")
Back to Journals » International Journal of Nanomedicine » Volume 16
Response Surface Optimization of Ultra-Elastic Nanovesicles Loaded with Deflazacort Tailored for Transdermal Delivery: Accentuated Bioavailability and Anti-Inflammatory Efficacy
Authors Ali AA, Hassan AH, Eissa EM , Aboud HM
Received 10 August 2020
Accepted for publication 19 December 2020
Published 25 January 2021 Volume 2021:16 Pages 591—607
DOI https://doi.org/10.2147/IJN.S276330
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Phong A Tran
Adel A Ali, Amira H Hassan, Essam M Eissa, Heba M Aboud
Department of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmacy, Beni-Suef University, Beni-Suef, Egypt
Correspondence: Heba M Aboud
Department of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmacy, Beni-Suef University, Beni-Suef, Egypt
Tel +20822162135
Fax +20822162136
Email [email protected]
Purpose: The aim of the present study was to develop deflazacort (DFZ) ultra-elastic nanovesicles (UENVs) loaded gel for topical administration to evade gastrointestinal adverse impacts accompanying DFZ oral therapy.
Methods: UENVs were elaborated according to D-optimal mixture design employing different edge activators as Span-60, Tween-85 and sodium cholate which were incorporated into the nanovesicles to improve the deformability of vesicles bilayer. DFZ-UENVs were formulated by thin-film hydration technique followed by characterization for different parameters including entrapment efficiency (%EE), particle size, in vitro release and ex vivo permeation studies. The composition of the optimized DFZ-UENV formulation was found to be DFZ (10 mg), Span-60 (30 mg), Tween-85 (30 mg), sodium cholate (3.93 mg), L-α phosphatidylcholine (60 mg) and cholesterol (30 mg). The optimum formulation was incorporated into hydrogel base then characterized in terms of physical parameters, in vitro drug release, ex vivo permeation study and pharmacodynamics evaluation. Finally, pharmacokinetic study in rabbits was performed via transdermal application of UENVs gel in comparison to oral drug.
Results: The optimum UENVs formulation exhibited %EE of 74.77± 1.33, vesicle diameter of 219.64± 2.52 nm, 68.88± 1.64% of DFZ released after 12 h and zeta potential of − 55.57± 1.04 mV. The current work divulged successful augmentation of the bioavailability of DFZ optimum formulation by about 1.37-fold and drug release retardation compared to oral drug tablets besides significant depression of edema, cellular inflammation and capillary congestion in carrageenan-induced rat paw edema model.
Conclusion: The transdermal DFZ-UENVs can achieve boosted bioavailability and may be suggested as an auspicious non-invasive alternative platform for oral route.
Keywords: deflazacort, ultra-elastic nanovesicles, D-optimal design, pharmacodynamics, pharmacokinetics
Introduction
The goal of corticosteroid therapy is to maximize the clinical benefit and to minimize the side effects. Corticosteroids exert a group of important anti-allergic and anti-inflammatory actions by different mechanisms including; inhibition of cytokine, inhibition of leucocyte priming in eosinophils and neutrophils, decreasing the vascular permeability, inhibition of metabolite of arachidonic acid and platelet activating factor release in addition to modulation of enzyme systems involved in inflammation.1 The conventional oral therapy by steroids like prednisone and prednisolone has various adverse effects in both short and long period of use.2
Deflazacort (DFZ) is a prednisolone derivative with prominent anti-inflammatory and immuno-suppressive effects. DFZ has been given for different conditions like rheumatoid arthritis, organ transplantation rejection, nephritic syndrome, obstructive pulmonary disease and for many other applications.3 DFZ is associated with lesser steroid-induced osteoporosis and growth retardation than other steroids.4 DFZ is present in the Egyptian market as Flazacor® tablets (6 and 30 mg) for oral administration.
From the results obtained from clinical studies concerning evaluation of both therapeutic activity and safety of corticosteroid treatment, DFZ has favorable therapeutic and safety profiles compared to other corticosteroids.5,6 Unfortunately, problems such as poor solubility, erratic absorption and many drug dosing are encountered with oral DFZ.7 Additionally, gastrointestinal symptoms are the most frequently reported adverse events in DFZ recipient.8,9
DFZ belongs to biopharmaceutics classification system (BCS) class II with an oral bioavailability of about 68% and short elimination half-life (t1/2) from 1.9 to 2.3 h.2 Different strategies have been developed with a focus on enhancing solubility, dissolution rate, and bioavailability of class II drugs.10
According to our knowledge, topical dosage forms of DFZ are not commercially available. Transdermal delivery of DFZ will be a non-invasive promising alternative route as well as can improve its delivery, effectiveness and avoid its oral side effects.
Ultra-elastic nanovesicles (UENVs) are one of the most promising vesicular systems that can enhance drug delivery through skin layers. They have been suggested to be more effective than conventional rigid vesicles as a vehicle for transdermal drug delivery. UENVs were firstly introduced by Cevc et al.11 who combined phosphatidylcholine with sodium cholate as an edge activator to form flexible membranes.
UENVs are vesicles with ultra-flexible lipid bilayer membranes which make them highly deformable so that they can penetrate through the tight pores in the epidermis.12 UENVs contain edge activators which make destabilization in the liposomal lipid bilayer leading to increase in the flexibility of liposomes. The drug delivery by using UENVs as vesicular carriers across the skin is more enhanced compared to that of conventional liposomes.13 They are believed to overcome the skin barrier by opening intercellular pathways as they deform themselves to fit through these channels. They have a very low pore penetration resistance and can squeeze themselves through skin pores five times to ten times smaller than their own diameter.14
UENVs can structurally modify stratum corneum (SC) by their constituting lipids in addition to deep penetration through the skin and acting as penetration enhancing carriers for many drugs with minimal risk of vesicular wall rupture compared to conventional vesicular carriers.15
The work in this study involved development of topical DFZ-UENVs as alternative to oral tablets to avoid its GIT side effects associated with its oral therapy. According to D-optimal designing, the optimum UENVs formulation was subjected to ex vivo and in vivo permeation studies with drug suspension. Histopathological evaluation was performed to elucidate the safety of the prepared carriers. Furthermore, the anti-inflammatory study was performed using the optimum UENVs formulation compared with free drug suspension in carrageenan-induced rat paw edema model.
Materials and Methods
Materials
Deflazacort was donated from Sedico pharmaceutical company (6th October City, Egypt), lecithin (Epikuron® 200 soya bean L-α phosphatidylcholine) was donated from Cargill (Minneapolis, MN, Germany). Sorbitan monostearate (Span-60), polyoxyethylene sorbitan trioleate (Tween-85), sodium cholate, cholesterol and carboxymethyl cellulose sodium were purchased from Sigma-Aldrich (St. Louis, MO, USA). Cellulose dialysis membrane with a molecular weight cutoff of 12,000–14,000 Da was purchased from SERVAPOR Company (Heidelberg, Germany). Carrageenan (SeaSpen PF NF) was a gift sample from FMC Biopolymers (Philadelphia Pennsylvania, USA). Etodolac was donated as a gift sample from Pharco pharmaceutical company (Alexandria, Egypt). All other chemicals and reagents used were of analytical grades and were used as received.
Statistical Design of the Study
In this study, D-optimal design was chosen because it would allow few runs while still being able to estimate all the factors of interest. Different edge activators amounts as quantitative independent variables include X1: Span-60 (mg), X2: Tween-85 (mg) and X3: sodium cholate (mg) while the dependent variables were Y1: particle size (nm), Y2: percent entrapment efficiency (%EE) and Y3: percent drug released after 12 h (Q12h). The ranges and levels of the defined factors are indicated in Table 1. Design-Expert® software (V.11, Stat-Ease, Inc., Minneapolis, USA) free trial version has been used for mathematical modeling and evaluation of the responses. A group of 15 runs were required to develop the appropriate models. The study parameters and runs of the design are presented in Table 1.
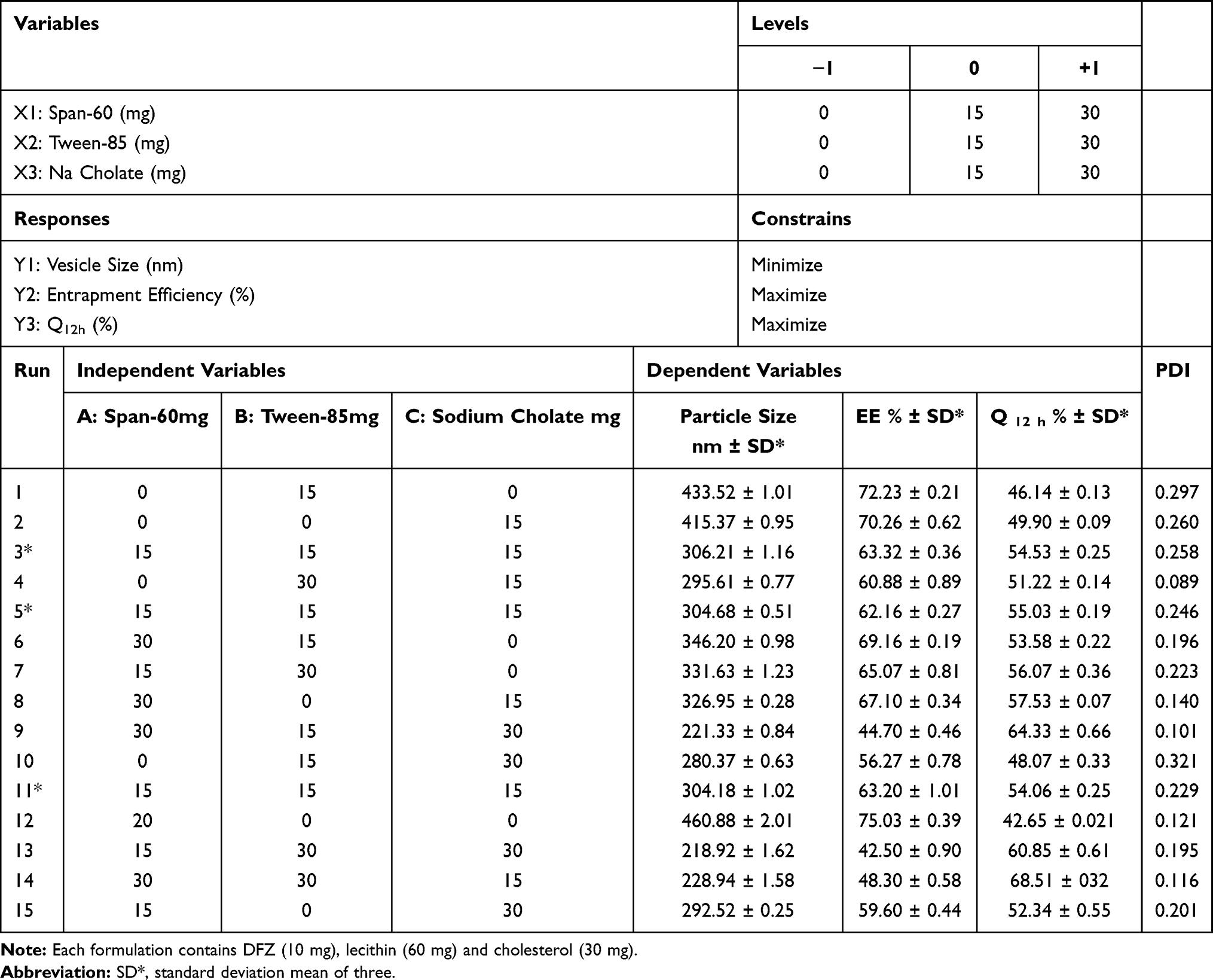 |
Table 1 Different Variables and Responses Used in D-Optimal Design of DFZ-Loaded UFNVs |
Preparation of DFZ-Loaded UENVs
According to thin film hydration method, UENVs were prepared as follows: DFZ (10 mg), lecithin (60 mg) and cholesterol (30 mg) in addition to different levels of edge activators (Span-60, Tween-85, sodium cholate) were dissolved in an amount of 10 mL of chloroform: methanol (1:1) mixture.
The clear solution was slowly evaporated at 40°C under pressure −60 Kpa using an evaporator (Stuart rotary evaporator, Wolf Laboratories, UK) connected to Stuart vacuum pump for 20 min under 80 rpm. The obtained homogenous film was then kept in a desiccator with vacuum for 4 h to ensure complete removal of trace working solvents. Hydration of the transparent film with 5 mL phosphate buffer solution (pH 5.5)16 was done and kept under 80 rpm for 60 min. For a significant size reduction, the prepared UENV dispersions were sonicated for 30 min in ultrasonic water bath (Sonix TV, North Charleston, SC). The suspensions were stored overnight in refrigerator at 4°C as stability purpose requirement.
Characterization of DFZ-UENVs
Determination of %EE
Percent EE of DFZ in the prepared UENVs was detected by measuring the amount of free DFZ in the dispersion medium.17 By centrifugation of 1 mL of each freshly prepared UENVs suspension using cooling centrifuge (SIGMA 3–30K, Germany) at 14,000 rpm at 4°C for 3 h to ensure complete settling of UENVs, the resultant supernatant was separated, then was appropriately diluted with 10% methanolic phosphate buffer solution (pH 5.5), filtered through 0.45 µm Millipore polycarbonate membrane filter (Whatman Ltd, Springfield Mill, UK) and analyzed for DFZ concentration spectrophotometrically at 248 nm using a UV spectrophotometer (JASCO, Japan). From the mean of three determinations, %EE was calculated using equation 1:
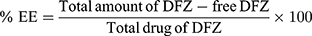
Determination of Vesicle Size
The freshly prepared UENVs suspension was diluted by deionized water (1:10) then added to the sample dispersion unit. Samples were vortexed to reduce the aggregation of the vesicles and measured by photon correlation spectroscopy (Zetasizer Nano ZS 7.11, Malvern Instruments, UK).18,19 UENVs particle size was determined in triplicate (n=3) and presented as mean ± SD.
In vitro Release Study of DFZ from the Prepared UENVs
For different formulations, an amount of settled UENVs equivalent to 10 mg DFZ was suspended in 5 mL phosphate buffer (pH 5.5) then placed in the donor part of Franz cells permeation apparatus (Orchid Scientific & Innovative India Pvt Ltd, India). A 50 mL of 10% methanolic phosphate buffer (pH 5.5) was placed in the receptor part to ensure a sink condition.20 The temperature of receptor part was 37±0.5°C and stirred at 100 rpm. The donor part was separated from the receptor part by presoaked cellulose dialysis membrane. After specific time periods 0.5, 1, 2, 3, 4, 5, 6, 8, 10 and 12 h, one mL sample was withdrawn, filtered through a millipore filter (0.45 µm) and DFZ content was detected spectrophotometrically at λmax 248 nm. The withdrawn volume was compensated with an equal fresh medium to maintain a constant volume. All experiments were performed in triplicate and the percent DFZ released was expressed using equation 2.
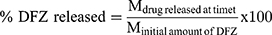
The drug release from DFZ suspension (10 mg DFZ suspended in 5 mL phosphate buffer solution pH 5.5) as a control was performed in the same manner and the data was used for comparison. The percent drug released after 12 h (Q12h) was determined and recorded as mean ± SD.
Optimization of DFZ-Loaded UENVs
Design-Expert® software was anticipated to select the optimized UENVs by employing the desirability function.21 The optimization process was deliberated to select a formulation with the greatest %EE and Q12h as well as the smallest particle size. The solution with desirability index near to one was elected. Finally, the optimum formulation was enrolled in further studies.
Morphological Study of the Optimized DFZ-UENVs
The morphology was investigated as follows; fresh DFZ-UENVs suspension was diluted with deionized water then loaded on a copper grid coated with carbon. For contrast enhancement negative stain of phosphotungstic (2% w/v) was applied to the prepared sample for 60 sec. The vesicles were allowed to dry on the copper grid then directly examined by TEM (JEM-1400 microscope; JEOL, Japan).22,23
Stability Study
The physical stability of the prepared optimum UENVs formulation was performed to evaluate the aggregation and leakage during storage. UENVs formulations were stored in vials at 4°C, 25°C and 37°C for 3 months. Samples were taken at different intervals. Particle size, zeta potential and %EE measurements were carried out over the period of the study. Moreover, the physical appearance of vesicular suspensions was examined visually for sedimentation.24,25
Preparation and Characterization of DFZ-UENVs Hydrogels
The optimum DFZ-UENVs formulation was incorporated into hydrogel base composed from 2% w/v CMC sodium using the cold method. CMC Na was added slowly to accurate volume of UENVs suspension (freshly prepared optimum formulation), 0.01% w/v of both methyl and propyl paraben were dissolved with continuous stirring till homogenous dispersion was obtained. The prepared hydrogel was kept at 4°C in refrigerator to allow complete gelling.26 The DFZ concentration in the prepared hydrogel was adjusted to be 10 mg/g.
In the same manner; plain hydrogel base and hydrogel containing free DFZ were prepared. Samples from the obtained hydrogels were taken to detect their drug content, pH, viscosity, mucoadhesive strength and organoleptic characteristics (texture, and transparency).27
Ex vivo Permeation Study of DFZ-UENVs Hydrogels
Male Wistar rats weighing 200–250 g were used in this study and the experimental procedure was in accordance with the guidelines of the Local Institutional Animal Ethics Committee of Beni-Suef University (Acceptance Serial No: REC-A-PhBSU-20003). The animals were anesthetized and euthanized by cervical dislocation.28 The shaved dorsal rat skin was used in this study. In order to obtain a skin free from adhering fat material, the dermal part of the skin was wiped with isopropanol cotton swab three times.29 The skin was kept in 0.9% w/v NaCl solution for 2 h, washed with deionized water, then became ready for permeation study.30
As previously mentioned in the in vitro release study, permeation studies were carried out as follows: Amounts of settled UENVs equivalent to 10 mg DFZ was reconstituted with 5 mL phosphate buffer (pH 5.5) then placed in the donor compartment of Franz diffusion permeation apparatus (Orchid Scientific & Innovative India Pvt Ltd, India). Samples were withdrawn after time periods of 0.5, 1, 2, 3, 4, 6, 8, 10, 12 and 24 h.31 The donor part was separated from the receptor part by cleaned rat skin. The cumulative amount of DFZ permeated after 24 h (Q24h) in µg/cm2 was graphically plotted as a function of time. The cumulative amount permeated was computed employing equation 3.
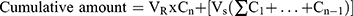
Taking into account the volume of the receptor phase (VR), the volume sampled at each time point (Vs) and the quantified concentration of the sample taken at the nth time point (Cn). The cumulative amount calculated at each time point applying equation 3 was further divided by the diffusion area of the Franz cells (in cm2).32 All experiments were carried out in triplicate and the average values were calculated.
Pharmacodynamics Study and Evaluation of Anti-Inflammatory Activity
All experiments were done in accordance with the guidelines of Local Institutional Animal Ethics Committee of Beni-Suef University (Acceptance Serial No: REC-A-PhBSU-20003). Anti-inflammatory effect of the prepared hydrogel formulations were evaluated by using the carrageenan-induced rat paw edema model.32–34 Wistar rats were used for the study. The animals of either sex weighing 200–250 g were divided into three groups of six rats each.
Edema induced was done by injection of Carrageenan solution (0.1 mL of 1% w/v in normal saline) into the plantar aponeurosis of the right hind paw. Thickness of the paw was measured before and 5 h after injection.35
Group A (control group) received plain gel, group B received free DFZ gel (10 mg/g) and group C received DFZ-UENVs gel (10 mg/g). Different gel formulations (0.25 g) were applied to the edematous paw and covered to prevent gel licking, the paw thickness was measured after 2 h from gel application36 by a caliber and the % edema inhibition was detected using the following formula:
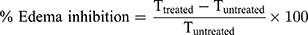
Where, Ttreated is the paw thickness after the topical DFZ treatment and Tuntreated is the paw thickness without treatment of DFZ topical gel. All measurements in the study were performed in triplicate. The experiments results were presented as a mean values ± SD. One-way ANOVA followed by Tukey-Kramer comparison test was done to determine the significance of difference (p value was less than 0.05).
Histopathological Study
A histopathological examination was performed for all groups. The rats were sacrificed by injecting an overdose of ketamine. The tissues of the right hind paw were excised and embedded in solidified paraffin blocks, cut, stained with stains (hematoxylin and eosin) then examined by optical microscope (Bio-med BML 2200, Japan) attached with a camera (Panasonic, WV-CP 240/G Suzhou, China).
Pharmacokinetic Study of DFZ Hydrogels
Animals
The pharmacokinetic study was carried out in accordance with the guidelines of the Local Institutional Animal Ethics Committee of Beni-Suef University (Acceptance Serial No: REC-A-PhBSU-20003). Three groups, each of three New Zealand white male rabbits (weights from 2–2.5 Kg) were used for examined formulations. Rabbits were fasted except for water for 24 h prior to the experiment. Animals were conscious and held in restrainers during dosing, blood sampling and the whole duration of the experiment.
Dosage and Dose Administration
Single dose of 5 mg DFZ per Kg was administered to animals.37 Group 1 received powdered traditional DFZ tablets suspended in purified water by oral route through oral gavage. Group 2 and 3 received topically the optimized DFZ-UENVs gel and free DFZ gel, respectively. Prior to gel application, the animal dorsal regions were shaved, cleaned with distilled water then dried by clean cotton pads. The gel was applied on over a surface area of 20 cm2.
Sample Collection
Two mL blood samples were collected from the ear (marginal ear vein) of the rabbits after specific time periods (0, 0.5, 1, 2, 4, 8, 12 and 24 h). The samples are stored into glass tubes (containing EDTA to avoid clotting), then centrifugation at 4000 rpm was done for 15 min for complete separation of the plasma. The separated plasma samples were kept at −20°C till analysis.
Chromatographic Conditions
The amount of DFZ in the samples was estimated using HPLC. The Agilent 1260 infinity (Germany) HPLC system is composed from diode array detector VL (G 1315D), preparative pump (G 1361A), thermostatted column compartment (G 1316A) in addition to 20 μL preparative auto sampler (G 2260A). Separation and quantification were carried out on C18 column 25 cm×4.6 mm (Zorbax Eclipse Plus, USA). The wavelength was set at 248 nm. The mobile phase was composed from acetonitrile: methanol: 0.067 M potassium dihydrogen phosphate (27:20:53 v/v/v) adjusted to pH 5.5 with 3 M NaOH and delivered at a flow rate of 0.75 mL/min. The mobile phase was prepared daily and degassed before use by ultrasonication. The elute was measured at 248 nm using Etodolac as an internal standard.38
Samples Preparation for Analysis
Plasma samples (500 μL) were mixed with 50 μL of Etodolac solution (10 μg/mL) and 450 μL of acetonitrile. The mixture was mixed for 10 sec, centrifuged for 10 min at 4000 rpm. Volume (20 μL) of supernatant was injected on the column for analysis.
Pharmacokinetic Analysis of the Data
Non-compartmental analysis was used to obtain the plasma pharmacokinetic parameters (Cmax, tmax, AUC0–24 and AUC0-∞) following administration of the three treatments were estimated for each rabbit using software WinNonLin (version 1.5, Scientific consulting, Inc., USA).
The area under the curve, AUC0–24 (ng.h/mL) was calculated using the trapezoidal rule from zero time to the last time of sampling. The AUC0-∞ (ng.h/mL) was calculated as AUC0-∞ =AUC0–24+Ct/k, where Ct is the concentration at time t, and k is the terminal elimination rate constant estimated by log-linear regression analysis. The apparent terminal elimination half-life (t1/2) was calculated as t1/2= 0.693/k.
The relative bioavailability (Frel) for both DFZ topical formulations can be finally assessed considering oral formulation (as a standard), from the equation 5:
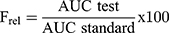
Statistical Analysis of the Data
Employing SPSS 16 (SPSS, Chicago, Illinois, USA) software computer program, all calculations were detected. Statistical analysis of the data was carried out using one-way ANOVA followed by the Tukey post-hoc test. The mean difference was considered statistically significant when p value is less than 0.05. All experiments were a mean of three results (n=3).
Results and Discussion
Study Design
The D-optimal design is an effective statistical technique of design of experiments (DOE) for optimizing complex mixture processes.39 By its advantageous characteristics in minimizing the variance of the parameters estimator through its ability in decreasing the norm of the covariance matrix, D-optimal designing was used in model parameters estimation in this study.40,41 Although D-optimal designing method is obviously a valuable criterion, it has undesirable feature of improving the estimation of one parameter at the expense of another.42 There is strong motivation to pose the hypothesis that use of a D-optimal designing will result in few number of experiments than that required by other traditional designs, without affecting the estimated parameters error and variance.
Characterization of the Prepared UENVs
UENVs are known to be flexible and ultra-deformable liposomes. They possess higher ability to penetrate SC than conventional liposomes. So, they are used for non-invasive drug delivery into or across the skin.43 Edge activators (ionic and non-ionic single chain surfactants) are incorporated in the vesicular formulations to destabilize the vesicles. They improve the deformability of vesicles bilayer as they decrease the interfacial tension.44 Different UENVs formulations were prepared by using different types of edge activators with different HLB values like Span-60 (HLB 4.7), Tween-85 (HLB 11) and sodium cholate (HLB 18).
Optimization of Formulation
Fifteen formulations were prepared according to the proposed experimental design and characterized for various responses as shown in Table 1. The relationships were mathematically established and the polynomial equation 6(a-c) coefficients fit well to the data, with R2 values in the range between 0.9482 and 0.9998 (p<0.05 in all the cases). Statistics of the model responses are summarized in Table 2. After comparing adjusted R2 and predicted R2 of different models, quadratic model showed a superior fit. As its parameters were comparatively higher than that of other models, in addition to precision for the quadratic model of the responses was the smallest.
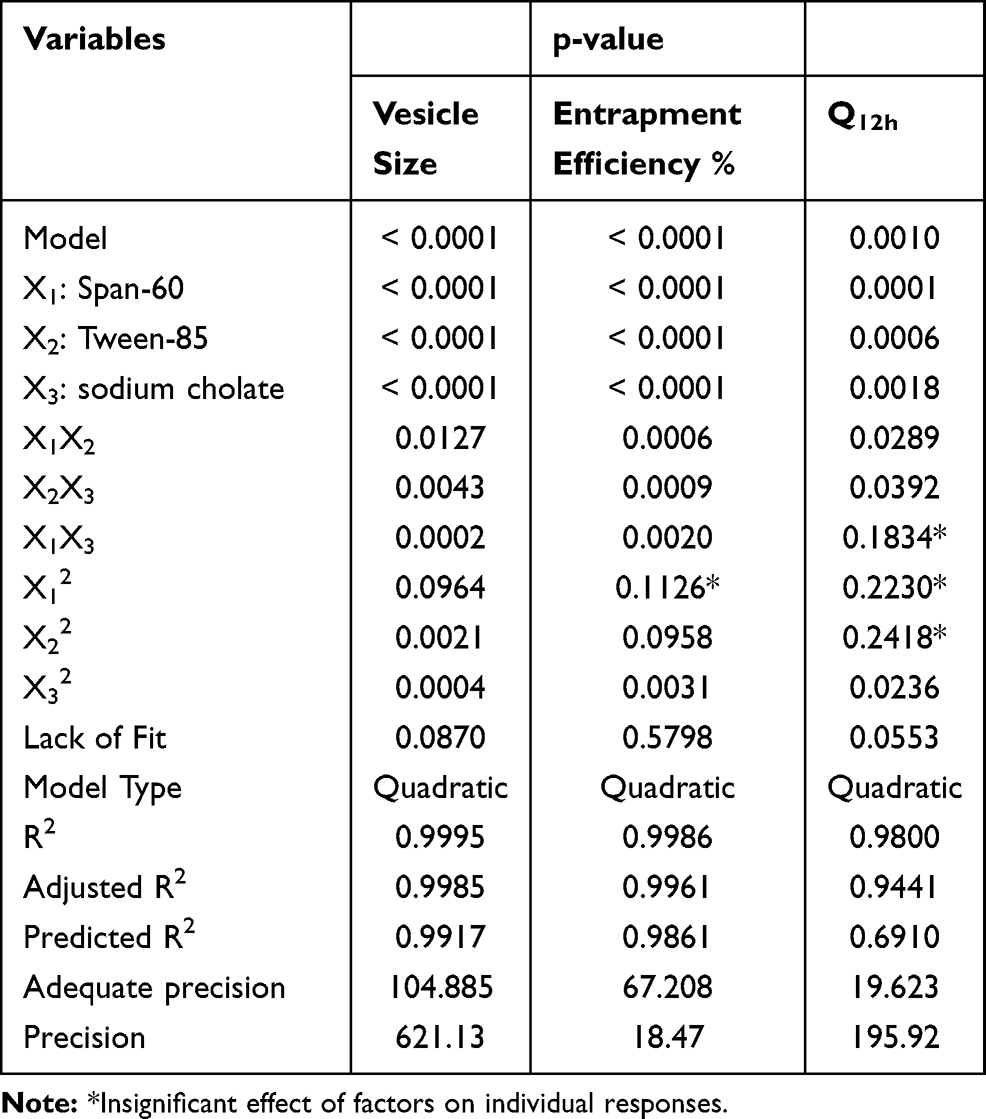 |
Table 2 The Different Model Terms and Their Significance as in the Final Model for Each Response |
From the ANOVA results listed in Table 2, it was observed that the main and interaction effects of model terms were statistically significant within significant model lack of fit. Also in Table 2, factor effects of D-optimal designing and associated p values for the responses are presented. If the effects are significantly deviated from zero and the p value is less than 0.05, a factor is considered to influence the response. For responses Y1: (Particle size) and Y2: (%EE), all the factors were significant, while for a response Y3: (Q12h) the factor: X1X3 was not significant, where p value was greater than 0.05.
Effects of Formulation Factors on the Mean Particle Size, %EE and Q12h
The polynomial equations obtained from the regression analysis are given below:
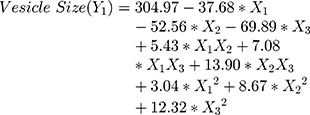
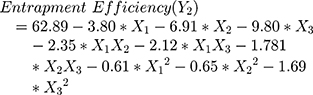
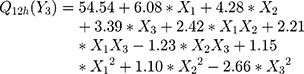
The response surface analysis was done to elucidate the effect of selected independent variables on the observed responses. Response surface plots are graphically represented in Figure 1A–F.
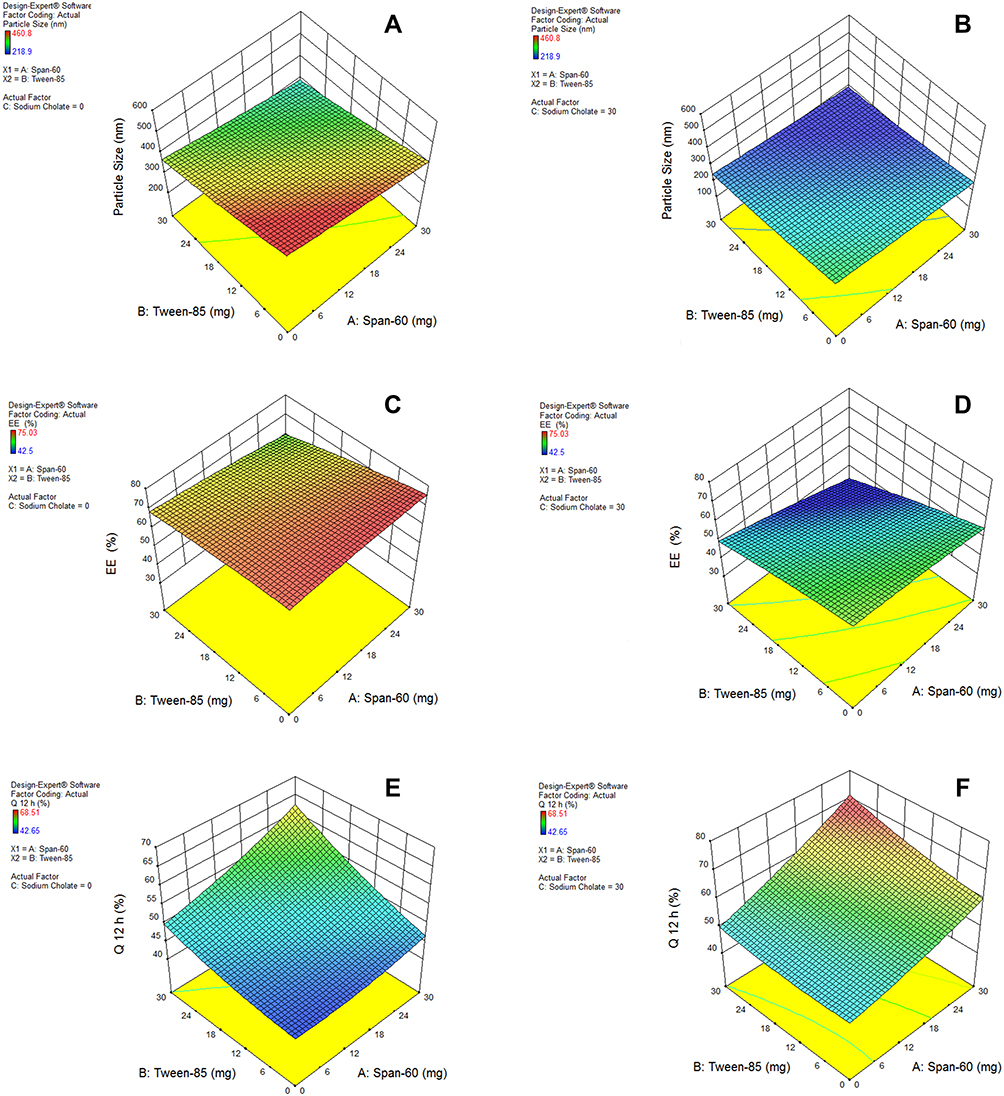 |
Figure 1 Response surface plots for the effects of Span-60 (X1) and Tween-85 (X2) concentrations at lower and upper limits of Sodium Cholate (X3) on: (A and B) particle size, (C and D) %EE and (E and F) Q12h (%) of the prepared UENVs formulations, respectively. |
Results showed that the type of edge activator X1, X2 and X3 had a significant impact on the size of DFZ-UENVs (p < 0.0015) and the particle size of the UENVs was in the range of 218.92 to 460.88 nm. Figure 1A and B depicts the response surface plot; it was noticed that an indirect relationship was obtained between UENVs particle size and edge activator concentration, the minimum diameter was observed at minimum concentrations of edge activators. The combination of Span-60 (HLB 4.7), Tween-85 (HLB 11) and sodium cholate (HLB 18) in the presence of cholesterol at higher concentrations led to vesicles with small size. It was concluded that at the higher surfactant concentration, the surfaces of the UENVs will be covered with surfactant molecules and therefore prevent them from aggregation.45,46
Also according to the previous findings in literature, increasing the surfactant HLB value results in the formation of large vesicles due to the increased surface free energy.47,48 Additionally, PDI values of all DFZ-UENVs fluctuated from 0.089 to 0.321 which could be perceived as a plausible mid-range elucidating homogeneity as well as uniform size distribution of the dispersions.21
Obtaining vesicles with good drug %EE is considered as the main target in the development of any vesicular carrier delivery system. A lot of experiments have been done by adding surfactants into vesicles lipid-bilayer to increase the encapsulation of both hydrophobic and hydrophilic drugs and to reduce drug leakage from the vesicular carriers.49–51 The %EE of the prepared UENVs was in the range of 42.50 to 75.03%.
Vesicular %EE depends on the vesicles stability which is mainly affected by the surfactant type incorporated in the lipid-bilayer. On the other hand, the surfactant type may affect the drug release and deposition through skin. In order to deliver enough amount of drug, a high drug entrapment within the vesicular structure is required.52
Cholesterol was incorporated in a fixed amount (30 mg) to all UENVs formulations in order to enhance the EE via increment of the viscosity of UENVs dispersion and triggering the flexible bilayer more rigid leading to stabilization of UENVs and reduction in vesicle permeability.53
Figure 1C and D shows the interaction effect and relationship between edge activator on the %EE as a response variable. A curvilinear plot was observed where with increasing the amount of edge activator, a linear decrease in the %EE was observed. Meanwhile, at low levels of edge activator, the %EE was ideal. This effect has been explained on the basis of the increased permeation of the UENVs through the membrane as a result of the surfactant molecules arrangement within the vesicular lipid-bilayer structure, forming pine holes in the membrane and thus, increasing its fluidity.54
Over more, the optimum concentration of edge activator relies on the phospholipid packing density and in turn the interaction between surfactant and phospholipid. Additionally, increasing the edge activators concentration, led to a decrease in %EE due to its competition on the drug loading within the lipid-bilayer, whenever it is known to have a higher ability to compete with the lipid layer.15,55,56
Conversely, some literatures have reported that increasing edge activators concentration will increase the number of vesicles leading to a high surface area of lipid-bilayer domain available to host hydrophobic drugs.57,58
The effect of HLB value of the edge activators on the %EE depends on where the drug is lipophilic or hydrophilic, many researchers suppose that using edge activators with a low HLB value could achieve the maximum entrapment of a lipophilic drug.30,59
From our results, at low level of sodium cholate (HLB 18), the %EE showed maximum values at higher levels of Span 60 (HLB 4.7) while at higher level of both sodium cholate (HLB 18) and Tween- 85 (HLB 11), the %EE showed minimum value.
From the study of Al-Mahallawi et al on the preparation of tenoxicam-loaded bilosomes, they found that vesicles containing Span-60 (HLB 4.7) had a significant high %EE. They attributed that to the high saturation of the surfactant alkyl chain length and (T°C), which in turn affected the tenoxicam %EE in the prepared bilosomes.60 Span-60 is a hydrophobic surfactant which is able to accommodate high concentration of DFZ into its hydrophobic region.61
The effect of different edge activators on Q12h of DFZ was illustrated in Figure 1E and F. It was noted that the maximum Q12h was obtained from higher concentrations of edge activators. The Q12h of different UENVs was in the range of 42.65 to 68.51%. UENVs with negative charge can easily permeate through skin and thus improving transdermal drug delivery.62 Therefore, nanovesicles with negatively charged surface may have effect on improvement of drug permeation through skin. This may be attributed to the elasticity and flexibility enhanced by the presence of Span-60 and cholate mixture.63
There is more than evidence that, the presence of edge activators could affect the pharmacokinetic and pharmacodynamic properties of vesicular delivery systems; such as improved drug release, skin permeability, time of circulation, and cellular uptake.
For example, in edge activators based liposomes; increasing the surfactant concentration will enhance ciprofloxacin release (encapsulated drug) from the carrier system. Conversely, ciprofloxacin release was dependent on the used type of edge activator while incorporating Tween-80 significantly enhanced the drug release.64 The same results were found in other studies, where the use of Tween-80 as edge activator in vesicular formulations increased the amount of the drug permeated through the skin.65,66
DFZ in vitro release profiles (data not mentioned) from different UENVs formulations showed a relatively high release of drug in the first 2 h, followed by a slow DFZ release rate in the next 6 h. This may be explained on the base of the high ordered lipid particles, which cannot reserve large amounts of DFZ.67
It was reported that the fast initial DFZ release rate was due to drug detachment from UENVs surface, while the slow DFZ release rate later was explained by the slow drug release from inner layers of the vesicles.68 DFZ release from control was noted to be faster than that from UENVs which may be a result of well-known reservoir effect of UENVs delivery systems that provided extended release profiles.69,70
Having the desirability value near to one, the optimum formulation was selected. The optimized DFZ-UENVs formulation was composed of DFZ (10 mg), Span-60 (30 mg), Tween-85 (30 mg), sodium cholate (3.93 mg), lecithin (60 mg) and cholesterol (30 mg).
The results of evaluation of optimum formulation showed %EE of 74.77±1.33%, particle size of 219.64±2.52 nm and 68.88±1.64% of DFZ released after 12 h in addition to zeta potential of −55.57±1.04 mV. Hence, it was selected for further evaluations.
The optimum DFZ-UENVs formulation was subjected to TEM study, in vivo study in addition to stability study. The transmission electron photomicrographs (Figure 2) of freshly prepared optimized formulation using negative stain of phosphotungstic (2% w/v) showed well-identified spherical vesicles which confirmed the vesicular characteristic of the prepared UENVs. The particle size obtained in the TEM photomicrograph was smaller compared to that obtained by photon correlation spectroscopy, which may be attributed to the drying step during preparation that usually causes shrinkage in vesicles and some sort of artifacts. In addition, the TEM photomicrograph are not reliable tool for particle size detections because the image may fall away from the mean of the particle size and may reside within the extremities of the particle size distribution.71
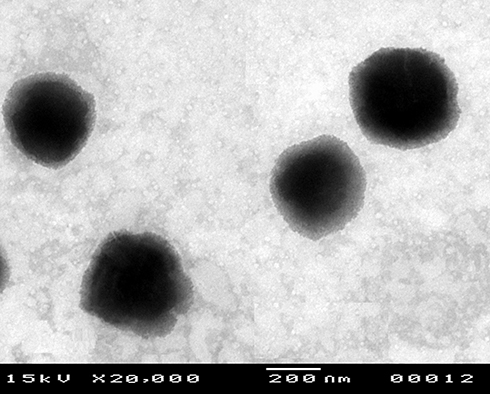 |
Figure 2 Transmission electron photomicrograph of the optimized DFZ-UENVs formulation. |
Stability of UENVs has an important role in development of transdermal drug delivery systems with high efficiency.72,73 Studying the stability of the optimized UENVs formulation was conducted by storing the formulation at different temperatures (4°C, 25°C and 37°C). The prepared UENVs were physically and chemically stable when stored in refrigerator. As shown in Table 3, there was no significant change noticed in physical appearance, particle size, %EE, Q12h (%) and zeta potential over the period of stability study at 4°C.
 |
Table 3 Values of Particle Size, Zeta Potential and EE (%) and Q12h (%) of the Optimized DFZ-UENVs Formulation After Storage for Three Months at 4°C, 25°C, and 37°C |
UENVs which were stored at 25°C and 37°C showed a significant increase in particle size due to vesicles aggregation. UENVs kept at 4°C contained higher DFZ % compared to those stored at higher temperatures. The %EE was decreased in UENVs which were stored at higher temperatures.12 This could be explained on the basis of enhanced fluidity of UENVs membrane or may be due to the effect of temperature on the gel to liquid phase transition of lipid-bilayers. There may be also possible degradation of the phospholipids constituting the UENVs membrane resulting in defects of membrane packing.27
The results revealed that storage of the UENVs vesicles in refrigerator temperature could minimize stability problems of UENVs. UENVs incorporation into hydrogel bases could enhance its stability. The explanation for this behavior may be due to the high viscosity of the gel bases that retards fusion of UENVs together.69
Characterization of DFZ-UENVs Hydrogels
The characterization parameters and general properties of the different prepared DFZ hydrogel formulations are listed in Table 4. All hydrogel formulations (plain gel, free DFZ gel and optimized DFZ-UENVs gel) were elegant in appearance and semisolid in consistency as required for topical administration. They also exhibited pseudoplastic with thixotropic characteristics (results not shown).
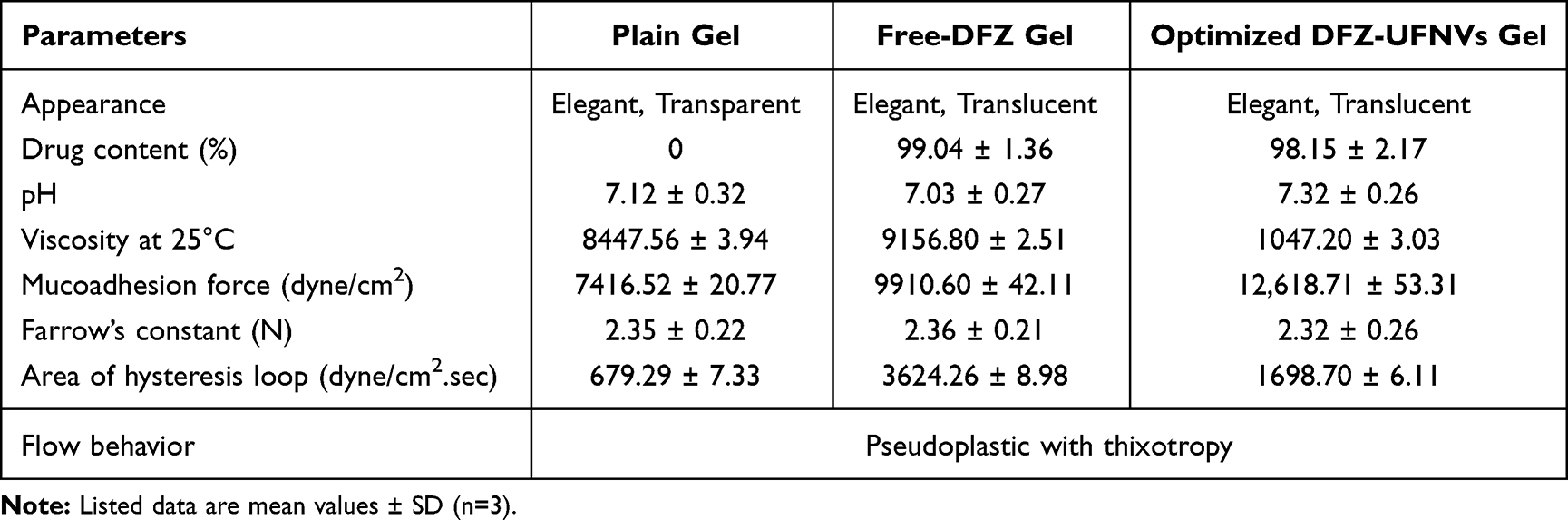 |
Table 4 Characterization and General Properties of the Prepared DFZ Hydrogel Formulations |
Pseudoplasticity results from colloidal network structure of the prepared hydrogel formulation that retains itself in the shear direction. So, viscosity decreases by increasing shear rate.
Thixotropic behavior of hydrogels was shown and the viscosity of prepared hydrogels was observed to be of lower value on the down curve than on the up curve at any rate of shear. This may be due to the breaking down occurring between neighboring polymer chains and in the entanglements between long polymer chain segments. The thixotropic behavior can be studied by measuring the loop area between up and down curves in resulted rheograms. In all prepared hydrogels, there were areas of loops in all rheograms indicating thixotropic behavior of prepared hydrogel formulations.74
Ex vivo Permeation Study of DFZ-UENVs Hydrogels
Permeation of drug molecules across biological membranes is a multi-step, multi-factorial phenomenon depending on various types of chemical, physical and biological interactions. However, the ex vivo permeation studies provide a valuable insight on the in vivo performance of many products.75
Study of ex vivo permeation of both free DFZ gel and DFZ-UENVs gel across rat skin for 24 h was performed and plot of the cumulative amount of DFZ-permeated against time is shown in Figure 3. The cumulative amount of DFZ permeated from the prepared DFZ-UENVs hydrogel formulation at 37°C was 14.69, 95.44 and 180.45 (µg/cm2) after 1, 6 and 12 h, respectively. While the amount of DFZ permeated from the hydrogel containing free drug was 4.62, 36.96 and 68.79 (µg/cm2) after 1, 2 and 12 h, respectively.
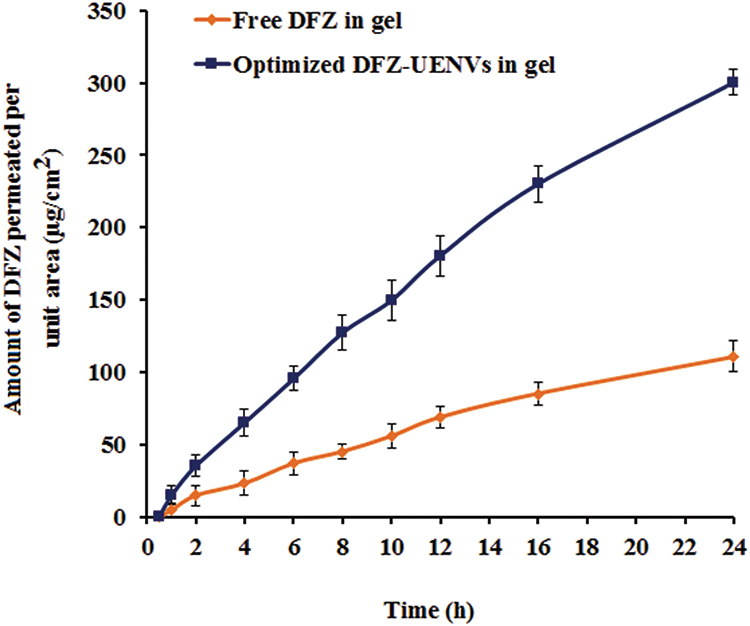 |
Figure 3 Ex vivo permeation profiles of DFZ from the optimized UENVs gel formulation compared to the free DFZ gel formulation (Data = Mean ± S.D, n =3). |
DFZ-UENVs gel showed significantly higher values in the amount permeated as compared to free DFZ gel that could be attributed to the composition of the incorporated UENVs and their effect as permeation enhancers through biological membranes.
Different mechanisms have been proposed for the enhanced skin delivery by UENVs carriers. The first mechanism is that these UENVs as carriers for DFZ can enter the SC in an intact state carrying bound DFZ molecules into the skin layers, under the effect of natural transcutaneous hydration gradient.59 This explains why UENVs were capable to deliver much larger amounts of DFZ to the skin layers compared to free DFZ gel. The second mechanism is the penetration enhancing ability of such vesicles, where vesicle bilayers enter SC then modify its intercellular lipids and in turn raise its weakness and fluidity.
As a result, drugs can further permeate solitary.76,77 In addition, phospholipids content in UENVs have an affinity for biological membranes, thus, the introduction of these vesicles with the lipid layers of the skin may also improve permeation ability of UENVs. Therefore, our conclusion is that both mechanisms resulted in enhanced skin delivery of DFZ by UENVs under non-occlusive conditions which is in harmony with previous reports.30,78,79
Pharmacodynamics Study and Evaluation of Anti-Inflammatory Activity
In vivo performance was evaluated indirectly by measuring anti-inflammatory activity of prepared hydrogel formulations in rats. Carrageenan injection causes a progressive increase in rat paw volume; this is attributed to two steps, the first step is the histamine and serotonin secretions, and the second step is prostaglandins and lysosomal bodies release, which are inflammation mediators susceptible to most anti-inflammatory drugs. The anti-inflammatory drug gel formulation should have fast inhibitory action without being removed from the site of application. The higher the inhibitory action, the higher pharmacological action is. As it has been reported by Mei et al, the drug formulations of small particle size showed higher acute anti-inflammatory action.33
Application of DFZ-UENVs gel resulted in greater mean edema inhibition compared to the conventional DFZ gel. These results may be attributed to the DFZ % released from conventional gel, was not sufficient for effective control of edema for long period of time. There was a significant difference between the DFZ groups and the control as reflected by a one way ANOVA with p < 0.05. From Figure 4, the % edema inhibition was found to be greater for DFZ-UENVs gel (a mean of 59.4% inhibition, after 90 min and 90.4% after 150 min) as compared to free DFZ gel which showed mean % edema inhibition of 15.6%, after 90 min and 36.43% after 150 min.
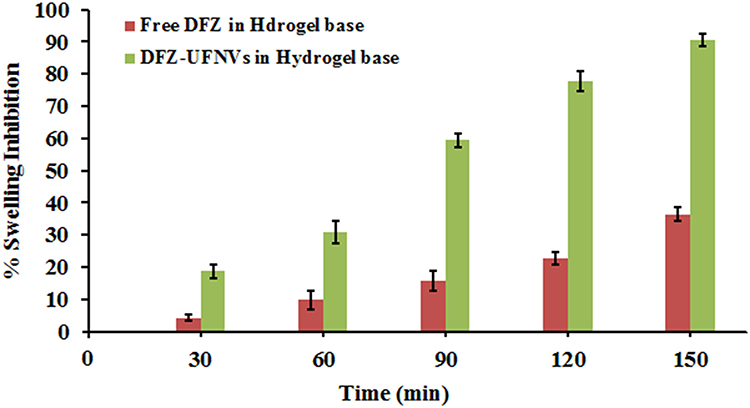 |
Figure 4 % Swelling inhibition of the optimized DFZ-UENVs gel in comparison with the free DFZ gel using the carrageenan induced rat paw edema model. (Data = Mean ± S.D, n =6). |
The improvement in the anti-inflammatory action of DFZ-UENVs gel formulation may be attributed to both higher DFZ release rate and enhanced skin permeability. Improved drug deposition from hydrogels containing DFZ-loaded UENVs and increased anti-inflammatory effect can be justified by aforementioned possible mechanisms. Also, nonionic edge activators used in UENVs hydrogel formulations can act as permeation enhancers as it had been previously explained.80
Cholesterol and hydrophobic surfactants in the UENVs offer extended surface area with occlusive film ability on the skin surface and thus decreasing water loss from skin layers. This resulted in an improvement of skin hydration and enhancement of DFZ penetration through skin.81
Histopathological Examination
Figure 5A–C shows histopathological sections of the studied groups. Investigation of the influence of formulations on skin irritation was carried out by topically applying each formulation. Figure 5A shows normal skin tissue with no edema (negative control) where the SC, dermis and epidermis layers were intact and normal. Figure 5B shows edematous skin tissue induced by carrageenan where dilation of lymphatic vessels and infiltration of mononuclear cells were noticed (black arrows) where the whole tissue was dispersed by edema and the SC with subsequent layers became thinner. From Figure 5C, DFZ-UENVs gel showed the higher anti-inflammatory activity among the tested formulations.
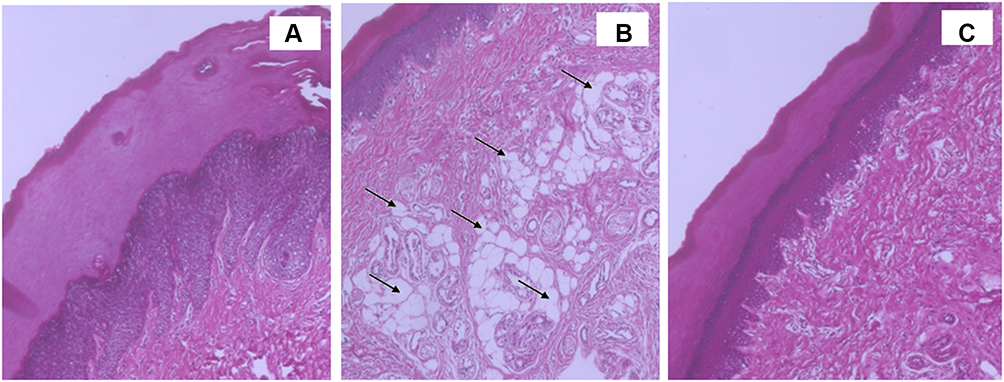 |
Figure 5 Light photomicrographs of morphology of rat’s skin after application of (A) control plain gel, (B) free DFZ gel and (C) optimized DFZ-UENVs gel. |
Pharmacokinetic Study of DFZ Hydrogels
The developed HPLC assay method was selective, reproducible and fully validated for determination of DFZ serum samples, with good linearity (1–200 ng/mL), accuracy and precision.
The average DFZ plasma concentration-time profiles following administration of oral solution, topical free DFZ gel and topical DFZ-UENVs gel are illustrated in Figure 6 and their corresponding pharmacokinetic characteristics are listed in Table 5.
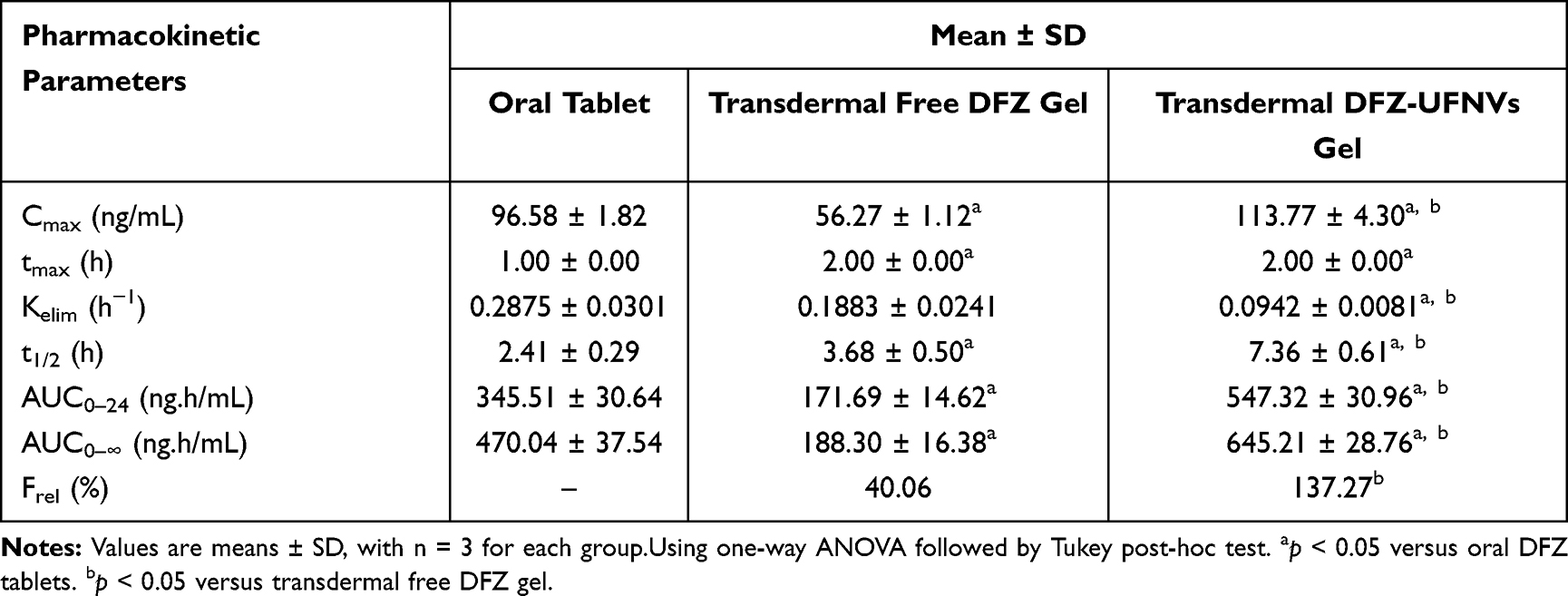 |
Table 5 Mean Pharmacokinetic Parameters for DFZ in Rabbit Plasma Following Administration of Oral DFZ Tablet, Transdermal Free DFZ Gel and Transdermal DFZ-UFNVs Gel |
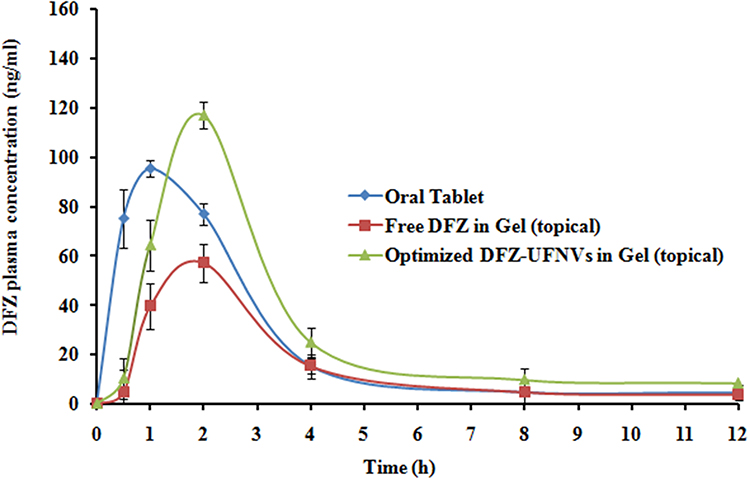 |
Figure 6 Mean DFZ concentrations (ng/ml) in rabbit plasma following administration of oral tablet suspension, topical free DFZ gel and topical optimized DFZ-UENVs gel formulation. (Data = Mean ± S.D, n =3). |
From the obtained results, DFZ was quickly absorbed and reach maximum plasma concentration (Cmax) of 96.58±1.82 ng/mL at 1 h after oral administration of the oral DFZ tablets, after which DFZ concentration was remarkably decreased in the following hours. Whereas the maximum concentrations of 56.27±1.12 ng/mL and 113.77±4.30 ng/mL were recorded at 2 h for both topical free DFZ gel and topical DFZ-UENVs gel, respectively. As compared to oral drug formulation, the Frel calculated from AUC0-∞ was found to be 40.06% and 137.27%, respectively for topical free DFZ gel and topical DFZ-UENVs gel which is considered a noticeable improvement in DFZ bioavailability. This bioavailability improvement can be referring to the topical administration that bypasses the exaggerated first-pass metabolism. While higher enhancement of topical DFZ-UENVs gel, compared to topical free DFZ gel, can be explained by the permeation enhancing effect of UENVs.
Also from the data in Table 5 and Figure 6, a significant increase in the drug half-life (t1/2) of topical DFZ-UENVs formulations was obtained. The topical DFZ-UENVs showed t1/2of 7.36 ± 0.61 h, while oral administration showed 2.41 ± 0.29 h. This indicates retardation in the DFZ release rate caused by the use of gel system. On the other hand, higher retardation may be due to the dual sustained effect caused by both UENVs and gel base. In conclusion, the significant increase in AUC0-∞ and Frel of UENVs formulation indicates boosted DFZ bioavailability.
Conclusion
From our investigation, improved anti-inflammatory efficacy of the prepared nanovesicles had been emphasized. DFZ-UENVs formulations were successfully prepared, characterized, and evaluated. The optimized UENVs formulation was incorporated in semisolid hydrogel dosage form. Transdermal DFZ-UENVs gel showed favorable rheological properties required for its topical application. DFZ-UENVs gel had a significantly enhanced skin permeation and anti-inflammatory activity in rats over free DFZ gel. Topical DFZ-UENVs formulation achieved an accentuated bioavailability with sustained drug release. This study showed promising delivery of DFZ through novel UENVs-based transdermal gel and could be suggested as a good alternative to oral DFZ dosage forms already existing in market.
Disclosure
The authors report no conflicts of interest for this work.
References
1. Schleimer RP. An overview of glucocorticoid anti-inflammatory actions. Eur J Clin Pharmacol. 1993;45(1):S3–S7. doi:10.1007/BF01844196
2. Joshi N, Rajeshwari K. Deflazacort. J Postgrad Med. 2009;55(4):296. doi:10.4103/0022-3859.58942
3. Corrêa G, Bellé LP, Bajerski L, et al. Development and validation of a reversed-phase HPLC method for the determination of deflazacort in pharmaceutical dosage forms. Chromatographia. 2007;65(9–10):591–594. doi:10.1365/s10337-007-0205-y
4. Markham A, Bryson HM. Deflazacort. Drugs. 1995;50(2):317–333. doi:10.2165/00003495-199550020-00008
5. Scremin A, Piazzon M, Silva MAS, et al. Spectrophotometric and HPLC determination of deflazacort in pharmaceutical dosage forms. Brazilian J Pharmaceutical Sci. 2010;46:281–287. doi:10.1590/S1984-82502010000200015
6. Parente L. Deflazacort: therapeutic index, relative potency and equivalent doses versus other corticosteroids. BMC Pharmacol Toxicol. 2017;18(1):1. doi:10.1186/s40360-016-0111-8
7. Sperandeo N, Kassuha D. Development and validation of a dissolution test for 6 mg deflazacort tablets. Sci Pharm. 2009;77(3):679–694. doi:10.3797/scipharm.090405
8. Griggs RC, Miller JP, Greenberg CR, et al. Efficacy and safety of deflazacort vs prednisone and placebo for Duchenne muscular dystrophy. Neurology. 2016;87(20):2123–2131. doi:10.1212/WNL.0000000000003217
9. Ferraris JR, Pasqualini T, Alonso G, et al. Effects of deflazacort vs. methylprednisone: a randomized study in kidney transplant patients. Pediatric Nephrology. 2007;22(5):734–741. doi:10.1007/s00467-006-0403-0
10. Patil SK, et al. Strategies for solubility enhancement of poorly soluble drugs. Int J Pharm Sci Rev Res. 2011;8(2):74–80.
11. Cevc G, et al. Ultra-High Efficiency of Drugs and Peptide Transfer Through the Intact Skin by Means of Novel Drug Carriers, Transfersomes. STS Publishing; 1993.
12. Aziz DE, Abdelbary AA, Elassasy AI. Fabrication of novel elastosomes for boosting the transdermal delivery of diacerein: statistical optimization, ex-vivo permeation, in-vivo skin deposition and pharmacokinetic assessment compared to oral formulation. Drug Deliv. 2018;25(1):815–826.
13. Singh D, Pradhan M, Nag M, et al. Vesicular system: versatile carrier for transdermal delivery of bioactives. Artif Cells, Nanomed Biotechnol. 2015;43(4):282–290. doi:10.3109/21691401.2014.883401
14. Abdel-Hafez SM, Hathout RM, Sammour OA. Curcumin-loaded ultradeformable nanovesicles as a potential delivery system for breast cancer therapy. Colloids Surf B Biointerfaces. 2018;167:63–72. doi:10.1016/j.colsurfb.2018.03.051
15. Ahad A, Aqil M, Kohli K, et al. Enhanced transdermal delivery of an anti-hypertensive agent via nanoethosomes: statistical optimization, characterization and pharmacokinetic assessment. Int J Pharm. 2013;443(1):26–38. doi:10.1016/j.ijpharm.2013.01.011
16. Schmid-Wendtner M-H, Korting HC. The pH of the skin surface and its impact on the barrier function. Skin Pharmacol Physiol. 2006;19(6):296–302. doi:10.1159/000094670
17. Abdelbary G, El-gendy N. Niosome-encapsulated gentamicin for ophthalmic controlled delivery. AAPS Pharmscitech. 2008;9(3):740–747. doi:10.1208/s12249-008-9105-1
18. Duangjit S, Opanasopit P, Rojanarata T, et al. Evaluation of meloxicam-loaded cationic transfersomes as transdermal drug delivery carriers. AAPS PharmSciTech. 2013;14(1):133–140. doi:10.1208/s12249-012-9904-2
19. Mahmoud MO, Aboud HM, Hassan AH, et al. Transdermal delivery of atorvastatin calcium from novel nanovesicular systems using polyethylene glycol fatty acid esters: ameliorated effect without liver toxicity in poloxamer 407-induced hyperlipidemic rats. J Control Release. 2017;254:10–22. doi:10.1016/j.jconrel.2017.03.039
20. Aboud HM, Ali AA, El-Menshawe SF, et al. Nanotransfersomes of carvedilol for intranasal delivery: formulation, characterization and in vivo evaluation. Drug Deliv. 2016;23(7):2471–2481. doi:10.3109/10717544.2015.1013587
21. El Menshawe SF, Nafady MM, Aboud HM, et al. Transdermal delivery of fluvastatin sodium via tailored spanlastic nanovesicles: mitigated Freund’s adjuvant-induced rheumatoid arthritis in rats through suppressing p38 MAPK signaling pathway. Drug Deliv. 2019;26(1):1140–1154. doi:10.1080/10717544.2019.1686087
22. Lei W, Yu C, Lin H, et al. Development of tacrolimus-loaded transfersomes for deeper skin penetration enhancement and therapeutic effect improvement in vivo. Asian j Pharmaceutical Sci. 2013;8(6):336–345. doi:10.1016/j.ajps.2013.09.005
23. Aboud HM, Hassan AH, Ali AA, et al. Novel in situ gelling vaginal sponges of sildenafil citrate-based cubosomes for uterine targeting. Drug Deliv. 2018;25(1):1328–1339. doi:10.1080/10717544.2018.1477858
24. Muppidi K, Pumerantz AS, Wang J, et al. Development and stability studies of novel liposomal vancomycin formulations. ISRN Pharm. 2012;2012:2012. doi:10.5402/2012/636743
25. Aboud HM, Mahmoud MO, Abdeltawab Mohammed M, et al. Preparation and appraisal of self-assembled valsartan-loaded amalgamated Pluronic F127/Tween 80 polymeric micelles: boosted cardioprotection via regulation of Mhrt/Nrf2 and Trx1 pathways in cisplatin-induced cardiotoxicity. J Drug Target. 2020;28(3):282–299. doi:10.1080/1061186X.2019.1650053
26. Abd-Allah FI, Dawaba HM, Ahmed A. Preparation, characterization, and stability studies of piroxicam-loaded microemulsions in topical formulations. Drug Discov Ther. 2010;4(4):267–275.
27. Wasankar SR, Faizi SM, Deshmuk AD. Formulation and development of liposomal gel for topical drug delivery system. Int J Pharmaceutical Sci Res. 2012;3(11):4461.
28. Escobar P, Vera AM, Neira LF, et al. Photodynamic therapy using ultradeformable liposomes loaded with chlorine aluminum phthalocyanine against L. (V.) braziliensis experimental models. Exp Parasitol. 2018;194:45–52. doi:10.1016/j.exppara.2018.09.016
29. Shakeel F, Baboota S, Ahuja A, et al. Skin permeation mechanism and bioavailability enhancement of celecoxib from transdermally applied nanoemulsion. J Nanobiotechnology. 2008;6(1):8. doi:10.1186/1477-3155-6-8
30. El Zaafarany GM, Awad GAS, Holayel SM, et al. Role of edge activators and surface charge in developing ultradeformable vesicles with enhanced skin delivery. Int J Pharm. 2010;397(1–2):164–172. doi:10.1016/j.ijpharm.2010.06.034
31. Moawad FA, Ali AA, Salem HF. Nanotransfersomes-loaded thermosensitive in situ gel as a rectal delivery system of tizanidine HCl: preparation, in vitro and in vivo performance. Drug Deliv. 2017;24(1):252–260. doi:10.1080/10717544.2016.1245369
32. Salem HF, Kharshoum RM, Sayed OM, et al. Formulation design and optimization of novel soft glycerosomes for enhanced topical delivery of celecoxib and cupferron by Box-Behnken statistical design. Drug Dev Ind Pharm. 2018;44(11):1871–1884. doi:10.1080/03639045.2018.1504963
33. Mei Z, Wu Q, Hu S, et al. Triptolide loaded solid lipid nanoparticle hydrogel for topical application. Drug Dev Ind Pharm. 2005;31(2):161–168. doi:10.1081/DDC-200047791
34. Özgüney IS, Karasulu HY, Kantarci G, et al. Transdermal delivery of diclofenac sodium through rat skin from various formulations. AAPS Pharmscitech. 2006;7(4):E39–E45. doi:10.1208/pt070488
35. Morris CJ. Carrageenan-induced paw edema in the rat and mouse. Inflammation Protocols. 2003;115–121.
36. Elkomy MH, Elmenshawe SF, Eid HM, et al. Topical ketoprofen nanogel: artificial neural network optimization, clustered bootstrap validation, and in vivo activity evaluation based on longitudinal dose response modeling. Drug Deliv. 2016;23(9):3294–3306. doi:10.1080/10717544.2016.1176086
37. Escudero A, Marín P, Cárceles CM, et al. Pharmacokinetics of deflazacort in rabbits after intravenous and oral administration and its interaction with erythromycin. J Vet Pharmacol Ther. 2018;41(1):e10–e15. doi:10.1111/jvp.12442
38. Özkan Y, Savaşer A, Taş Ç, et al. Drug dissolution studies and determination of deflazacort in pharmaceutical formulations and human serum samples by RP‐HPLC. J Liq Chromatogr Relat Technol. 2003;26(13):2141–2156. doi:10.1081/JLC-120022399
39. Holm R, Jensen I, Sonnergaard J. Optimization of self-microemulsifying drug delivery systems (SMEDDS) using a D-optimal design and the desirability function. Drug Dev Ind Pharm. 2006;32(9):1025–1032. doi:10.1080/03639040600559024
40. Berger MP, Wong W-K. Applied Optimal Designs. John Wiley & Sons; 2005.
41. Abdel-Hafez SM, Hathout RM, Sammour OA. Towards better modeling of chitosan nanoparticles production: screening different factors and comparing two experimental designs. Int J Biol Macromol. 2014;64:334–340. doi:10.1016/j.ijbiomac.2013.11.041
42. Franceschini G, Macchietto S. Model-based design of experiments for parameter precision: state of the art. Chem Eng Sci. 2008;63(19):4846–4872. doi:10.1016/j.ces.2007.11.034
43. Hussain A, Singh S, Sharma D, et al. Elastic liposomes as novel carriers: recent advances in drug delivery. Int J Nanomedicine. 2017;12:5087. doi:10.2147/IJN.S138267
44. Aggarwal N, Goindi S. Preparation and evaluation of antifungal efficacy of griseofulvin loaded deformable membrane vesicles in optimized guinea pig model of Microsporum canis—Dermatophytosis. Int J Pharm. 2012;437(1):277–287. doi:10.1016/j.ijpharm.2012.08.015
45. Chaudhary H, Kohli K, Kumar V. Nano-transfersomes as a novel carrier for transdermal delivery. Int J Pharm. 2013;454(1):367–380. doi:10.1016/j.ijpharm.2013.07.031
46. Singh S, et al. The role of surfactants in the formulation of elastic liposomal gels containing a synthetic opioid analgesic. Int J Nanomedicine. 2016;11:1475.
47. Aggarwal D, Garg A, Kaur IP. Development of a topical niosomal preparation of acetazolamide: preparation and evaluation. J Pharmacy Pharmacol. 2004;56(12):1509–1517. doi:10.1211/0022357044896
48. Abdelkader H, et al. Preparation of niosomes as an ocular delivery system for naltrexone hydrochloride: physicochemical characterization. Die Pharmazie Int J Pharmaceutical Sci. 2010;65(11):811–817.
49. Ali MH, Moghaddam B, Kirby DJ, et al. The role of lipid geometry in designing liposomes for the solubilisation of poorly water soluble drugs. Int J Pharm. 2013;453(1):225–232. doi:10.1016/j.ijpharm.2012.06.056
50. Bunker A, Magarkar A, Viitala T. Rational design of liposomal drug delivery systems, a review: combined experimental and computational studies of lipid membranes, liposomes and their PEGylation. Biochimica Et Biophysica Acta (BBA)-Biomembranes. 2016;1858(10):2334–2352. doi:10.1016/j.bbamem.2016.02.025
51. Bnyan R, Khan I, Ehtezazi T, et al. Surfactant effects on lipid-based vesicles properties. J Pharm Sci. 2018;107(5):1237–1246. doi:10.1016/j.xphs.2018.01.005
52. Abd-Elal RM, Shamma RN, Rashed HM, et al. Trans-nasal zolmitriptan novasomes: in-vitro preparation, optimization and in-vivo evaluation of brain targeting efficiency. Drug Deliv. 2016;23(9):3374–3386. doi:10.1080/10717544.2016.1183721
53. Elnaggar YS, et al. Lecithin-based nanostructured gels for skin delivery: an update on state of art and recent applications. J Controlled Release. 2014;180:10–24. doi:10.1016/j.jconrel.2014.02.004
54. Mishra D, et al. Elastic liposomes mediated transdermal delivery of an anti-hypertensive agent: propranolol hydrochloride. J Pharm Sci. 2007;96(1):145–155. doi:10.1002/jps.20737
55. El-Refaie WM, Elnaggar YSR, El-Massik MA, et al. Novel curcumin-loaded gel-core hyaluosomes with promising burn-wound healing potential: development, in-vitro appraisal and in-vivo studies. Int J Pharm. 2015;486(1–2):88–98. doi:10.1016/j.ijpharm.2015.03.052
56. Malakar J, Sen SO, Nayak AK, et al. Formulation, optimization and evaluation of transferosomal gel for transdermal insulin delivery. Saudi Pharmaceutical j. 2012;20(4):355–363. doi:10.1016/j.jsps.2012.02.001
57. El-Laithy HM, Shoukry O, Mahran LG. Novel sugar esters proniosomes for transdermal delivery of vinpocetine: preclinical and clinical studies. European j Pharmaceutics Biopharmaceutics. 2011;77(1):43–55. doi:10.1016/j.ejpb.2010.10.011
58. Zheng W-S, et al. Preparation and quality assessment of itraconazole transfersomes. Int J Pharm. 2012;436(1–2):291–298. doi:10.1016/j.ijpharm.2012.07.003
59. Jain S, et al. Transfersomes—a novel vesicular carrier for enhanced transdermal delivery: development, characterization, and performance evaluation. Drug Dev Ind Pharm. 2003;29(9):1013–1026. doi:10.1081/DDC-120025458
60. Al-mahallawi AM, Abdelbary AA, Aburahma MH. Investigating the potential of employing bilosomes as a novel vesicular carrier for transdermal delivery of tenoxicam. Int J Pharm. 2015;485(1):329–340. doi:10.1016/j.ijpharm.2015.03.033
61. Ruckmani K, Sankar V. Formulation and optimization of zidovudine niosomes. AAPS Pharmscitech. 2010;11(3):1119–1127. doi:10.1208/s12249-010-9480-2
62. Mekkawy A, Fathy M, El-Shanawany S. Formulation and in vitro evaluation of fluconazole topical gels. British Journal of Pharmaceutical Research. 2013;3:293–313. doi:10.9734/BJPR/2013/2775
63. Al-mahallawi AM, Khowessah OM, Shoukri RA. Nano-transfersomal ciprofloxacin loaded vesicles for non-invasive trans-tympanic ototopical delivery: in-vitro optimization, ex-vivo permeation studies, and in-vivo assessment. Int J Pharm. 2014;472(1–2):304–314. doi:10.1016/j.ijpharm.2014.06.041
64. Cipolla D, et al. Modifying the release properties of liposomes toward personalized medicine. J Pharm Sci. 2014;103(6):1851–1862. doi:10.1002/jps.23969
65. Elsayed MM, Abdallah OY, Naggar VF, et al. Deformable liposomes and ethosomes: mechanism of enhanced skin delivery. Int J Pharm. 2006;322(1–2):60–66. doi:10.1016/j.ijpharm.2006.05.027
66. Zylberberg C, Matosevic S. Pharmaceutical liposomal drug delivery: a review of new delivery systems and a look at the regulatory landscape. Drug Deliv. 2016;23(9):3319–3329. doi:10.1080/10717544.2016.1177136
67. Mokhtar M, Sammour OA, Hammad MA, et al. Effect of some formulation parameters on flurbiprofen encapsulation and release rates of niosomes prepared from proniosomes. Int J Pharm. 2008;361(1):104–111. doi:10.1016/j.ijpharm.2008.05.031
68. Panwar P, et al. Preparation, characterization, and in vitro release study of albendazole-encapsulated nanosize liposomes.. Int J Nanomedicine. 2010;5(101):8. doi:10.2147/ijn.s8030
69. Ghanbarzadeh S, Arami S. Enhanced transdermal delivery of diclofenac sodium via conventional liposomes, ethosomes, and transfersomes. Biomed Res Int. 2013;2013:1–7. doi:10.1155/2013/616810
70. Shaji J, Lal M. Preparation, Optimization and evaluation of transferosomal formulation for enhanced transdermal delivery of a COX-2 Inhibitor. Int J Pharmacy Pharmaceutical Sci. 2014;6(1):467–477.
71. Naguib SS, Hathout RM, Mansour S. Optimizing novel penetration enhancing hybridized vesicles for augmenting the in-vivo effect of an anti-glaucoma drug. Drug Deliv. 2017;24(1):99–108. doi:10.1080/10717544.2016.1233588
72. Khalil RM, Abdelbary A, Kocova El-Arini S, et al. Evaluation of bilosomes as nanocarriers for transdermal delivery of tizanidine hydrochloride: in vitro and ex vivo optimization. J Liposome Res. 2019;29(2):171–182. doi:10.1080/08982104.2018.1524482
73. El-Alim SHA, Kassem AA, Basha M, et al. Comparative study of liposomes, ethosomes and transfersomes as carriers for enhancing the transdermal delivery of diflunisal: in vitro and in vivo evaluation. Int J Pharm. 2019;563:293–303. doi:10.1016/j.ijpharm.2019.04.001
74. Varma VNSK, Maheshwari PV, Navya M, et al. Calcipotriol delivery into the skin as emulgel for effective permeation. Saudi Pharmaceutical J. 2014;22(6):591–599. doi:10.1016/j.jsps.2014.02.007
75. Ammar HO, Salama HA, Ghorab M, et al. Nanoemulsion as a potential ophthalmic delivery system for dorzolamide hydrochloride. AAPS Pharmscitech. 2009;10(3):808. doi:10.1208/s12249-009-9268-4
76. Sharma VK, Sarwa KK, Mazumder B. Fluidity enhancement: a critical factor for performance of liposomal transdermal drug delivery system. J Liposome Res. 2014;24(2):83–89. doi:10.3109/08982104.2013.847956
77. Wakaskar RR. General overview of lipid–polymer hybrid nanoparticles, dendrimers, micelles, liposomes, spongosomes and cubosomes. J Drug Target. 2018;26(4):311–318. doi:10.1080/1061186X.2017.1367006
78. Cosco D, et al. Ultradeformable liposomes as multidrug carrier of resveratrol and 5-fluorouracil for their topical delivery. Int J Pharm. 2015;489(1–2):1–10. doi:10.1016/j.ijpharm.2015.04.056
79. El Menshawe SF, et al. A novel nanogel loaded with chitosan decorated bilosomes for transdermal delivery of terbutaline sulfate: artificial neural network optimization, in vitro characterization and in vivo evaluation. Drug Deliv Transl Res. 2020;10(2):471–485.
80. Mali N, Darandale S, Vavia P. Niosomes as a vesicular carrier for topical administration of minoxidil: formulation and in vitro assessment. Drug Deliv Transl Res. 2013;3(6):587–592. doi:10.1007/s13346-012-0083-1
81. Balakrishnan P, Shanmugam S, Lee WS, et al. Formulation and in vitro assessment of minoxidil niosomes for enhanced skin delivery. Int J Pharm. 2009;377(1):1–8. doi:10.1016/j.ijpharm.2009.04.020
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.