invest. 2019;49(5):e13090. doi:10.1111/eci.13090.
8. Wilson L, Saseen JJ. Gouty arthritis: a review of acute management and prevention. Pharmacotherapy. 2016;36(8):906–922. doi:10.1002/phar.1788
9. So AK, Martinon F. Inflammation in gout: mechanisms and therapeutic targets. Nat Rev Rheumatol. 2017;13(11):639–647. doi:10.1038/nrrheum.2017.155
10. Elliott EI, Sutterwala FS. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol Rev. 2015;265(1):35–52. doi:10.1111/imr.12286
11. Jo EK, Kim JK, Shin DM, Sasakawa C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol Immunol. 2016;13(2):148–159. doi:10.1038/cmi.2015.95
12. Kingsbury SR, Conaghan PG, McDermott MF. The role of the NLRP3 inflammasome in gout. J Inflamm Res. 2011;4:39–49. doi:10.2147/JIR.S11330
13. Klück V, Jansen TLTA, Janssen M, et al. Dapansutrile, an oral selective NLRP3 inflammasome inhibitor, for treatment of gout flares: an open-label, dose-adaptive, proof-of-concept, phase 2a trial. Lancet Rheumatol. 2020;2(5):e270–e280. doi:10.1016/s2665-9913(20)30065-5
14. Chu J, Thomas LM, Watkins SC, Franchi L, Núñez G, Salter RD. Cholesterol-dependent cytolysins induce rapid release of mature IL-1beta from murine macrophages in a NLRP3 inflammasome and cathepsin B-dependent manner. J Leukoc Biol. 2009;86(5):1227–1238. doi:10.1189/jlb.0309164
15. He Y, Hara H, Núñez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci. 2016;41(12):1012–1021. doi:10.1016/j.tibs.2016.09.002
16. Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11(2):136–140. doi:10.1038/ni.1831
17. Shimada K, Crother TR, Karlin J, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36(3):401–414. doi:10.1016/j.immuni.2012.01.009
18. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221–225. doi:10.1038/nature09663
19. Kavas GO, Ayral PA, Elhan AH. The effects of resveratrol on oxidant/antioxidant systems and their cofactors in rats. Adv Clin Exp Med. 2013;22(2):151–155.
20. de la Lastra CA, Villegas I. Resveratrol as an anti-inflammatory and anti-aging agent: mechanisms and clinical implications. Mol Nutr Food Res. 2005;49(5):405–430. doi:10.1002/mnfr.200500022
21. Bertelli AA, Ferrara F, Diana G, et al. Resveratrol, a natural stilbene in grapes and wine, enhances intraphagocytosis in human promonocytes: a co-factor in antiinflammatory and anticancer chemopreventive activity. Int J Tissue React. 1999;21(4):93–104.
22. Rauf A, Imran M, Butt MS, Nadeem M, Peters DG, Mubarak MS. Resveratrol as an anti-cancer agent: a review. Crit Rev Food Sci Nutr. 2018;58(9):1428–1447. doi:10.1080/10408398.2016.1263597
23. Shi YW, Wang CP, Liu L, et al. Antihyperuricemic and nephroprotective effects of resveratrol and its analogues in hyperuricemic mice. Mol Nutr Food Res. 2012;56(9):1433–1444. doi:10.1002/mnfr.201100828
24. Chen H, Zheng S, Wang Y, et al. The effect of resveratrol on the recurrent attacks of gouty arthritis. Clin Rheumatol. 2014. doi:10.1007/s10067-014-2826-5
25. Yang QB, He YL, Zhong XW, Xie WG, Zhou JG. Resveratrol ameliorates gouty inflammation via upregulation of sirtuin 1 to promote autophagy in gout patients. Inflammopharmacology. 2019;27(1):47–56. doi:10.1007/s10787-018-00555-4
26. Chen X, Lu J, An M, Ma Z, Zong H, Yang J. Anti-inflammatory effect of resveratrol on adjuvant arthritis rats with abnormal immunological function via the reduction of cyclooxygenase-2 and prostaglandin E2. Mol Med Rep. 2014;9(6):2592–2598. doi:10.3892/mmr.2014.2070
27. El-Ghazaly MA, Fadel NA, Abdel-Naby DH, Abd El-Rehim HA, Zaki HF, Kenawy SA. Amelioration of adjuvant-induced arthritis by exposure to low dose gamma radiation and resveratrol administration in rats. Int J Radiat Biol. 2020;96(7):857–867. doi:10.1080/09553002.2020.1748911
28. Zhou Q, Lin FF, Liu SM, Sui XF. Influence of the total saponin fraction from Dioscorea nipponica Makino on TLR2/4-IL1R receptor singnal pathway in rats of gouty arthritis. J Ethnopharmacol. 2017;206:274–282. doi:10.1016/j.jep.2017.04.024
29. Coderre TJ, Wall PD. Ankle joint urate arthritis in rats provides a useful tool for the evaluation of analgesic and anti-arthritic agents. Pharmacol Biochem Behav. 1988;29(3):461–466. doi:10.1016/0091-3057(88)90004-4
30. Chung YH, Kim HY, Yoon BR, Kang YJ, Lee WW. Suppression of Syk activation by resveratrol inhibits MSU crystal-induced inflammation in human monocytes. J Mol Med (Berl). 2019;97(3):369–383. doi:10.1007/s00109-018-01736-y
31. Martin WJ, Walton M, Harper J. Resident macrophages initiating and driving inflammation in a monosodium urate monohydrate crystal-induced murine peritoneal model of acute gout. Arthritis Rheum. 2009;60(1):281–289. doi:10.1002/art.24185
32. Mian W, Zhang M, Ma Y, et al. Chaetocin attenuates gout in mice through inhibiting HIF-1α and NLRP3 inflammasome-dependent IL-1β secretion in macrophages. Arch Biochem Biophys. 2019;670:94–103. doi:10.1016/j.abb.2019.06.010
33. Zaiss MM, Maslowski KM. In vitro inflammasome assay. Bio-Protocol. 2014;4(11):e1142. doi:10.21769/BioProtoc.1142.
34. Pietrosimone KM, Jin M, Poston B, Liu P. Collagen-induced arthritis: a model for murine autoimmune arthritis. Bio Protoc. 2015;5(20):e1626. doi:10.21769/bioprotoc.1626
35. Dalbeth N, Merriman TR, Stamp LK. Gout. Lancet. 2016;388(10055):2039–2052. doi:10.1016/S0140-6736(16)00346-9
36. Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440(7081):237–241. doi:10.1038/nature04516
37. Gicquel T, Robert S, Loyer P, et al. IL-1β production is dependent on the activation of purinergic receptors and NLRP3 pathway in human macrophages. FASEB J. 2015;29(10):4162–4173. doi:10.1096/fj.14-267393
38. Zheng SC, Zhu XX, Xue Y, et al. Role of the NLRP3 inflammasome in the transient release of IL-1β induced by monosodium urate crystals in human fibroblast-like synoviocytes. J Inflamm (Lond). 2015;12:30. doi:10.1186/s12950-015-0070-7
39. Robinson PC, Horsburgh S. Gout: joints and beyond, epidemiology, clinical features, treatment and co-morbidities. Maturitas. 2014;78(4):245–251. doi:10.1016/j.maturitas.2014.05.001
40. Inokuchi T, Moriwaki Y, Tsutsui H, et al. Plasma interleukin (IL)-18 (interferon-gamma-inducing factor) and other inflammatory cytokines in patients with gouty arthritis and monosodium urate monohydrate crystal-induced secretion of IL-18. Cytokine. 2006;33(1):21–27. doi:10.1016/j.cyto.2005.11.010
41. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. 2019;19(8):477–489. doi:10.1038/s41577-019-0165-0
42. Joosten LA, Netea MG, Mylona E, et al. Engagement of fatty acids with Toll-like receptor 2 drives interleukin-1β production via the ASC/caspase 1 pathway in monosodium urate monohydrate crystal-induced gouty arthritis. Arthritis Rheum. 2010;62(11):3237–3248. doi:10.1002/art.27667
43. Jiang L, Zhang L, Kang K, et al. Resveratrol ameliorates LPS-induced acute lung injury via NLRP3 inflammasome modulation. Biomed Pharmacother. 2016;84:130–138. doi:10.1016/j.biopha.2016.09.020
44. He Q, Li Z, Wang Y, Hou Y, Li L, Zhao J. Resveratrol alleviates cerebral ischemia/reperfusion injury in rats by inhibiting NLRP3 inflammasome activation through Sirt1-dependent autophagy induction. Int Immunopharmacol. 2017;50:208–215. doi:10.1016/j.intimp.2017.06.029
45. Tschopp J, Schroder K. NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production? Nat Rev Immunol. 2010;10(3):210–215. doi:10.1038/nri2725
46. Ashrafi G, Schwarz TL. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013;20(1):31–42. doi:10.1038/cdd.2012.81
47. Chen G, Han Z, Feng D, et al. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol Cell. 2014;54(3):362–377. doi:10.1016/j.molcel.2014.02.034
48. Zhong Z, Umemura A, Sanchez-Lopez E, et al. NF-κB restricts inflammasome activation via elimination of damaged mitochondria. Cell. 2016;164(5):896–910. doi:10.1016/j.cell.2015.12.057
49. Saitoh T, Akira S. Regulation of inflammasomes by autophagy. J Allergy Clin Immunol. 2016;138(1):28–36. doi:10.1016/j.jaci.2016.05.009
50. Wu J, Li X, Zhu G, Zhang Y, He M, Zhang J. The role of Resveratrol-induced mitophagy/autophagy in peritoneal mesothelial cells inflammatory injury via NLRP3 inflammasome activation triggered by mitochondrial ROS. Exp Cell Res. 2016;341(1):42–53. doi:10.1016/j.yexcr.2016.01.014
51. He L, Zhou Q, Huang Z, et al. PINK1/Parkin-mediated mitophagy promotes apelin-13-induced vascular smooth muscle cell proliferation by AMPKα and exacerbates atherosclerotic lesions. J Cell Physiol. 2019;234(6):8668–8682. doi:10.1002/jcp.27527
52. Nguyen TN, Padman BS, Lazarou M. Deciphering the molecular signals of PINK1/Parkin mitophagy. Trends Cell Biol. 2016;26(10):733–744. doi:10.1016/j.tcb.2016.05.008
53. Lazarou M, Sliter DA, Kane LA, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524(7565):309–314. doi:10.1038/nature14893
54. Schlesinger N. Difficult-to-treat gouty arthritis: a disease warranting better management. Drugs. 2011;71(11):1413–1439. doi:10.2165/11592290-000000000-00000
55. Shin WY, Shim DW, Kim MK, et al. Protective effects of Cinnamomum cassia (Lamaceae) against gout and septic responses via attenuation of inflammasome activation in experimental models. J Ethnopharmacol. 2017;205:173–177. doi:10.1016/j.jep.2017.03.043
56. Huang D, Chen Y, Chen W, et al. Anti-inflammatory effects of the extract of Gnaphalium affine D. Don in vivo and in vitro. J Ethnopharmacol. 2015;176:356–364. doi:10.1016/j.jep.2015.11.010
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.