")
Back to Journals » Breast Cancer: Targets and Therapy » Volume 12
Resistance and Overcoming Resistance in Breast Cancer
Authors Luque-Bolivar A , Pérez-Mora E, Villegas VE , Rondón-Lagos M
Received 5 July 2020
Accepted for publication 15 September 2020
Published 11 November 2020 Volume 2020:12 Pages 211—229
DOI https://doi.org/10.2147/BCTT.S270799
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Pranela Rameshwar
Andrea Luque-Bolivar,1 Erika Pérez-Mora,1 Victoria Eugenia Villegas,2 Milena Rondón-Lagos1
1School of Biological Sciences, Universidad Pedagógica y Tecnológica de Colombia, Tunja 150003, Colombia; 2Biology Program, Faculty of Natural Sciences, Universidad del Rosario, Bogotá 111221, Colombia
Correspondence: Victoria Eugenia Villegas
Biology Program, Faculty of Natural Sciences, Universidad del Rosario, Bogotá 111221, Colombia
Tel/Fax +57-1-297-0200 Ext 4029
Email [email protected]
Milena Rondón-Lagos
School of Biological Sciences, Universidad Pedagógica y Tecnológica de Colombia, Tunja 150003, Colombia
Tel/Fax +57-8-7420-8263
Email [email protected]
Abstract: The incidence and mortality of breast cancer (BC) have increased in recent years, and BC is the main cause of cancer-related death in women worldwide. One of the most significant clinical problems in the treatment of patients with BC is the development of therapeutic resistance. Therefore, elucidating the molecular mechanisms involved in drug resistance is critical. The therapeutic decision for the management of patients with BC is based not only on the assessment of prognostic factors but also on the evaluation of clinical and pathological parameters. Although this has been a successful approach, some patients relapse and/or eventually develop resistance to treatment. This review is focused on recent studies on the possible biological and molecular mechanisms involved in both response and resistance to treatment in BC. Additionally, emerging treatments that seek to overcome resistance and reduce side effects are also described. A greater understanding of the mechanisms of action of treatments used in BC might contribute not only to the enhancement of our understanding of the mechanisms involved in the development of resistance but also to the optimization of the existing treatment regimens.
Keywords: breast cancer, endocrine therapy, chemotherapy, resistance, emerging treatments
Introduction
Cancer is a common disease and represents one of the biggest health problems in the world and a significant global concern. The incidence and mortality rates of breast cancer (BC) have increased in recent years, and BC is currently the leading cause of cancer death in women worldwide.1 Decision making for the treatment of patients with BC is primarily based on the assessment of clinical and pathological parameters. In particular, the immunohistochemical evaluation of prognostic factors, such as estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor 2 (HER2) are critical for tumor subtype classification and histological grade, which play an important role in determining therapeutic strategies. For example, patients with ER-positive (ER+) tumors receive endocrine therapy, and a small fraction of these patients also receive chemotherapy. Patients with HER2-positive tumors (HER2 enriched or HER2+) are treated with antibodies directed against HER2 or small-molecule inhibitors in combination with chemotherapy. Patients with triple-negative tumors receive mainly chemotherapy.2–4
BC is a heterogeneous and complex disease in which each patient has unique morphological and molecular features, rather than a disease in which only a few genes, proteins, and/or signaling pathways contribute to disease progression in a simple, independent, and autonomous manner. Studies have shown that patients with the same type of BC can show differential responses to treatment, which further indicates the high heterogeneity in this disease. Despite the great technological advances that have enhanced our understanding of human cancers as heterogeneous diseases, current clinicopathological and molecular parameters leave a significant number of patients at risk of over-treatment and side effects.
Currently, drug-resistant BC is treated by selecting other drugs, without understanding the molecular mechanisms involved in the resistance of a given case. Better understanding of the mechanisms involved in the development of resistance might not only reduce the adverse effects of treatment but also lead to the development of new strategies for improving diagnosis and prognosis and achieving a better response to therapy. This review focuses on recent studies on the biological and molecular mechanisms of response and resistance to treatment in BC.
BC
According to the International Agency for Research on Cancer, particularly the GLOBOCAN program, 2.1 million cases of BC were estimated in 2018 compared with 12.7 million in 2008,1,3 accounting for nearly one in four cancer cases in women.4 This increase in incidence may be explained by the growth and aging of the world population and the adoption of cancer-promoting lifestyles.
The development of molecular techniques, such as RNA sequencing, has allowed the determination of gene expression profiles, identification of tumor heterogeneity, and molecular classification of BC. Thus, six subtypes have been proposed based on the expression of ER, PR, estrogen-associated genes (ESR1, GATA3, FOXA1), and genes associated with the induction of proliferation, such as HER2 and other genes located in the region of the HER2 amplicon on chromosome 17.5 These six subtypes include the following: luminal A and luminal B, which are ER+ tumors, with expression of epithelial markers; HER2+ or HER2 enriched tumors, which show overexpression of the HER2 gene; normal-like, with an expression profile that is similar to non-cancerous breast tissue; basal-like/triple negative BC (TNBC), which are characterized by lack of expression of ER, PR and HER2; and claudin-low, which is enriched in epithelial-to-mesenchymal transition (EMT) features.6 Some authors only consider five of the six breast cancer subtypes mentioned above and exclude the low claudin subtype.
Treatment of BC
BC is a heterogeneous disease in which each patient has individual characteristics, which has led to the search for new markers to improve not only the diagnosis but also the prognosis and to achieve a better treatment response. Currently, strategies for the treatment of BC depend on the tumor subtype, and the selected treatments are directed to specific targets that are functionally altered in each subtype. For example, endocrine therapy is used for tumors with positive hormone receptors (ER and PR) (luminal A and luminal B),7,8 however, some patients also require chemotherapy. For HER2+ tumors (luminal B and HER2+), treatment involves the use of monoclonal antibodies that recognize the extracellular domain of HER2 (trastuzumab) and inhibitors of the tyrosine kinase domain of HER2 (such as lapatinib), or even RNAi-mediated silencing,9 as well as endocrine therapy in cases with positive hormone receptors. Although chemotherapy is usually only given for TNBC, several molecular targets are being explored for this BC subtype, including epidermal growth factor receptor (EGFR), androgen receptor (AR), poly (ADP-ribose) polymerase (PARP), and vascular endothelial growth factor (VEGF).
Generally, conventional therapeutic treatments for the management of BC patients have included endocrine therapy, targeted therapy and chemotherapy (Figure 1). However, in recent years, other treatments have been explored, including cyclin-dependent kinase 4/6 (CDK4/6) inhibitors, microRNAs (miRNAs), immunotherapy, clustered regularly interspaced short palindromic repeats (CRISPR), tyrosine kinase inhibitors (TKIs), nanotechnological approaches, drug repurposing, and electrochemotherapy (ECT) (Figure 1). These new strategies may offer additional benefits to conventional treatments in terms of overcoming resistance and decreasing side effects.
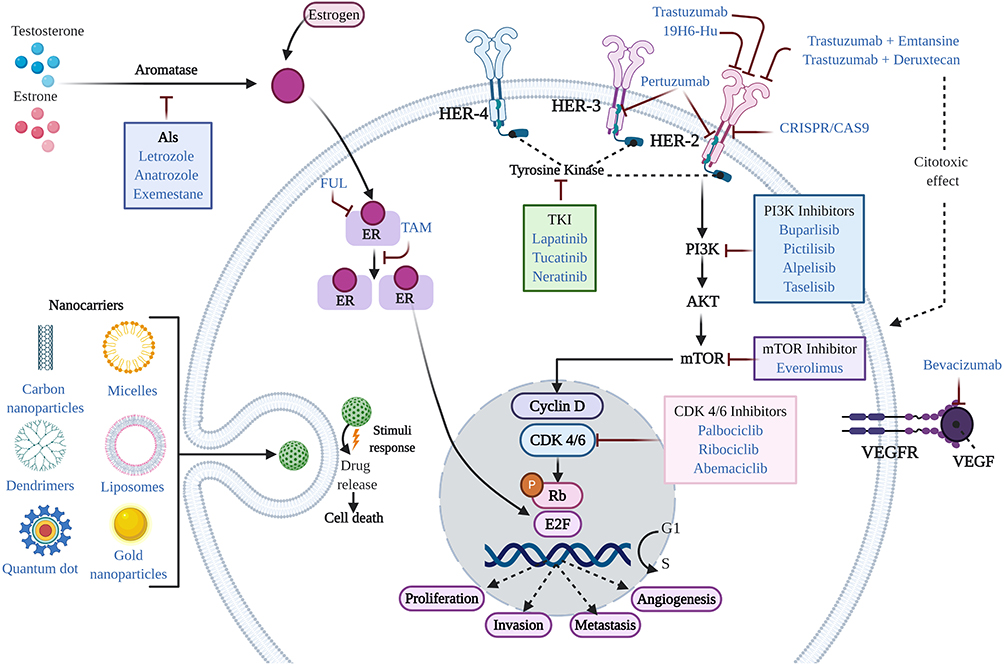 |
Figure 1 Mechanism of action of agents used in the treatment of BC. In general, conventional therapeutic treatments for the management of BC patients include endocrine therapy, targeted therapy, and chemotherapy. Standard therapy for the treatment of ER+ BC is typically based on the use of endocrine therapy (TAM, FUL, and AIs). Additional treatments include CDK4/6 inhibitors, PI3K inhibitors, and drug repurposing. Treatments approved for HER2+ BC include humanized antibodies (trastuzumab, pertuzumab, and 19H6-Hu), PI3K inhibitors, antibody-drug conjugates (trastuzumab emtansine and trastuzumab deruxtecan), TKI inhibitors, mTOR inhibitors, nanotechnological approaches (nanocarriers), and CRISPR. Treatments for TNBC include chemotherapy and immunotherapy (bevacizumab), among others. |
Resistance and Overcoming Resistance in ER+ BC
Standard therapy for the treatment of ER+ BC is typically based on the use of endocrine therapy. This therapy includes the use of selective ER modulators, such as tamoxifen (TAM),10 selective ER downregulators (fulvestrant, FUL), and aromatase inhibitors (AIs)11 (Figure 1). Additional treatments include CDK4/6 inhibitors,12 PI3K inhibitors, and drug repurposing.
Endocrine Therapy
TAM
TAM is an non-steroidal anti-estrogen with partial estrogen-agonist activity that is widely used in the treatment of ER+ BC (Figure 1).13,14 Although patients with ER- tumors do not typically respond to these treatments, studies have shown that between 5% and 10% of these patients still benefit from therapy with TAM.13,15,16 The response to TAM is often of limited duration, suggesting that patients treated with TAM develop resistance over time.17,18 However, the mechanism through which this resistance occurs remains unknown. Several studies in patients with BC in the metastatic phase have reported that although >50% of patients with ER+ BC respond to TAM therapy, 40% of patients receiving TAM as adjuvant therapy eventually relapse and die from their illness.17 Resistance is thus the main problem with endocrine therapy.
Although endocrine therapy with TAM has been highly successful, 20%–30% of patients develop therapeutic resistance.14,17,19 Several mechanisms have been suggested that can lead to the development of resistance to endocrine therapy and include genetic and epigenetic factors.20,21 Among the main mechanisms leading to TAM resistance are:
Mutations in the ESR1 gene have been associated with acquired resistance from prolonged exposure to endocrine therapy with anti-estrogens.22 The specific point mutations in ESR1 that are associated with resistance lead to the loss of ER expression and the development of resistance to treatment.23 Such modifications cause further proliferation and tumor progression in the absence of hormonal stimulation.23
One of the molecular mechanisms underlying the resistance to endocrine therapy involves polymorphisms in cytochrome P450 family 2 subfamily D member 6 (CYP2D6), a member of the cytochrome P450 family that converts TAM into active metabolites (4-hydroxy tamoxifen-4OH-TAM). Such polymorphisms lead to the expression of enzymes with different levels of activity that result in reduced responses to TAM.24 (Figure 2A)
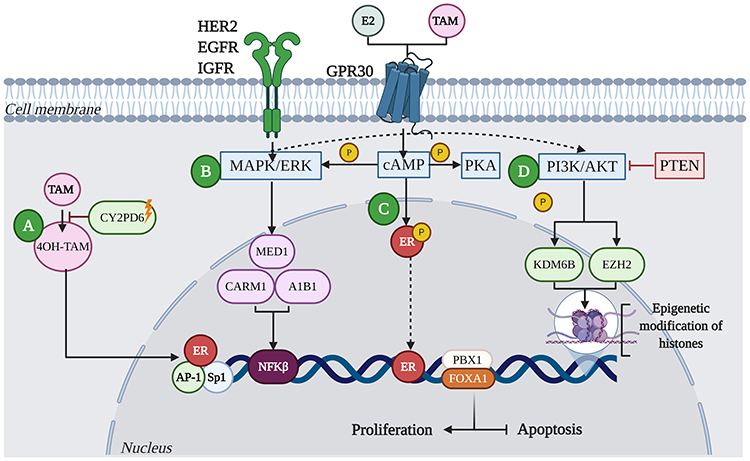 |
Figure 2 Mechanisms of resistance to endocrine therapy with tamoxifen (TAM) in breast cancer. (A) Resistance in BC is characterized by the deficiency of CYP2D6, which reduces the metabolism of 4OH-TAM. (B) Acquired resistance to TAM involves alterations in translation signals by increasing the expression and activity of tyrosine kinase receptor family proteins, such as HER2, EGFR, IGFR and GPR30. These events lead to aberrant activation of cAMP/PKA, MAPK/ERK and PI3K/AKT signaling pathways. Activation of these kinase pathways can result in phosphorylation of ER and its co-activators such as A1B1, MED1, or CARM1, leading to the activation of proliferation and inhibition of apoptosis. (C) Deregulation of ER increases not only HER2-mediated signaling, but the activation of transcription factors such as Sp1, AP-1 and NFκB, promoting oncogene transcription. Likewise, factors such as FOXA1 and PBX1 can recruit ER to specific genomic sites. (D) Other mechanisms that can lead to resistance to TAM involve the activation of alternate signaling pathways; for example, mutations in the tumor suppressor protein, PTEN, has been shown to increase phosphorylation of PI3K/AKT in ER+ tumors, leading to therapeutic resistance. Additionally, aberrant phosphorylation of proteins that modify histones, such as KDM6B and EZH2, can lead to resistance to endocrine therapy from the reactivation of genes. |
Alterations in translation signals lead to increased expression and activity of the tyrosine kinase family receptors, such as human EGFR, insulin-like growth factor 1 receptor (IGFR), and G protein-coupled estrogen receptor (GPR30).25 These events result in aberrant activation of cyclic adenosine monophosphate/protein kinase A (cAMP/PKA), mitogen-activated protein kinase (MAPK/ERK), and phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT) signaling pathways.26 Activation of these kinases leads to phosphorylation of ER and its co-activators such as collagen-binding A-domains of integrins (A1B1), mediator complex subunit 1 (MED1), or the coactivator-associated arginine methyltransferase 1 (CARM1), leading to the activation of proliferation and inhibition of apoptosis.27 (Figure 2B).
In endocrine resistance, expression of factors such as fork head box protein A1 (FOXA1) and PBX homeobox 1 (PBX1) is often altered, leading to the altered or aberrant expression of ER28 (Figure 2C). ER deregulation increases not only HER2-mediated signaling but also the activation of transcription factors such as SP1, AP-1, and nuclear factor kappa B (NFκB), which promotes the transcription of oncogenes.29
Other mechanisms that can lead to TAM resistance include the activation of alternative signaling pathways that prevent and/or exceed blocking of estrogen signaling induced by TAM. For example, mutations in the tumor suppressor protein, phosphatase and tensin homolog (PTEN), leads into uncontrolled transduction of the PI3K signal in ER+ tumors, leading to treatment resistance30,31 (Figure 2D). Alternatively, tumors can acquire TAM resistance through the regulation of AKT.32 Additionally, tumor cells can achieve resistance by decreasing the concentration of active TAM through the metabolism of an altered TAM.21 Furthermore, activation of PI3K/AKT pathway causes phosphorylation of demethyltransferase proteins [lysine demethylase 6B (KDM6B)] and methyltransferase proteins [similar to enhancer of zeste 2 polycomb repressive complex 2 subunit (EZH2)], which modify histones; this epigenetic mechanism leads to the development of endocrine therapy resistance from the reactivation of genes (Figure 2D).
FUL
FUL is an ER+ regulator with low cytotoxicity that, contrary to TAM, acts as an antagonist by binding, blocking and degrading ER and inhibiting ER transcriptional activity33 (Figure 1). Therefore, its action is more powerful than that of TAM.34 FUL has recently been approved for use in combination with Piqray (alpelisib),35 which acts as a selective inhibitor of PI3K.36 The effectiveness of this combination therapy (FUL + Piqray) was determined in the SOLAR-1 trial, in which postmenopausal women and men with advanced or metastatic BC with ER+/PR+/HER2- and mutations in the PIK3CA gene who were treated with the combination showed significantly prolonged progression-free survival.37
AIs
AIs are one of the major treatment options for ER+ BC in postmenopausal patients. AIs stop estrogen production in postmenopausal women by blocking the aromatase enzyme, which converts androgen hormone to small amounts of estrogen. Anastrozole, exemestane and letrozole are AIs that are used in all clinical stages of BC worldwide (Figure 1). Anastrozole and letrozole are non-steroidal AIs, while exemestane is a steroidal AI.38 Although these three compounds have been shown to suppress overall plasma and tissue estrogens by >90%, some studies suggest that letrozole is the most potent of the nonsteroidal compounds.39
In metastatic BC, these AIs can be used sequentially and cause new responses in selected patients after resistance to the first treatment. For example, metastatic BC patients can benefit from a steroidal AI (exemestane) when they relapse after initial treatment with a non-steroidal AI (anastrozole or letrozole).39–41 However, information about the benefit of using a non-steroidal AI after a steroid AI is scarce. Based on the results in metastatic BC after progression with anastrozole or letrozole therapy, treatment with exemestane alone or in combination with an mTOR inhibitor such as everolimus has been recommended.38
CDK4/6 Inhibitors
The addition of CDK4/6 inhibitors to standard endocrine therapy has improved outcomes in first and last line therapy settings.12 CDK4/6 inhibitors are anticancer drugs that have been mainly investigated in the last decade. The anti-carcinogenic effect of CDK4/6 inhibitors is based on their ability to block the progression of the cell cycle from G1 phase to S phase through blocking the activity of the cyclin D-CDK4/6 holoenzyme, thus limiting the proliferation of sensitive tumor cells.42 Currently, three selective CDK4/6 inhibitors, palbociclib, ribociclib, and abemaciclib, have been approved by the Food and Drug Administration (FDA) for use in the treatment of ER+/HER2- BC43 (Figure 1). The use of these inhibitors in combination with AI as first-line therapy or with FUL as second-line therapy in ER+/HER2- metastatic BC has shown improved progression-free survival in Phase III trials.12,44,45 However, abemaciclib is the only inhibitor among the three CDK4/6 inhibitors that has received FDA approval as monotherapy in ER+/HER2- metastatic BC, indicating its high potential as a single agent.46,47
Several phase III trials, such as the PALOMA-3 trial (FUL + palbociclib/placebo),48 the MONARCH-2 trial (FUL + abemaciclib/placebo),44 and the MONALEESA-3 trial (FUL with or without ribociclib),49 have demonstrated the efficacy of the combination of FUL and CDK4/6 inhibitors. In the PALOMA-3 trial, the addition of palbociclib to FUL resulted in a prolongation of overall survival of 6.9 months among patients with advanced ER+/HER2- BC who showed disease progression after prior endocrine therapy.50 In the MONARCH-2 and MONALEESA-3 clinical trials, the combination of FUL with abemaciclib or ribociclib, respectively, resulted in an improvement in both progression-free survival and overall survival.50,51
Despite the benefits of CDK4/6 inhibitors, some adverse effects have been observed, including hematological toxicities, mainly neutropenia, and non-hematological toxicities such as fatigue, nausea, vomiting, stomatitis, alopecia, skin rash, diarrhea, decreased appetite, and infections. However, such toxicities are uncomplicated and manageable with dose interruption or reduction.52
Recent studies have also shown that CDK4/6 inhibitors have similar efficacy when combined with an AI in the first-line treatment of ER+ metastatic BC and are superior to monotherapy with FUL or AI, regardless of tumor characteristics.12 These findings confirm that CDK4/6 inhibitors improve survival outcomes for ER+ BC patients when incorporated with AI in both the first and last lines.47
PI3K Inhibitors
The PI3K/AKT/mammalian target of rapamycin (mTOR) pathway has been demonstrated to be involved in secondary endocrine resistance in ER+ BC,53 and thus inhibition of this pathway is a promising approach to overcome resistance. Therapeutic blocking of this signaling pathway includes the use of PI3K inhibitors (PI3Ki) such as pan-PI3K inhibitors, which include buparlisib and pictilisib54 (Figure 1). Buparlisib is an orally available pan-PI3Ki and the most clinically advanced agent in this class. The efficacy of this PI3Ki has been confirmed in several clinical trials (NCT01339442, NCT01610284), in which the combination of buparlisib with FUL in ER+ BC patients increased prolonged progression-free survival compared with FUL alone.55 Pictilisib is an oral pan-PI3Ki of multiple PI3K isoforms. The combination of pictilisib and FUL in patients with postmenopausal metastatic BC previously treated with an AI was evaluated in a randomized Phase II clinical trial (FERGI, NCT01437566). The trial results showed no significant difference in progression-free survival with FUL + pictilisib compared with FUL + placebo. According to the authors, such results could be due to the higher toxicity from the combination treatment, including rash, diarrhea, transaminitis, and fatigue, which limits the administered dose of pictilisib.56 Based on the above results, the development of pictilisib in this setting was discontinued. Additionally, the mTOR inhibitor everolimus (Afinitor) (Figure 1) overcomes resistance to hormone therapy by controlling the AKT/mTOR signaling pathway.57 Furthermore, recent studies showed the benefit of everolimus with CDK4/6 inhibitor treatment in ER+ metastatic BC patients.58
Drug Repurposing
Drug reuse or repositioning refers to the process of seeking new medical treatments within available medications, rather than developing new medications.59 This strategy reduces the approval time for drug use and increases the success of clinical development. One example of a drug that has been repurposed for BC treatment is raloxifene, which has been used for the treatment and prevention of postmenopausal osteoporosis. Raloxifene was approved by the FDA in 2010 for the primary chemoprevention of BC,60 and one study showed that raloxifene was as effective as TAM in reducing the risk of BC.61 In fact, raloxifene was associated with a reduced risk of invasive BC in postmenopausal women.62 Other repurposed drugs with possible anticancer activity in BC are listed in Table 1.
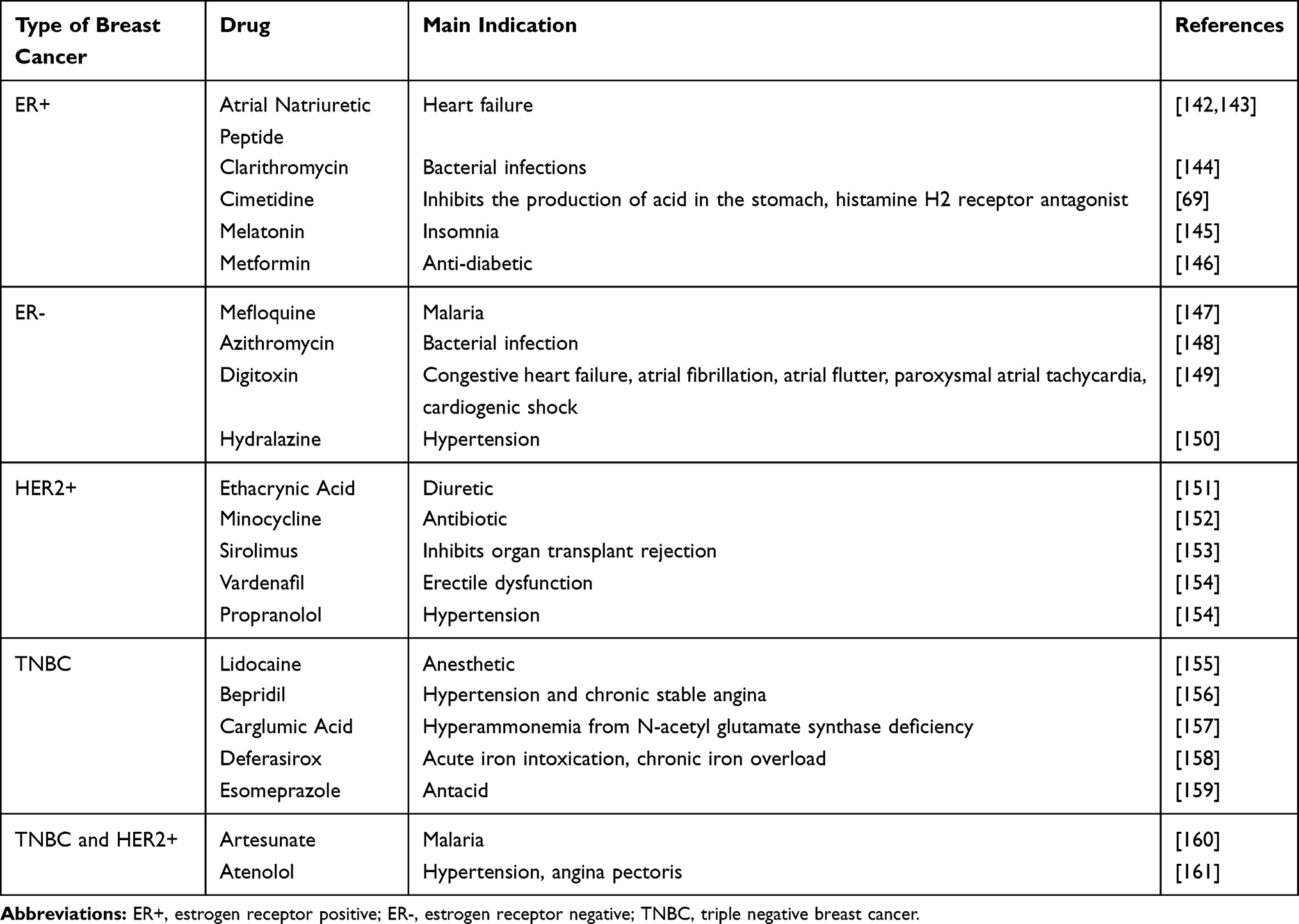 |
Table 1 Some Repurposed Drugs for Breast Cancer Treatment |
In addition, anticancer activity have also been evidenced with the use of metformin, which is used for the treatment of type II diabetes; aspirin, an anti-coagulant;63 clopidogrel, which is used for post-myocardial infarction;64 and fingolimod, a drug for multiple sclerosis.65 Although repurposed drugs have shown more benefits than conventional treatments, some professionals oppose the use of repurposed drugs.66 However, numerous studies have demonstrated the efficiency and effectiveness of repurposed drugs in reducing resistance and toxicity in patients, especially drug combinations of reused drugs and conventional drugs for BC.
Resistance and Overcoming Resistance in HER2+ BC
Drugs approved for the treatment of HER2+ BC include humanized antibodies, PI3K inhibitors, antibody-drug conjugates (ADCs), and TKIs. Additionally, several drugs for the treatment of HER2+ BC, including ADCs, are currently being reviewed by the FDA for possible approval.
Humanized Monoclonal Antibodies (mAbs)
Currently available humanized antibodies for the treatment of HER2+ BC include trastuzumab, pertuzumab, and 19H6-Hu.
Trastuzumab
Trastuzumab specifically binds to the extracellular domain of HER2 to prevent the antibody-dependent cellular cytotoxicity,67–69 inhibiting the downstream signaling of HER270 and negatively regulating HER249,71,72 (Figure 1).The introduction of trastuzumab to therapeutic regimens for patients with HER2+ BC has improved both the response and clinical outcome of these patients. However, not all patients with HER2+ BC respond to treatment and some develop resistance. The mechanisms related to resistance to trastuzumab therapy include the truncated form of HER2 (P95HER2),73 masking by epitopes,74,75 activation of the PI3K/AKT/mTOR signaling pathway,76 and FCGRIIa polymorphisms,77 among others.
In some BC patients, the HER2 protein (185 kDa) gradually loses its extracellular domain through proteolytic detachment and the remaining 95 kDa fragment (p95HER2) associated with the membrane acquires constitutive activity.78 The p95HER2 truncated form of HER2 lacks the extracellular domain, which is the binding site for trastuzumab; however, because the intracellular domain is intact (which shows strong kinase activity), some studies have associated p150 HER2 with clinical resistance to trastuzumab.73 Compared with tumors expressing the full-length HER2, p95HER2-expressing tumors have worse prognosis and are at an increased risk of metastasis.73
A mechanism of obstruction of the connection point between trastuzumab and HER2 corresponds to the masking of epitopes, with proteins Mucin 4 (MUC 4) and hyaluronic complex polymer (CD44) being involved in this mechanism.74 When MUC4 and CD44 are active, they alter the HER2 binding point,74,75 leading to a decrease in trastuzumab binding by 20%.75
The PI3K signaling pathway plays a crucial role in growth and cell survival. Somatic missense mutations in the PIK3CA gene are common in patients with HER2+ BC.76 In addition to contributing to neoplastic transformation,26 these mutations lead to alterations in the PI3K/AKT/mTOR pathway and reduced efficacy of trastuzumab therapy. Therefore, additional diagnostic testing for planning customized treatment, such as detecting PIK3CA gene mutations, have recently been approved and are used to establish personalized therapies.79
Immune effector cells, such as natural killer cells or macrophages, can recognize and bind trastuzumab through their receptors. Some of the receptors, called Fc-gamma receptors, including FCGRIIa, seem to be crucial for the clinical response to trastuzumab. Thus, the presence of polymorphisms in these receptors can modulate the response to trastuzumab and result in the development of resistance.77
Pertuzumab
Pertuzumab is a second-generation recombinant humanized monoclonal antibody that binds to the extracellular dimerization domain II of HER2, preventing its heterodimerization with HER1, HER3, HER4 and IGF-1R31,80 and thus inhibiting cell proliferation (Figure 1). Pertuzumab has been associated with both increased progression-free survival for patients with metastatic BC81 and better outcomes for patients with early BC. However, in patients with non-visceral metastases or small primary tumors with negative nodes, the therapeutic benefit of pertuzumab is relatively small.82
19H6-Hu
Based on the low response rates of BC patients to trastuzumab therapy, new treatment strategies were explored. The new anti-HER2 antibody (19H6-Hu), which enhances the anti-tumor efficacy of trastuzumab and pertuzumab with a distinct mechanism of action, has shown promising efficacy. 19H6-Hu is a novel humanized anti-HER2 monoclonal antibody that binds to the HER2 extracellular domain with high affinity (Figure 1) and inhibited the proliferation of multiple HER2+ BC cell lines as a single agent or in combination with trastuzumab. One study showed that 19H6-Hu in combination with trastuzumab was more effective at blocking phosphorylation of ERK1/2, AKT (S473) and HER2 (Y1248) in HER2+ BC cells compared with trastuzumab alone or in combination with pertuzumab.83
PI3K Inhibitors
PI3K inhibitors are used in combination with AIs for the treatment of metastatic BC.84 However, most PI3K inhibitors are more toxic than beneficial. The FDA approved more selective, less toxic, and more effective inhibitors for the treatment of metastatic BC in patients with PIK3CA gene mutation, such as alpelisib and taselisib85 (Figure 1). Additionally, another study reported that PI3K/AKT/mTOR inhibitors in combination with trastuzumab or trastuzumab and paclitaxel were efficient and safe for patients and showed beneficial anti-tumor activities.86
ADCs
ADCs are a means of delivering cytotoxic drugs specifically to cancer cells. The mechanism of action of ADCs involves the delivery and subsequent internalization of the ADC and the release of highly active, free cytotoxic agents into cancer cells, ultimately leading to cell death. ADCs used in the treatment of advanced BC include trastuzumab emtansine (T-DM1)87 and trastuzumab deruxtecan88 (Figure 1). T-DM1 is a conjugate of trastuzumab and a cytotoxic drug (DM1, derived from maytansine) that is effective and generally well tolerated when administered as a single agent. The efficacy of this ADC has been demonstrated in randomized trials.89 Although the superior efficacy of T-DM1 compared with trastuzumab or trastuzumab plus chemotherapy has been reported in the treatment of metastatic BC, most patients treated with T-DM1 eventually show disease progression,87 and some HER2+ BCs do not respond or only respond minimally to T-DM1.87 One study showed that altered traffic/metabolism of T-DM1 is one of the predominant mechanisms associated with resistance to T-DM1, and a greater understanding of such mechanisms is necessary to develop strategies to overcome T-DM1 resistance.90
Another ADC used in the treatment of BC is trastuzumab deruxtecan. This conjugate comprises an anti-HER2 antibody, a cleavable tetrapeptide-based linker, and a cytotoxic topoisomerase I inhibitor. Although the use of this ADC in a pretreated patient population with HER2+ metastatic BC showed durable antitumor activity, the efficacy of this ADC in HER2+ metastatic BC patients previously treated with TDM-1 needs to be confirmed.88 Trastuzumab deruxtecan is also being evaluated for the treatment of metastatic BC in patients with low HER2 expression, for whom current available therapies targeting HER2 are ineffective. The results of trastuzumab deruxtecan treatment in these patients have shown a response rate of 44.2%.91
Currently, several HER2-directed ADCs are under clinical investigation for both HER2 amplified and HER2 expressing but not amplified BCs, including ARX788 and XMT-1522.ARX788, a novel next-generation anti-HER2 ADC containing an anti-HER2 monoclonal antibody site-specifically conjugated to amberstatin,92,93 has shown anti-tumor effects and rapid tumor regression in murine xenograft models of the HER2+ BC cell lines BT474 and HCC1954.92 Furthermore, ARX788 showed a stronger inhibitory effect than T-DM1 on T-DM1-responsive BC cells and caused complete tumor regression in a trastuzumab-resistant BC xenograft model derived from JIMT-1 cells.94 Two Phase 1 trials on of ARX-788 (clinicaltrials.gov identifiers: NCT02512237 and NCT03255070) are ongoing but the results have not yet been published.95 XMT-1522 is a ADC containing a human IgG1 anti-HER2 monoclonal antibody (HT-19) that binds to domain IV of HER2 to an epitope that is distinct from the trastuzumab binding site.96 Notably, the HT-19 antibody does not compete with trastuzumab or pertuzumab for binding to HER2.97 Clinical studies on a panel of 25 tumor cell lines differentially expressing HER2 showed that XMT-1522 was more potent than T-DM1 in inhibiting cell proliferation.97 Additionally, in a HER2 BC BT474 xenograft model and a patient-derived HER2 xenograft model, treatment with XMT-1522 induced complete tumor regression at doses of 2 mg/kg and 1 mg/kg, respectively.97 Benefits of XMT-1522 therapy were also observed in a BC xenograft model, in which a significant inhibition of tumor growth was observed.98 Antitumor efficacy, with complete response, was also observed in some mice after the combined treatment of XMT-1522 with the anti-PD1 monoclonal antibody; the response was better when the two drugs were administered sequentially (XMT-1522 followed by anti-PD1 monoclonal antibody).95 The use of XMT-1522 is currently being investigated in patients with advanced BC expressing HER2 progressing on standard therapy (clinicaltrials.gov identifier: NCT02952729).
Other ADCs currently under preclinical and clinical research for the treatment of BC include A166, ALT-P7, DHES0815A, DS-8201a, RC48, SYD985, and MEDI4276. However, to the best of our knowledge, there are no published data available for A166, ALT-P7, and DHES0815A at the time of writing.
TKIs
TKIs used to treat HER2+ BC include lapatinib, neratinib, and tucatinib (Figure 1). Lapatinib, a potent and reversible small molecule TKI that binds both HER2 and EGFR, inhibits the growth of trastuzumab-resistant HER2+ tumor cells.99 Neratinib, an irreversible small-molecule TKI of HER1, HER2 and HER4,100 has been shown to be effective as a single agent in the treatment of HER2+ metastatic BC pretreated with trastuzumab.101
Tucatinib is an investigational, oral TKI that is highly selective for the kinase domain of HER2 with minimal inhibition of EGFR.102 Combinations between tucatinib and monoclonal antibodies (such as trastuzumab) have been recently developed for the treatment of HER2+ metastatic BC patients. For instance, a non-randomized open-label phase 1b study found that the combination of tucatinib with trastuzumab and capecitabine showed encouraging antitumor activity in patients with HER2+ BC, including those with brain metastases. However, side effects such as diarrhea, nausea, palmo-plantar erythrodysesthesia syndrome, fatigue, and vomiting were observed.103 Additionally, in heavily pretreated patients with HER2+ metastatic BC, the combination of tucatinib plus trastuzumab and capecitabine resulted in better progression-free survival and overall survival outcomes.104
Resistance and Overcoming Resistance in TNBC
Treatment of TNBC mainly involves the use of chemotherapy. Additionally, given the immunogenicity of TNBC, this type of cancer can respond to immunotherapy. New therapeutic options are currently being developed for this disease, including ECT, androgen antagonists such as bicalutamide and abiraterone acetate,105 and nanosomal docetaxel lipid suspension (NDLS)–based chemotherapy.106
Chemotherapy
Despite the associated short- and long-term risks, chemotherapy remains essential to prevent recurrence in many patients with advanced BC. Chemotherapy is the only systemic therapy with proven efficacy in TNBC and an important complement to endocrine therapy or HER2-targeted therapy in patients with hormone receptor-positive (ER+ and PR+) BC.2 In fact, the first-line implementation of taxanes has been associated with optimization of the immune status of BC patients and therefore with a good clinical response. Optimization of immune status has been associated with increased activity of natural killer and lymphokine-activated killer cells, with increased levels of interleukin-6 (IL-6) and reduced levels of IL-1 and tumor necrosis factor.107 Chemotherapeutics include the use of alkylating agents, antimetabolites, anti-tumor antibiotics, topoisomerase inhibitors, TKIs, and mitotic inhibitors.108
Resistance to chemotherapy is a result of chronic exposure to chemotherapy and increases the chances of recurrence and accelerated metastasis.109 Therefore, resistance is a major limitation for the successful treatment of BC. However, the exact mechanisms leading to such resistance remain unclear.110 Several mechanisms associated with the resistance to chemotherapy are described below.
Membrane Glycoproteins That Act as Effusion Pumps
Membrane glycoproteins that act as effusion pumps are ATP hydrolysis–dependent proteins that actively transport different substrates across the cell membrane. The ATP-dependent transporters or ABC transporters (ATP-binding cassette) include multidrug resistance proteins (MRP) such as the permeability glycoprotein (P-GP). These transport proteins are involved in the most widely recognized drug resistance mechanism.111 However, recent studies show that FUL reverses resistance to doxorubicin (DOX) mediated by glycoprotein transporters in hormone receptor-negative BC cells. The reversal of resistance occurs through the sensitization of tumor cells potentiating DOX cytotoxicity by increased intracellular drug, and further activation of apoptosis and cell cycle arrest34 (Figure 3A).
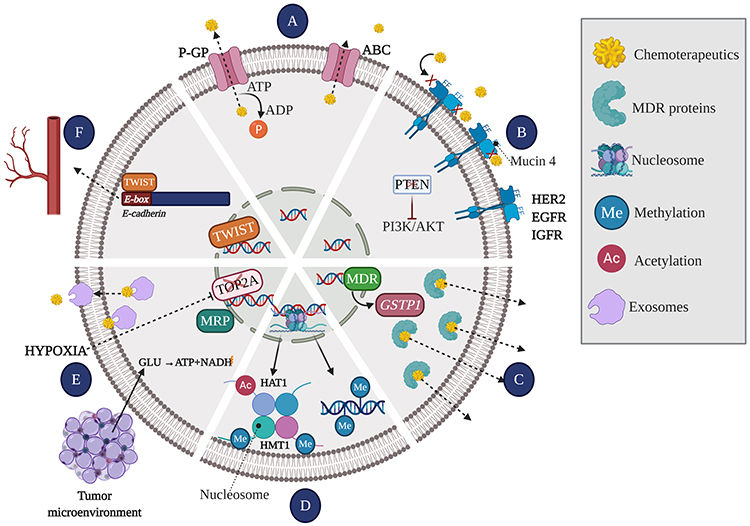 |
Figure 3 Mechanisms leading to the generation of resistance to chemotherapy. (A) Effusion pumps: Membrane glycoproteins act as pumps extracting the drug efflux of intracellular space, thus reducing drug concentration and efficacy. (B) Receptor affinity: The number and affinity of receptors present on the membrane determine the effectiveness of the drug, allowing it to be paired or not. (C) The enzyme system involved in the deactivation of anti-cancer drugs, and therefore involved in the process of drug resistance, includes multi-drug resistance gene (MDR1) and glutathione S transferase Pi (GSTP1). While GSTP1 decreases the concentration and the effective life of drugs, resulting in drug inefficiency, MDR1 gene is significantly overexpressed in multidrug resistance phenotype. (D) Epigenetic mechanisms: DNA methylation and histone modifications [acetylation (Ac) or methylation (Me)] lead to the silencing of tumor suppressor genes and/or overexpression of oncogenes. (E) Tumor microenvironment: the tumor microenvironment creates the optimal niche for the development of cancer cells. In conditions such as hypoxia, cancer cells interact with stromal and immune system cells through exosomes from which they acquire nutrients to use other metabolic pathways, leading to the overexpression of the MRP and deactivating and extracting the drug from inside the cell. Additionally, hypoxia is a trigger for tumor resistance to chemotherapy, since it leads to both decreased DNA topoisomerase II alpha (TOP2A) expression and upregulation of MRP. (F) Angiogenesis: The TWIST1 transcription factor can recognize the E-box gene sequence on the promoter of E-cadherin and depress its transcription, thereby leading to decreased cell adhesion and promoting angiogenesis. |
Receptor Affinity
The amount and affinity of receptors present in the membrane determine the effectiveness of the drug, allowing it to be paired or not (Figure 3B).
Enzyme System That Deactivates Anticancer Drugs
The enzyme system involved in the deactivation of anti-cancer drugs, and therefore involved in the process of drug resistance,111,112 includes multi-drug resistance gene (MDR1) and glutathione S transferase Pi (GSTP1). While GSTP1 decreases the concentration and the effective life of drugs, resulting in drug inefficiency,113 MDR1 gene is significantly overexpressed in multidrug resistance phenotype114 (Figure 3C).
Epigenetic Mechanisms
Epigenetic mechanisms, such as DNA methylation and histone modifications, play a key role in the regulation of gene expression (silencing of tumor suppressor genes and/or overexpression of oncogenes)110,115 (Figure 3D). Alterations in epigenetic mechanism have not only been associated with the acquisition of resistance but are also involved in tumor progression and metastasis.109 The application of epigenetic therapies, such as hydralazine and valproic acid, has been reported to achieve a significant reversal of acquired resistance to chemotherapy.110 In fact, it has been indicated that the molecular effects exerted by valproic acid and hydralazine include the inhibition of histone deacetylases, the demethylation of DNA and the reactivation of genes in primary tumors of patients with BC, increasing the efficacy of chemotherapy.116
Tumor Microenvironment
The tumor microenvironment includes stromal cells (fibroblasts, vascular cells, and immune system cells), soluble factors (such as growth factors, transcription factors, hormones, and cytokines), extracellular matrix, signaling molecules, hypoxia (which facilitates the release of exosomes), and mechanical signals (exosomes).109,117 In addition to creating favorable niches for metastasis, these factors facilitate the transfer miRNA, cytokines, and P-GP from resistant cells, altering the gene expression of sensitive cells, thereby increasing their survival.112,117,118 Tumors are usually exposed to hypoxic conditions; therefore, to obtain energy, they must rely on glycolysis, which turns chemotherapeutic drugs ineffective by the increased expression of metabolic enzymes. In fact, Tsuruo et al119 reported that hypoxia is a trigger for tumor resistance to chemotherapy, since it leads to both decreased DNA topoisomerase II alpha (TOP2A) expression and upregulation of MRP expression (Figure 3E).
Angiogenesis
The twist family bHLH transcription factor 1 (TWIST1) plays a key role in angiogenesis. TWIST1 positively regulates EMT,109 invasion, and metastasis.118 The TWIST1 transcription factor can recognize the E-box gene sequence on the promoters of E-cadherin and depress its transcription, thereby leading to decreased cell adhesion and promoting angiogenesis.113 In addition, was reported that up-regulation of TWIST1 by NF-κB contributes to the chemoresistance113,115 (Figure 3F).
Pharmacokinetics
Pharmacokinetics are associated with the reduction of intracellular drug accumulation because of increased drug efflux, causing the generation of reactive oxygen species and detoxification mediated by exosomes.110,112,120 Pharmacodynamics may confer chemoresistance because it leads to alterations of the drug target,110 repression of tumor suppressor genes, impaired DNA damage repair, acquisition of characteristics similar to cancer stem cells, cellular changes induced by EMT for angiogenesis, and tumor microenvironment conditions.113 The significant reduction in survival rates for repeated relapse patients is the most concerning issue associated with pharmacokinetics.
Resistance to Treatment with Anthracyclines: DOX
Anthracyclines, such as DOX, are one of the most widely used drugs in the treatment of metastatic BC as a single-agent therapy. Anthracyclines are DNA damaging agents and inhibit TOP2A. DOX intercalates into DNA, preventing TOP2A binding and causing replication fork blockage,117 which eventually leads to apoptosis.10 DOX is used as the first-line treatment in BC and primarily for the treatment of advanced BC with an overall response rate of 30%–50%,120 alone or in combination therapy with paclitaxel,110 docetaxel (DOC), cyclophosphamide, 5-fluorouracil, trifluridine, or vorinostat. Despite its extensive use, DOX is an antitumor drug with high cytotoxicity.110 The cardiotoxicity mechanisms of DOX remain to be explained; however, formation of DNA adducts and free radicals have been reported as potential mechanisms.120
Diverse hypotheses have been formulated around DOX resistance, including resistance related to BC tumor subtypes,121 resistance related to the altered expression of specific proteins, including NFκB and small modifier proteins like ubiquitin (SUMO), and resistance related to epigenetic modifications. High levels of IL-6, IL-8, IL-1β, transforming growth factor beta (TGF-β), and the prion protein (PrPc) have also been associated with resistance to DOX.117
Resistance to Treatment with Taxanes: Docetaxel (DOC)
DOC is an antineoplastic agent; its mechanism of action involves inhibiting cell division by stabilizing microtubules, inhibiting tubulin depolymerization, and killing tumor cells.108 DOC is one of the most active drugs available for the treatment of metastatic BC.122 DOC is systematically applied in patients who do not respond or develop resistance to chemotherapy with anthracyclines (such as DOX).3,108 The risks and long-term benefits of chemotherapy are poorly understood because many studies do not report long-term results or do not monitor therapeutic resistance.120 However, in recent prospective trials on neoadjuvant chemotherapy, taxanes along with anthracyclines have been observed to increase life expectancy by metastasis-free survival in more aggressive tumors.118
Resistance to DOC has been mainly associated with miRNA transport and membrane glycoproteins via exosomes, which also promotes high drug efflux from the cell interior to the exterior.112 DOC resistance is highly influenced by the presence of cytokines. For example, IL-8 leads to increased NFκB and is involved in the activation of survival pathways such as those mediated by tumor necrosis factor receptor 2 (TNFR2), whereas high levels of IL-17A lead to increased cell proliferation and thus resistance.117
Immunotherapy
Considering that TNBC has the highest frequency of mutations and therefore the highest possibility of expressing immunogenic neoantigens, this BC subtype is considered to have the highest probability of responding to immunotherapy.123 Cancer immunotherapy aims to overcome the ability of tumor cells to resist the endogenous immune response by stimulating the patient’s immune system. Immunotherapy mainly involves the use of mAbs that are selectively directed to a specific target involved in tumor cell proliferation.124 Among the mAbs used in BC, the synthetic antigen sialyl-Tn has been proven to be safe and highly efficient because it triggers a large immune response.125
Other mAbs include bevacizumab, an antibody that inhibits the activation of VEGF receptor, thereby increasing the rate of drug response and progression-free survival. Other mAbs, such as aflibercept (a recombinant fusion protein) and pazopanib (a TKI) have shown good results in the inhibition of BC tumor cell migration and metastasis. Some mAbs substantially improve the effects of DOX in tumor cells expressing high levels of EGFR.126
In recent years, anti-PD-1 receptor antibodies have been considered incredibly promising in the field of cancer immunotherapy. The PD-1 receptor and its ligands PD-L1/PD-L2 belong to the family of immune control proteins and act as co-inhibitory factors to modulate the response of T cells and to prevent chronic autoimmune inflammation.126,127 Anti-PD-1 mAbs (such as pembrolizumab and nivolumab), which are known as immune checkpoint inhibitors, block the PD-1 receptor or PD-L1 and PD-L2 ligands expressed by cancer cells and prevent their binding; this leads to an inhibition of the immune modulatory signal and allows T cells to remain active against tumors. However, a limited efficacy of these mAbs (targeting PD-1 and PD-L1) has been reported, mainly owing to the induction of apoptosis in patients with metastatic BC because of the low proportion of lymphocytes in infiltrating tumors. Therefore, a combination of immunotherapy and targeted therapies such as angiogenesis inhibitors is used for BC patients.126,127
ECT
ECT was developed as a treatment option for TNBC patients, given the absence of viable therapeutic targets in this BC subtype. ECT involves the efficient delivery of natural bioactive molecules with anti-cancer effects via a biophysical means and is based on the combined use of electroporation together with chemotherapy drugs to increase absorption. Recent research has demonstrated the efficacy of ECT and showed that ECT can dramatically increase the intracellular concentration and cytotoxicity of several FDA-approved drugs, such as bleomycin and cisplatin, leading to a complete tumor response with minimal drug doses.128 Additional studies in BC cell lines showed that ECT increased the cytotoxicity of curcumin treatment by 7-fold,129 increased apoptosis in MDA-MB-231 cells, and minimally affected the viability of non-cancerous MCF10A cells.129,130 A recent study demonstrated the anticancer effects of ECT against the MDA-MB-231 cell line, which is representative of the TNBC subtype.131 These effects include the suppression of key proteins involved in the proliferation, differentiation, migration, survival, and apoptosis of cancer cells.
Emerging Treatments
In recent years, emerging treatments are being developed that could offer additional benefits to conventional treatments that are mainly related to overcoming resistance and decreased side effects. These treatments involve nanotechnological approaches (Figure 1), CRISPR, exosomes, and miRNAs.
Nanotechnological Approaches
Nanotechnological approaches are being developed to minimize the toxicity in healthy tissues generated by targeted therapy while maintaining the efficacy of the treatments. These approaches are based on a nanoparticle delivery system (nanocarriers) that increases the drug-site contact time and the elimination time and reduces the resistance to the drug. Examples of nanocarriers in BC therapy include nanoparticles, polymeric micelles, dendrimers, carbon nanotubes, liposomes, and quantum dots107,112,113 (Figure 1).
The NDLS-based chemotherapy system was recently developed. NDLS is a new formulation of lipid-based, polysorbate 80 and ethanol-free formulation of docetaxel developed to overcome toxicity problems.132 The function of NDLS is based on its ability to infiltrate and become entrapped in the damaged tumor vasculature and collagen material of necrotic tumor tissue, leading to increased drug retention (leading to an enhanced permeability and retention effect).106 NDLS showed comparable efficacy and tolerability to conventional DOC in the treatment of metastatic BC in both prospective and retrospective studies.132 A recent study demonstrated that NDLS-based chemotherapy was effective and well tolerated in the treatment of patients with BC in all settings (neoadjuvant, adjuvant, and metastatic).106 The efficacy of NDLS is currently being prospectively evaluated in patients with TNBC (ClinicalTrials.gov identifier: NCT03671044).
CRISPR
Given the wide variety of morphological and molecular features of tumor cells, the identification of new therapeutic targets to prolong survival and limit the acquisition of resistance in patients with BC is crucial. Implementation of genomics edition by CRISPR system has provided a glimpse of broad cellular heterogeneity and the opportunity to act on tumor cells with mutant alleles,133,134 such as alterations in EGFR, KRAS, BAP1, BRAF, BRCA1, and BRCA2, or with replicative immortality without affecting normal cells.133,135 The CRISPR system (CRISPR/Cas9) has been established in preclinical trials as an effective tool to specifically attack and/or resensitize tumor cells.136
Potential targets have been identified in BC through CRISPR/Cas9, including microtubule affinity regulating kinase 4 (MARK4) and fermitin family homolog-2 (FERMT2), which are related to metastasis; microtubule-associated serine/threonine kinase (MASTL), which is involved in cell proliferation; the tyrosine protein kinase FYN that is involved in drug resistance; and cyclin-dependent kinase 7 (CDK7), which is negatively associated with tumor size and grade.135 CRISPR/Cas9 has also been used to generate changes in exons of the HER2 gene. Such changes lead to the expression of a truncated HER2 protein that prevents dimerization of the transmembrane domain, leading to inhibition of signaling pathways such as MAPK/ERK and PI3K/AKT, as well as cell proliferation and tumorigenicity.134 As with immunotherapy, CRISPR/Cas9 can also target stromal cells in the tumor microenvironment to generate mutations in VEGF and VEGFR2, thereby inhibiting stromal cell migration activity.136
Exosomes
Exosomes, an important class of extracellular vesicles, transport and distribute biological macromolecules, such as proteins, nucleic acids, and anti-tumor drugs, and are considered promising therapeutic strategies for targeted drug delivery in BC.137 The therapeutic potential of exosomes as nano-transporters of DOX has been considered as a strategy to overcome resistance to chemotherapy.138 The use of exosomes could allow the targeted administration of drugs, overcoming several limitations associated with conventional nanoparticles, including cytotoxicity, drug modification, greater synergistic effects, and biocompatibility, among others.107,112,113
miRNAs
Several miRNAs have been identified as biomarkers for prognosis and treatment planning in BC.139 miRNAs also play a crucial role in the regulation of BC cell sensitivity to chemotherapy.140,141 For example, miR326 and miR-451 negatively regulate the expression of MDR genes, including MDR-1, to overcome resistance. MDR-1, which encodes the P-GP transporter, multi-drug resistance proteins (MRPs), and the BC resistance protein (BCRP), which transport both hydrophilic and hydrophobic substrates, are particularly important for chemotherapy.111 Over 80% of the currently used anticancer agents are transported by P-GP, and thus its overexpression can lead to multi-drug resistance.113 Several reports have identified miRNAs that play important functions in BC (Table 2).
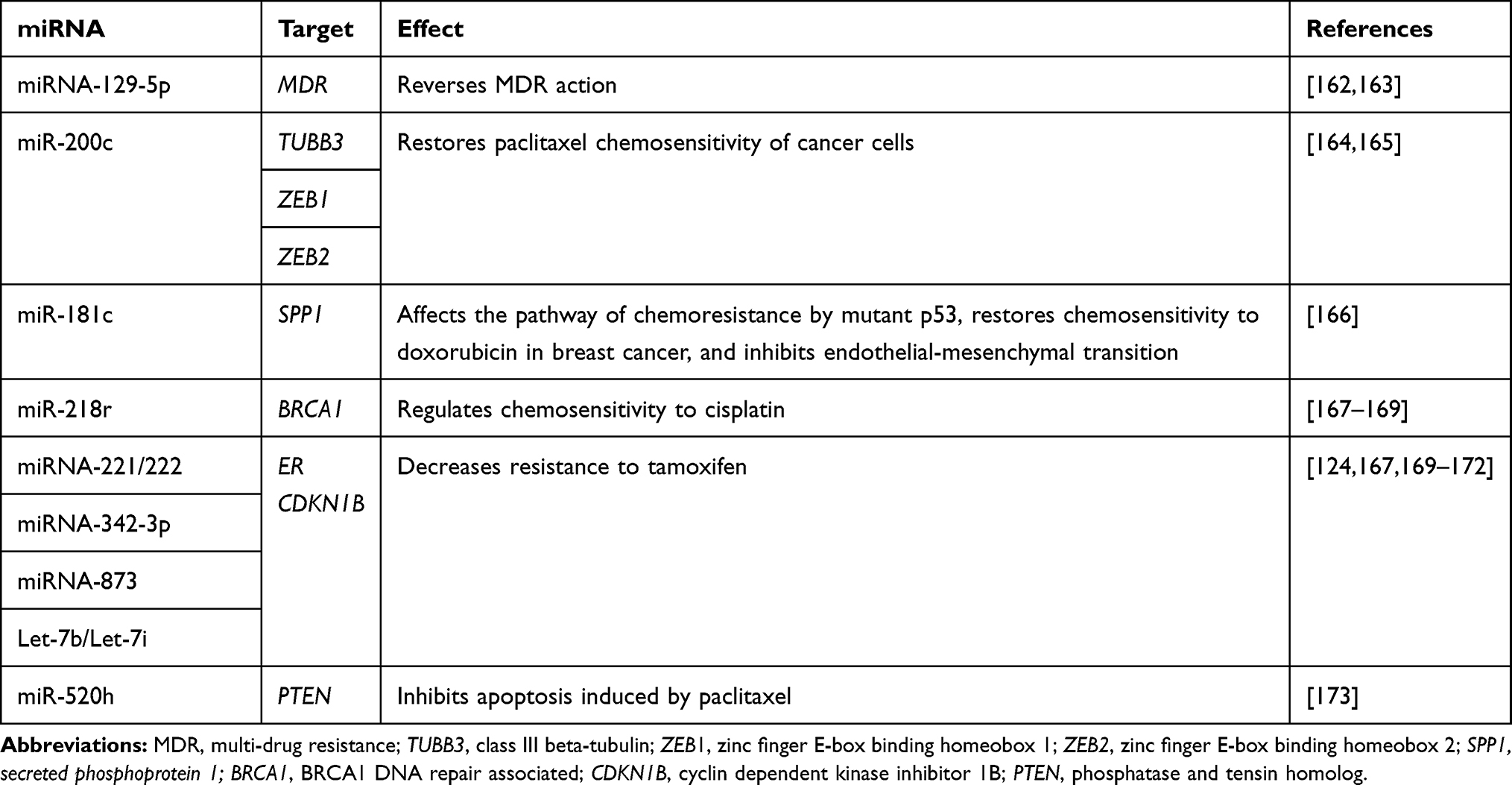 |
Table 2 miRNAs in the Response to Breast Cancer Therapy |
Conclusions
Cancer is a common disease and represents one of the biggest health problems in the world, with BC being the leading cause of cancer death in women. One of the most significant clinical problems in the treatment of BC is the development of drug resistance. Therefore, the identification of the possible biological and molecular mechanisms involved in such resistance could help the development of new therapeutic targets. Recent research has shown that the application of combined therapies and use of emerging therapies may be effective for overcoming resistance and minimizing side effects, respectively.
Acknowledgments
We thank the Universidad del Rosario for the payment of the edition and publication of the article. The authors also thank Gabrielle White Wolf, PhD, from Edanz Group (https://en-author-services.edanzgroup.com/ac) for editing a draft of this manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
1. International Agency for Research on Cancer. Global cancer observatory. Cancer Today; 2018. Available from: https://gco.iarc.fr/.
2. Waks A, Winer E. Breast cancer treatment: a review. JAMA. 2019;321(3):288–300. doi:10.1001/jama.2018.19323
3. International Agency for Research on Cancer. Global cancer observatory. Latest world cancer statistics – GLOBOCAN 2012: estimated cancer incidence, mortality and prevalence Worldwide in 2012; 2013. Available from: https://www.iarc.fr/news-events/latest-world-cancer-statistics-globocan-2012-estimated-cancer-incidence-mortality-and-prevalence-worldwide-in-2012/.
4. Bray F, Ferlay J, Laversanne M, et al. Cancer I ncidence in F ive C ontinents: inclusion criteria, highlights from Volume X and the global status of cancer registration. Int J Cancer. 2015;137(9):2060–2071. doi:10.1002/ijc.29670
5. Harbeck N, Cortes J, Gnant M, et al. Breast cancer. Nat Rev. 2019;5(66). doi:10.1038/s41572-019-0111-2
6. Prat A, Parker JS, Karginova O, et al. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010;12(5):R68. doi:10.1186/bcr2635
7. Parisot JP, Hu XF, DeLuise M, Zalcberg JR. Altered expression of the IGF-1 receptor in a tamoxifen-resistant human breast cancer cell line. Br J Cancer. 1999;79(5–6):693–700. doi:10.1038/sj.bjc.6690112
8. Berry D, Muss H, Thor A, et al. HER-2/neu and p53 expression versus tamoxifen resistance in estrogen receptor–positive, node-positive breast cancer. J Clin Oncol. 2000;18(20):3471–3479. doi:10.1200/JCO.2000.18.20.3471
9. Tai W, Mahato R, Cheng K. The role of HER2 in cancer therapy and targeted drug delivery. J Control Release. 2010;146(3):264–275. doi:10.1016/j.jconrel.2010.04.009
10. Early Breast Cancer Trialists’ Collaborative Group. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005;365(9472):1687–1717. doi:10.1016/S0140-6736(05)66544-0.
11. Spring LM, Gupta A, Reynolds KL, et al. Neoadjuvant endocrine therapy for estrogen receptor–positive breast cancer: a systematic review and meta-analysis. JAMA Oncol. 2016;2(11):1477–1486. doi:10.1001/jamaoncol.2016.1897
12. Rossi V, Berchialla P, Giannarelli D, et al. Should all patients with HR-positive HER2-negative metastatic breast cancer receive CDK 4/6 inhibitor as first-line based therapy? A network meta-analysis of data from the PALOMA 2, MONALEESA 2, MONALEESA 7, MONARCH 3, FALCON, SWOG and FACT trials. Cancers (Basel). 2019;11(11):1661. doi:10.3390/cancers11111661
13. Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lancet. 2011;378(9793):771–784. doi:10.1016/S0140-6736(11)60993-8.
14. Droog M, Beelen K, Linn S, Zwart W. Tamoxifen resistance: from bench to bedside. Eur J Pharmacol. 2013;717(1–3):47–57. doi:10.1016/j.ejphar.2012.11.071
15. Abe O, Abe R, Enomoto K, et al. Tamoxifen for early breast cancer: an overview of the randomised trials. Lancet. 1998;351(9114):1451–1467. doi:10.1016/S0140-6736(97)11423-4
16. Early Breast Cancer Trialists’ Collaborative Group. Systemic treatment of early breast cancer by hormonal, cytotoxic, or immune therapy. 133 randomised trials involving 31,000 recurrences and 24,000 deaths among 75,000 women.. Lancet. 1992;339(8784):1–15.
17. Colleoni M, Gelber S, Goldhirsch A, et al. Tamoxifen after adjuvant chemotherapy for premenopausal women with lymph node-positive breast cancer: International Breast Cancer Study Group Trial 13-93. J Clin Oncol off J Am Soc Clin Oncol. 2006;24(9):1332–1341. doi:10.1200/JCO.2005.03.0783
18. Kumar R, Zakharov MN, Khan SH, et al. The dynamic structure of the estrogen receptor. J Amino Acids. 2011;2011:1–7. doi:10.4061/2011/812540
19. Kedia-Mokashi N, Makawy AEL, Saxena M, Balasinor NH. Chromosomal aberration in the post-implantation embryos sired by tamoxifen treated male rats. Mutat Res - Genet Toxicol Environ Mutagen. 2010;703(2):169–173. doi:10.1016/j.mrgentox.2010.08.016
20. Bianco S, Gévry N. Endocrine resistance in breast cancer: from cellular signaling pathways to epigenetic mechanisms. Transcription. 2012;3(4):165–170. doi:10.4161/trns.20496
21. Nass N, Kalinski T. Tamoxifen resistance: from cell culture experiments towards novel biomarkers. Pathol Res Pract. 2015;211(3):189–197. doi:10.1016/j.prp.2015.01.004
22. Zundelevich A, Dadiani M, Kahana-Edwin S, et al. ESR1 mutations are frequent in newly diagnosed metastatic and loco-regional recurrence of endocrine-treated breast cancer and carry worse prognosis. Breast Cancer Res. 2020;22(1):16. doi:10.1186/s13058-020-1246-5
23. Li S, Shen D, Shao J, et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep. 2013;4(6):1116–1130. doi:10.1016/j.celrep.2013.08.022
24. McGraw J, Waller D. Cytochrome P450 variations in different ethnic populations. Expert Opin Drug Metab Toxicol. 2012;8(3):371–382. doi:10.1517/17425255.2012.657626
25. Ignatov A, Ignatov T, Weißenborn C, et al. G-protein-coupled estrogen receptor GPR30 and tamoxifen resistance in breast cancer. Breast Cancer Res Treat. 2011;128(2):457–466. doi:10.1007/s10549-011-1584-1
26. Kang S, Bader A, Vogt P. Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc Natl Acad Sci U S A. 2005;102(3):802–807. doi:10.1073/pnas.0408864102
27. Zhao W, Zhang Q, Kang X, Jin S, Lou C. AIB1 is required for the acquisition of epithelial growth factor receptor-mediated tamoxifen resistance in breast cancer cells. Biochem Biophys Res Commun. 2009;380(3):699–704. doi:10.1016/j.bbrc.2009.01.155
28. Razavi P, Chang MT, Xu G, et al. The genomic landscape of endocrine-resistant advanced breast cancers. Cancer Cell. 2018;34(3):427–438.e6. doi:10.1016/j.ccell.2018.08.008
29. Huang D, Yang F, Wang Y, Guan X. Mechanisms of resistance to selective estrogen receptor down-regulator in metastatic breast cancer. Biochim Biophys Acta - Rev Cancer. 2017;1868(1):148–156. doi:10.1016/j.bbcan.2017.03.008
30. Li J, Yen C, Liaw D, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275(5308):1943–1947. doi:10.1126/science.275.5308.1943
31. Miller TW, Pérez-Torres M, Narasanna A, et al. Loss of Phosphatase and tensin homologue deleted on chromosome 10 engages ErbB3 and insulin-like growth factor-I receptor signaling to promote antiestrogen resistance in breast cancer. Cancer Res. 2009;69(10):4192–4201. doi:10.1158/0008-5472.CAN-09-0042
32. Raha P, Thomas S, Munster PN. Epigenetic modulation: a novel therapeutic target for overcoming hormonal therapy resistance. Epigenomics. 2011;3(4):451–470. doi:10.2217/epi.11.72
33. Noriega-Reyes MY, Langley McCarron E. Estrogen receptor corregulators and their implication in breast cancer. Cancerology. 2008;3:29–40.
34. Huang Y, Jiang D, Sui M, Wang X, Fan W. Fulvestrant reverses doxorubicin resistance in multidrug-resistant breast cell lines independent of estrogen receptor expression. Oncol Rep. 2017;37:705–712. doi:10.3892/or.2016.5315
35. U.S Food and Drug Administration. FDA approves first PI3K inhibitor for breast cancer. Press Announcements; 2019.
36. Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation, and therapeutic targeting. Nat Rev Cancer. 2015;15(1):7–24. doi:10.1038/nrc3860
37. André F, Ciruelos EM, Rubovszky G, et al. Alpelisib (ALP) + fulvestrant (FUL) for advanced breast cancer (ABC): results of the Phase 3 SOLAR-1 trial. En:
38. Bahrami N, Chang G, Kanaya N, et al. Changes in serum estrogenic activity during neoadjuvant therapy with letrozole and exemestane. J Steroid Biochem Mol Biol. 2020;200:105641. doi:10.1016/j.jsbmb.2020.105641
39. Geisler J, Helle H, Ekse D, et al. Letrozole is superior to anastrozole in suppressing breast cancer tissue and plasma estrogen levels. Clin Cancer Res. 2008;14(19):6330–6335. doi:10.1158/1078-0432.CCR-07-5221
40. Carlini P, Michelotti A, Ferretti G, et al. Clinical evaluation of the use of exemestane as further hormonal therapy after nonsteroidal aromatase inhibitors in postmenopausal metastatic breast cancer patients. Cancer Invest. 2007;25(2):102–105. doi:10.1080/07357900701224789
41. Chin YS, Beresford MJ, Ravichandran D, Makris A. Exemestane after non-steroidal aromatase inhibitors for post-menopausal women with advanced breast cancer. Breast. 2007;16(4):436–439. doi:10.1016/j.breast.2007.02.002
42. Sharpless NE, Sherr CJ. Forging a signature of in vivo senescence. Nat Rev Cancer. 2015;15(7):397–408. doi:10.1038/nrc3960
43. Spring LM, Wander SA, Zangardi M, Bardia A. CDK 4/6 inhibitors in breast cancer: current controversies and future directions. Curr Oncol Rep. 2019;21(3):25. doi:10.1007/s11912-019-0769-3
44. Goetz MP, Toi M, Campone M, et al. MONARCH 3: abemaciclib as initial therapy for advanced breast cancer. J Clin Oncol. 2017;35(32):3638–3646. doi:10.1200/JCO.2017.75.6155
45. Sledge GW, Toi M, Neven P, et al. MONARCH 2: abemaciclib in combination with fulvestrant in women with HR+/HER2-advanced breast cancer who had progressed while receiving endocrine therapy. J Clin Oncol. 2017;35(25):2875–2884. doi:10.1200/JCO.2017.73.7585
46. Dickler MN, Tolaney SM, Rugo HS, et al. MONARCH 1, a phase II study of abemaciclib, a CDK4 and CDK6 inhibitor, as a single agent, in patients with refractory HR+/HER2− metastatic breast cancer. Clin Cancer Res. 2017;23(17):5218–5224. doi:10.1158/1078-0432.CCR-17-0754
47. Chong QY, Kok ZH, Bui NLC, et al. A unique CDK4/6 inhibitor: current and future therapeutic strategies of abemaciclib. Pharmacol Res. 2020;156:104686. doi:10.1016/j.phrs.2020.104686
48. Finn RS, Crown JP, Lang I, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised Phase 2 study. Lancet Oncol. 2015;16(1):25–35. doi:10.1016/S1470-2045(14)71159-3
49. Hortobagyi GN, Stemmer SM, Burris HA, et al. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N Engl J Med. 2016;375(18):1738–1748. doi:10.1056/NEJMoa1609709
50. Turner NC, Slamon DJ, Ro J, et al. Overall survival with palbociclib and fulvestrant in advanced breast cancer. N Engl J Med. 2018;379(20):1926–1936. doi:10.1056/NEJMoa1810527
51. Killock D. CDK4/6 inhibitors prolong OS. Nat Rev Clin Oncol. 2019;1.
52. Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov. 2015;14(2):130–146. doi:10.1038/nrd4504
53. Miller TW, Hennessy BT, González-Angulo AM, et al. Hyperactivation of phosphatidylinositol-3 kinase promotes escape from hormone dependence in estrogen receptor–positive human breast cancer. J Clin Invest. 2010;120(7):2406–2413. doi:10.1172/JCI41680
54. Verret B, Cortes J, Bachelot T, Andre F, Arnedos M. Efficacy of PI3K inhibitors in advanced breast cancer. Ann Oncol. 2019;30(Supplement_10):x12–x20. doi:10.1093/annonc/mdz381
55. Ma CX, Luo J, Naughton M, et al. A Phase I trial of BKM120 (Buparlisib) in combination with fulvestrant in postmenopausal women with estrogen receptor–positive metastatic breast cancer. Clin Cancer Res. 2016;22(7):1583–1591. doi:10.1158/1078-0432.CCR-15-1745
56. Krop IE, Mayer IA, Ganju V, et al. Pictilisib for oestrogen receptor-positive, aromatase inhibitor-resistant, advanced or metastatic breast cancer (FERGI): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2016;17(6):811–821. doi:10.1016/S1470-2045(16)00106-6
57. Hurvitz SA, Peddi PF. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. Breast Dis. 2013;24(1):79–81. doi:10.1016/j.breastdis.2013.01.007
58. Dhakal A, Antony Thomas R, Levine EG, et al. Outcome of everolimus-based therapy in hormone-receptor-positive metastatic breast cancer patients after progression on palbociclib. Breast Cancer Basic Clin Res. 2020;14:1178223420944864. doi:10.1177/1178223420944864
59. Langedijk J, Mantel-Teeuwisse AK, Slijkerman DS, Schutjens M-HDB. Drug repositioning and repurposing: terminology and definitions in literature. Drug Discov Today. 2015;20(8):1027–1034. doi:10.1016/j.drudis.2015.05.001
60. D’Amelio P, Isaia GC. The use of raloxifene in osteoporosis treatment. Expert Opin Pharmacother. 2013;14(7):949–956. doi:10.1517/14656566.2013.782002
61. Waters EA, McNeel TS, Stevens WM, Freedman AN. Use of tamoxifen and raloxifene for breast cancer chemoprevention in 2010. Breast Cancer Res Treat. 2012;134(2):875–880. doi:10.1007/s10549-012-2089-2
62. Lippman ME, Cummings SR, Disch DP, et al. Effect of raloxifene on the incidence of invasive breast cancer in postmenopausal women with osteoporosis categorized by breast cancer risk. Clin Cancer Res. 2006;12(17):5242–5247. doi:10.1158/1078-0432.CCR-06-0688
63. Shah RR, Stonier PD. Repurposing old drugs in oncology: opportunities with clinical and regulatory challenges ahead. J Clin Pharm Ther. 2019;44(1):6–22.
64. Denslow A, Świtalska M, Jarosz J, et al. Clopidogrel in a combined therapy with anticancer drugs—effect on tumor growth, metastasis, and treatment toxicity: studies in animal models. PLoS One. 2017;12(12):e0188740. doi:10.1371/journal.pone.0188740
65. White C, Alshaker H, Cooper C, Winkler M, Pchejetski D. The emerging role of FTY720 (Fingolimod) in cancer treatment. Oncotarget. 2016;7(17):23106. doi:10.18632/oncotarget.7145
66. Pantziarka P, Bouche G, Meheus L, Sukhatme V, Sukhatme VP, Vikas P. The repurposing drugs in oncology (ReDO) project. Ecancermedicalscience. 2014;8. doi:10.3332/ecancer.2014.485
67. Barok M, Isola J, Pályi-Krekk Z, et al. Trastuzumab causes antibody-dependent cellular cytotoxicity–mediated growth inhibition of submacroscopic JIMT-1 breast cancer xenografts despite intrinsic drug resistance. Mol Cancer Ther. 2007;6(7):2065–2072. doi:10.1158/1535-7163.MCT-06-0766
68. Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med. 2000;6(4):443–446. doi:10.1038/74704
69. Pantziarka P, Bouche G, Meheus L, Sukhatme V, Sukhatme VP. Repurposing drugs in oncology (ReDO) - mebendazole as an anti-cancer agent. Ecancermedicalscience. 2014;8(1). doi:10.3332/ecancer.2014.443
70. Nagata Y, Lan K, Zhou X, et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6(2):117–127. doi:10.1016/j.ccr.2004.06.022
71. Moasser M. Targeting the function of the HER2 oncogene in human cancer therapeutics. Oncogene. 2007;26(46):6577–6592. doi:10.1038/sj.onc.1210478
72. Shi Y, Fan X, Meng W, Deng H, Zhang N, An Z. Engagement of immune effector cells by trastuzumab induces HER2/ERBB2 downregulation in cancer cells through STAT1 activation. Breast Cancer Res. 2014;16(2):R33. doi:10.1186/bcr3637
73. Scaltriti M, Rojo F, Ocaña A, et al. Expression of p95HER2, a truncated form of the HER2 receptor, and response to anti-HER2 therapies in breast cancer. J Natl Cancer Inst. 2007;99(8):628–638. doi:10.1093/jnci/djk134
74. Nahta R, Yu D, Hung MC, Hortobagyi GN, Esteva FJ. Mechanisms of disease: understanding resistance to HER2-targeted therapy in human breast cancer. Nat Clin Pract Oncol. 2006;3(5):269–280. doi:10.1038/ncponc0509
75. Nagy P, Friedländer E, Tanner M, et al. Decreased accessibility and lack of activation of ErbB2 in JIMT-1, a herceptin-resistant, MUC4-expressing breast cancer cell line. Cancer Res. 2005;65(2):473–482.
76. Campbell I, Russell S, Choong D, et al. Mutation of the PIK3CA gene in ovarian and breast cancer. Cancer Res. 2004;64(21):7678–7681. doi:10.1158/0008-5472.CAN-04-2933
77. Warmerdam P, Van de Winkel J, Vlug A, Westerdaal N, Capel P. A single amino acid in the second Ig-like domain of the human Fc gamma receptor II is critical for human IgG2 binding. J Immunol. 1991;147(4):1338–1343.
78. Christianson T, Doherty J, Lin Y, et al. NH2-terminally truncated HER-2/neu protein: relationship with shedding of the extracellular domain and with prognostic factors in breast cancer. Cancer Res. 1998;58(22):5123–5129.
79. U.S. Food and Drug Administration. FDA approves first PI3K inhibitor for breast cancer. FDA news release; 2019. Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-first-pi3k-inhibitor-breast-cancer.
80. Quandt D, Fiedler E, Boettcher D, Marsch WC, Seliger B. B7-h4 expression in human melanoma: its association with patients’ survival and antitumor immune response. Clin Cancer Res. 2011;17(10):3100–3111.
81. Swain SM, Baselga J, Kim S-B, et al. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N Engl J Med. 2015;372(8):724–734. doi:10.1056/NEJMoa1413513
82. Ishii K, Morii N, Yamashiro H. Pertuzumab in the treatment of HER2-positive breast cancer: an evidence-based review of its safety, efficacy, and place in therapy. Core Evid. 2019;14:51–70. doi:10.2147/ce.s217848
83. Zhang X, Chen J, Weng Z, et al. A new anti-HER2 antibody that enhances the anti-tumor efficacy of trastuzumab and pertuzumab with a distinct mechanism of action. Mol Immunol. 2020;119:48–58. doi:10.1016/j.molimm.2020.01.009
84. Qin H, Liu L, Sun S, et al. The impact of PI3K inhibitors on breast cancer cell and its tumor microenvironment. PeerJ. 2018;6:e5092. doi:10.7717/peerj.5092
85. Tran B, Bedard PL. Luminal-B breast cancer and novel therapeutic targets. Breast Cancer Res. 2011;13(6):221. doi:10.1186/bcr2904
86. Tong CWS, Wu M, Cho W, To KKW. Recent advances in the treatment of breast cancer. Front Oncol. 2018;8:227. doi:10.3389/fonc.2018.00227
87. Barok M, Joensuu H, Isola J. Trastuzumab emtansine: mechanisms of action and drug resistance. Breast Cancer Res. 2014;16(2):1–12. doi:10.1186/bcr3621
88. Modi S, Saura C, Yamashita T, et al. Trastuzumab deruxtecan in previously treated HER2-positive breast cancer. N Engl J Med. 2020;382(7):610–621. doi:10.1056/NEJMoa1914510
89. Begovac M QTc prolongation in patients treated with trastuzumab and ado-trastuzumab-emtazine; 2019. Available from: https://repozitorij.mef.unizg.hr/islandora/object/mef:2463.
90. Hunter FW, Barker HR, Lipert B, et al. Mechanisms of resistance to trastuzumab emtansine (T-DM1) in HER2-positive breast cancer. Br J Cancer. 2019;122(5):603–612. doi:10.1158/1535-7163.MCT-17-0296
91. Modi S, Tsurutani J, Tamura K, et al. Trastuzumab deruxtecan (DS-8201a) in subjects with HER2-low expressing breast cancer: updated results of a large phase 1 study. Cancer Res. 2019;79(4Suppl):P6–17.
92. Skidmore L, Sakamuri S, Knudsen N, et al. ARX788, a site-specific anti-HER2 antibody drug conjugate, demonstrates potent and selective activity in HER2 low and T-DM1 resistant breast and gastric cancers. Mol Cancer Ther. 2020:
93. Newman DJ, Cragg GM. Current status of marine-derived compounds as warheads in anti-tumor drug candidates. Mar Drugs. 2017;15(4):99. doi:10.3390/md15040099
94. Barok M, Le Joncour V, Martins A, et al. ARX788, a novel anti-HER2 antibody-drug conjugate, shows anti-tumor effects in preclinical models of trastuzumab emtansine-resistant HER2-positive breast cancer and gastric cancer. Cancer Lett. 2020;473:156–163. doi:10.1016/j.canlet.2019.12.037
95. Rinnerthaler G, Gampenrieder SP, Greil R. HER2 directed antibody-drug-conjugates beyond T-DM1 in breast cancer. Int J Mol Sci. 2019;20(5):1115. doi:10.3390/ijms20051115
96. Yurkovetskiy A, Gumerov D, Ter-Ovanesyan E, et al. Non-clinical pharmacokinetics of XMT-1522, a HER2 targeting auristatin-based antibody drug conjugate. Cancer Res. 2017;77(13):48. doi:10.1158/1538-7445.AM2017-48
97. Bergstrom DA, Bodyak N, Park PU, et al. Abstract P4-14-28: XMT-1522 induces tumor regressions in pre-clinical models representing HER2-positive and HER2 low-expressing breast cancer. 2016. doi:10.1158/1538-7445.SABCS15-P4-14-28
98. Traore T, Khattar M. Abstract lb-294: synergy of an anti-HER2 ADC TAK-522 (XMT-1522) in combination with anti-PD1 monoclonal antibody (MAB) in a syngeneic breast cancer model expressing human HER2. 2018. doi:10.1158/1538-7445.AM2018-LB-294
99. Murphy CG, Modi S. HER2 breast cancer therapies: a review. Biol Targets Ther. 2009;3:289–301. doi:10.2147/BTT.S3479
100. Rabindran SK, Discafani CM, Rosfjord EC, et al. Antitumor activity of HKI-272, an orally active, irreversible inhibitor of the HER-2 tyrosine kinase. Cancer Res. 2004;64(11):3958–3965. doi:10.1158/0008-5472.CAN-03-2868
101. Li M, Hirano K, Ikeda Y, et al. Triglyceride deposit cardiomyovasculopathy: a rare cardiovascular disorder. Orphanet J Rare Dis. 2019;14(1):1–9. doi:10.1186/s13023-019-1087-4
102. Moulder SL, Borges VF, Baetz T, et al. Phase I study of ONT-380, a HER2 inhibitor, in patients with HER2+-advanced solid tumors, with an expansion cohort in HER2+ metastatic breast cancer (MBC). Clin Cancer Res. 2017;23(14):3529–3536. doi:10.1158/1078-0432.CCR-16-1496
103. Murthy R, Borges VF, Conlin A, et al. Tucatinib with capecitabine and trastuzumab in advanced HER2-positive metastatic breast cancer with and without brain metastases: a non-randomised, open-label, phase 1b study. Lancet Oncol. 2018;19(7):880–888. doi:10.1016/S1470-2045(18)30256-0
104. Murthy RK, Loi S, Okines A, et al. Tucatinib, trastuzumab, and capecitabine for HER2-positive metastatic breast cancer. N Engl J Med. 2020;382(7):597–609. doi:10.1056/NEJMoa1914609
105. Bonnefoi H, Grellety T, Tredan O, et al. A phase II trial of abiraterone acetate plus prednisone in patients with triple-negative androgen receptor positive locally advanced or metastatic breast cancer (UCBG 12-1). Ann Oncol. 2016;27(5):812–818. doi:10.1093/annonc/mdw067
106. Subramanian S, Prasanna R, Biswas G, et al. Nanosomal docetaxel lipid suspension-based chemotherapy in breast cancer: results from a multicenter retrospective study. Breast Cancer Targets Ther. 2020;12:77–85. doi:10.2147/BCTT.S236108
107. Loibl S, Furlanetto J. Targeting the immune system in breast cancer: hype or hope?: TILs and newer immune-based therapies being evaluated for HER2+ and TNBC. Curr Breast Cancer Rep. 2015;7(4):203–209. doi:10.1007/s12609-015-0193-0
108. Verweij J, Clavel M, Chevalier B. Paclitaxel (TaxolTM) and docetaxel (TaxotereTM): not simply two of a kind. Ann Oncol. 1994;5(6):495–505. doi:10.1093/oxfordjournals.annonc.a058903
109. Norouzi S, Gorgi Valokala M, Mosaffa F, Zirak MR, Zamani P, Behravan J. Crosstalk in cancer resistance and metastasis. Crit Rev Oncol Hematol. 2018;132:145–153. doi:10.1016/j.critrevonc.2018.09.017
110. Ponnusamy L, Mahalingaiah PKS, Chang YW, Singh KP. Reversal of epigenetic aberrations associated with the acquisition of doxorubicin resistance restores drug sensitivity in breast cancer cells. Eur J Pharm Sci. 2018;123:56–69. doi:10.1016/j.ejps.2018.07.028
111. Tang Y, Wang Y, Kiani MF, Wang B. Classification, treatment strategy, and associated drug resistance in breast cancer. Clin Breast Cancer. 2016;16(5):335–343. doi:10.1016/j.clbc.2016.05.012
112. Yu D, Wu Y, Shen H, et al. Exosomes in development, metastasis and drug resistance of breast cancer. Cancer Sci. 2015;106(8):959–964. doi:10.1111/cas.12715
113. Ji X, Lu Y, Tian H, Meng X, Wei M, Cho WC. Chemoresistance mechanisms of breast cancer and their countermeasures. Biomed Pharmacother. 2019;114:108800. doi:10.1016/j.biopha.2019.108800
114. Jing X, Zhang H, Hu J, et al. β-arrestin 2 is associated with multidrug resistance in breast cancer cells through regulating MDR1 gene expression. Int J Clin Exp Pathol. 2015;8(2):1354–1363.
115. Gao X, Wu Y, Qiao L, Feng X. SENP2 suppresses NF-κB activation and sensitizes breast cancer cells to doxorubicin. Eur J Pharmacol. 2019;854:179–186. doi:10.1016/j.ejphar.2019.03.051
116. Dueñas-Gonzalez A, Coronel J, Cetina L, González-Fierro A, Chavez-Blanco A, Taja-Chayeb L. Hydralazine-valproate: a repositioned drug combination for the epigenetic therapy of cancer. Expert Opin Drug Metab Toxicol. 2014;10(10):1433–1444. doi:10.1517/17425255.2014.947263
117. Tan C, Hu W, He Y, et al. Cytokine-mediated therapeutic resistance in breast cancer. Cytokine. 2018;108:151–159. doi:10.1016/j.cyto.2018.03.020
118. DeMichele A, Yee D, Esserman L. Mechanisms of resistance to neoadjuvant chemotherapy in breast cancer. N Engl J Med. 2017;377(23):2287–2289. doi:10.1056/NEJMcibr1711545
119. Tsuruo T, Naito M, Tomida A, et al. Molecular targeting therapy of cancer: drug resistance, apoptosis and survival signal. Cancer Sci. 2003;94(1):15–21. doi:10.1111/j.1349-7006.2003.tb01345.x
120. Hasim MS, Nessim C, Villeneuve PJ, Vanderhyden BC, Dimitroulakos J. Activating transcription factor 3 as a novel regulator of chemotherapy response in breast cancer. Transl Oncol. 2018;11(4):988–998. doi:10.1016/j.tranon.2018.06.001
121. Davis T, van Niekerk G, Peres J, Prince S, Loos B, Engelbrecht AM. Doxorubicin resistance in breast cancer: a novel role for the human protein AHNAK. Biochem Pharmacol. 2018;148:174–183. doi:10.1016/j.bcp.2018.01.012
122. Crown J. A review of the efficacy and safety of docetaxel as monotherapy in metastatic breast cancer. Semin Oncol. 1999;26(1supl. 3):5–9.
123. Jazieh K, Bell R, Agarwal N, Abraham J. Novel targeted therapies for metastatic breast cancer. Ann Transl Med. 2020;8(14):907. doi:10.21037/atm.2020.03.43
124. Ghaderi F, Ahmadvand S, Ramezani A, Montazer M, Ghaderi A. Production and characterization of monoclonal antibody against a triple negative breast cancer cell line. Biochem Biophys Res Commun. 2018;505(1):181–186. doi:10.1016/j.bbrc.2018.09.087
125. Maruthanila VL, Elancheran R, Kunnumakkara AB, Kabilan S, Kotoky J. Recent development of targeted approaches for the treatment of breast cancer. Breast Cancer. 2017;24(2):191–219. doi:10.1007/s12282-016-0732-1
126. Esteva FJ, Hubbard-Lucey VM, Tang J, Pusztai L. Immunotherapy and targeted therapy combinations in metastatic breast cancer. Lancet Oncol. 2019;20(3):e175–e186. doi:10.1016/S1470-2045(19)30026-9
127. Le Du F, Perrin C, Brunot A, et al. Therapeutic innovations in breast cancer. Presse Med. 2019;48(10):1131–1137. doi:10.1016/j.lpm.2019.04.005
128. Miklavčič D, Mali B, Kos B, Heller R, Serša G. Electrochemotherapy: from the drawing board into medical practice. Biomed Eng Online. 2014;13(1):1–20. doi:10.1186/1475-925X-13-29
129. Mittal L, Aryal UK, Camarillo IG, Raman V, Sundararajan R. Effective electrochemotherapy with curcumin in MDA-MB-231-human, triple negative breast cancer cells: a global proteomics study. Bioelectrochemistry. 2020;131:107350. doi:10.1016/j.bioelechem.2019.107350
130. Mittal L, Raman V, Camarillo IG, Garner AL, Sundararajan R. Viability and cell cycle studies of metastatic triple negative breast cancer cells using low voltage electrical pulses and herbal curcumin. Biomed Phys Eng Express. 2019;5(2):25040. doi:10.1088/2057-1976/aaf2c3
131. Mittal L, Camarillo IG, Varadarajan GS, Srinivasan H, Aryal UK, Sundararajan R. High-throughput, label-free quantitative proteomic studies of the anticancer effects of electrical pulses with turmeric silver nanoparticles: an in vitro model study. Sci Rep. 2020;10(1):1–18. doi:10.1038/s41598-020-64128-8
132. Ahmad A, Sheikh S, Taran R, et al. Therapeutic efficacy of a novel nanosomal docetaxel lipid suspension compared with taxotere in locally advanced or metastatic breast cancer patients. Clin Breast Cancer. 2014;14(3):177–181. doi:10.1016/j.clbc.2013.09.011
133. Moses C, Garcia-Bloj B, Harvey AR, Blancafort P. Hallmarks of cancer: the CRISPR generation. Eur J Cancer. 2018;93:10–18. doi:10.1016/j.ejca.2018.01.002
134. Wang H, Sun W. CRISPR-mediated targeting of HER2 inhibits cell proliferation through a dominant negative mutation. Cancer Lett. 2017;385:137–143. doi:10.1016/j.canlet.2016.10.033
135. Liu B, Saber A, Haisma HJ. CRISPR/Cas9: a powerful tool for identification of new targets for cancer treatment. Drug Discov Today. 2019;24(4):955–970. doi:10.1016/j.drudis.2019.02.011
136. Yi L, Li J. CRISPR-Cas9 therapeutics in cancer: promising strategies and present challenges. Biochim Biophys Acta Rev Cancer. 2016;1866(2):197–207. doi:10.1016/j.bbcan.2016.09.002
137. Mughees M, Kumar K, Wajid S. Exosome vesicle as a nano-therapeutic carrier for breast cancer. J Drug Target. 2020;8:1–28. doi:10.1080/1061186X.2020.1808001
138. Yong T, Zhang X, Bie N, et al. Tumor exosome-based nanoparticles are efficient drug carriers for chemotherapy. Nat Commun. 2019;10(1). doi:10.1038/s41467-019-11718-4
139. Nassar FJ, Nasr R, Talhouk R. MicroRNAs as biomarkers for early breast cancer diagnosis, prognosis and therapy prediction. Pharmacol Ther. 2017;172:34–49. doi:10.1016/j.pharmthera.2016.11.012
140. Çalışkan M, Güler H, Bozok Çetintaş V. Current updates on microRNAs as regulators of chemoresistance. Biomed Pharmacother. 2017;95:1000–1012. doi:10.1016/j.biopha.2017.08.084
141. Tomar D, Yadava AS, Kumar D, Bhadauriya G, Kundu GC. Non-coding RNAs as potential therapeutic targets in breast cancer. Biochim Biophys Acta Gene Regul Mech. 2019;863(4):194. doi:10.1016/j.bbagrm.2019.04.005
142. Houssen ME, Ghazy HF, Farag K, et al. Serum atrial natriuretic peptide: a suspected biomarker of breast cancer. Contemp Oncol. 2017;21(1):54. doi:10.5114/wo.2017.66657
143. Aleck K, Hallman K, Quigley M, et al. Effects of atrial natriuretic peptide on p53 and estrogen receptor in breast cancer cells. BioResearch. 2017;6(1):141–150. doi:10.1089/biores.2017.0009
144. Van Nuffel AMT, Sukhatme V, Pantziarka P, Meheus L, Sukhatme VP, Bouche G. Repurposing drugs in oncology (ReDO) - clarithromycin as an anti-cancer agent. Ecancermedicalscience. 2015;9:1–26. doi:10.3332/ecancer.2015.513
145. Griffin F, Marignol L. Therapeutic potential of melatonin for breast cancer radiation therapy patients. Int J Radiat Biol. 2018;94(5):472–477. doi:10.1080/09553002.2018.1446227
146. Amaral MEA, Nery LR, Leite CE, de Azevedo WF, Campos MM. Pre-clinical effects of metformin and aspirin on the cell lines of different breast cancer subtypes. Invest New Drugs. 2018;36(5):782–796. doi:10.1007/s10637-018-0568-y
147. Sharma N, Thomas S, Golden EB, et al. Inhibition of autophagy and induction of breast cancer cell death by mefloquine, an antimalarial agent. Cancer Lett. 2012;326(2):143–154. doi:10.1016/j.canlet.2012.07.029
148. Lamb R, Ozsvari B, Lisanti CL, et al. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: treating cancer like an infectious disease. Oncotarget. 2015;6(7):4569. doi:10.18632/oncotarget.3174
149. Ishida J, Konishi M, Ebner N, Springer J. Repurposing of approved cardiovascular drugs. J Transl Med. 2016;14(1):269. doi:10.1186/s12967-016-1031-5
150. Graça I, Sousa EJ, Costa-Pinheiro P, et al. Anti-neoplastic properties of hydralazine in prostate cancer. Oncotarget. 2014;5(15):5950–5964. doi:10.18632/oncotarget.1909
151. Liu B, Huang X, Hu Y, et al. Ethacrynic acid improves the antitumor effects of irreversible epidermal growth factor receptor tyrosine kinase inhibitors in breast cancer. Oncotarget. 2016;7(36):58038. doi:10.18632/oncotarget.10846
152. Niu G, Liao Z, Cai L, Wei R, Sun L. The combined effects of celecoxib and minocycline hydrochloride on inhibiting the osseous metastasis of breast cancer in nude mice. Cancer Biother Radiopharm. 2008;23(4):469–476. doi:10.1089/cbr.2008.0475
153. Hu J, Ljubimova JY, Inoue S, et al. Phosphodiesterase type 5 inhibitors increase Herceptin transport and treatment efficacy in mouse metastatic brain tumor models. PLoS One. 2010;5(4):e10108. doi:10.1371/journal.pone.0010108
154. Pantziarka P, Bryan BA, Crispino S, Dickerson EB. Propranolol and breast cancer—a work in progress. Ecancermedicalscience. 2018;12:1–6. doi:10.3332/ecancer.2018.ed82
155. Chamaraux-Tran TN, Mathelin C, Aprahamian M, et al. Antitumor effects of lidocaine on human breast cancer cells: an in vitro and in vivo experimental trial. Anticancer Res. 2018;38(1):95–105. doi:10.21873/anticanres.12196
156. Park S-H, Chung YM, Ma J, Yang Q, Berek JS, Hu MCT. Pharmacological activation of FOXO3 suppresses triple-negative breast cancer in vitro and in vivo. Oncotarget. 2016;7(27):42110. doi:10.18632/oncotarget.9881
157. Chen C-T, Chen Y-C, Yamaguchi H, Hung M-C. Carglumic acid promotes apoptosis and suppresses cancer cell proliferation in vitro and in vivo. Am J Cancer Res. 2015;5(12):3560.
158. Tury S, Assayag F, Bonin F, et al. The iron chelator deferasirox synergises with chemotherapy to treat triple‐negative breast cancers. J Pathol. 2018;246(1):103–114. doi:10.1002/path.5104
159. Goh W, Sleptsova-Freidrich I, Petrovic N. Use of proton pump inhibitors as adjunct treatment for triple-negative breast cancers. An introductory study. J Pharm Pharm Sci. 2014;17(3):439–446. doi:10.18433/j34608
160. Greenshields AL, Fernando W, Hoskin DW. The anti-malarial drug artesunate causes cell cycle arrest and apoptosis of triple-negative MDA-MB-468 and HER2-enriched SK-BR-3 breast cancer cells. Exp Mol Pathol. 2019;107:10–22. doi:10.1016/j.yexmp.2019.01.006
161. Talarico G, Orecchioni S, Dallaglio K, et al. Aspirin and atenolol enhance metformin activity against breast cancer by targeting both neoplastic and microenvironment cells. Sci Rep. 2016;6:18673. doi:10.1038/srep18673
162. Zhang L, Wang X, Chen P. MiR-204 down regulates SIRT1 and reverts SIRT1-induced epithelial-mesenchymal transition, anoikis resistance and invasion in gastric cancer cells. BMC Cancer. 2013;13:1–9. doi:10.1186/1471-2407-13-290
163. Zeng H, Wang L, Wang J, et al. microRNA-129-5p suppresses Adriamycin resistance in breast cancer by targeting SOX2. Arch Biochem Biophys. 2018;651:52–60. doi:10.1016/j.abb.2018.05.018
164. Cochrane D, Spoelstra N, Howe E, Nordeen S, Richer J. MicroRNA-200c mitigates invasiveness and restores sensitivity to microtubule-targeting chemotherapeutic agents. Mol Cancer Ther. 2009;8(5):1055–1066. doi:10.1158/1535-7163.MCT-08-1046
165. Bian X, Liang Z, Feng A, Salgado E, Shim H. HDAC inhibitor suppresses proliferation and invasion of breast cancer cells through regulation of miR-200c targeting CRKL. Biochem Pharmacol. 2018;147:30–37. doi:10.1016/j.bcp.2017.11.008
166. Han B, Huang J, Han Y, et al. The microRNA miR-181c enhances chemosensitivity and reduces chemoresistance in breast cancer cells via down-regulating osteopontin. Int J Biol Macromol. 2019;125:544–556. doi:10.1016/j.ijbiomac.2018.12.075
167. Gao L, Guo Q, Li X, et al. MiR-873/PD-L1 axis regulates the stemness of breast cancer cells. EBioMedicine. 2019;41:395–407. doi:10.1016/j.ebiom.2019.02.034
168. Setijono SR, Park M, Kim O, Kim Y, Won Cho K, Jung Song S. miR-218 and miR-129 regulate breast cancer progression by targeting Lamins. Biochem Biophys Res Commun. 2018;496(3):826–833. doi:10.1016/j.bbrc.2018.01.146
169. Muluhngwi P, Klinge CM. Identification of miRNAs as biomarkers for acquired endocrine resistance in breast cancer. Mol Cell Endocrinol. 2017;456:76–86. doi:10.1016/j.mce.2017.02.004
170. Kagepura Thammaiah C, Jayaram S. Role of let-7 family microRNA in breast cancer. Non-Coding RNA Res. 2016;1(1):77–82. doi:10.1016/j.ncrna.2016.10.003
171. Li B, Lu Y, Yu L, et al. miR-221/222 promote cancer stem-like cell properties and tumor growth of breast cancer via targeting PTEN and sustained Akt/NF-κB/COX-2 activation. Chem Biol Interact. 2017;277:33–42. doi:10.1016/j.cbi.2017.08.014
172. Eissa S, Matboli M, Sharawy A, El-Sharkawi F. Prognostic and biological significance of microRNA-221 in breast cancer. Gene. 2015;574(1):163–167. doi:10.1016/j.gene.2015.08.004
173. Geng W, Song H, Zhao Q, et al. MiR-520h stimulates drug resistance to paclitaxel by targeting the OTUD3-PTEN axis in breast cancer. Biomed Res Int. 2020;2020:1–11. doi:10.1155/2020/9512793
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.