")
Back to Journals » OncoTargets and Therapy » Volume 12
Response and acquired resistance to savolitinib in a patient with pulmonary sarcomatoid carcinoma harboring MET exon 14 skipping mutation: a case report
Authors Han S, Fang J, Lu S , Wang L , Li J, Cheng M , Ren Y, Su W
Received 29 March 2019
Accepted for publication 16 July 2019
Published 6 September 2019 Volume 2019:12 Pages 7323—7328
DOI https://doi.org/10.2147/OTT.S210365
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Geoffrey Pietersz
Sen Han,1 Jian Fang,1 Shun Lu,2 Linfang Wang,3 Jing Li,3 Min Cheng,3 Yongxin Ren,3 Weiguo Su3
1Department of Thoracic Oncology Ii, Peking University Cancer Hospital & Institute, Beijing, People’s Republic of China; 2Shanghai Lung Cancer Center, Shanghai Chest Hospital, Shanghai Jiao Tong University, Shanghai, People’s Republic of China; 3Hutchison MediPharma Limited, Shanghai, People’s Republic of China
Correspondence: Jian Fang
Department of Thoracic Oncology II, Peking University Cancer Hospital & Institute, 52 Fucheng Road, Haidian District, Beijing 100142, People’s Republic of China
Tel +86 108 819 6492
Fax +86 10 8 819 6478
Email [email protected]
Background: Pulmonary sarcomatoid carcinoma (PSC) is a rare and poorly differentiated type of non-small cell lung cancer (NSCLC) with specific characteristics, which usually presents a challenge in clinical practice. Mesenchymal–epithelial transition (MET) gene has been identified as a promising target for treatments in the past few years. Here, we report a case of a patient with PSC harboring MET exon 14 mutation, who responded to a novel MET inhibitor – savolitinib.
Case presentation: A 75-year-old male patient with symptoms of cough, dyspnea and intermittent chest pain was diagnosed with sarcomatoid carcinoma. The tumor involved the right lung, the right hilum and multiple lesions in the right pleura, indicating a clinical disease stage IV. Next-generation sequencing of lung biopsy specimen indicated a MET exon 14 skipping mutation (NM_000245:c.3028+3A>G), with a variant allele frequency of 73.9%. The patient achieved a rapid and durable partial response with the initiation of savolitinib administration (600 mg, orally, once daily). The progression-free survival in this patient was 36 weeks. There were no ≥grade 3 adverse events reported and there was no dose reduction during treatment. Following savolitinib treatment, the allele frequency of MET exon 14 mutation in plasma circulating tumor DNA decreased with the reduction in tumor size. At the time of disease progression, fibroblast growth factor receptor 1 (FGFR1), EGFR and KRAS gene amplification were newly identified in tumor biopsy sample.
Conclusion: This patient with PSC harboring MET exon 14 skipping mutation achieved significant clinical benefit with savolitinib treatment. Emergence of FGFR1, EGFR and KRAS gene amplification at the time of disease progression was likely responsible for the resistance to savolitinib in this case.
Keywords: targeted therapy, tumor response, acquired resistance, lung cancer
Background
Pulmonary sarcomatoid carcinoma (PSC) is a rare and poorly differentiated type of non-small-cell lung cancer (NSCLC), which accounts for less than 1% of all lung cancers.1 Both carcinomatous and sarcomatous elements coexist in the tumor tissue. According to the WHO classification, PSC includes the following categories: spindle cell carcinoma (a carcinoma consisting of almost only epithelial spindle cells), giant cell carcinoma (a carcinoma almost purely consisting of tumor giant cells), pleomorphic carcinoma (a poorly differentiated NSCLC which is composed of at least 10% spindle and/or giant cells), carcinosarcoma (a malignant tumor consisting of a mixture of NSCLC and true sarcoma) and pulmonary blastoma (a biphasic tumor composed of fetal adenocarcinoma and primitive mesenchymal stroma).2 Compared with other histologic subtypes, PSC behaves in an aggressive fashion and is associated with poor prognosis, even at an early stage.3,4 While disease symptoms generally resemble the other types of NSCLC, PSC is associated with certain clinical factors such as older age at diagnosis (mostly in the sixth/seventh decades), smoker predominance and bulky tumors.5–7 It has been reported that a substantial proportion of patients with PSC have advanced disease at initial diagnosis.8,9 Current evidence shows that the median survival time in patients with advanced PSC is worse than for other histologic subtypes of NSCLC. According to the analysis of National Cancer Database and the Surveillance, Epidemiology, and End Results database, the median survival time for stage IV disease ranges from 3.0 to 5.4 months.1,8 In addition to the highly progressive clinical behavior of PSC, the high rate of resistance to platinum-based chemotherapy could be another reason for the poor prognosis of this disease.10 The rarity of PSC, leading to the small sample size of genomic profiling investigations, brings difficulties for molecule characterization of this disease. Variable frequency of targetable driver mutation, such as the EGFR gene or anaplastic lymphoma kinase (ALK) gene, has been reported in PSC. For example, Liu et al reported no EGFR mutation in 36 PSC samples,11 while Kaira et al identified approximately 20% (3/17) EGFR mutation.12 Compared to EGFR and ALK, a higher frequency of mesenchymal–epithelial transition (MET) gene alteration in PSC has been reported in the literature.11,13
MET is the tyrosine kinase receptor for hepatocyte growth factor and plays an important role in embryogenesis, tumor growth and metastasis. MET gene alterations include amplification, activating point mutation and MET exon 14 skipping. MET exon 14 is responsible for encoding intracellular MET juxtamembrane domain, which is required for the recruitment of the ubiquitin ligase and mediates the degradation of MET protein by ubiquitin pathway.14 Somatic splice sites alterations at MET exon 14, resulting in exon skipping and the loss of MET exon 14, would increase MET stability and enhance the oncogenic potential of MET gene. MET exon 14 skipping has been reported in approximately 1–3% of NSCLC cases,15 but a higher incidence (ranging from 9.5% to 22%) has been observed in the PSC subtype.11,13 MET exon 14 skipping appears mutually exclusive of other targetable genetic alterations, such as EGFR, ALK and ROS1.11 Evidence from early clinical trials of MET inhibitors, such as crizotinib, cabozantinib and tepotinib, shows a good response to these agents by NSCLC patients with MET exon 14 skipping,16 suggesting that MET exon 14 is an attractive target for the treatment of lung cancer in these patients. Savolitinib (also known as AZD6094, HMPL-504 and volitinib) is a novel, oral, potent and highly selective MET inhibitor. In preclinical studies, savolitinib displayed potent activity in vitro against MET kinase and the downstream signaling targets.17 Savolitinib is currently being investigated in several solid tumors, including renal cancer, NSCLC and gastric cancer. Here, we present a case of a patient with PSC harboring MET exon 14 skipping mutation treated with savolitinib in a Phase II clinical trial.
Case report
A 75-year-old male patient presented to the Peking University Cancer Hospital & Institute in September 2017, with symptoms of cough, dyspnea and intermittent chest pain. The patient had been a heavy smoker for 40 years, having smoked 40 cigarettes per day. CT revealed a lesion with the longest diameter of 5.6 cm in the right inferior lobe, enlarged lymph nodes in the right hilum and multiple metastatic lesions in the right pleura. Pathologic examination of the lung lesion biopsy indicated stage IV (T3N1M1) sarcomatoid carcinoma, possibly transformed from adenocarcinoma. Next-generation sequencing (NGS) of the lung biopsy specimen indicated a MET exon 14 skipping mutation (NM_000245:c.3028+3A>G), with a variant allele frequency (VAF) of 73.9%, which could potentially cause alternative splicing of the MET protein. Furthermore, NGS showed MET amplification, TP53 (V173M) mutation, FBXW7 (G459R) mutation, but no EGFR, KRAS, ALK or ROS1 gene alterations were detected. The patient refused to receive chemotherapy.
After signing a written informed consent, the patient was enrolled in a Phase II clinical trial of savolitinib treating patients with unresectable or metastatic PSC or other NSCLC harboring MET exon 14 skipping mutation (NCT02897479), which had been approved by the ethical committee of the hospital. In November 2017, the patient started treatment with savolitinib, 600 mg orally, once daily. After 6 weeks of treatment, the first tumor evaluation showed a partial response (target lesion shrunk from 5.6 cm at baseline to 3 cm) (Figure 1), based on the Response Evaluation Criteria in Solid Tumors version 1.1 and the patient’s Eastern Cooperative Oncology Group performance status, which improved from 1 to 0. This partial response was confirmed in the next tumor evaluation at week 12. All reported treatment-related adverse events (AEs) were grade 1, including white blood cell count decrease, nausea, diarrhea and rash. The severity of AEs was between grade 1 and grade 2, as per the Common Terminology Criteria for Adverse Events version 4.03. The patient had a dose interruption of 5 days due to pyrexia (unrelated to treatment). No dose reduction occurred. After 36 weeks of treatment, the patient had disease progression with target lesion increasing to 3.8 cm, which led to treatment discontinuation. The progression-free survival with savolitinib treatment for this patient was 36 weeks.
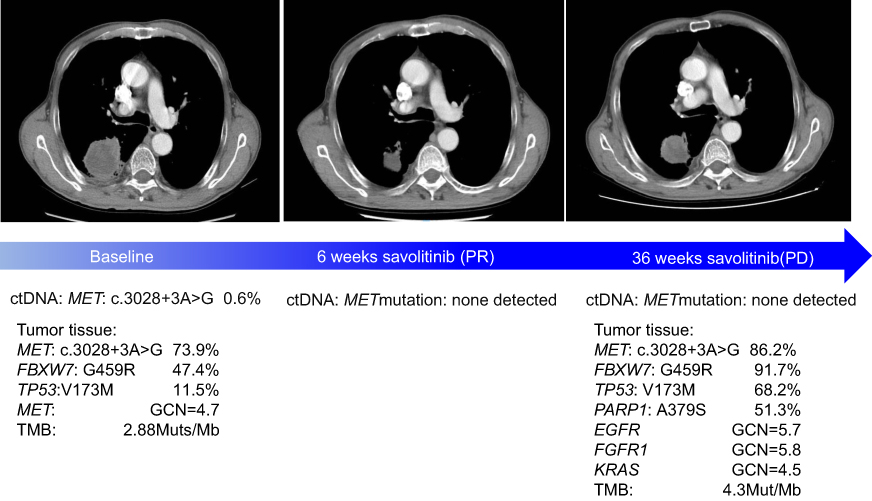 |
Figure 1 Radiological assessments of tumor response and molecular profiling in ctDNA and tumor tissue.Abbreviation: ctDNA, circulating tumor DNA. |
The patient consented to participate in the biomarker exploration of this study. The plasma circulating tumor DNA (ctDNA) samples were serially obtained from baseline until disease progression and tumor biopsy sample was collected at disease progression. An NGS panel targeting 422 cancer-relevant genes was used to investigate the DNA alterations in the tumor biopsy and plasma samples.18
At baseline, prior to savolitinib treatment, the patient’s ctDNA showed a low level of MET exon 14 mutation (c.3028+3A>G) with VAF of 0.6%. After 6 weeks of treatment, with the tumor size reduction, the VAF of MET mutation dropped to below the limit of detection and remained undetected in subsequent cycles at weeks 18, 24, 30 and even at time of progression (week 36) (Figure 1). Except for MET exon 14 mutation, no other somatic mutation was found in ctDNA samples. The tumor biopsy performed at disease progression showed the same mutation sites in MET, TP53 and FBXW7 genes found prior to savolitinib treatment. In addition, moderate levels of gene copy numbers of EGFR (GCN =5.7), FGFR1 (GCN =5.8) and KRAS (GCN =4.5) were newly identified by NGS at disease progression and were further confirmed using fluorescence in situ hybridization technology (Figure 2). This result suggests that the acquired gene amplification might be associated with resistance to savolitinib. Interestingly, MET amplification was not detected by NGS in tumor biopsy specimen at disease progression (Figures 1 and 2).
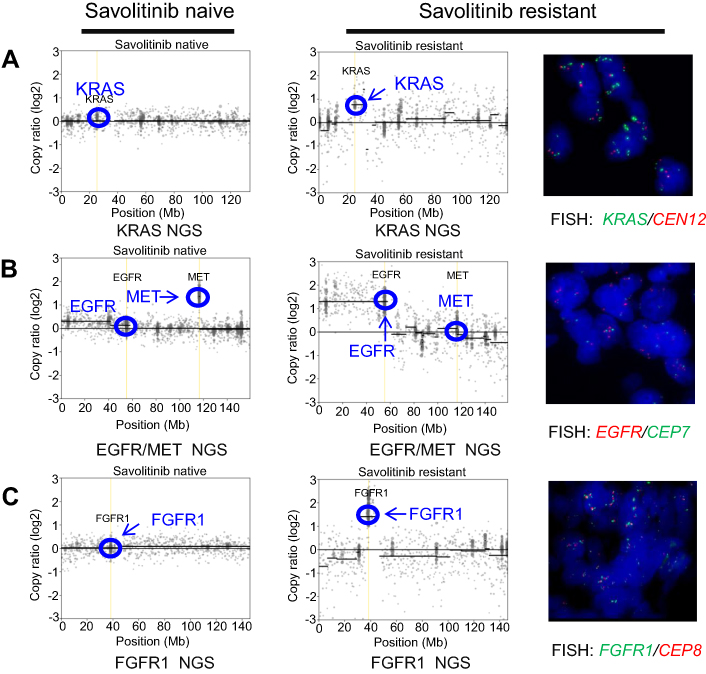 |
Figure 2 Copy number profile of tumor tissue. Left column: baseline; Right column: after 36 weeks at time of progression.Notes: (A) Next-generation sequencing (NGS, left panels) copy number plots of the KRAS locus on chromosome 12 from savolitinib-naïve and savolitinib-resistant tumors. Fluorescence in situ hybridization (FISH) for KRAS (red) and centromere 12 (CEN12, green) is shown for savolitinib-resistant tumor. (B) NGS copy number plots of EGFR/MET locus on chromosome 7 from savolitinib-naïve and savolitinib-resistant tumors. FISH for EGFR (red) and centromere 7 (CEP7, green) is shown for savolitinib-resistant tumor. (C) NGS copy number plots of FGFR1 locus on chromosome 8 from savolitinib-naïve and savolitinib-resistant tumors. FISH for FGFR1 (green) and centromere 8 (CEP8, red) is shown for savolitinib-resistant tumor.Abbreviations: NGS, next-generation sequencing; EGFR, epidermal growth factor receptor; MET, mesenchymal–epithelial transition; FISH, fluorescence in situ hybridization; CEN12, centromere 12; FGFR, fibroblast growth factor receptor; CEP7, chromosome enumeration probe 7; CEP8, chromosome enumeration probe 8. |
In September 2018, after discontinuation of savolitinib, crizotinib 250 mg, orally, twice daily was initiated, as second-line treatment. After 8 weeks of treatment, the patient experienced disease progression with an increase in the primary lung lesion, significant increase in pleural effusion and new lesions in adrenal glands, identified in December 2018. He suffered from decreased appetite, fatigue and hypoalbuminemia. In January 2019, the patient died of cachexia.
Discussion
To our knowledge, this is the first case report describing a clinical response to savolitinib in a patient with PSC harboring MET exon 14 skipping mutation. The rapid and durable response in this case demonstrated the potent inhibition of MET by savolitinib, accompanied by the general performance status improvement after treatment. Treatment tolerability was acceptable in this case, without reported grade 3 or greater AEs and no need for dose reduction. Previous reports showed antitumor activity of MET inhibitors (eg, crizotinib, and capmatinib) in lung adenocarcinomas, also including the PSC subtype. Our case further demonstrated that PSC also responds to a MET inhibitor, if the target is effectively suppressed, indicating that MET exon 14 might be a driver gene in NSCLC regardless of histopathology subtypes. In this case, MET exon 14 alteration was the only somatic mutation detected in ctDNA samples and the VAF decreased significantly when a partial response was achieved. This trend of VAF change was concordant with the tumor size changes, which might serve as a probe of response in the clinical setting. The lack of VAF increase in MET mutation at the time of disease progression might be explained by the development of acquired resistance driving tumor growth independent of MET gene status, whilst MET was still effectively suppressed by exposure to savolitinib.
Two major mechanisms of resistance have been reported in crizotinib-treated NSCLC patients carrying MET exon 14 mutations: one is the “on-target” mechanism, which is the development of secondary MET alteration in kinase domain, such as D1228N and Y1230S, and the other is “by-pass tract activation”, such as KRAS amplification.19 In our case, the “by-pass tract activation” was observed: concurrent FGFR1, EGFR and KRAS amplification appearing at disease progression. KRAS amplification was reported in preclinical studies to induce constitutive activation of the Ras/mitogen activated protein kinase (MAPK) signaling and consequently confer resistance to crizotinib.19 EGFR amplification was also identified in some crizotinib-resistance cases19; however, no further functional studies to link this gene alteration with the response to MET inhibitors. To our knowledge, this is the first report that suggests that FGFR1 amplification might be associated with acquired resistance to a selective MET inhibitor in NSCLC. Further investigation is warranted to test whether these genetic alterations occur in the same tumor cell or different tumor cells and whether FGFR1, EGFR or MAPK pathway inhibition could restore the sensitivity to savolitinib treatment. Interestingly, in our case, TP53 gene mutation was detected both before treatment and at the disease progression. TP53 is the most common gene mutation in multiple cancer types, including PSC. In general, tumors harboring TP53 mutations often demonstrate high levels of chromosome instability,20 especially the mutations located in the DNA-binding domain, like V173M in our case. We hypothesized that chromosome instability might be associated with multiple gene amplifications, such as EGFR, KRAS and FGFR1, during savolitinib treatment, consequently resulting in acquired resistance. Further research into this association is needed.
There are few reported studies which investigated subsequent treatment following the disease progression on a MET inhibitor. Compared to type I MET inhibitor (eg, crizotinib, capmatinib and savolitinib), type II MET inhibitors (eg, cabozantinib, glesatinib) differ in their binding modes and selectivity profiles and tend to have activity against multiple kinases.21 An in vitro study demonstrated that MET D1228 and Y1230 mutations were moderately resistant to crizotinib and strongly resistant to both savolitinib and capmatinib, while they maintained sensitivity to type II tyrosine kinase inhibitors (TKIs).22 A case of a patient with MET exon 14 skipping mutation who experienced durable tumor response with cabozantinib treatment after disease progression on crizotionib has recently been reported.23 These observations need to be further validated in clinical trials, to test the efficacy of type II MET inhibitors in NSCLC population resistant to type I MET inhibitors.
Another approach to overcome acquired resistance may be combining MET inhibitors with other targeted therapies, such as TKIs targeting EGFR, FGFR or MAPK pathway inhibitors suggested in our case. A combination of a MET inhibitor and immunotherapy is another option for further investigation. A recent study reported a high level of PD-L1 expression in 46% (25/54) of NSCLC harboring MET exon 14 alterations.24 MET overexpression accompanied by high PD-L1 expression has been observed in other solid tumors.25 Based on these observations, further research is warranted to explore the optimal treatment combinations for patients with NSCLC refractory to prior treatment with MET inhibitors. Furthermore, the added toxicity of these combinations needs to be well characterized and managed in the clinical trial setting.
Conclusion
In summary, we report a clinical response to savolitinib in a PSC patient carrying MET exon 14 skipping mutation. Our observation suggests that savolitinib might be of clinical benefit to PSC patients with MET exon 14 skipping mutation; however, the efficacy and safety of savolitinib need to be further validated in clinical trials. Resistance to treatment with savolitinib in this case was likely due to the emergence of FGFR1, EGFR and KRAS gene amplification; however, further studies are needed to confirm the functions of these gene alterations and their impacts on the efficacy of savolitinib.
Ethics statement
This phase 2 clinical trial of savolitinib in lung cancer (NCT02897479) was approved by the ethical committee of Peking University Cancer Hospital & Institute. The patient signed a written informed consent to participate in the trial.
Consent for publication
Documented written consent for the publication of the data was obtained from the patient. The sponsor agreed to the publication of this case report.
Acknowledgments
This work was supported by Hutchison MediPharma Limited.
Author contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
Shun Lu received speakers’ fees from AstraZeneca and Roche, served in a consulting/advisory role for AstraZeneca, Boehringer Ingelheim, Roche, Simcere and Hutchison MediPharma Limited, and received research funding from AstraZeneca, Hutchison MediPharma and Roche. Linfang Wang, Jing Li, Min Cheng, Yongxin Ren, and Weiguo Su are employees of Hutchison MediPharma Limited. Weiguo Su received stock from Hutchison Chi-Med. The phase 2 clinical trial of savolitinib in lung cancer (NCT02897479) is sponsored by Hutchison MediPharma Limited. The authors report no other conflicts of interest in this work.
References
1. Yendamuri S, Caty L, Pine M, et al. Outcomes of sarcomatoid carcinoma of the lung: a Surveillance, Epidemiology, and End Results database analysis. Surgery. 2012;152(3):397–402. doi:10.1016/j.surg.2012.05.007
2. Travis WD, Brambilla E, Nicholson AG, et al. The 2015 World Health Organization classification of lung tumors: impact of genetic, clinical and radiologic advances since the 2004 classification. J Thorac Oncol. 2015;10(9):1243–1260. doi:10.1097/JTO.0000000000000630
3. Martin LW, Correa AM, Ordonez NG, et al. Sarcomatoid carcinoma of the lung: a predictor of poor prognosis. Ann Thorac Surg. 2007;84(3):973–980. doi:10.1016/j.athoracsur.2007.03.099
4. Roesel C, Terjung S, Weinreich G, et al. Sarcomatoid carcinoma of the lung: a rare histological subtype of non-small cell lung cancer with a poor prognosis even at earlier tumour stages. Interact Cardiovasc Thorac Surg. 2017;24(3):407–413. doi:10.1093/icvts/ivw281
5. Gu L, Xu Y, Chen Z, Pan Y, Lu S. Clinical analysis of 95 cases of pulmonary sarcomatoid carcinoma. Biomed Pharmacother. 2015;76:134–140. doi:10.1016/j.biopha.2015.10.009
6. Ung M, Rouquette I, Filleron T, et al. Characteristics and clinical outcomes of sarcomatoid carcinoma of the lung. Clin Lung Cancer. 2016;17(5):391–397. doi:10.1016/j.cllc.2016.03.001
7. Maneenil K, Xue Z, Liu M, et al. Sarcomatoid carcinoma of the lung: the Mayo clinic experience in 127 patients. Clin Lung Cancer. 2018;19(3):e323–e33. doi:10.1016/j.cllc.2017.12.008
8. Steuer CE, Behera M, Liu Y, et al. Pulmonary sarcomatoid carcinoma: an analysis of the National Cancer Database. Clin Lung Cancer. 2017;18(3):286–292. doi:10.1016/j.cllc.2016.11.016
9. Rahouma M, Kamel M, Narula N, et al. Pulmonary sarcomatoid carcinoma: an analysis of a rare cancer from the Surveillance, Epidemiology, and End Results database. Eur J Cardiothorac Surg. 2018;53(4):828–834. doi:10.1093/ejcts/ezx417
10. Vieira T, Girard N, Ung M, et al. Efficacy of first-line chemotherapy in patients with advanced lung sarcomatoid carcinoma. J Thorac Oncol. 2013;8(12):1574–1577. doi:10.1097/JTO.0b013e318287c562
11. Liu X, Jia Y, Stoopler MB, et al. Next-generation sequencing of pulmonary sarcomatoid carcinoma reveals high frequency of actionable MET gene mutations. J Clin Oncol. 2016;34(8):794–802. doi:10.1200/JCO.2015.62.0674
12. Kaira K, Horie Y, Ayabe E, et al. Pulmonary pleomorphic carcinoma: a clinicopathological study including EGFR mutation analysis. J Thorac Oncol. 2010;5(4):460–465. doi:10.1097/JTO.0b013e3181ce3e3c
13. Kim EK, Kim KA, Lee CY, et al. Molecular diagnostic assays and clinicopathologic implications of MET exon 14 skipping mutation in non-small-cell lung cancer. Clin Lung Cancer. 2019;20(1):e123–e32. doi:10.1016/j.cllc.2018.10.004
14. Ma PC, Kijima T, Maulik G, et al. c-MET mutational analysis in small cell lung cancer: novel juxtamembrane domain mutations regulating cytoskeletal functions. Cancer Res. 2003;63(19):6272–6281.
15. Vuong HG, Ho ATN, Altibi AMA, Nakazawa T, Katoh R, Kondo T. Clinicopathological implications of MET exon 14 mutations in non-small cell lung cancer - a systematic review and meta-analysis. Lung Cancer. 2018;123:76–82. doi:10.1016/j.lungcan.2018.07.006
16. Miranda O, Farooqui M, Siegfried JM. Status of agents targeting the HGF/c-MET axis in lung cancer. Cancers (Basel). 2018;10(9):
17. Gavine PR, Ren Y, Han L, et al. Volitinib, a potent and highly selective c-MET inhibitor, effectively blocks c-MET signaling and growth in c-MET amplified gastric cancer patient-derived tumor xenograft models. Mol Oncol. 2015;9(1):323–333. doi:10.1016/j.molonc.2015.05.003
18. Yang Z, Yang N, Ou Q, et al. Investigating novel resistance mechanisms to third-generation EGFR tyrosine kinase inhibitor osimertinib in non-small cell lung cancer patients. Clin Cancer Res. 2018;24(13):3097–3107. doi:10.1158/1078-0432.CCR-17-2310
19. Bahcall M, Awad MM, Sholl LM, et al. Amplification of wild-type KRAS imparts resistance to crizotinib in MET exon 14 mutant non-small cell lung cancer. Clin Cancer Res. 2018;24(23):5963–5976. doi:10.1158/1078-0432.CCR-18-0876
20. Eischen CM. Genome stability requires p53. Cold Spring Harb Perspect Med. 2016;6(6):a026096. doi:10.1101/cshperspect.a026096
21. Yap TA, Popat S. Targeting MET exon 14 skipping alterations: has lung cancer MET its match? J Thorac Oncol. 2017;12(1):12–14. doi:10.1016/j.jtho.2016.09.002
22. Bahcall M, Sim T, Paweletz CP, et al. Acquired METD1228V mutation and resistance to MET inhibition in lung cancer. Cancer Discov. 2016;6(12):1334–1341. doi:10.1158/2159-8290.CD-16-0686
23. Klempner SJ, Borghei A, Hakimian B, Ali SM, Ou SI. Intracranial activity of cabozantinib in MET exon 14-positive NSCLC with brain metastases. J Thorac Oncol. 2017;12(1):152–156. doi:10.1016/j.jtho.2016.09.002
24. Sabari JK, Leonardi GC, Shu CA, et al. PD-L1 expression, tumor mutational burden, and response to immunotherapy in patients with MET exon 14 altered lung cancers. Ann Oncol. 2018;29(10):2085–2091. doi:10.1093/annonc/mdx807
25. Kammerer-Jacquet SF, Medane S, Bensalah K, et al. Correlation of c-MET expression with PD-L1 expression in metastatic clear cell renal cell carcinoma treated by sunitinib First-Line Therapy. Target Oncol. 2017;12(4):487–494. doi:10.1007/s11523-017-0498-1
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.