")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 11 » Issue 1
Resolvin D1 prevents smoking-induced emphysema and promotes lung tissue regeneration
Authors Kim K, Park TS , Kim Y, Lee JS, Oh YM , Lee SD, Lee SW
Received 9 November 2015
Accepted for publication 7 March 2016
Published 27 May 2016 Volume 2016:11(1) Pages 1119—1128
DOI https://doi.org/10.2147/COPD.S100198
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Richard Russell
Kang-Hyun Kim,1 Tai Sun Park,2,3 You-Sun Kim,1,3 Jae Seung Lee,2,3 Yeon-Mok Oh,2,3 Sang-Do Lee,2,3 Sei Won Lee2,3
1Asan Institute for Life Sciences, 2Department of Pulmonology and Critical Care Medicine, Clinical Research Center for Chronic Obstructive Airway Diseases, Asan Medical Center, 3Department of Pulmonology and Critical Care Medicine, University of Ulsan College of Medicine, Seoul, Republic of Korea
Purpose: Emphysema is an irreversible disease that is characterized by destruction of lung tissue as a result of inflammation caused by smoking. Resolvin D1 (RvD1), derived from docosahexaenoic acid, is a novel lipid that resolves inflammation. The present study tested whether RvD1 prevents smoking-induced emphysema and promotes lung tissue regeneration.
Materials and methods: C57BL/6 mice, 8 weeks of age, were randomly divided into four groups: control, RvD1 only, smoking only, and smoking with RvD1 administration. Four different protocols were used to induce emphysema and administer RvD1: mice were exposed to smoking for 4 weeks with poly(I:C) or to smoking only for 24 weeks, and RvD1 was injected within the smoking exposure period to prevent regeneration or after completion of smoking exposure to assess regeneration. The mean linear intercept and inflammation scores were measured in the lung tissue, and inflammatory cells and cytokines were measured in the bronchoalveolar lavage fluid.
Results: Measurements of mean linear intercept showed that RvD1 significantly attenuated smoking-induced lung destruction in all emphysema models. RvD1 also reduced smoking-induced inflammatory cell infiltration, which causes the structural derangements observed in emphysema. In the 4-week prevention model, RvD1 reduced the smoking-induced increase in eosinophils and interleukin-6 in the bronchoalveolar lavage fluid. In the 24-week prevention model, RvD1 also reduced the increased neutrophils and total cell counts induced by smoking.
Conclusion: RvD1 attenuated smoking-induced emphysema in vivo by reducing inflammation and promoting tissue regeneration. This result suggests that RvD1 may be useful in the prevention and treatment of emphysema.
Keywords: resolvin D1, COPD, emphysema
Introduction
COPD is a major global health problem, with a prevalence of 5%–25% among adults worldwide.1 COPD is among the top ten most common chronic health conditions and is associated with a marked restriction in daily activity and substantial utilization of health care resources.2 COPD is commonly treated with bronchodilators, which have shown beneficial effects in several clinical trials;3,4 however, bronchodilators act on airways, not lung parenchyma, so the clinical response to treatment is the weakest, among COPD phenotypes, in emphysema, which mainly involves lung parenchyma.5 To reduce hyperinflated lung, lung volume reduction therapy is currently undergoing trial; however, this approach can be applied only to a subset of patients.6,7 Furthermore, no treatment restores damaged lung parenchyma. Considering the paucity of treatments for emphysema, novel approaches are urgently needed.
The resolution of inflammation is now considered a dynamic program that is partially dependent on the equilibrium of leukocyte ingress and egress at inflamed sites.8 Resolvin D1 (RvD1), a novel lipid derived from docosahexaenoic acid (DHEA), showed potent anti-inflammatory and proresolving properties in preclinical models of peritonitis,9 colitis,10 and dermal inflammation,11 and recently in aspiration pneumonia12 and cigarette smoking-induced lung inflammation.13
The main pathogenesis of emphysema is irreversible lung damage from chronic inflammation induced by smoking. Imbalance between protease and antiprotease is a dominant hypothesis.14 Macrophages and neutrophils are main sources of proteases in lungs, and the degree of inflammation induced by these cells correlated with the severity of airflow obstruction.15 According to a hypothesis from a UK study, repeated airway infection and hypersecretion of mucus are causes of COPD. In addition, respiratory tract infection is an important cause of acute exacerbation and progression of this disease.16 We hypothesized that RvD1, which mediates the resolution of lung inflammation, may prevent emphysema and restore lung tissue by reducing irreversible injury. To test this hypothesis, the effect of RvD1 was assessed in a smoking-induced emphysema model.
Materials and methods
Emphysema mouse model
All the animals used in this study were specific-pathogen-free female C57BL/6 mice (Orient Bio, Seongnam, Republic of Korea) that were of age 8 weeks. All were kept on a 12-hour light and 12-hour dark cycle with free access to food and water. The lung damage from smoking and inflammation is the main pathogenesis of emphysema. Animal models of emphysema were induced by two well-established methods based on this pathogenesis. First, mice were exposed to cigarette smoke for 4 weeks with administration of 50 μg (1 μg/μL) of poly(I:C) via nasal aspiration at 3 weeks and 4 weeks (4-week model).17 Second, mice were exposed to cigarette smoke for 24 weeks (24-week model). Cigarette smoke exposure was performed using 12 commercial cigarettes (8.0 mg of tar, 0.80 mg/cigarette, Eighty Eight Lights; KT&G, Daejeon, Republic of Korea) per day according to a protocol previously described18 with modification. In brief, ten to 12 mice were settled in an inhalation box (50 ×40 ×30 cm) connected to a pump and exposed for 10 minutes to mainstream cigarette smoke generated simultaneously from four cigarettes. The mice remained in the box for an additional 10 minutes after the cigarettes had burned. The box was then ventilated to remove the cigarette smoke, and the mice breathed normal room air for 5 minutes. Two additional exposures were performed in the same manner in a day. Control (CTL) or RvD1 only (RE) animals inhaled only clean room air in the cages. The Institutional Animal Care and Use Committee of the Asan Medical Center approved this study (2012-01-176), and all the experiments were performed in accordance with the committee guidelines. Animal care and treatment were guided according to the 8th edition of the Guide for the Care and Use of Laboratory Animals.
Experimental design
Four different administration protocols of RvD1 (Caymen, Ann Arbor, MI, USA; 100 ng or phosphate buffered saline [PBS] of the same volume) were performed. In the first protocol, RvD1 or PBS was injected in the tail vein 3 weeks and 4 weeks after the start of smoking exposure in the 4-week model. One day after the completion of the 4 weeks smoking exposure, mice were sacrificed (4-week prevention model; Figure 1A). In the second protocol, RvD1 or PBS was injected in the tail vein 4 weeks and 5 weeks after the start of smoking exposure in the 4-week model. Two weeks after the completion of the 4-week smoking exposure, mice were sacrificed (4-week regeneration model; Figure 1B). In the third protocol, RvD1 or PBS was injected at 18 weeks, 20 weeks, 22 weeks, and 24 weeks after the start of smoking exposure in the 24-week model. One day after the completion of the 24-week smoking exposure, mice were sacrificed (24-week prevention model; Figure 1C). In the fourth protocol, RvD1 or PBS was injected at 0 week, 2 weeks, 4 weeks, and 6 weeks after the completion of smoking exposure in the 24-week model. Eight weeks after the completion of the 24-week smoking exposure, mice were sacrificed (24-week regeneration model; Figure 1D). Each model included four groups: CTL, RE, smoking only (SM), and smoking exposure and RvD1 (SR).
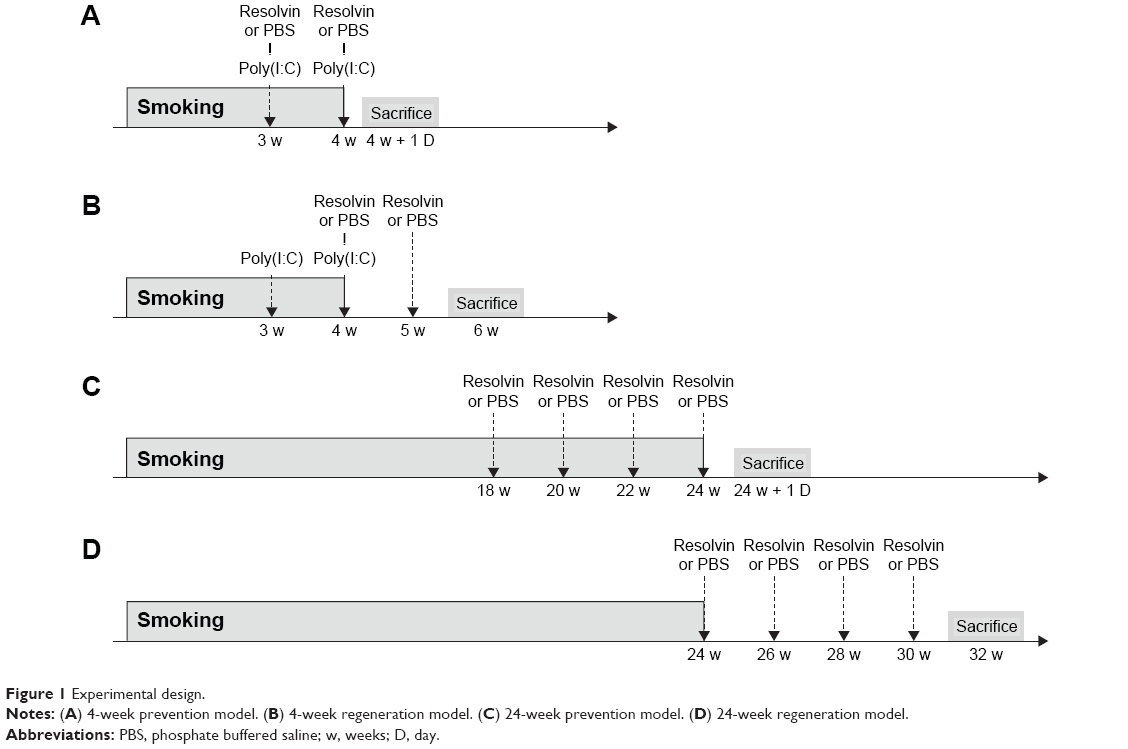 | Figure 1 Experimental design. |
Bronchoalveolar lavage
Mice were sacrificed using 40 mg/kg of Zoletil (Virbac, Carros, France) and 10 mg/kg of Rompun (Bayer AG, Leverkusen, Germany). The trachea was catheterized and perfused with two 0.6 mL and one 0.7 mL aliquot of cold saline. The cellular and liquid fractions of bronchoalveolar lavage (BAL) fluid were separated by centrifugation (2,200 rpm at 4°C for 5 minutes). The cell pellet was suspended, seeded onto a slide, and stained with Diff-Quick (Sysmex, Kobe, Japan). The BAL liquid fractions were stored in aliquots at -80°C pending cytokine measurement.
Quantification of cytokines in cell culture supernatant and BAL fluid
The levels of interleukin-6 (IL-6) in the culture supernatants and BAL fluids were measured with a commercially available ELISA kit (Merck Millipore, Billerica, MA, USA) using fluorescently labeled microsphere beads and a Luminex 100/200™ (Luminex Co., Austin, TX, USA) reader.
Histologic analysis
The left main bronchus was ligated, and the right lung was inflated with 0.5% low temperature melting agarose at a pressure of 15 cmH2O.19 The lungs were then fixed with 3.5% formalin, embedded in paraffin, cut into 5 μm thick sections, and stained with hematoxylin and eosin. The mean linear intercept (MLI) was measured as described previously.20 In brief, the average interalveolar septal wall distances were determined by the number of interruptions in 1 mm lines of alveolar wall. Any line that crossed a large vessel or bronchus was excluded from evaluation. Four or more lines were drawn in each field, and at least three random fields were examined per mouse. The degree of peribronchial and perivascular inflammations was scored on a subjective scale of 0 (no) to 4 (severe) in a blind manner. Each scoring was performed with the comparison of the standardized figures presenting grades. The inflammation score was defined as the sum of the peribronchial and perivascular scores (0–8).21,22 Histologic analyses were performed in different mice than those used for BAL.
Statistical analysis
All values were represented as mean ± standard deviation. Differences between multiple groups were analyzed by one-way analysis of variance with Tukey’s post tests or two-way analysis of variance with Bonferroni post tests. Significance was defined by a P-value of 0.05. All statistical calculations were conducted in GraphPad Prism Version 5.03 for Windows (GraphPad Software, Inc., La Jolla, CA, USA).
Results
Inhibition of cigarette smoke induced emphysema development by RvD1
We first evaluated the effects of RvD1 using four different experimental protocols as described previously. In all the four models, cigarette smoke induced lung parenchymal destruction and airspace enlargement leading to increased MLI. RvD1 administration attenuated lung destruction significantly. In the 4-week prevention and regeneration models, the MLIs of the SM and SR groups were 50.75±4.09 μm vs 43.52±2.89 μm (P<0.05) and 49.42±2.82 μm vs 42.94±3.24 μm (P<0.01), respectively. In the 24-week prevention and regeneration models, the MLIs of the SM and SR groups were 52.46±4.17 μm vs 45.25±1.47 (P<0.01) and 49.22±4.51 μm vs 40.44±3.02 (P<0.01), respectively. The MLIs of the SR groups were not significantly different from those of the CTL groups in all of these protocols (Figure 2).
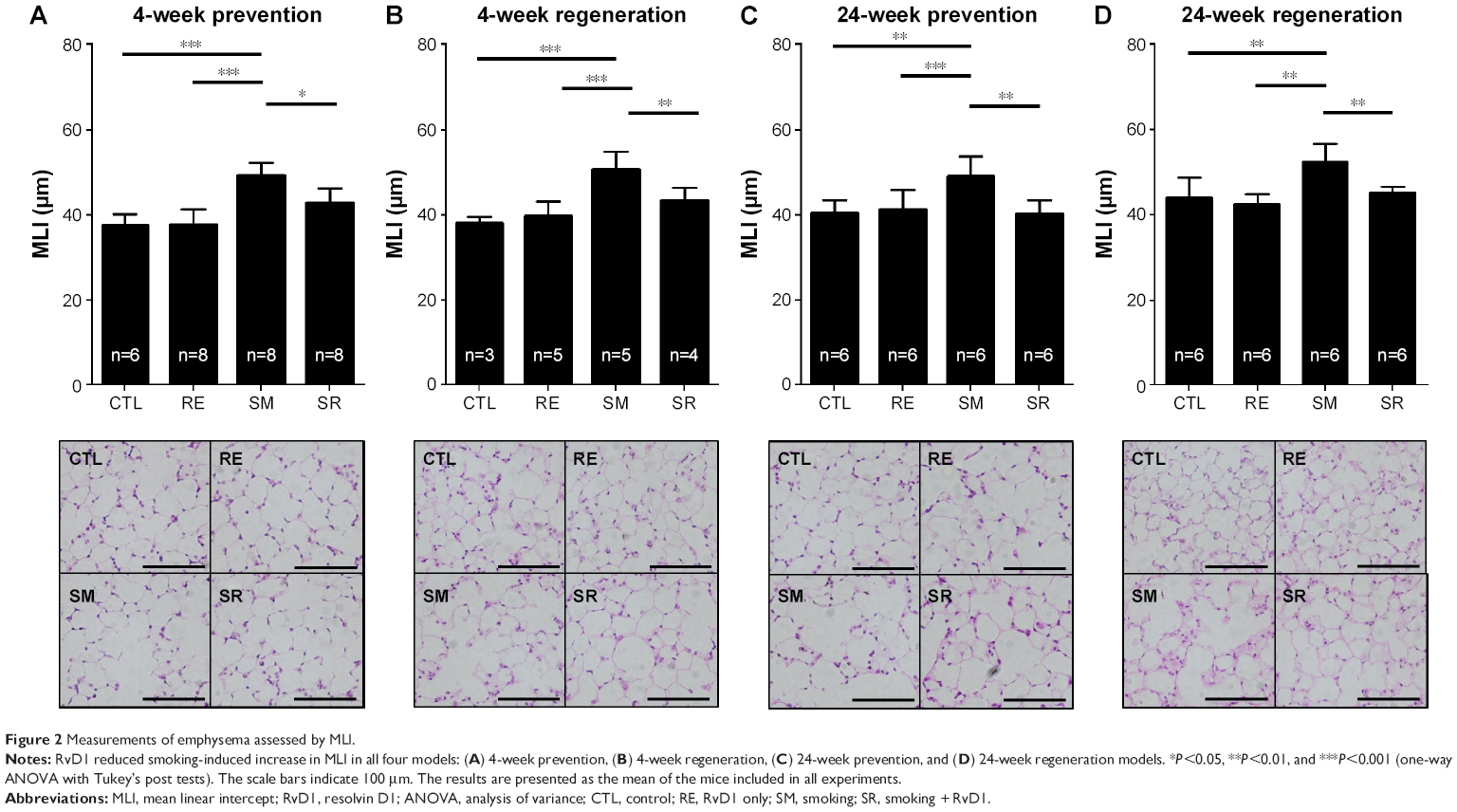 | Figure 2 Measurements of emphysema assessed by MLI. |
Reduction of peribronchial and perivascular inflammatory cell infiltration by RvD1
Inflammatory cell infiltration contributes to structural derangements in COPD via the release of proteases and reactive oxygen species.23,24 Therefore, we tested the effect of RvD1 on inflammatory cell infiltration induced by smoking. In all models, smoking caused inflammatory cell infiltration in perivascular and peribronchial areas. RvD1 inhibited inflammatory cell infiltration consistently in all the four models. Histologic score in the SR group showed a significant decrease in smoking-induced inflammatory cell infiltration. No difference in histologic scores was observed between the CTL and RE groups (Figure 3).
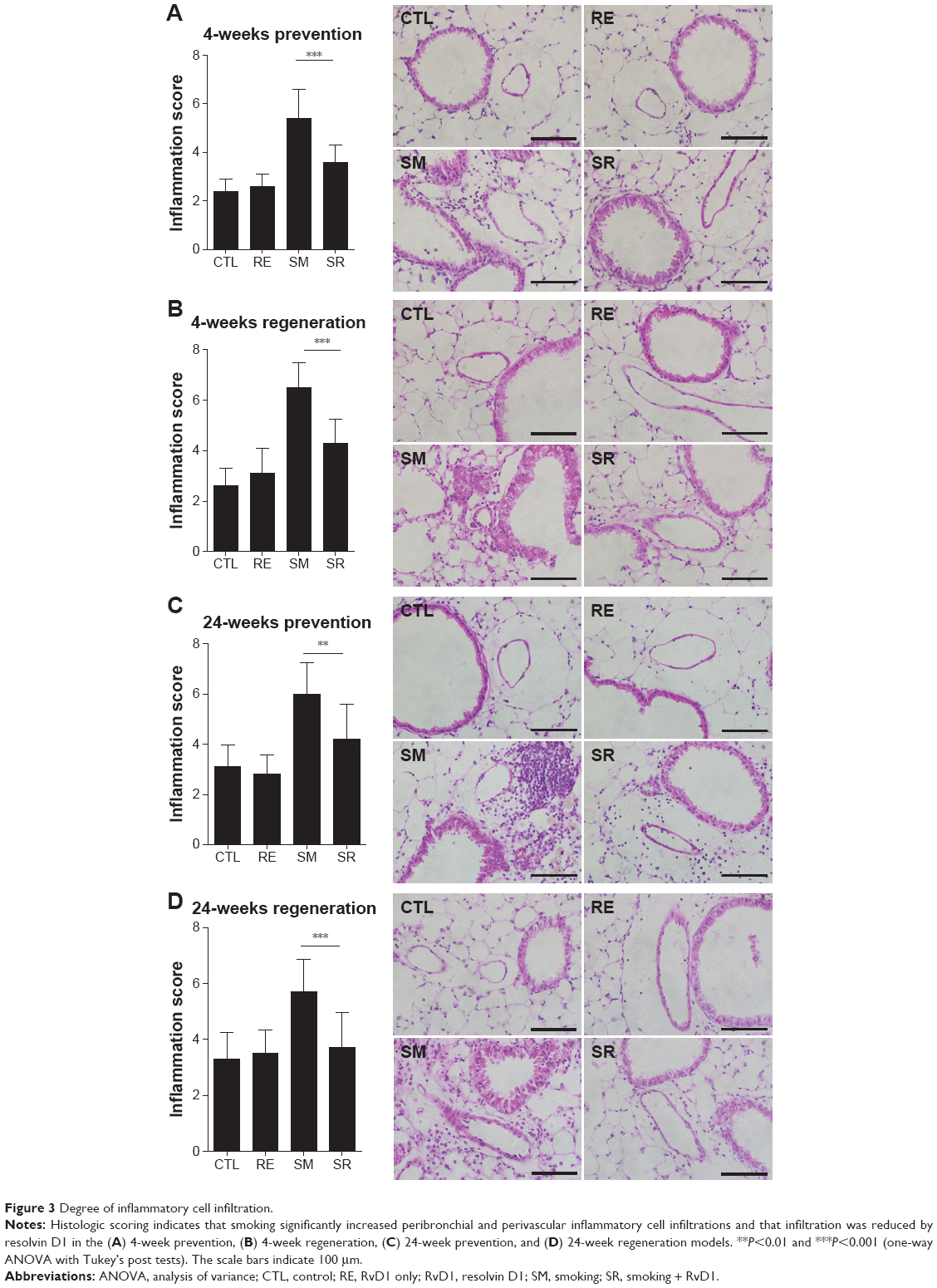 | Figure 3 Degree of inflammatory cell infiltration. |
RvD1 reduced eosinophil counts and IL-6 in the BAL fluid in the 4-week prevention model
Chronic exposure to smoking significantly increased total cell counts, eosinophil counts, and neutrophil counts in the BAL fluid compared to the CTL group in the 4-week prevention model. RvD1 reduced eosinophil counts significantly in the SR group compared to the SM group (9.79±1.16 [×105]/mL vs 5.47±1.54 [×105]/mL, P<0.001). Neutrophil counts were also reduced in the SR group compared to the SM group without reaching statistical significance (2.32±1.00 [×105]/mL vs 1.63±0.80 [×105]/mL, P>0.05). No significant difference in total cell counts was observed between the SM and SR groups. In the 4-week prevention model, IL-6 was also increased in the BAL fluids of the SM group compared to those of the CTL group, and the IL-6 level was decreased significantly in the SR group compared to that in the SM group (1,008.02±247.65 pg/mL vs 598.51±161.94 pg/mL, P<0.01; Figure 4).
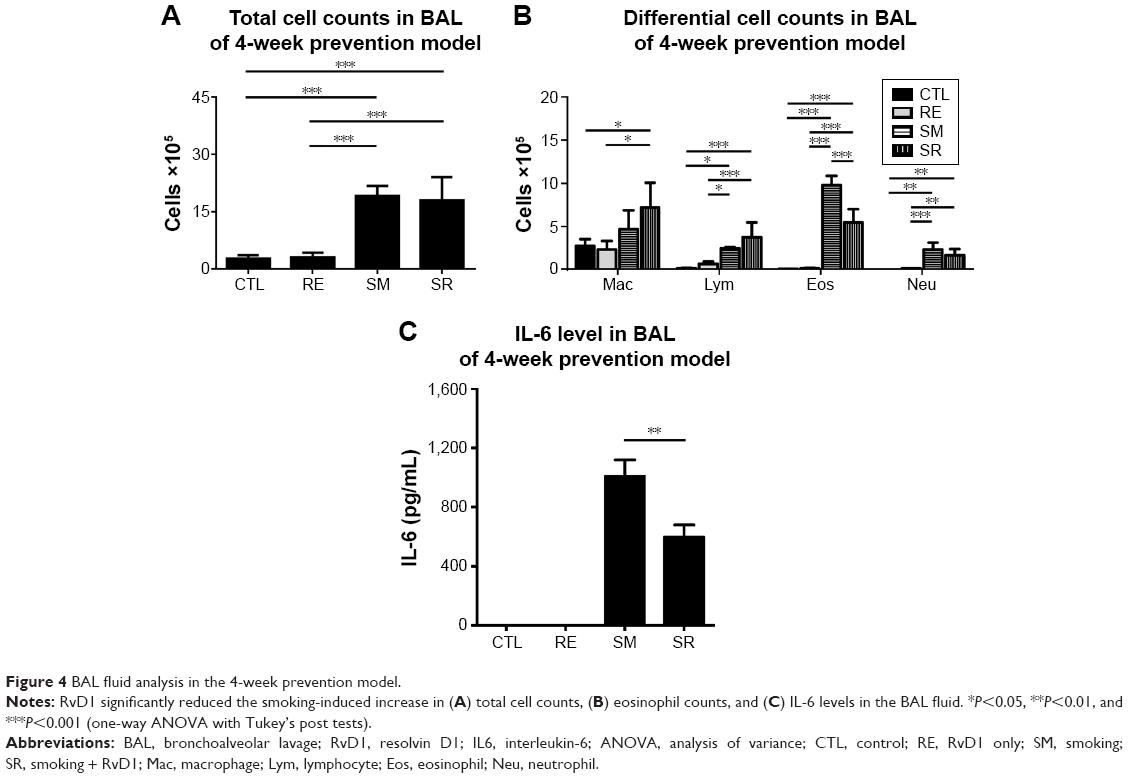 | Figure 4 BAL fluid analysis in the 4-week prevention model. |
RvD1 reduced total cell counts and neutrophil counts in the BAL fluid in the 24-week prevention model
Chronic exposure to smoking also significantly increased total cell counts, eosinophil counts, and neutrophil counts in the BAL fluid compared to those in the CTL group in the 24-week prevention model. RvD1 reduced the total cell count and neutrophil count significantly in the SR group compared to those in the SM group (total cell count: 4.19±0.47 cells [×105]/mL vs 3.28±0.75 cells [×105]/mL, P<0.05; neutrophil count: 0.87±0.34 cells [×105]/mL vs 0.45±0.14 cells [×105]/mL, P<0.001). No significant difference in the eosinophil count was observed between the SM and SR groups (0.15±0.03 cells [×105]/mL vs 0.17±0.06 cells [×105]/mL, P>0.05). In the 24-week prevention model, IL-6 levels of the BAL fluid were reduced in the SR group compared to those in the SM group without reaching statistical significance (Figure 5). IL-6 was not detected in the 24-week regeneration models (data not shown).
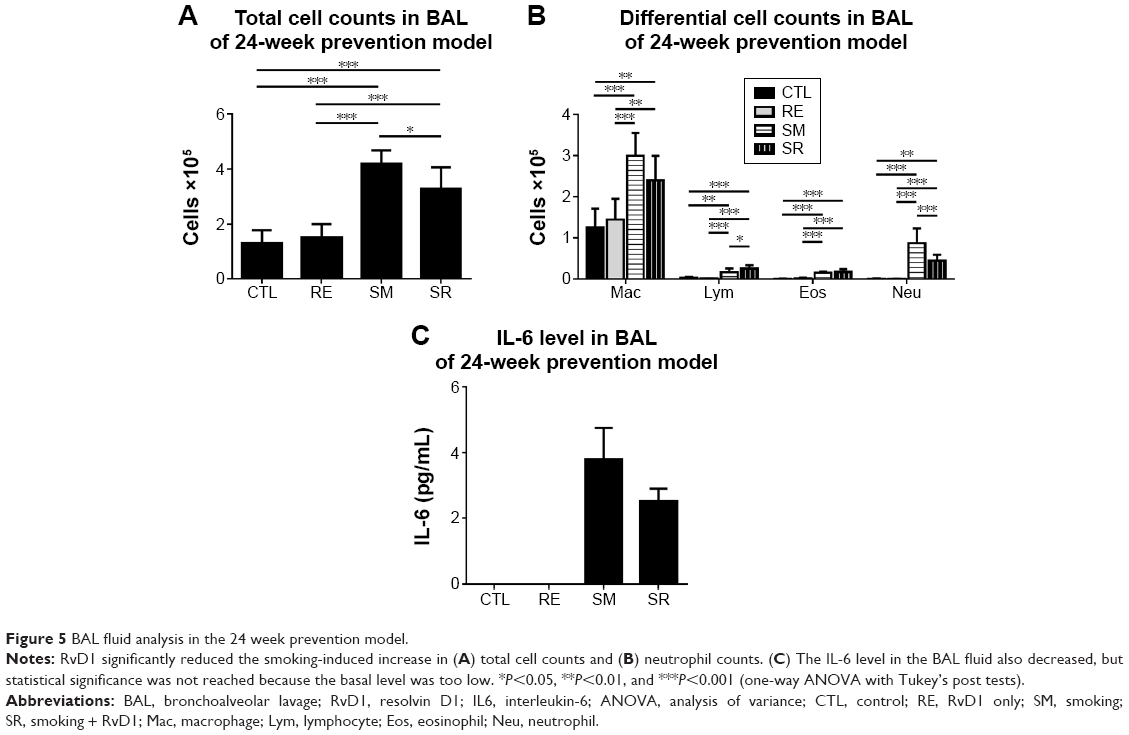 | Figure 5 BAL fluid analysis in the 24 week prevention model. |
Discussion
This study showed that RvD1 can protect against the alveolar destruction induced by chronic exposure to smoking. RvD1 attenuated the inflammation caused by cigarette smoking. These results indicate that RvD1 can prevent smoking-induced lung damage and has possibility to regenerate lung tissue.
RvD1 is a lipid that promotes the resolution of inflammation, which is quite important to return to homeostasis. Unresolved chronic inflammation can cause tissue damage and fibrosis, followed by decreased function. RvD1 has already shown benefits in pulmonary infectious and allergic disease models by reducing inflammation. Resolvin E1 protects against acid-initiated acute lung injury and pneumonia by blocking neutrophil trafficking and proinflammatory mediator release.12 RvD1 attenuates lung inflammation in lipopolysaccharide-induced acute lung injury by suppressing nuclear factor-kappa B activation.25 Resolvin E1 inhibits bronchial hyperresponsiveness, mucus secretion, and eosinophil recruitment in an ovalbumin-sensitized asthma model.26 A recent report showed that RvD1 resolves cigarette smoke-induced lung inflammation in the acute phase.13 In human alveolar macrophages, resolvin decreased inflammatory cytokines and enhanced phagocytosis.27 Considering this body of evidence, resolvin may provide therapeutic benefit in emphysema, an inflammatory disease induced by chronic exposure to smoking, and a recent study supports this.28 Meanwhile, there were no reports of therapeutic resolvin administration in emphysema in human beings. The present study provides the novel evidence that the beneficial effect of resolvin on smoking-induced emphysema is consistent in various models, and it can be the basis for future human trials.
Although little is known about the role of resolvin in human beings, several indirect lines of evidence support beneficial effects in respiratory diseases. Enteric feeding of supplements enriched in omega-3 polyunsaturated fatty acids (a precursor of resolvin) improves clinical outcomes in acute respiratory distress syndrome by reducing time to liberation from mechanical ventilation and discharge from the intensive care unit.29 A high intake of eicosapentaenoic acid (EPA) and DHEA was inversely correlated with the risk of COPD in smokers, suggesting a protective effect against smoking.30 EPA and DHEA supplementation also significantly attenuates the allergen-induced asthmatic response.31 Japanese workers, who have a higher fish consumption than their American counterparts, have a lower incidence of chronic bronchitis and a higher mean forced expiratory volume in 1 second than their American counterparts despite equivalent levels of cigarette smoking.32
RvD1 attenuated the MLI increased by smoking in the 4-week and 24-week models and also decreased inflammatory cell counts in the BAL fluid; however, BAL fluids differed in their composition of inflammatory cells: differences in eosinophil counts were prominent in the 4-week models, whereas differences in neutrophil counts were observed in the 24-week models. New-onset smoking is a significant risk factor for acute eosinophilic pneumonia,33 which is characterized by acute and organizing diffuse alveolar damage with eosinophil infiltration.34 Acute eosinophilic pneumonia is the only well-established respiratory disease that develops in the initiation phase of smoking; therefore, eosinophil accumulation may be an important pathogenic feature of the inflammatory process in this phase. This suggests that the duration, time, and method of smoking can make a difference in alveolar cell composition. During chronic exposure to smoking, neutrophils are the main contributors to lung destruction, especially in emphysema.35,36 Cigarette smoke disrupts normal neutrophil apoptosis and efferocytosis. Impairment of neutrophil clearance can lead to irreversible local tissue damage via production of mediators, including neutrophil elastase and reactive oxygen species.37 Considering the result of our study, resolvin can attenuate lung damage by reducing key inflammatory cells in both the initial and late phases of smoking. RvD1 also decreased the smoking-induced increase in IL-6; however, no statistical significance was reached in the 24-week model. Although smoking increases neutrophils in the BAL fluid over time,38 the increase in inflammatory cytokines usually falls off after the acute phase of inflammation, which persists for several days.39 The elevation in inflammatory cytokines such as IL-6 by chronic exposure to smoking does not seem high enough to reach statistical significance. Other cytokines such as interleukin-8 and interleukin-1 beta can have a role in the recruitment of neutrophils in chronic smoking. Meanwhile, the inflammation score suggested that chronic inflammatory change still persisted even after the recruitment of inflammatory cells has reduced. These histologic changes from chronic exposure to smoking might include fibrosis, bronchial wall thickening, or vascular remodeling.
To restore destroyed and injured lung is the ultimate goal of emphysema treatment, but this is not achieved by the current treatment. Pharmacologic treatment of COPD involves bronchodilators, which aim to decrease airway resistance; this treatment shows limited effectiveness against decreased elastic recoil and hyperinflation, which is the main pathogenesis of emphysema.5 Smoking-related inflammation persists for several years even after smoking cessation, and COPD not only persists but may also continue to worsen.40 A limited number of anti-inflammatory therapies, including a phosphodiesterase-4 inhibitor and a steroid, are available; however, long-term steroid use carries risks of adverse effects that potentially outweigh benefits,41 and phosphodiesterase-4 inhibitors benefit mainly in chronic bronchitis.42 In this context, our findings have implications for the treatment of emphysema and future clinical trials. Proresolving lipid mediators such as resolvin and its precursors, including EPA and DHEA, might be therapeutically useful to protect from and reduce lung injury in diseases induced by chronic smoke exposure such as COPD. Resolvin has a significant clinical potential because it promotes inflammation resolution and tissue repair without being immunosuppressive.
However, we should keep in mind that several kinds of molecules that improved emphysema in animal models failed to show efficacy in human beings. Simvastatin inhibits cigarette smoking-induced emphysema in rats,22 and several retrospective studies suggested that statins could also be beneficial in human beings.43,44 Unfortunately, no reduction in COPD exacerbation was observed in a large randomized trial.45 Several other treatments, including vitamin C,46 angiotensin receptor blockade,47 and stem cells,48 attenuated smoke-induced emphysema in animals, but these effects have not been reproduced in human beings yet. Therefore, proresolving lipids should be tested in human trials. For a successful human study, the appropriate target population should be selected and the most effective administration protocols should be used. It is not clear whether nutritional supplementation with fish oil or omega-3 polyunsaturated fatty acids is enough or whether injections of more refined forms, such as resolvin or protectin, is necessary.
Conclusion
RvD1 attenuated smoking-induced emphysema and showed possibility to regenerate lung tissue by reducing inflammatory cells and cytokines. Although the exact mechanism should be elucidated further, these findings indicate that resolvin may play a role in the treatment of cigarette smoking-induced COPD.
Acknowledgment
This study was supported by a grant (2013-553) from the Asan Institute for Life Sciences, Asan Medical Center, Seoul, Republic of Korea.
Author contributions
All authors contributed toward data collection, statistical analysis, drafting and critically revising the paper, gave final approval of the version to be published, and agreed to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
GOLD [homepage on the Internet]. Global Strategy for the Diagnosis, Management and Prevention of COPD; 2015. Available from: http://www.goldcopd.org/. Accessed March 26, 2016. | ||
Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006;3(11):e442. | ||
Calverley PM, Anderson JA, Celli B, et al. Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. N Engl J Med. 2007;356(8):775–789. | ||
Tashkin DP, Celli B, Senn S, et al. A 4-year trial of tiotropium in chronic obstructive pulmonary disease. N Engl J Med. 2008;359(15):1543–1554. | ||
Lee JH, Lee YK, Kim EK, et al. Responses to inhaled long-acting beta-agonist and corticosteroid according to COPD subtype. Respir Med. 2010;104(4):542–549. | ||
Fishman A, Martinez F, Naunheim K, et al. A randomized trial comparing lung-volume-reduction surgery with medical therapy for severe emphysema. N Engl J Med. 2003;348(21):2059–2073. | ||
Sciurba FC, Ernst A, Herth FJ, et al. A randomized study of endobronchial valves for advanced emphysema. N Engl J Med. 2010;363(13):1233–1244. | ||
Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8(5):349–361. | ||
Sun YP, Oh SF, Uddin J, et al. Resolvin D1 and its aspirin-triggered 17R epimer. Stereochemical assignments, anti-inflammatory properties, and enzymatic inactivation. J Biol Chem. 2007;282(13):9323–9334. | ||
Arita M, Yoshida M, Hong S, et al. Resolvin E1, an endogenous lipid mediator derived from omega-3 eicosapentaenoic acid, protects against 2,4,6-trinitrobenzene sulfonic acid-induced colitis. Proc Natl Acad Sci U S A. 2005;102(21):7671–7676. | ||
Serhan CN. Resolution phase of inflammation: novel endogenous anti-inflammatory and proresolving lipid mediators and pathways. Annu Rev Immunol. 2007;25:101–137. | ||
Seki H, Fukunaga K, Arita M, et al. The anti-inflammatory and proresolving mediator resolvin E1 protects mice from bacterial pneumonia and acute lung injury. J Immunol. 2010;184(2):836–843. | ||
Hsiao HM, Sapinoro RE, Thatcher TH, et al. A novel anti-inflammatory and pro-resolving role for resolvin D1 in acute cigarette smoke-induced lung inflammation. PLoS One. 2013;8(3):e58258. | ||
Shapiro SD. Proteolysis in the lung. Eur Respir J Suppl. 2003;44:30s–32s. | ||
Saetta M, Turato G, Maestrelli P, Mapp CE, Fabbri LM. Cellular and structural bases of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2001;163(6):1304–1309. | ||
Sethi S. Bacterial infection and the pathogenesis of COPD. Chest. 2000;117(5 Suppl 1):286S–291S. | ||
Kang MJ, Lee CG, Lee JY, et al. Cigarette smoke selectively enhances viral PAMP- and virus-induced pulmonary innate immune and remodeling responses in mice. J Clin Invest. 2008;118(8):2771–2784. | ||
Huh JW, Kim SY, Lee JH, et al. Bone marrow cells repair cigarette smoke-induced emphysema in rats. Am J Physiol Lung Cell Mol Physiol. 2011;301(3):L255–L266. | ||
Halbower AC, Mason RJ, Abman SH, Tuder RM. Agarose infiltration improves morphology of cryostat sections of lung. Lab Invest. 1994;71(1):149–153. | ||
Thurlbeck WM. Measurement of pulmonary emphysema. Am Rev Respir Dis. 1967;95(5):752–764. | ||
McKay A, Leung BP, McInnes IB, Thomson NC, Liew FY. A novel anti-inflammatory role of simvastatin in a murine model of allergic asthma. J Immunol. 2004;172(5):2903–2908. | ||
Lee JH, Lee DS, Kim EK, et al. Simvastatin inhibits cigarette smoking-induced emphysema and pulmonary hypertension in rat lungs. Am J Respir Crit Care Med. 2005;172(8):987–993. | ||
Churg A, Wang RD, Tai H, et al. Macrophage metalloelastase mediates acute cigarette smoke-induced inflammation via tumor necrosis factor-alpha release. Am J Respir Crit Care Med. 2003;167(8):1083–1089. | ||
Carnevali S, Petruzzelli S, Longoni B, et al. Cigarette smoke extract induces oxidative stress and apoptosis in human lung fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2003;284(6):L955–L963. | ||
Liao Z, Dong J, Wu W, et al. Resolvin D1 attenuates inflammation in lipopolysaccharide-induced acute lung injury through a process involving the PPARgamma/NF-kappaB pathway. Respir Res. 2012;13:110. | ||
Haworth O, Cernadas M, Yang R, Serhan CN, Levy BD. Resolvin E1 regulates interleukin 23, interferon-gamma and lipoxin A4 to promote the resolution of allergic airway inflammation. Nat Immunol. 2008;9(8):873–879. | ||
Croasdell A, Thatcher TH, Kottmann RM, et al. Resolvins attenuate inflammation and promote resolution in cigarette smoke-exposed human macrophages. Am J Physiol Lung Cell Mol Physiol. 2015;309(8):L888–L901. | ||
Hsiao HM, Thatcher TH, Colas RA, Serhan CN, Phipps RP, Sime PJ. Resolvin D1 reduces emphysema and chronic inflammation. Am J Pathol. 2015;185(12):3189–3201. | ||
Gadek JE, DeMichele SJ, Karlstad MD, et al. Effect of enteral feeding with eicosapentaenoic acid, gamma-linolenic acid, and antioxidants in patients with acute respiratory distress syndrome. Enteral Nutrition in ARDS Study Group. Crit Care Med. 1999;27(8):1409–1420. | ||
Shahar E, Folsom AR, Melnick SL, et al. Dietary n-3 polyunsaturated fatty acids and smoking-related chronic obstructive pulmonary disease. Atherosclerosis Risk in Communities Study Investigators. N Engl J Med. 1994;331(4):228–233. | ||
Arm JP, Horton CE, Spur BW, Mencia-Huerta JM, Lee TH. The effects of dietary supplementation with fish oil lipids on the airways response to inhaled allergen in bronchial asthma. Am Rev Respir Dis. 1989;139(6):1395–1400. | ||
Comstock GW, Stone RW, Sakai Y, Matsuya T, Tonascia JA. Respiratory findings and urban living. Arch Environ Health. 1973;27(3):143–150. | ||
Shorr AF, Scoville SL, Cersovsky SB, et al. Acute eosinophilic pneumonia among US Military personnel deployed in or near Iraq. JAMA. 2004;292(24):2997–3005. | ||
Tazelaar HD, Linz LJ, Colby TV, Myers JL, Limper AH. Acute eosinophilic pneumonia: histopathologic findings in nine patients. Am J Respir Crit Care Med. 1997;155(1):296–302. | ||
Hogg JC, Chu F, Utokaparch S, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350(26):2645–2653. | ||
Shapiro SD, Goldstein NM, Houghton AM, Kobayashi DK, Kelley D, Belaaouaj A. Neutrophil elastase contributes to cigarette smoke-induced emphysema in mice. Am J Pathol. 2003;163(6):2329–2335. | ||
Abboud RT, Vimalanathan S. Pathogenesis of COPD. Part I. The role of protease-antiprotease imbalance in emphysema. Int J Tuberc Lung Dis. 2008;12(4):361–367. | ||
D’Hulst AI, Vermaelen KY, Brusselle GG, Joos GF, Pauwels RA. Time course of cigarette smoke-induced pulmonary inflammation in mice. Eur Respir J. 2005;26(2):204–213. | ||
Perez-Rial S, del Puerto-Nevado L, Terron-Exposito R, Giron-Martinez A, Gonzalez-Mangado N, Peces-Barba G. Role of recently migrated monocytes in cigarette smoke-induced lung inflammation in different strain of mice. PLoS One. 2013;8(9):e72975. | ||
Decramer M, Janssens W, Miravitlles M. Chronic obstructive pulmonary disease. Lancet. 2012;379(9823):1341–1351. | ||
Joo MJ, Au DH, Fitzgibbon ML, Lee TA. Inhaled corticosteroids and risk of pneumonia in newly diagnosed COPD. Respir Med. 2010;104(2):246–252. | ||
Rennard SI, Calverley PM, Goehring UM, Bredenbroker D, Martinez FJ. Reduction of exacerbations by the PDE4 inhibitor roflumilast – the importance of defining different subsets of patients with COPD. Respir Res. 2011;12:18. | ||
Alexeeff SE, Litonjua AA, Sparrow D, Vokonas PS, Schwartz J. Statin use reduces decline in lung function: VA Normative Aging Study. Am J Respir Crit Care Med. 2007;176(8):742–747. | ||
Mancini GB, Etminan M, Zhang B, Levesque LE, FitzGerald JM, Brophy JM. Reduction of morbidity and mortality by statins, angiotensin-converting enzyme inhibitors, and angiotensin receptor blockers in patients with chronic obstructive pulmonary disease. J Am Coll Cardiol. 2006;47(12):2554–2560. | ||
Criner GJ, Connett JE, Aaron SD, et al. Simvastatin for the prevention of exacerbations in moderate-to-severe COPD. N Engl J Med. 2014;370(23):2201–2210. | ||
Koike K, Ishigami A, Sato Y, et al. Vitamin C prevents cigarette smoke-induced pulmonary emphysema in mice and provides pulmonary restoration. Am J Respir Cell Mol Biol. 2014;50(2):347–357. | ||
Podowski M, Calvi C, Metzger S, et al. Angiotensin receptor blockade attenuates cigarette smoke-induced lung injury and rescues lung architecture in mice. J Clin Invest. 2012;122(1):229–240. | ||
Schweitzer KS, Johnstone BH, Garrison J, et al. Adipose stem cell treatment in mice attenuates lung and systemic injury induced by cigarette smoking. Am J Respir Crit Care Med. 2011;183(2):215–225. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.