")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 12
Repression of Toll-like receptor-4 by microRNA-149-3p is associated with smoking-related COPD
Authors Shen W, Liu J, Zhao G, Fan M, Song G, Zhang Y, Weng Z, Zhang Y
Received 17 November 2016
Accepted for publication 27 December 2016
Published 22 February 2017 Volume 2017:12 Pages 705—715
DOI https://doi.org/10.2147/COPD.S128031
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Russell
Wen Shen,1,* Jia Liu,2,* Guohou Zhao,1 Minjuan Fan,1 Gao Song,3 Yang Zhang,1 Zhiying Weng,3 You Zhang4
1Department of Respiratory Medicine, 2Department of Experimental Zoology, The Second Affiliated Hospital of Kunming Medical University, 3School of Pharmaceutical Science, Kunming Medical University, 4Department of Hematology, The Second Affiliated Hospital of Kunming Medical University, Kunming, Yunnan Province, People’s Republic of China
*These authors contributed equally to this work
Background: Smoking is the leading cause of COPD. Exploring molecular markers and understanding the pathogenic mechanisms of smoking-related COPD are helpful for early clinical diagnosis and treatment of the disease. This study aims to identify specific circulating microRNAs (miRNAs) from the blood of COPD patients with a long history of smoking.
Methods: Blood samples from four different groups were collected, and miRNA microarray was performed. Differential expression of miRNAs was verified by quantitative polymerase chain reaction. In vitro, THP-1 cells were cultured and stimulated with cigarette smoke extract (CSE) or transfected with miR-149-3p inhibitor/mimics. Protein levels of Toll-like receptor 4 (TLR-4) and nuclear factor κB (NF-κB) were detected using Western blot and immunofluorescence. Interleukin (IL)-1β and tumor necrosis factor (TNF)-α levels were determined by an enzyme-linked immunosorbent assay.
Results: miRNA profiling revealed that the expression of 56 miRNAs was changed between the four groups. Expression of miR-149-3p in group C (non-smoker non-COPD) was higher than in group S (smoker non-COPD), S-COPD (smoker with stable COPD) and AE-COPD (smoker with acute exacerbation COPD). CSE stimulation down-regulated the expression of miR-149-3p and up-regulated the TLR-4 and NF-κB levels in THP-1 cells. Transfecting miR-149-3p inhibitors in THP-1 cells also increased the expression of its target genes. Furthermore, overexpression of miR-149-3p inhibited the TLR-4/NF-κB signaling pathways and reduced the secretion of IL-1β and TNF-α.
Conclusion: This study found that smoking can induce differential expression of circulating miRNAs, such as down-regulation of miR-149-3p. Reducing miR-149-3p may increase the inflammatory response in COPD patients through the regulation of the TLR-4/NF-κB signaling pathway.
Keywords: smoking, COPD, microRNA-149-3p, Toll-like receptor 4, nuclear factor κB
Introduction
COPD is a major cause of illness and death worldwide.1 According to the World Health Organization (WHO), ~3 million people in the world die as a consequence of COPD every year.2 COPD involves chronic inflammation of the lungs, particularly in the peripheral airways and parenchyma, which increases during periods of acute exacerbation. It is also associated with systemic inflammation, which may contribute to or worsen several comorbidities and may be derived from the “spill-over” of inflammatory mediators from the peripheral lungs.3 The development of COPD is multifactorial, and the risk factors are both genetic and environmental.4 Even though traffic and other outdoor pollution, secondhand smoke (SHS), biomass smoke and dietary factors are all associated with COPD, cigarette smoking is regarded as one of the most important contributors to COPD.5
Increasing evidence showing that smoking is a cause of COPD has been growing for >40 years and has been extensively reviewed in three US Surgeon General’s Reports.6–8 The estimated fraction of COPD mortality attributable to smoking was 54% for men 30–69 years of age and 52% for men 70 years of age or older.9 Exposure to SHS, which contains potent respiratory irritants, also leads to chronic airway inflammation and obstruction. A study from the People’s Republic of China found that self-reported cumulative lifetime SHS exposure at home and work was related to a greater risk of COPD, as defined by spirometry (Global initiative for chronic Obstructive Lung Disease [GOLD] stage 1 or greater).10 Another study showed that living with a smoker was associated with a greater risk for COPD.11
MicroRNAs (miRNAs) are small non-coding RNA molecules that modulate the levels of specific genes and proteins.12 Identifying the expression patterns of miRNAs in COPD may enhance our understanding of the mechanisms underlying COPD. Several studies have investigated the effects of cigarette smoke on miRNA expression.13,14 A total of 70 miRNAs and 2,667 messenger RNAs (mRNAs) were differentially expressed from lung tissues obtained from patients with COPD and smokers without COPD.15 In recent years, mechanisms of many miRNAs related to COPD, such as miR-145,16,17 miR-146a,18 miR-106b19 and miR-223,20 have been reported. This study also found that 140 miRNAs that were derived from COPD patients and compared to healthy controls were significant and 14 miRNAs that were derived from patients with lung cancer and compared to COPD patients were significant.21 It is important to discover more candidate markers to distinguish between the different stages or severity in COPD patients for diagnosis and treatment. However, miRNAs caused by smoking in COPD patients still need further research. Circulating miRNAs as stable blood-based markers can be applied to cancer detection,22–24 coronary artery disease detection,25 liver injury evaluation26 and COPD diagnosis.27 Several studies found that skeletal muscle-specific miRNAs can be detected in the blood of COPD patients.28,29 In addition, another study reported that genotypes of miR-149 were associated with the overall survival of lung cancer patients.30 To explore differentially expressed circulating miRNAs caused by smoking in CODP patients, we collected blood samples of smoking-related COPD patients at different stages of the disease and performed miRNA microarray. Finally, we chose one of the differentially expressed miRNAs, miR-149-3p, and studied its mechanism through further in vitro experiments.
Materials and methods
Ethics statement
The use of human blood samples for this study was approved by the Medical Ethics Committee of The Second Affiliated Hospital of Kunming Medical University. Written consent was received from all participants in this study at the time of sample collection.
Clinical samples
Blood samples from four groups of patients were collected at The Second Affiliated Hospital of Kunming Medical University and stored in liquid nitrogen for further use. Each of the four groups contained 20 blood samples: as control, non-smoker non-COPD (group C); smoker non-COPD (group S); smoker with stable COPD (group S-COPD); smoker with acute exacerbation COPD (group AE-COPD).
Cell culture and transient transfection
The murine monocytic cell line THP-1 (ATCC, Rockville, MD, USA) was cultured by Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum, 2 mM/L glutamine, 100 units/mL penicillin and 0.1 mg/mL streptomycin, in a humidified incubator (5% CO2 in air) at 37°C. Cigarette smoke extract (CSE) was prepared by a method modified from a previously described protocol.31 Briefly, one cigarette was bubbled through 25 mL of DMEM at a constant rate, and this solution was considered as 100% concentration of CSE. The 100% CSE was freshly generated for each experiment and diluted to a final working concentration (10%) and used within 30 min. To analyze the effect of miR-149-3p, cells were passed into six-well plates (3×107 cells/plate), and after ~24 h at 37°C, they were transfected with miR-149-3p mimics, inhibitors or negative control (NC) with Lipofectamine 2000. Subsequent processing was completed according to the study protocol.
RNA extraction and miRNA microarray
Trizol (Thermo Fisher Scientific, Waltham, MA, USA) was used to extract the total RNAs from blood samples or cells. Then mirVana miRNA Isolation Kit (Ambion, Austin, TX, USA) was used to purify small RNAs in accordance with the manufacturer’s protocol. The concentration and purity of RNAs were determined by optical density 260/280 readings using spectrophotometer (NanoDrop ND-2000). By using the RNA 6000 Nano Lab-on-a-Chip kit and the Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA), RNA integrity was determined by electrophoresis.
Microarray assay was performed by KangchengBio Corporation, Shanghai, the People’s Republic of China. The RNA extract from six blood samples in each group was used for microassay analysis by miRCURY LNA™ microRNA Array according to the manufacturer’s instruction. Threshold values of ≥1.5- and ≤−1.5-fold change were used to identify the differentially expressed genes. The data were log 2 transformed and median centered by genes using the Adjust Data function of Mev (Multiexperiment Viewer) software (Dana-Farber Cancer Institute, Boston, MA, USA) and then further analyzed with hierarchical clustering with average linkage.
Quantitative polymerase chain reaction (qPCR) analysis
Total RNAs of 60 blood samples (20 in each group) and cells under different treatments were isolated and purified. The samples with OD260/280 ratio >1.8 were reversely transcribed using a GoScropt™ Reverse Transcription System Kit (Promega, Madison, WI, USA) according to the manufacturer’s instructions. The expressions of selected miRNA were analyzed using qPCR with a Go Taq®qPCR Master Mix kit (Promega) on StepOne Plus PCR System (Applied Biosystems, Carlsbad, CA, USA). glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control, and 2−ΔΔCT method was used to calculate the expression of miRNA. The primers for qPCR analysis are given in Table 1.
 | Table 1 Primers information for PCR used in this study |
Western blot
Cells were lysed in radio-immunoprecipitation assay buffer. Protein concentrations were determined using the BCA Protein Assay Kit (Thermo Fisher Scientific). Protein samples were boiled, then separated on polyacrylamide gels and transferred to polyvinylidene fluoride membrane (EMD Millipore, Billerica, MA, USA). Membranes were incubated overnight at 4°C with anti-Toll-like receptor 4 (TLR-4) (Abcam; diluted 1:500) and anti-nuclear factor κB (NF-κB) p65 (Abcam; diluted 1:5,000), or anti-GAPDH antibody (Abcam; diluted 1:5,000). GAPDH was used as an internal control. After washing with Tris-HCl Tween buffer solution, the blots were incubated with a horseradish peroxidase (HRP)-conjugated secondary antibody (Santa Cruz; diluted 1:2,000) at room temperature for 1 h. The blots were visualized by Pierce ECL Plus Western Blotting Substrate (Pierce) following the manufacturer’s instructions. Immunoreactive bands were detected with enhanced chemiluminescent HRP substrate (Millipore).
Immunofluorescence
Cells were fixed with 4% paraformaldehyde solution for 20 min at room temperature and then washed with phosphate-buffered saline (PBS). Triton X-100 (0.5%) in PBS was used for permeabilization of cells. Then, the cells were blocked with 10% fetal calf serum (FCS) for 1 h at room temperature and incubated with anti-TLR-4 (Abcam; diluted 1:200) and anti-NF-κB p65 (Abcam; diluted 1:500) overnight at 4°C. After washing with PBS, the cells were incubated with secondary antibody. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole for 5 min at room temperature. Images of the staining were taken with a fluorescence microscope (Thermo Fisher Scientific).
Enzyme-linked immunosorbent assay (ELISA)
Levels of tumor necrosis factor-α (TNF-α) and interleukin (IL)-1β in treated cells were measured using an antigen-based sandwich ELISA. Mouse TNF-α ELISA Kit (Elabscience, E-EL-M0037c) and Mouse IL-1β ELISA Kit (Elabscience, E-EL-M0049c) were used, and the absorbance was measured at 450 nm using a microplate spectrophotometer (Thermo, Multiskan GO).
Statistical analysis
Data of miRNAs expression from the four groups were analyzed using Bonferroni’s multiple comparison test. Differences between groups in in vivo experiment were determined by an unpaired Student’s t-test method using the Prism (GraphPad Software Inc., La Jolla, CA, USA). A P-value <0.05 was considered to be statistically significant.
Results
Smoking-induced different expression levels of miRNAs including down-regulation of miR-149-3p
To explore differentially expressed circulating miRNAs caused by smoking in COPD patients, we collected blood samples from group C, S, S-COPD and AE-COPD. We performed total RNA extraction and miRNA microarray. Among 689 types of human miRs that could be detected by the probes, the expression levels of 56 miRNAs were changed between the four groups (Figure 1A). Based on the data of fold changing (unpublished data), six members, including miR-3202, miR-26a-5p, miR-451b, miR-96-5p, miR-301a-5p and miR-149-3p, were selected for qPCR validation. We observed that miR-3202, miR-26a-5p, miR-451b and miR-149-3p, were down-regulated in the S, S-COPD and AE-COPD groups. miR-96-5p was reduced in the blood samples of group S compared to group C, while in the S-COPD and AE-COPD groups, it showed significantly higher expression. Besides, miR-301a-5p exhibited low expression levels only in COPD smokers with acute exacerbation period (Figure 1B).
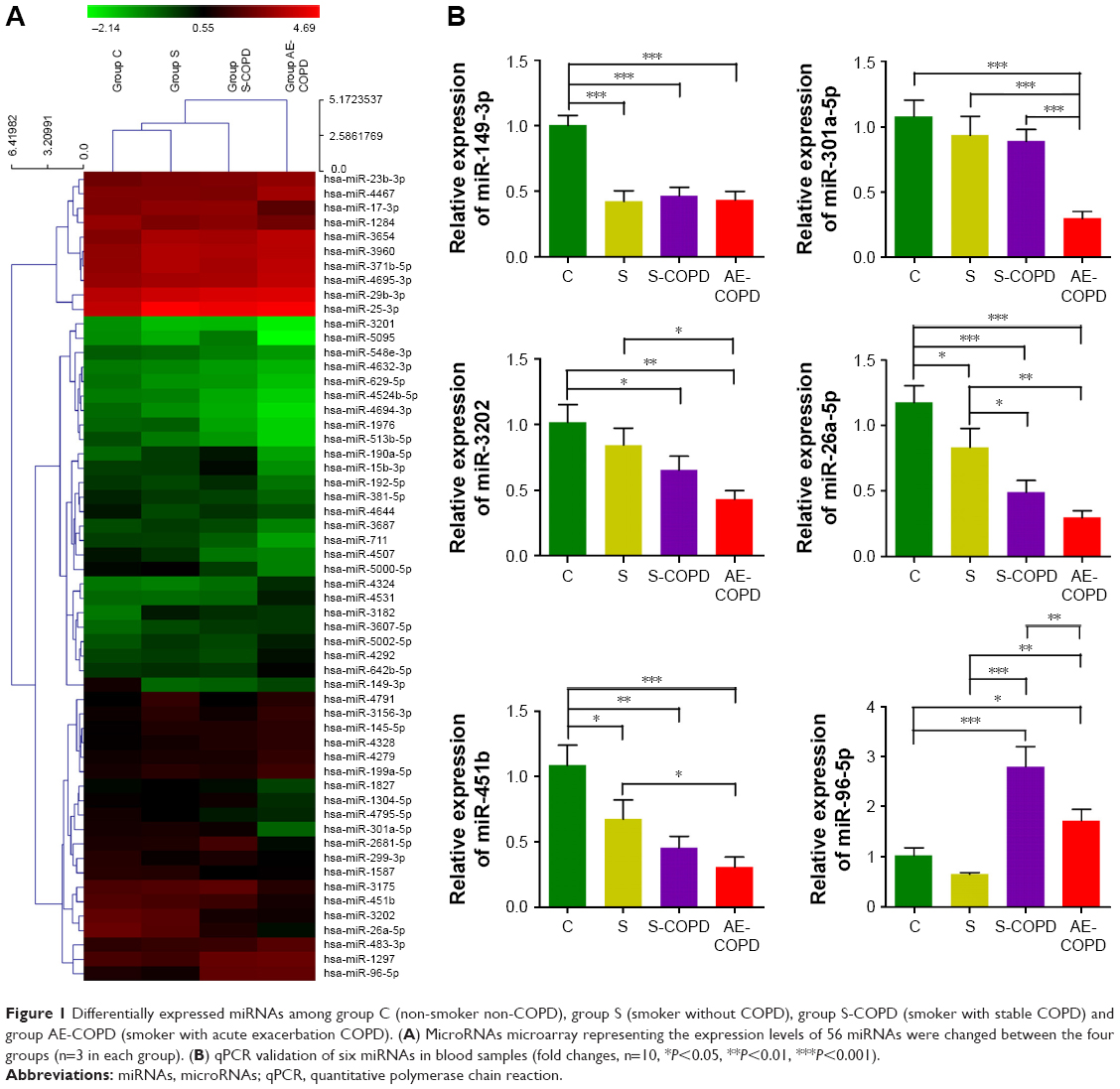 | Figure 1 Differentially expressed miRNAs among group C (non-smoker non-COPD), group S (smoker without COPD), group S-COPD (smoker with stable COPD) and group AE-COPD (smoker with acute exacerbation COPD). (A) MicroRNAs microarray representing the expression levels of 56 miRNAs were changed between the four groups (n=3 in each group). (B) qPCR validation of six miRNAs in blood samples (fold changes, n=10, *P<0.05, **P<0.01, ***P<0.001). |
Decreasing miR-149-3p caused up-regulation of TLR-4 in THP-1 cells stimulated by CSE
As miR-149-3p was down-regulated in the three smoking groups, we performed CSE stimulation treatment on the THP-1 cell line. Concentration of IL-1β and TNF-α was significantly increased in the CSE group compared with the NC group (Figure 2A). qPCR results show that miR-149-3p was significantly reduced by CSE stimulation (Figure 2B). Through bioinformatics prediction, we found that TLR-4 may be a target gene of miR-149-3p. The direct regulating effect of miR-149-3p on TLR-4 was verified by a dual luciferase assay. Reporter assay revealed that overexpression of miR-149-3p significantly suppressed the luciferase activity of wild TLR-4 3′ untranslated region (UTR) plasmid in THP-1 cells (P<0.05) without change in luciferase activity of mutant 3′UTR TLR-4 plasmid (Figure 2C and D). Moreover, CSE stimulation caused up-regulation of TLR-4 at both the mRNA and the protein levels (Figure 2E and F). The proteins downstream of TLR-4, mainly NF-κB p65, showed increased expressions in the CSE group. Consistent with these observations, immunofluorescence results showed similar changes in TLR-4 and NF-κB p65 protein levels (Figure 3A and B).
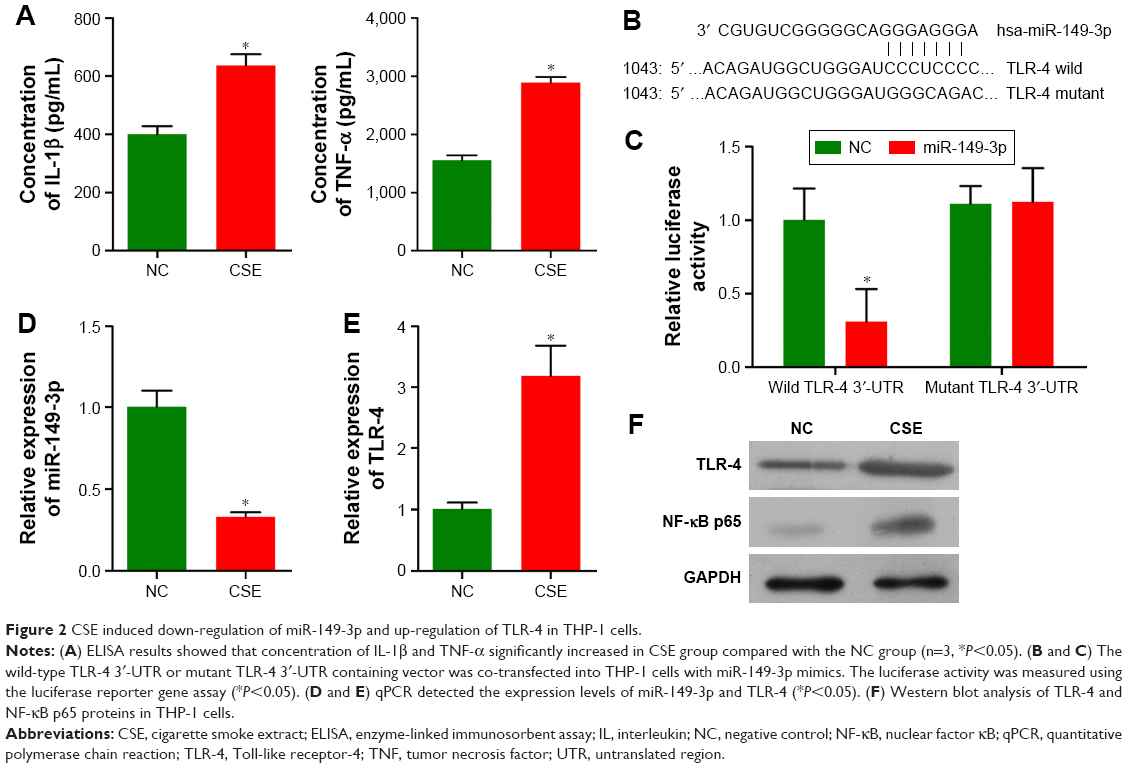 | Figure 2 CSE induced down-regulation of miR-149-3p and up-regulation of TLR-4 in THP-1 cells. |
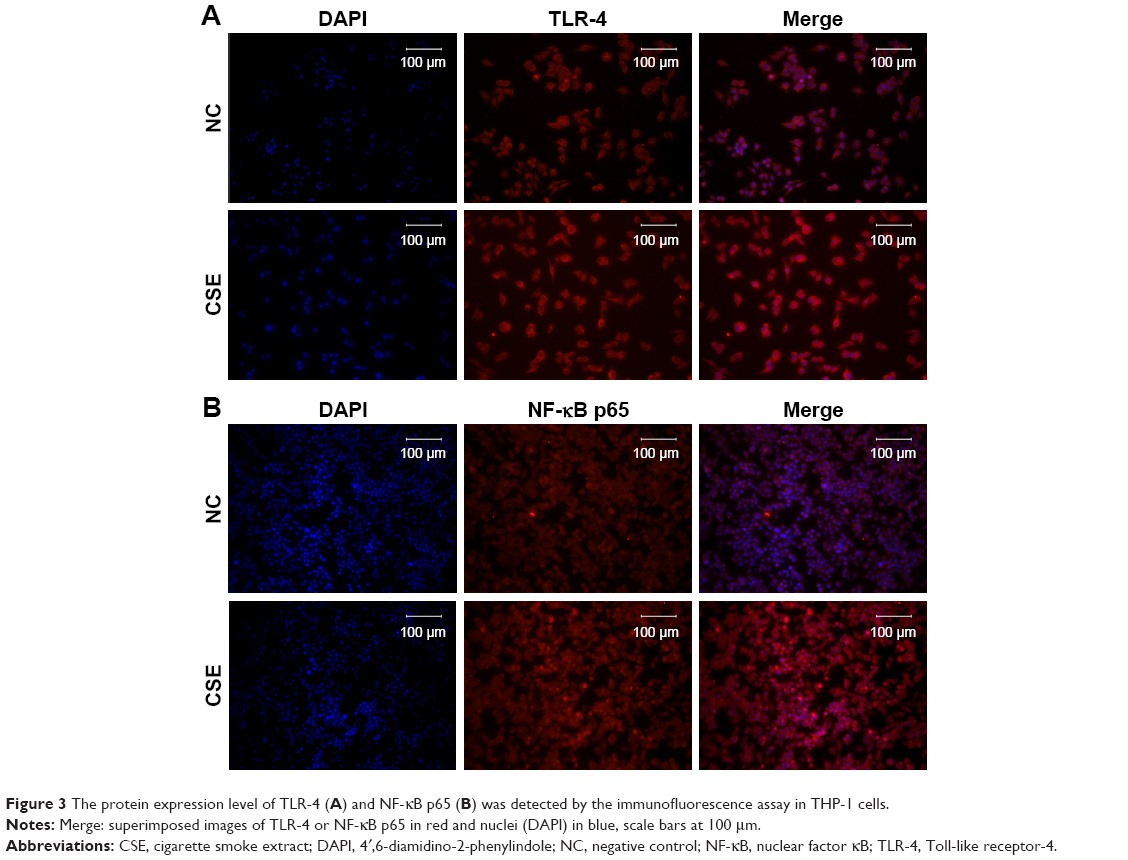 | Figure 3 The protein expression level of TLR-4 (A) and NF-κB p65 (B) was detected by the immunofluorescence assay in THP-1 cells. |
Transfection of miR-149-3p inhibitors activated TLR-4/NF-κB p65 signaling pathway
To validate the down-regulation of miR-149-3p, we induced the activation of TLR-4/NF-κB p65 signaling pathway. miR-149-3p inhibitors were transfected into THP-1 cells. ELISA assay revealed that IL-1β and TNF-α significantly increased in the miR-149-3p inhibitor group compared with the NC group (Figure 4A). The mRNA level of TLR-4 was increased (Figure 4B), and protein levels of TLR-4 and NF-κB p65 were up-regulated in miR-149-3p inhibitor group compared to the NC group (Figure 4C). The immunofluorescence assay further confirmed that miR-149-3p inhibitors induced the activation of TLR-4/NF-κB p65 signaling pathway (Figure 5A and B).
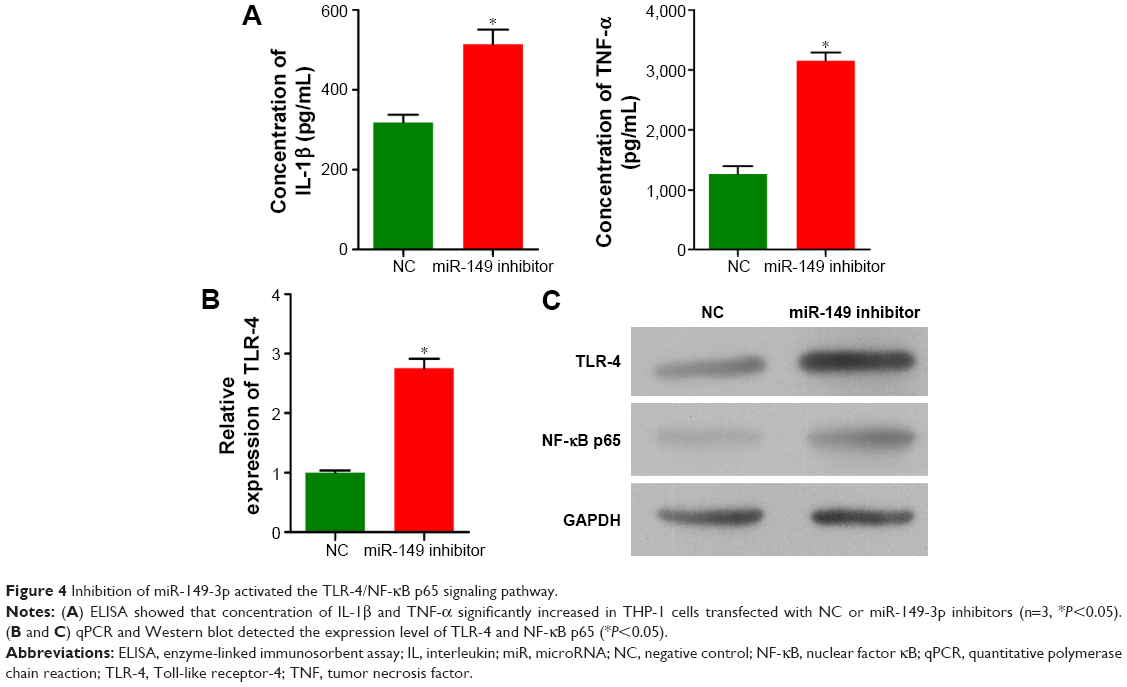 | Figure 4 Inhibition of miR-149-3p activated the TLR-4/NF-κB p65 signaling pathway. |
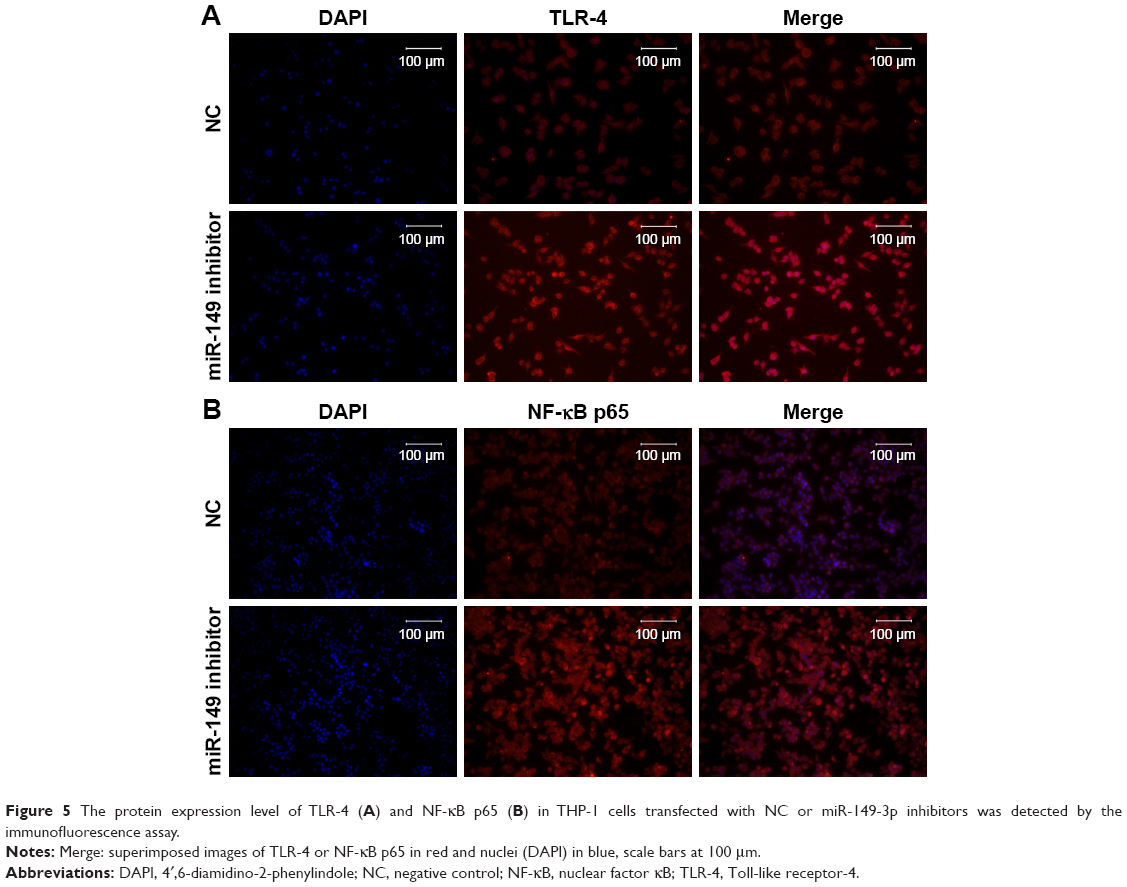 | Figure 5 The protein expression level of TLR-4 (A) and NF-κB p65 (B) in THP-1 cells transfected with NC or miR-149-3p inhibitors was detected by the immunofluorescence assay. |
Transfection of miR-149-3p mimics reversed the expression of TLR-4 in CSE-stimulated THP-1 cells
To examine whether CSE affected the generation of inflammatory cytokines, we passed the THP-1 cells through miR-149-3p. THP-1 cells were infected with miR-149-3p mimics after CSE stimulation treatment. As shown in Figure 6A, miR-149-3p was increased in the transfected miR-149-3p mimics group as compared with the NC group. The ELISA assay demonstrated a significant decrease in IL-1β and TNF-α (Figure 6B) levels. The qPCR analysis showed that TLR-4 mRNA expression levels decreased in transfected miR-149-3p mimics group, and Western blots showed suppression of TLR-4/NF-κB p65 signaling pathway induced by miR-149-3p overexpression (Figure 6C and D). Images of immunofluorescence assay further confirmed this result (Figure 7A and B).
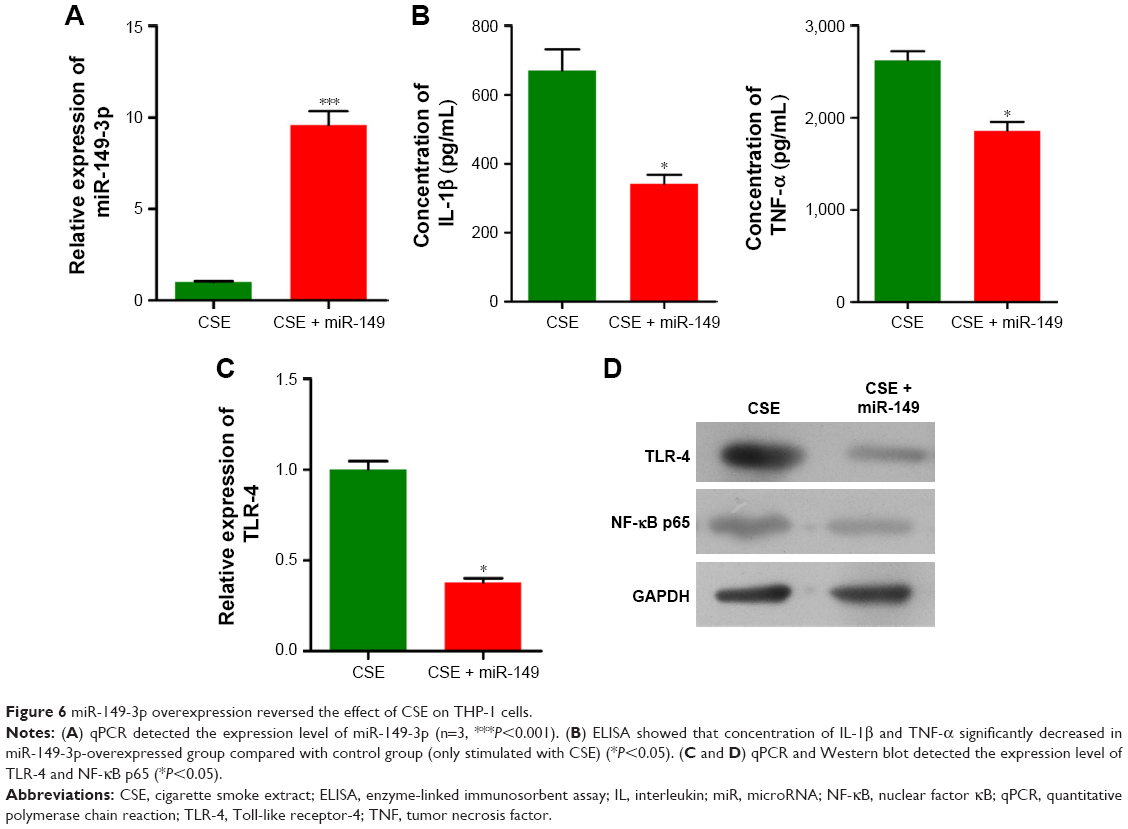 | Figure 6 miR-149-3p overexpression reversed the effect of CSE on THP-1 cells. |
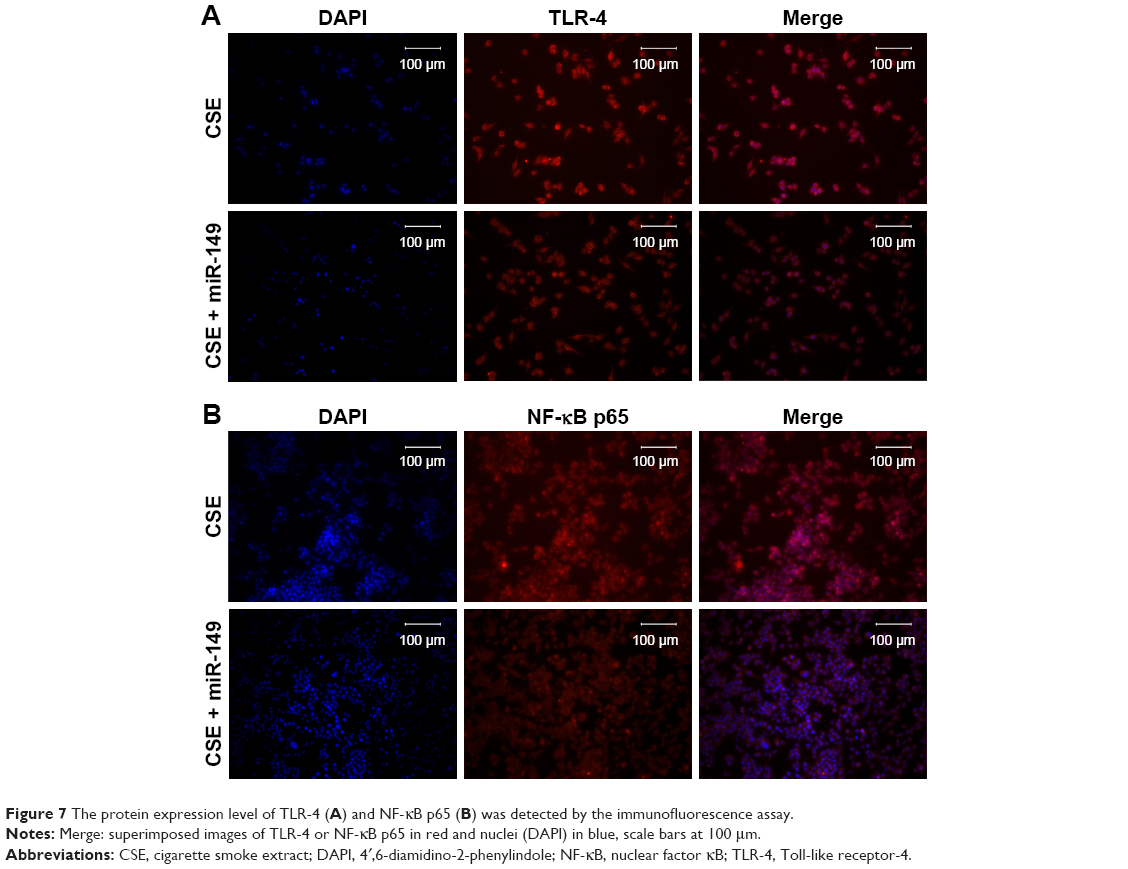 | Figure 7 The protein expression level of TLR-4 (A) and NF-κB p65 (B) was detected by the immunofluorescence assay. |
Discussion
We conducted a comprehensive analysis of miRNA in whole-blood samples from four different groups: non-smokers, smokers, catabatic period smokers with catabatic period COPD, and smokers with acute exacerbation period COPD. Our results revealed 56 differentially expressed miRNAs between the four groups. Validation with qPCR found that miR-3202, miR-26a-5p, miR-451b and miR-149-3p were significantly down-regulated in the three smoking groups compared with the non-smoking group. miR-96-5p expression was reduced in the blood of the normal smoking group compared to the normal non-smoking group, while in the two COPD groups, the expression of miR-96-5p was significantly higher. Besides, miR-301a-5p showed low expression levels only in the acute exacerbation period in smoking COPD patients. A comprehensive analysis of the miRNAs showed many of the same miRNAs that have been reported to play an important role in the occurrence and development of COPD. For example, our miRNA microarray showed that miR-145 was increased in blood samples in COPD patients compared with normal people, and Perry’s study suggested that miR-145 negatively regulates pro-inflammatory cytokine release from airway smooth muscle cells in COPD by targeting SMAD3.16 The role of miR-26a has been investigated in the modulation of an inflammatory response in cultured microglia, and it was found that miR-26a modulated inflammatory response induced by TLR-4 stimulation.32 In addition, another study found that miR-149 is down-regulated in osteoarthritic chondrocytes, and this decrease seems to be correlated to increased expression of pro-inflammatory cytokines such as TNF-α, IL-1β and IL-6.33 TNF-α induces the expression of genes associated with endothelial dysfunction through p38MAPK-mediated down-regulation of miR-149.34 Furthermore, miR-149 is considered as a tumor suppressor to various tumors, such as renal cell carcinoma,35 colorectal carcinoma36 and hepatocellular carcinoma.37 miR-149 is expressed in many tissues and thus plays different roles.
In this study, miR-149 was decreased in the smoking group compared with the non-smoking group. Down-regulation of miR-149 probably plays a key role in the pro-inflammatory cascade in lungs and bronchi. It has been shown that the C-containing genotypes of miR-149 rs2292832 are associated with a better overall survival of non-small-cell lung cancer,30 while another study reported that there was no association between miR-149 rs2292832 polymorphism with lung cancer risk in Chinese non-smoking females.38 Therefore, miR-149 expressed in lungs and bronchi may be affected by smoking. To explore this mechanism further, we predicted a target gene of miR-149 through 3′UTR sequence analysis on Targetscan, miRanda and DIANA-microT and performed simulated experiments in THP-1 cells. Simulation of CSE resulted in a decrease in miR-149 expression and an increase in the expression of its target gene, TLR-4. Furthermore, transfection of miR-149 inhibitors induces the up-regulation of TLR-4. Finally, miR-149 overexpression reverses the effect of CSE on THP-1 cells. We speculated that the miR-149 negatively regulates the inflammatory response in THP-1 cells by targeting TLR-4. Previous reports found that miR-149 might be a key player of the immune modulator for TLR/MyD88 signaling pathway in macrophages.39,40 TLR-4 is regulated by members of the let-7 miRNA family. The induction of let-7e expression could decrease the TLR-4 expression on the cell surface of liver-derived cholangiocytes.41
Our study shows that the change in miR-149 expression caused the alteration of TLR-4 and NF-κB p65 protein levels. Many studies have investigated the expression of TLRs on both immune and epithelial cells in COPD.42–44 Nadigel et al45 demonstrated that CD8+ T cells exposed to cigarette smoke condensate increased TLR-4 and TLR-9 levels and also increased cytokine production. Another study found that acute cigarette exposure results in lipopolysaccharides-independent TLR4 activation, leading to IL-1 production and IL-1R1 signaling, which is crucial for cigarette smoke-induced inflammation leading to COPD with emphysema.46 Geraghty et al47 found that mice exposed to acute levels of cigarette smoke exhibited increased TLR-4 expression and TLR-4 signaling, through MyD88 and IRAK1, which play a predominant role in matrix metalloproteinase 1 (MMP-1) induction. We observed TLR-4, NF-κB p65, IL-1β and TNF-α levels after miR-149 overexpression or knockdown. Results revealed that miR-149 suppresses inflammation through targeting TLR-4.
Conclusion
Our study demonstrated that smoking can induce differential expression of circulating miRNAs, such as down-regulation of miR-149-3p. Reducing miR-149-3p may increase the inflammatory response in COPD patients through the regulation of TLR-4/NF-κB signaling.
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (No 81402991) and Yunnan Provincial Science and Technology Department (No 20141A033).
Author contributions
Conceived and designed the paper: WS, JL and YZ. Collected the data: WS, JL, GZ, MF, GS and YZ. Analyzed the data: WS and JL. Wrote the paper: WS, ZW and YZ. All authors read and approved the final manuscript. All authors contributed toward data analysis, drafting and revising the paper and agreed to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Vestbo J, Hurd SS, Agustí AG, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2007;187(176):532–555. | ||
Diaz-Guzman E, Mannino DM. Epidemiology and prevalence of chronic obstructive pulmonary disease. Clin Chest Med. 2014;35(1):7–16. | ||
Balasubramanian VP, Varkey B. Chronic obstructive pulmonary disease: effects beyond the lungs. Curr Opin Pulm Med. 2006;12(2):106–112. | ||
Wang J, Satya S, Kuhle JW. Tobacco smoking and environmental risk factors for chronic obstructive pulmonary disease. Clin Chest Med. 2014;35(1):17–27. | ||
Eisner MD, Anthonisen N, Coultas D, et al. An official American Thoracic Society public policy statement: novel risk factors and the global burden of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;182(5):693–718. | ||
National Center for Chronic Disease Prevention and Health Promotion (US) Office on Smoking and Health. The Health Consequences of Smoking – 50 Years of Progress: A Report of the Surgeon General. Atlanta, GA: Centers for Disease Control and Prevention (US); 2014. | ||
U.S. Department of Health and Human Services. The Health Consequences of Smoking: A Report of the Surgeon General. Atlanta, GA: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health. 2004. | ||
Moritsugu KP. The 2006 Report of the Surgeon General: the health consequences of involuntary exposure to tobacco smoke. American Journal of Preventive Medicine. 2007;32(6):542–543. | ||
Ezzati M, Lopez AD. Estimates of global mortality attributable to smoking in 2000. Lancet. 2003;362(9387):847–852. | ||
Yin P, Jiang CQ, Cheng KK, et al. Passive smoking exposure and risk of COPD among adults in China: the Guangzhou Biobank Cohort Study. Lancet. 2007;370(9589):751–757. | ||
Sezer H, Akkurt I, Guler N, Marakoğlu K, Berk S. A case-control study on the effect of exposure to different substances on the development of COPD. Ann Epidemiol. 2006;16(1):59–62. | ||
Bartel DP. MicroRNA target recognition and regulatory functions. Cell. 2009;136(2):215–233. | ||
Izzotti A, Calin GA, Arrigo P, Steele VE, Croce CM, De Flora S. Downregulation of microRNA expression in the lungs of rats exposed to cigarette smoke. FASEB J. 2009;23(3):806–812. | ||
Izzotti A, Calin GA, Steele VE, Croce CM, De Flora S. Relationships of microRNA expression in mouse lung with age and exposure to cigarette smoke and light. FASEB J. 2009;23(23):3243–3250. | ||
Ezzie ME, Crawford M, Cho JH, et al. Gene expression networks in COPD: microRNA and mRNA regulation. Thorax. 2011;67(2):122–131. | ||
O’Leary L, Sevinç K, Papazoglou IM, et al. Airway smooth muscle inflammation is regulated by microRNA-145 in COPD. FEBS Lett. 2016;590(9):1324–1334. | ||
O’Leary L, Tildy B, Papazoglou I, Adcock I, Fan C, Perry M. miRNA-145 & SMAD3 expression in patients with COPD. Eur Respir J. 2014;44:3342. | ||
Zago M, Souza ARD, Hecht E, et al. The NF-κB family member RelB regulates microRNA miR-146a to suppress cigarette smoke-induced COX-2 protein expression in lung fibroblasts. Toxicol Lett. 2014;226(2):107–116. | ||
Soeda S, Ohyashiki JH, Ohtsuki K, Umezu T, Setoguchi Y, Ohyashiki K. Clinical relevance of plasma miR-106b levels in patients with chronic obstructive pulmonary disease. Int J Mol Med. 2013;31(3):533–539. | ||
Leuenberger C, Schuoler C, Mignan C, et al. microRNA-223 suppresses histone deacetylase 2 in chronic obstructive pulmonary disease. Eur Respir J. 2015;46(suppl):59. | ||
Leidinger P, Keller A, Borries A, et al. Specific peripheral miRNA profiles for distinguishing lung cancer from COPD. Lung Cancer. 2011;74(1):41–47. | ||
Heneghan HM, Miller N, Lowery AJ, Sweeney KJ, Newell J, Kerin MJ. Circulating microRNAs as novel minimally invasive biomarkers for breast cancer. Ann Surg. 2010;251(3):499–505. | ||
Tsujiura M, Ichikawa DS, Shiozaki A, et al. Circulating microRNAs in plasma of patients with gastric cancers. Br J Cancer. 2010;102(7):1174–1179. | ||
Ke Z, Zhang CY. Circulating microRNAs: a novel class of biomarkers to diagnose and monitor human cancers. Med Res Rev. 2012;32(2):326–348. | ||
Fichtlscherer S, De RS, Fox H, et al. Circulating microRNAs in patients with coronary artery disease. Circ Res. 2010;107(5):677–684. | ||
Lewis PJS, Dear J, Platt V, et al. Circulating microRNAs as potential markers of human drug-induced liver injury. Hepatology. 2011;54(5):1767–1776. | ||
Donaldson A, Lewis A, Natanek S, Man W, Kemp P, Polkey M. Investigating circulating microRNAs as potential biomarkers of quadriceps weakness in COPD. Eur Respir J. 2011;38:206. | ||
Donaldson AVJ, Lewis A, Natanek A, Man WD, Kemp P, Polkey MI. S49 Increased skeletal muscle-specific microRNA-1 in the blood of COPD patients. Thorax. 2011;66(suppl 4):A25–A25. | ||
Donaldson A, Natanek SA, Lewis A, et al. Increased skeletal muscle-specific microRNA in the blood of patients with COPD. Thorax. 2013;68(12):1140–1149. | ||
Lingzi X, Zhihua Y, Xuelian L, et al. Genetic variants in microRNAs predict non-small cell lung cancer prognosis in Chinese female population in a prospective cohort study. Oncotarget. Epub 2016 Nov 4. | ||
Krimmer DI, Burgess JK, Wooi TK, Black JL, Oliver BG. Matrix proteins from smoke-exposed fibroblasts are pro-proliferative. Am J Respir Cell Mol Biol. 2011;46(1):34–39. | ||
Kumar A, Bhatia HS, de Oliveira AC, Fiebich BL. microRNA-26a modulates inflammatory response induced by toll-like receptor 4 stimulation in microglia. J Neurochem. 2015;135(6):1189–1202. | ||
Santini P, Politi L, Vedova PD, Scandurra R, Scottod AA. The inflammatory circuitry of miR-149 as a pathological mechanism in osteoarthritis. Rheumatol Int. 2014;34(5):711–716. | ||
Palmieri D, Capponi S, Geroldi A, Mura M, Mandich P, Palombo D. TNFα induces the expression of genes associated with endothelial dysfunction through p38MAPK-mediated down-regulation of miR-149. Biochem Biophys Res Commun. 2013;443(1):246–251. | ||
Jin L, Li Y, Liu J, et al. Tumor suppressor miR-149-5p is associated with cellular migration, proliferation and apoptosis in renal cell carcinoma. Mol Med Rep. 2016;13(6):5386–5392. | ||
Liu X, Xie T, Mao X, Xue L, Chu X, Chen L. MicroRNA-149 increases the sensitivity of colorectal cancer cells to 5-fluorouracil by targeting forkhead box transcription factor FOXM1. Cell Physiol Biochem. 2016;39(2):617–629. | ||
Lin L, Zhang YD, Chen ZY, Chen Y, Ren CP. The clinicopathological significance of miR-149 and PARP-2 in hepatocellular carcinoma and their roles in chemo/radiotherapy. Tumor Biol. 2016;37(9): 12339–12346. | ||
Li H, Ren Y, Xia L, et al. Association of microRNA-149 polymorphism with lung cancer risk in Chinese non-smoking female: a case-control study. PLoS One. 2016;11(9):e0163626. | ||
Xu G, Zhang Z, Xing Y, et al. MicroRNA-149 negatively regulates TLR-triggered inflammatory response in macrophages by targeting MyD88. J Cell Biochem. 2014;115(5):919–927. | ||
Schnitger AKD, Machova A, Mueller RU, et al. Listeria monocytogenes infection in macrophages induces vacuolar-dependent host miRNA response. PLoS One. 2004;510(5):502–513. | ||
Chen XM, Splinter PL, O’Hara SP, Larusso NF. A cellular micro-RNA, let-7i, regulates Toll-like receptor 4 expression and contributes to cholangiocyte immune responses against Cryptosporidium parvum infection. J Biol Chem. 2007;282(39):28929–28938. | ||
Pons J, Sauleda J, Regueiro V, et al. Expression of Toll-like receptor 2 is up-regulated in monocytes from patients with chronic obstructive pulmonary disease. Respir Res. 2006;7(1):64. | ||
Macredmond RE, Greene CM, Dorscheid DR, Mcelvaney NG, O’Neill SJ. Epithelial expression of TLR4 is modulated in COPD and by steroids, salmeterol and cigarette smoke. Respir Res. 2007;8(1):84. | ||
Zuany-Amorim C, Hastewell J, Walker C. Toll-like receptors as potential therapeutic targets for multiple diseases. Nat Rev Drug Discov. 2002;1(10):797–807. | ||
Nadigel J, Préfontaine D, Baglole CJ, et al. Cigarette smoke increases TLR4 and TLR9 expression and induces cytokine production from CD8+ T cells in chronic obstructive pulmonary disease. Respir Res. 2011;12(10):1214–1216. | ||
Doz E, Noulin N, Boichot E, et al. Cigarette smoke-induced pulmonary inflammation is TLR4/MyD88 and IL-1R1/MyD88 signaling dependent. J Immunol. 2008;180(2):1169–1178. | ||
Geraghty P, Dabo AJ, D’Armiento J. TLR4 protein contributes to cigarette smoke-induced matrix metalloproteinase-1 (MMP-1) expression in chronic obstructive pulmonary disease. J Biol Chem. 2011;286(34):30211–30218. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.