")
Back to Journals » International Journal of Nephrology and Renovascular Disease » Volume 7
Renal manifestations of genetic mitochondrial disease
Authors O'Toole J
Received 24 October 2013
Accepted for publication 14 November 2013
Published 31 January 2014 Volume 2014:7 Pages 57—67
DOI https://doi.org/10.2147/IJNRD.S37887
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
John F O'Toole
Department of Internal Medicine, Division of Nephrology, MetroHealth Medical System, Case Western Reserve University School of Medicine, Cleveland, OH, USA
Abstract: Mitochondrial diseases can be related to mutations in either the nuclear or mitochondrial genome. Childhood presentations are commonly associated with renal tubular dysfunction, but renal involvement is less commonly reported outside of this age-group. Mitochondrial diseases are notable for the significant variability in their clinical presentation and the broad spectrum of genes implicated in their etiology. These features contribute to the challenges of establishing a definitive diagnosis and understanding the pathogenetic mechanisms leading to kidney involvement in these diseases. Here, we review the deoxyribonucleic acid variants in the mitochondrial and nuclear genomes that have been associated with a kidney phenotype, and examine some of the possible pathogenic mechanisms that may contribute to the expression of a renal phenotype.
Keywords: genetics, kidney, mitochondria
Introduction
According to the endosymbiont hypothesis, mitochondria have evolved from the incorporation of an α-proteobacterium into a primordial cell, eventually leading to the formation of modern eukaryotes.1 They are well known as the cornerstone of intracellular energy production, but also have additional functions critical to the cell, including apoptosis, anapleurotic reactions, and a role in innate immunity. The mitochondria are comprised of an inner and outer membrane, and the mammalian mitochondrial proteome is composed of over 1,000 proteins.2 The large majority of these are encoded by nuclear genes and imported into the mitochondria after cytosolic translation. A major source of cellular energy is the respiratory chain, which is composed of over 80 protein subunits assembled into five respiratory complexes that are embedded in the inner mitochondrial membrane. A unique feature of mitochondria is that they contain their own genome, a 16,569-base-pair circular, double-stranded deoxyribonucleic acid (DNA) molecule.3 Thirteen proteins are encoded on the mitochondrial genome, and all are subunits of the respiratory chain; seven are components of complex I, one is a component of complex III, three are components of complex IV, and two are components of complex V.3 In addition, the mitochondrial genome also contains two ribosomal ribonucleic acid (rRNA) molecules and 22 transfer RNA (tRNA) molecules required for the translation of the 13 mitochondrially encoded protein subunits. Multiple copies of the mitochondrial genome are contained within each mitochondria, and each cell may contain thousands of copies of the mitochondrial genome.4 Mitochondria along with the mitochondrial genome are maternally inherited5 and are passed from the mother to offspring without paternal contribution. Since the mitochondria are assembled from gene products encoded on both the nuclear and mitochondrial genomes, genetic variation in either genome may contribute to mitochondrial function, and the inheritance pattern will depend upon which genome carries the pathogenic variants.
Mitochondrial diseases are a clinically and genetically heterogeneous group of disorders. The most dominant clinical manifestations have been related to neurologic and muscular phenotypes; however, as the awareness and recognition of mitochondrial diseases has increased, the spectrum of clinical manifestations has expanded to include other organ systems, such as the heart, liver, gastrointestinal tract, and the kidney.6 The renal manifestations that have been reported in the setting of mitochondrial dysfunction include renal tubular dysfunction, interstitial nephritis, glomerular pathology, and in rare cases cystic disease. Mitochondrial diseases are often classified by the constellation of clinical symptoms present in an individual. In some cases, a clinically defined mitochondrial syndrome has been associated with multiple distinct genetic etiologies. Alternatively, the same genetic lesion has been observed in association with variable clinical presentations. Renal manifestations have been reported in individuals with some of these well-recognized mitochondrial disease syndromes, such as MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like symptoms), MERRF (myoclonus, epilepsy with ragged red fibers), Pearson syndrome, Kearns–Sayre syndrome, and Leigh syndrome. In other cases, renal disease has been reported as the only disease manifestation or with clinical features that do not fit neatly into a defined syndrome, but have been associated with nuclear or mitochondrial DNA mutations affecting mitochondrial proteins.
Here, we review mitochondrial DNA mutations and mutations in nuclear genes encoding mitochondrial proteins that have been associated with kidney disease, and examine some of the pathogenetic mechanisms that may contribute to a renal phenotype.
Mitochondrial DNA mutations associated with a renal phenotype
Variation that occurs in the mitochondrial genome may include base substitutions, deletions or duplications. Publicly available databases of mitochondrial DNA (mtDNA) variation provide a useful resource for researchers and clinicians and are easily accessible (http://www.mitomap.org/MITOMAP7 and http://www.mtdb.igp.uu.se8). Variation that occurs in the mitochondrial genome can be characterized with a view to its functional effect. While some variants may be neutral or adaptive, others are deleterious and therefore designated mutations.7 When mutations occur in the 13 proteins encoded on the mitochondrial genome, they are expected to exert their effect through decrements in oxidative phosphorylation, as these are subunits of the respiratory chain. Similarly, mitochondrial DNA mutations occurring in the rRNA or tRNA molecules may impair the translation of the 13 mitochondrially encoded protein subunits, also negatively impacting respiratory chain function.
The expression of a clinical phenotype due to mitochondrial DNA mutation is dependent upon several factors. Some factors, such as gene–gene or gene–environment interactions, contribute to disease expression of mtDNA mutations just as they do for mutations in the nuclear genome. Other factors that contribute to the clinical phenotype of mtDNA mutations are unique to the mitochondrial genome. The fact that each cell may contain thousands of mtDNA molecules is central to the unique properties of mtDNA mutations. When all of the mtDNA molecules in a cell are identical, the mtDNA population is referred to as homoplastic. On the other hand, if the mtDNA population of a cell is composed of more than one species of mtDNA molecules (eg, some of these molecules carry an mtDNA mutation and some do not), this population is referred to as heteroplasmic. As the fraction of mutant mtDNA increases in a cell or tissue, the chance that a clinical phenotype will occur in that tissue also increases, likely related to defects in oxidative phosphorylation.9 A related contributor to phenotypic variability is the reliance of different tissues upon oxidative metabolism. This results in tissue-specific thresholds for defects in oxidative phosphorylation, where tissues with high metabolic activity and heavy reliance on oxidative metabolism manifest clinical phenotypes at lower levels of mutant mtDNA heteroplasmy. Since mitochondria and the mtDNA they contain must be distributed with each cell division, this creates the possibility for the unequal distribution of mutant mtDNA when it exists in a heteroplasmic population during cell division. Therefore, oocytes may have variable mtDNA loads and the mutant mtDNA molecules may be unevenly divided throughout embryonic development, leading to variability in the tissue distribution of mutant mtDNA between mother and offspring, as well as between siblings.
MELAS and the m.A3243G base transition
MELAS is a mitochondrial syndrome defined by the clinical features of mitochondrial encephalopathy with dementia or seizures, and stroke-like episodes in individuals less than 40 years of age, myopathy with ragged red fibers on muscle biopsy, and lactic acidosis.10 MELAS is due to mutations of the mitochondrial genome and therefore follows a maternal inheritance pattern. While at least 29 mutations have been reported in association with MELAS, 80% of cases are associated with a base transition in the mtRNALeu gene – m.A3243G.10 As with many mitochondrial diseases, there is a substantial degree of clinical variability amongst affected individuals, even in the presence of the same mitochondrial DNA mutation, and this is likely related to factors noted above.
Although renal disease is well known to occur in association with MELAS, it is not common. In a case series of 110 individuals diagnosed with MELAS syndrome, renal involvement was noted to be present in two individuals.11 By far the most common mtDNA mutation reported in association with MELAS and a kidney phenotype is the base transition m.A3243G in the mtRNALeu gene (Figure 1). Although not a classic criterion defining MELAS syndrome, deafness has frequently been observed in individuals with renal involvement and often predates the onset of renal involvement.12 In addition to individuals with MELAS syndrome, the m.A3243G mutation has also been detected in individuals with maternally inherited diabetes with deafness, and is present in about 1% of diabetic individuals in Europe and Japan.13 Due to this association with deafness and the occasional difficulty of ascertaining a clear inheritance pattern in some small kindred, the diagnosis of Alport’s syndrome may be considered in these individuals.13 However, hematuria is usually absent and ultrastructural renal histopathology typically lacks the glomerular basement-membrane disruption characteristic of Alport’s syndrome. Some reports have described the progression of symptoms from deafness, diabetes, and renal involvement to progress to more classic features of MELAS over time,14 suggesting that the clinical features are more consistent with a disease spectrum rather than distinct syndromes.15 The progressive nature of clinical features may be related to a replication advantage that has been reported for mtDNA molecules carrying the m.A3243G mutation,16 leading to the progressive accumulation of mutant mtDNA and eventually surpassing the threshold levels for each tissue.
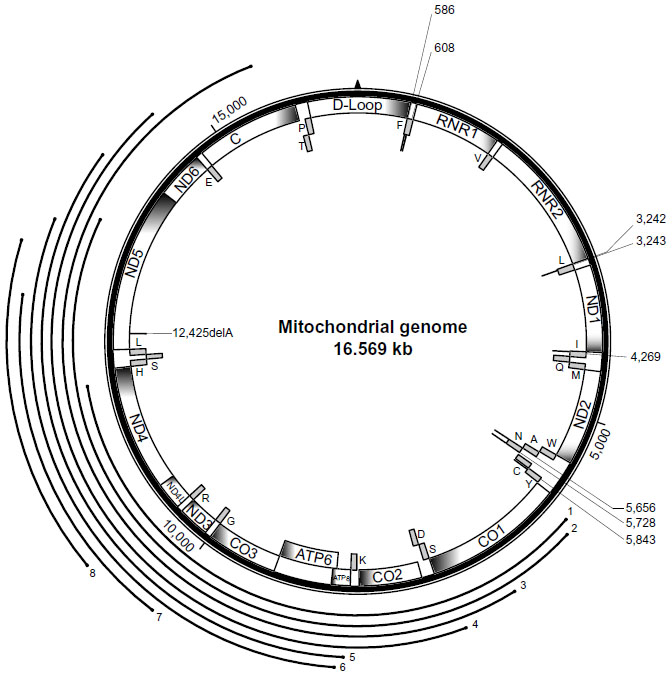 | Figure 1 Mitochondrial genome with the positions of mutations associated with a kidney phenotype. A schematic of the circular 16,569 base pair human mitochondrial genome is shown with the position of 13 protein-coding mitochondrial genes (ND1, ND2, CO1, CO2, ATP8, ATP6, CO3, ND3, ND4L, ND4, ND5, ND6, CYTB), two ribosomal ribonucleic acid (RNA) molecules (RNR1, RNR2) and the noncoding D-loop, depicted as labeled boxes on the interior of the circle. The position of the 22 mitochondrial transfer RNAs (tRNAs) is shown with the solid-gray boxes with the single-letter amino acid designation shown for the respective tRNA. The position of the mitochondrial transfer deoxyribonucleic acid (mtDNA) single-base mutations are shown with a solid-bold line on the interior of the circle connected to a light line indicating the base position of the mutation. Large mitochondrial DNA deletions are represented by bold lines labeled 1–8 on the exterior of the circle phenotype. |
Rare case reports have described additional mitochondrial point mutations associated with kidney phenotypes (Table 1); these include focal segmental glomerulosclerosis (FSGS)17–19 or interstitial fibrosis with progressive renal insufficiency.20–22 The majority of mtDNA point mutations reported to date have been associated with proteinuria, a kidney phenotype typical of glomerular injury, and renal histopathology has demonstrated focal or global glomerulosclerosis on kidney biopsy. Renal tubular dysfunction and tubulointerstitial nephritis are less common findings on kidney biopsy. While kidney disease due to mitochondrial point mutations has occasionally been reported in the absence of extrarenal features,13,23 it more commonly occurs in the setting of MELAS syndrome14,24–28 or deafness, which usually occurs with diabetes.11,12,26,29–35 Point mutations that occur in a protein-coding region of mtDNA in association with a kidney phenotype are very rare.36 A single-base deletion in the complex I subunit NADH dehydrogenase 5, has been reported to occur in a single individual with glomerulocystic kidney disease and renal failure.
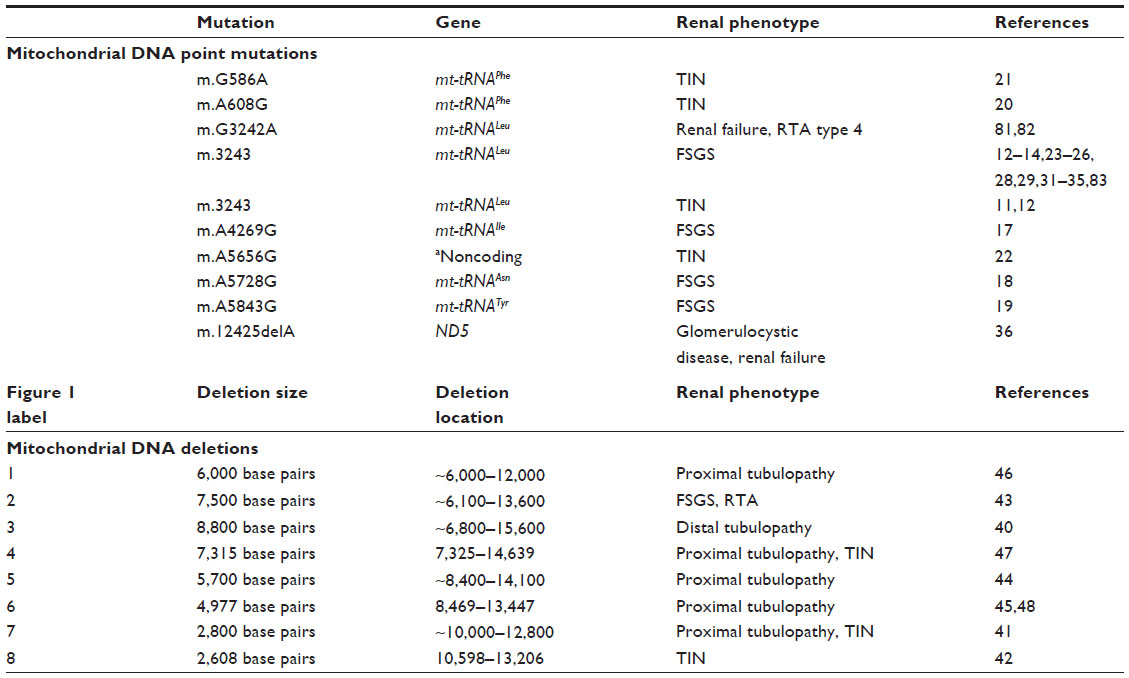 | Table 1 Mitochondrial DNA mutations associated with a kidney phenotype |
The pathogenetic mechanisms underlying the renal phenotypes associated with mtDNA point mutations, and specifically m.A3243G, are not entirely clear. The presence of abnormal mitochondria in electron microscopic images of kidney biopsies suggests that mitochondrial dysfunction is central to disease pathogenesis and may be related to defects in oxidative phosphorylation and energy metabolism.37 In vitro analysis of the mitochondrial tRNALeu m.A3243G mutation causes defects in protein synthesis and mitochondrial respiration as the mutant mtDNA approaches homoplasmic status.38 However, cells were protected from these defects if they retained even a small amount of normal mtDNA (ie, >6% of total mtDNA lacking the mutation). Others have speculated that since the m.A3243G mutation is associated with the dramatic vascular phenotype of stroke-like episodes in the context of MELAS syndrome, renal hemodynamic alterations may underlie the pathogenesis of the FSGS lesion observed in association with the m.A3243G mutation.26
Mitochondrial DNA deletions
Interestingly, the mitochondrial syndromes and kidney phenotypes associated with mtDNA deletions are distinct from those observed with mtDNA point mutations. The two mitochondrial disease syndromes reported with mtDNA deletions and a renal phenotype are Kearns–Sayre and Pearson syndromes. Kearns–Sayre syndrome typically includes chronic progressive external ophthalmoplegia, ptosis, and pigmentary retinopathy in individuals less than 20 years of age. Pearson syndrome occurs in infancy, and characteristically includes sideroblastic anemia and exocrine pancreatic dysfunction. Both of these syndromes have been described with additional clinical features that may include a renal phenotype. Renal tubular dysfunction is the kidney phenotype most often reported with mtDNA deletions (Table 1), but interstitial nephritis39–41 and glomerular pathologies42 have also been described. Kearns–Sayre39,42 and Pearson syndromes43 have been described with unique but overlapping mtDNA deletions (Figure 1), and one report describes an individual presenting with symptoms of Pearson Syndrome but developing features of Kearns–Sayre syndrome over time.44 The renal tubular defects associated with mtDNA deletions may affect the proximal40,43–47 or distal renal tubular segments.39 The rarity of reports that include mtDNA deletions and a kidney phenotype make it difficult to draw any genotype–phenotype correlations, and the factors that contribute to the variety and penetrance of clinical features associated with mtDNA deletions may be related to the inheritance and distribution of mtDNA, as discussed earlier. It is interesting to note that there is little overlap between the mtDNA deletions and the mtDNA point mutations; one case with a single-base deletion and a renal phenotype has been reported in the segments of mtDNA involving larger deletions36 (Figure 1), but whether this lack of overlap contributes to the disease penetrance or renal phenotype is currently unknown.
Nuclear-encoded genes for mitochondrial proteins with renal phenotype
The mitochondrial proteome is composed of over 1,000 proteins,2 but only 13 of these are encoded by the mitochondrial genome.3 This underlines the importance of the nuclear genome to the maintenance and proper function of the mitochondria, and mutations in several nuclear genes encoding mitochondrial proteins have been reported. The 13 proteins encoded by genes on the mitochondrial genome are all subunits of the respiratory chain. Therefore, mtDNA mutations may be expected to exert their effect on mitochondrial function through defects in oxidative phosphorylation and energy metabolism. Mutations in nuclear-encoded mitochondrial proteins may lead to defects in energy metabolism, but may also cause decrements in additional mitochondrial functions, including mitochondrial protein translation and posttranslational modification, protection from oxidant stress, and mitochondrial DNA maintenance. Based upon the current literature, mutations in nuclear genes encoding mitochondrial proteins are an uncommon yet intriguing cause of kidney disease. The variability in the renal phenotype of mutations in nuclear genes encoding mitochondrial proteins is not certain at present, and in some ways is more puzzling than the variability seen in association with mutations in the mitochondrial genome. While mtDNA heteroplasmy and unequal distribution of mutant mtDNA during cell division and development will not contribute to disease expression of nuclear-encoded genes, several factors are likely to contribute to the renal disease phenotype, including gene–gene interactions, gene–environment interactions, and tissue-specific thresholds for mitochondrial defects.
In the following sections, we review a spectrum of nuclear genes encoding mitochondrial proteins that have been associated with kidney phenotypes. These genes are discussed in the following sections organized by their mitochondrial function.
Coenzyme Q10 biosynthesis
Mutations in the enzymes required for the coenzyme Q10 biosynthetic pathway more reliably include a renal phenotype than genetic lesions in any other nuclear-encoded mitochondrial pathway or protein complex. The reasons for this are currently uncertain and include a number of intriguing possibilities. The biosynthetic pathway of human coenzyme Q has largely been inferred from the study of Saccharomyces cerevisiae, where the genes required for coenzyme Q biosynthesis were initially characterized and human orthologues were subsequently identified using sequence homology.48 Coenzyme Q10 is well known as an electron carrier in the mitochondrial respiratory chain, accepting electrons from respiratory chain entry sites, including complexes I, II, and the electron transfer flavoprotein then donating electrons to its downstream electron acceptor – complex III. Several additional functional roles have been attributed to coenzyme Q10, including that of a lipid-soluble antioxidant.48,49 In humans, coenzyme Q10 is assembled from a benzoquinone ring conjugated to a chain of isoprenoid units, usually ten in humans. To date, mutations in four nuclear-encoded genes of the coenzyme Q biosynthetic pathway – PDSS2, COQ2, COQ6, and COQ950 – have been identified in individuals with a renal phenotype in an autosomal-recessive inheritance pattern.
Most mutations in the enzymes of the coenzyme Q biosynthetic pathway identified thus far are associated with proteinuric glomerular disease (Table 2). While a single case presenting with a neonatal diagnosis of crescentic glomerulonephritis in an individual with COQ2 mutations has been reported, individuals more typically present with steroid-resistant nephrotic syndrome.51–54 Similarly, individuals with PDSS2 and COQ6 mutations have also presented with proteinuric kidney disease. The single reported case of PDSS2 mutations,55 as well as 13 individuals’ COQ6 mutations, were all diagnosed with nephrotic syndrome before 6 years of age.56 An exception to the glomerular phenotype observed with mutations in other coenzyme Q biosynthetic pathway genes is the single kindred with homozygous mutation in COQ9.50 The phenotype reported with COQ9 mutations included renal tubular dysfunction in addition to cerebral and cerebellar atrophy, seizures, and lactic acidosis.
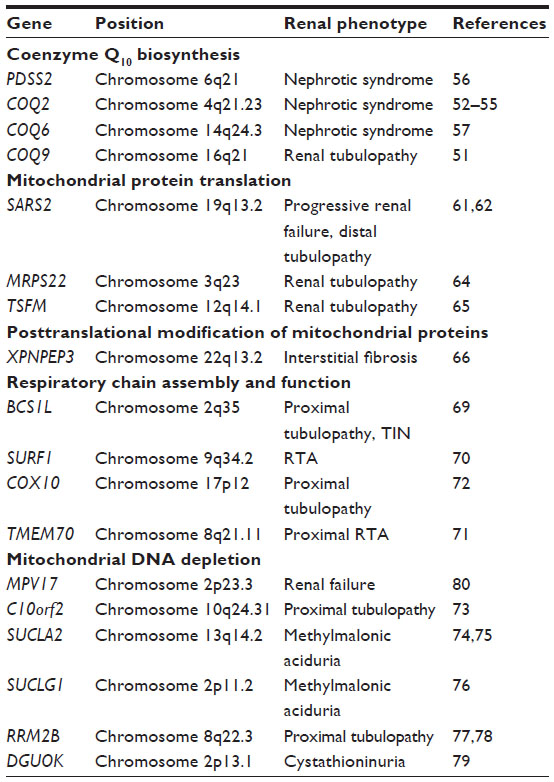 | Table 2 Nuclear genes encoding mitochondrial proteins associated with a kidney phenotype |
The relative consistency of the renal phenotype observed with mutations in the coenzyme Q biosynthetic pathway is interesting, yet the underlying mechanism is uncertain. While primary coenzyme Q10 deficiencies are associated with decrements in oxidative phosphorylation, it is interesting to note that isolated renal disease has been reported in the absence of extrarenal manifestations54 or with limited extrarenal involvement.56 The absence of clinical manifestations in tissues (eg, nervous and muscle tissues) more heavily reliant on oxidative phosphorylation for energy metabolism and typical of mitochondrial disease suggests that defects in oxidative phosphorylation may not completely explain the disease pathology. Even within the kidney, the podocyte is not regarded as a cell type that is heavily dependent upon respiratory metabolism to maintain proper function. Rather, the renal tubules are generally recognized to have substantial cellular energy demands necessary for solute transport, and much of this cellular energy is supplied by respiratory metabolism, as reflected by the high density of mitochondria in the renal tubules. Since these mutations are encoded on nuclear genes and are expected to be present in all cells, it is reasonable to question whether the podocyte or the renal tubular cell is the kidney cell type most critically related to the renal pathology. Genetic studies in mouse models have established that loss of Pdss2 function in the podocyte is sufficient to recapitulate the kidney phenotype observed in individuals with genetic causes of primary coenzyme Q10 deficiency.57 While decrements in oxidative phosphorylation may contribute to podocyte damage and the nephrotic proteinuria observed with primary coenzyme Q10 deficiencies, there may also be a role for the antioxidant function of coenzyme Q10 or in de novo pyrimidine biosynthesis.53
Interestingly, a recent report examined the role of PDSS2 in a cohort of individuals with focal segmental glomerulosclerosis. A haplotype defined by two noncoding single-nucleotide polymorphisms in the PDSS2 gene was identified at an increased frequency in European American populations with FSGS, but not African American populations with FSGS.58 Another interesting observation was that lymphoblastoid cell lines derived from individuals with FSGS had decreased coenzyme Q10 content compared to cell lines from control populations without kidney disease, but this did not correlate with the PDSS2 haplotype. These results suggest that proteinuric kidney disease may contribute to a secondary form of coenzyme Q10 deficiency that may occur through epigenetic mechanisms. Alternatively, individuals maintaining lower coenzyme Q10 levels due to undefined mechanisms may be predisposed to the development of proteinuric kidney disease related to FSGS. While the podocyte seems to be the critical cell mediating the proteinuric kidney disease in the setting of genetic causes of primary coenzyme Q10 deficiency, the functional role or roles of coenzyme Q10 that are lacking in these diseases remain to be conclusively established. It should also be pointed out that additional genetic causes of primary coenzyme Q10 deficiency have been identified, which lack a renal phenotype. The reason that these components of the coenzyme Q10 biosynthetic pathway do not cause kidney disease is unknown. An expanded understanding of this pathway in humans and its differences across tissues and cell types may provide insights into the critical functional role of coenzyme Q10 in maintaining proper podocyte function.
Mitochondrial protein translation
Since the mtDNA encodes 13 polypeptide subunits of the respiratory chain, an intramitochondrial mechanism for the synthesis of these proteins is required. The mtDNA contains 22 tRNAs, one for each amino acid, except leucine and serine, which have two mitochondrial tRNAs. In contrast, all of the aminoacyl tRNA synthetases are encoded in the nuclear genome. Mutations in many of the aminoacyl tRNA synthetases have been identified, reviewed elsewhere,59 and the phenotypic effect of mutations in each of these genes demonstrates a remarkable degree of tissue specificity. Interestingly, of the 20 tRNA synthetases that aminoacylate mitochondrially encoded tRNAs, only one has been associated with a kidney phenotype. Mutations in the SARS2 gene, encoding the mitochondrial seryl-tRNA synthetase, have been identified in individuals with infantile onset of multiorgan disease dominated by renal failure and tubular dysfunction.60 The clinical phenotype is characterized by hyperuricemia, pulmonary hypertension, renal failure, and alkalosis. Of the 22 mitochondrially encoded tRNAs, two are serine tRNAs – mt-tRNASerAGY and mt-tRNASerUCN – and are both acylated by the SARS2 gene product. The original mutation identified in the setting of HUPRA only affected the aminoacylation of mt-tRNASerAGY, where it was reduced to only 10%–20% of normal values and was not acetylated, whereas the abundance and aminoacylation of mt-tRNASerUCN was comparable to normal levels.60 A subsequent report identified a different homozygous mutation in the same C-terminal globular domain of SARS2 in another individual presenting with HUPRA syndrome.61 A channeling hypothesis has been proposed to explain the reduction of mt-tRNASerAGY observed in the setting of SARS2 mutations.62 This hypothesis states that intramitochondrial RNA that is not bound to protein is rapidly degraded, and this functions as a mechanism to maintain the proper levels of mitochondrial tRNAs. Since the SARS2 mutation described is expected to impair the interaction of SARS2 with mt-tRNASerAGY and leave this tRNA uncharged, it is expected to accelerate the degradation of mt-tRNASerAGY.
In addition to the renal failure observed in affected individuals, it is interesting to note that the renal tubular functional defects are reflective of distal tubular dysfunction, as opposed to proximal tubular defects, which have been more commonly reported in the context of mitochondrial disease.6 As the mutations in SARS2 have been associated with a selective loss of the mt-tRNASerAGY, it is tempting to consider this tRNA gene as a strong candidate for mitochondrial mutations that would lead to a kidney phenotype; however, mutations reported to date do not include a renal phenotype.
While the mitochondrial genome encodes two rRNA molecules, all of the mitochondrial ribosomal protein subunits required for the translation of the 13 mitochondrial proteins are encoded by nuclear genes and imported into the mitochondria. Mutations in the ribosomal protein MRPS22 have been associated with renal tubulopathy in an individual with generalized edema, hypotonia, and cardiomyopathy.63 Biochemical activities of the respiratory chain complexes I, III, IV, and V were reduced to between 8% and 30% of normal values in muscle from one affected individual, whereas complex II, which is composed entirely of nuclear-encoded subunits and therefore not dependent upon the mitochondrial protein synthetic machinery, was within normal limits. Interestingly, the abundance of the 12S rRNA encoded on the mitochondrial genome was found to be only 10% of normal, leading the authors to speculate that rRNAs that fail to be incorporated into the ribosome are preferentially degraded, as previously described in the channeling hypothesis discussed earlier.
Finally, mutations in TSFM encoding a mitochondrial translation factor have been associated with encephalomyopathy, cardiomyopathy, and hepatic failure, and one of four individuals was also diagnosed with renal tubular dysfunction that was not further characterized.64 These results again highlight the broad clinical variability in the presentation of genetically mediated mitochondrial disease. Several distinct components of the mitochondrial translational machinery have been associated with a renal phenotype, most commonly characterized by renal tubular dysfunction with or without progressive renal insufficiency.
Posttranslational processing of mitochondrial proteins
Mutations in XPNPEP3, a mitochondrial aminopeptidase, were identified in two independent kindred65 with a renal phenotype characterized by tubular atrophy with interstitial fibrosis and progressive renal insufficiency. One of these kindred with an early truncating mutation presented with evidence of multiorgan system involvement and complex I deficiency in muscle biopsy. Studies examining the mitochondrial function of the S. cerevisiae XPNPEP3 orthologue suggest that it participates in the posttranslational modification of proteins after import into the mitochondria,66 and loss of function may result in activation of the target of rapamycin.67
Respiratory chain assembly and function
Mutations in several other nuclear-encoded genes have been reported in individuals with spectrum respiratory chain defects, including decreased activity of complex I,65 complex III,68 complex IV,69 and complex V.70 Mutations in the mitochondrial chaperone BCS1L have been associated with complex III-deficiency encephalopathy, liver failure, and proximal tubulopathy, or rarely interstitial nephritis presenting in the neonatal period.68 SURF1 encodes a protein necessary for the assembly or maintenance of complex IV, and mutations have been associated with Leigh’s disease and complex IV defects. Rarely, proximal or distal renal tubular acidosis has been associated with SURF1 mutations.69 Mutations in COX10, an additional complex IV assembly factor, have also been identified in individuals with proximal tubular dysfunction in the setting of ataxia, ptosis, pyramidal syndrome, seizures, hypotonia, and lactic acidosis.71 Complex V deficiency presenting with hypotonia, cardiomyopathy, and lactic acidosis has been associated with mutations in TMEM70.70 A series of 25 individuals with TMEM70 mutations found five individuals to have echogenic kidneys with loss of corticomedullary differentiation on renal ultrasound performed in the neonatal period, and two of these also had a proximal renal tubular acidosis.70
Mitochondrial DNA-depletion syndromes
The mitochondrial DNA-depletion syndromes represent a group of disorders characterized by the loss of mtDNA copy number in the cells of affected tissues. The majority of individuals presenting with these syndromes do not have a renal phenotype; however, a few individuals with a kidney phenotype have been identified with an mtDNA-depletion syndrome related to several genes (Table 2). The renal phenotype is most characteristic of renal tubular dysfunction,72–78 but renal failure has also been described.79
Conclusion
Kidney disease is an uncommon but important clinical feature of mitochondrial diseases. Inherited mitochondrial diseases may be transmitted through nuclear or mitochondrial DNA, and the mode of inheritance has important implications for the clinical expression of disease. Genetic and environmental modifiers may contribute to the clinical variability observed in mitochondrial disease transmitted through nuclear or mitochondrial DNA. In addition, the disease expression of mutations transmitted through mitochondrial DNA may also be subject to variability related to heteroplasmy, unequal distribution through cell division, and tissue-specific thresholds for mitochondrial dysfunction. Together, all of these features contribute to the dramatic clinical variability observed in genetic mitochondrial disease.
Mutations in a large number of genes encoded on both the mitochondrial and the nuclear genomes have been identified in individuals with mitochondrial disease. The mitochondria have a well-known role in cellular energy metabolism; therefore, it is not surprising that many mutations associated with mitochondrial disease have a negative impact on this function. Mitochondrial DNA mutations are expected to exert their effect through defects in oxidative phosphorylation, and this may also be true of mutations in many nuclear-encoded mitochondrial proteins. A careful examination of mitochondrial diseases associated with a kidney phenotype suggests that pathogenic mechanisms beyond defects in energy metabolism may also be important, and may include the antioxidant properties of coenzyme Q or defects in cell signaling with mitochondrial damage. Future research is needed to determine how these and other pathogenetic mechanisms may contribute to the expression of clinical disease.
Kidney disease is a complex and intriguing aspect of mitochondrial disorders. Many genetic causes of mitochondrial dysfunction have already been identified, and with the advance of sequencing technologies, this number is certain to grow. An improved understanding of these genetic causes and the mechanisms through which mitochondrial dysfunction contributes to kidney disease is the first step toward effective diagnostics and treatments.
Disclosure
The author reports no conflicts of interest in this work.
References
Gray MW. Mitochondrial evolution. Cold Spring Harb Perspect Biol. 2012;4:a011403. | |
Pagliarini DJ, Calvo SE, Chang B, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. | |
Anderson S, Bankier AT, Barrell BG, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. | |
García-Rodríguez LJ. Appendix 1. Basic properties of mitochondria. Methods Cell Biol. 2007;80:809–812. | |
Giles RE, Blanc H, Cann HM, Wallace DC. Maternal inheritance of human mitochondrial DNA. Proc Natl Acad Sci U S A. 1980;77:6715–6719. | |
Niaudet P, Rotig A. The kidney in mitochondrial cytopathies. Kidney Int. 1997;51:1000–1007. | |
Ruiz-Pesini E, Lott MT, Procaccio V, et al. An enhanced MITOMAP with a global mtDNA mutational phylogeny. Nucleic Acids Res. 2007;35:D823–D828. | |
Ingman M, Gyllensten U. mtDB: Human Mitochondrial Genome Database, a resource for population genetics and medical sciences. Nucleic Acids Res. 2006;34:D749–D751. | |
Hayashi J, Ohta S, Kikuchi A, Takemitsu M, Goto Y, Nonaka I. Introduction of disease-related mitochondrial DNA deletions into HeLa cells lacking mitochondrial DNA results in mitochondrial dysfunction. Proc Natl Acad Sci U S A. 1991;88:10614–10618. | |
Hirano M, Pavlakis SG. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes (MELAS): current concepts. J Child Neurol. 1994;9:4–13. | |
Hirano M, Konishi K, Arata N, et al. Renal complications in a patient with A-to-G mutation of mitochondrial DNA at the 3243 position of leucine tRNA. Intern Med. 2002;41:113–118. | |
Guery B, Choukroun G, Noël LH, et al. The spectrum of systemic involvement in adults presenting with renal lesion and mitochondrial tRNA(Leu) gene mutation. J Am Soc Nephrol. 2003;14:2099–2108. | |
Jansen JJ, Maassen JA, van der Woude FJ, et al. Mutation in mitochondrial tRNALeu(UUR) gene associated with progressive kidney disease. J Am Soc Nephrol. 1997;8:1118–1124. | |
Lederer SR, Klopstock T, Schiffl H. MELAS: a mitochondrial disorder in an adult patient with a renal transplant. Wien Klin Wochenschr. 2010;122:363–365. | |
Windpessl M, Wallner M. m.3243A>G: many faces of one single point mutation. Wien Klin Wochenschr. 2010;122:601–602. | |
Yoneda M, Chomyn A, Martinuzzi A, Hurko O, Attardi G. Marked replicative advantage of human mtDNA carrying a point mutation that causes the MELAS encephalomyopathy. Proc Natl Acad Sci U S A. 1992;89:11164–11168. | |
Taniike M, Fukushima H, Yanagihara I, et al. Mitochondrial tRNA(Ile) mutation in fatal cardiomyopathy. Biochem Biophys Res Commun. 1992;186:47–53. | |
Meulemans A, Seneca S, Lagae L, et al. A novel mitochondrial transfer RNA(Asn) mutation causing multiorgan failure. Arch Neurol. 2006;63:1194–1198. | |
Scaglia F, Vogel H, Hawkins EP, Vladutiu GD, Liu LL, Wong LJ. Novel homoplasmic mutation in the mitochondrial tRNATyr gene associated with atypical mitochondrial cytopathy presenting with focal segmental glomerulosclerosis. Am J Med Genet A. 2003;123A:172–178. | |
Tzen CY, Tsai JD, Wu TY, et al. Tubulointerstitial nephritis associated with a novel mitochondrial point mutation. Kidney Int. 2001;59:846–854. | |
D’Aco KE, Manno M, Clarke C, Ganesh J, Meyers KE, Sondheimer N. Mitochondrial tRNA(Phe) mutation as a cause of end-stage renal disease in childhood. Pediatr Nephrol. 2013;28:515–519. | |
Zsurka G, Ormos J, Ivanyi B, et al. Mitochondrial mutation as a probable causative factor in familial progressive tubulointerstitial nephritis. Hum Genet. 1997;99:484–487. | |
Hotta O, Inoue CN, Miyabayashi S, Furuta T, Takeuchi A, Taguma Y. Clinical and pathologic features of focal segmental glomerulosclerosis with mitochondrial tRNALeu(UUR) gene mutation. Kidney Int. 2001;59:1236–1243. | |
Piccoli GB, Bonino LD, Campisi P, et al. Chronic kidney disease, severe arterial and arteriolar sclerosis and kidney neoplasia: on the spectrum of kidney involvement in MELAS syndrome. BMC Nephrol. 2012;13:9. | |
Mochizuki H, Joh K, Kawame H, et al. Mitochondrial encephalomyopathies preceded by de-Toni-Debre-Fanconi syndrome or focal segmental glomerulosclerosis. Clin Nephrol. 1996;46:347–352. | |
Nakamura S, Yoshinari M, Doi Y, et al. Renal complications in patients with diabetes mellitus associated with an A to G mutation of mitochondrial DNA at the 3243 position of leucine tRNA. Diabetes Res Clin Pract. 1999;44:183–189. | |
Yanagihara C, Oyama A, Tanaka M, Nakaji K, Nishimura Y. An autopsy case of mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes syndrome with chronic renal failure. Intern Med. 2001;40:662–665. | |
Lau KK, Yang SP, Haddad MN, Butani L, Makker SP. Mitochondrial encephalopathy with lactic acidosis and stroke-like episodes syndrome with hypothyroidism and focal segmental glomerulosclerosis in a paediatric patient. Int Urol Nephrol. 2007;39:941–946. | |
Damian MS, Hertel A, Seibel P, et al. Follow-up in carriers of the ‘MELAS’ mutation without strokes. Eur Neurol. 1998;39:9–15. | |
Mima A, Shiota F, Matsubara T, et al. An autopsy case of mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) with intestinal bleeding in chronic renal failure. Ren Fail. 2011;33:622–625. | |
Cheong HI, Chae JH, Kim JS, et al. Hereditary glomerulopathy associated with a mitochondrial tRNA(Leu) gene mutation. Pediatr Nephrol. 1999;13:477–480. | |
Kurogouchi F, Oguchi T, Mawatari E, et al. A case of mitochondrial cytopathy with a typical point mutation for MELAS, presenting with severe focal-segmental glomerulosclerosis as main clinical manifestation. Am J Nephrol. 1998;18:551–556. | |
Lowik MM, Hol FA, Steenbergen EJ, Wetzels JF, van den Heuvel LP. Mitochondrial tRNALeu(UUR) mutation in a patient with steroid-resistant nephrotic syndrome and focal segmental glomerulosclerosis. Nephrol Dial Transplant. 2005;20:336–341. | |
Doleris LM, Hill GS, Chedin P, et al. Focal segmental glomerulosclerosis associated with mitochondrial cytopathy. Kidney Int. 2000;58:1851–1858. | |
Yamagata K, Muro K, Usui J, et al. Mitochondrial DNA mutations in focal segmental glomerulosclerosis lesions. J Am Soc Nephrol. 2002;13:1816–1823. | |
Alston CL, Morak M, Reid C, et al. A novel mitochondrial MTND5 frameshift mutation causing isolated complex I deficiency, renal failure and myopathy. Neuromuscul Disord. 2010;20:131–135. | |
Sunada Y. Glomerulopathy in MELAS syndrome. Intern Med. 2001;40:561–562. | |
Chomyn A, Martinuzzi A, Yoneda M, et al. MELAS mutation in mtDNA binding site for transcription termination factor causes defects in protein synthesis and in respiration but no change in levels of upstream and downstream mature transcripts. Proc Natl Acad Sci U S A. 1992;89:4221–4225. | |
Goto Y, Itami N, Kajii N, Tochimaru H, Endo M, Horai S. Renal tubular involvement mimicking Bartter syndrome in a patient with Kearns-Sayre syndrome. J Pediatr. 1990;116:904–910. | |
Szabolcs MJ, Seigle R, Shanske S, Bonilla E, DiMauro S, D’Agati V. Mitochondrial DNA deletion: a cause of chronic tubulointerstitial nephropathy. Kidney Int. 1994;45:1388–1396. | |
Rotig A, Goutieres F, Niaudet P, et al. Deletion of mitochondrial DNA in patient with chronic tubulointerstitial nephritis. J Pediatr. 1995;126:597–601. | |
Eviatar L, Shanske S, Gauthier B, et al. Kearns-Sayre syndrome presenting as renal tubular acidosis. Neurology. 1990;40:1761–1763. | |
Majander A, Suomalainen A, Vettenranta K, et al. Congenital hypoplastic anemia, diabetes, and severe renal tubular dysfunction associated with a mitochondrial DNA deletion. Pediatr Res. 1991;30:327–330. | |
McShane MA, Hammans SR, Sweeney M, et al. Pearson syndrome and mitochondrial encephalomyopathy in a patient with a deletion of mtDNA. Am J Hum Genet. 1991;48:39–42. | |
Campos Y, Garcia-Silva T, Barrionuevo CR, Cabello A, Muley R, Arenas J. Mitochondrial DNA deletion in a patient with mitochondrial myopathy, lactic acidosis, and stroke-like episodes (MELAS) and Fanconi’s syndrome. Pediatr Neurol. 1995;13:69–72. | |
Au KM, Lau SC, Mak YF, et al. Mitochondrial DNA deletion in a girl with Fanconi‘s syndrome. Pediatr Nephrol. 2007;22:136–140. | |
Niaudet P, Heidet L, Munnich A, et al. Deletion of the mitochondrial DNA in a case of de Toni-Debre-Fanconi syndrome and Pearson syndrome. Pediatr Nephrol. 1994;8:164–168. | |
Quinzii CM, Hirano M. Coenzyme Q and mitochondrial disease. Dev Disabil Res Rev. 2010;16:183–188. | |
Schmelzer C, Döring F. Micronutrient special issue: coenzyme Q(10) requirements for DNA damage prevention. Mutat Res. 2012;733:61–68. | |
Duncan AJ, Bitner-Glindzicz M, Meunier B, et al. A nonsense mutation in COQ9 causes autosomal-recessive neonatal-onset primary coenzyme Q10 deficiency: a potentially treatable form of mitochondrial disease. Am J Hum Genet. 2009;84:558–566. | |
Quinzii C, Naini A, Salviati L, et al. A mutation in para-hydroxybenzoate-polyprenyl transferase (COQ2) causes primary coenzyme Q10 deficiency. Am J Hum Genet. 2006;78:345–349. | |
Mollet J, Giurgea I, Schlemmer D, et al. Prenyldiphosphate synthase, subunit 1 (PDSS1) and OH-benzoate polyprenyltransferase (COQ2) mutations in ubiquinone deficiency and oxidative phosphorylation disorders. J Clin Invest. 2007;117:765–772. | |
López-Martin JM, Salviati L, Trevisson E, et al. Missense mutation of the COQ2 gene causes defects of bioenergetics and de novo pyrimidine synthesis. Hum Mol Genet. 2007;16:1091–1097. | |
Diomedi-Camassei F, Di GS, Santorelli FM, et al. COQ2 nephropathy: a newly described inherited mitochondriopathy with primary renal involvement. J Am Soc Nephrol. 2007;18:2773–2780. | |
Lopez LC, Schuelke M, Quinzii CM, et al. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am J Hum Genet. 2006;79:1125–1129. | |
Heeringa SF, Chernin G, Chaki M, et al. COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. J Clin Invest. 2011;121:2013–2024. | |
Peng M, Falk MJ, Haase VH, et al. Primary coenzyme Q deficiency in Pdss2 mutant mice causes isolated renal disease. PLoS Genet. 2008;4:e1000061. | |
Gasser DL, Winkler CA, Peng M, et al. Focal segmental glomerulosclerosis is associated with a PDSS2 haplotype and, independently, with a decreased content of coenzyme Q10. Am J Physiol Renal Physiol. 2013;305:F1228–F1238. | |
Konovalova S, Tyynismaa H. Mitochondrial aminoacyl-tRNA synthetases in human disease. Mol Genet Metab. 2013;108:206–211. | |
Belostotsky R, Ben-Shalom E, Rinat C, et al. Mutations in the mitochondrial seryl-tRNA synthetase cause hyperuricemia, pulmonary hypertension, renal failure in infancy and alkalosis, HUPRA syndrome. Am J Hum Genet. 2011;88:193–200. | |
Rivera H, Martín-Hernández E, Delmiro A, et al. A new mutation in the gene encoding mitochondrial seryl-tRNA synthetase as a cause of HUPRA syndrome. BMC Nephrol. 2013;14:195. | |
Belostotsky R, Frishberg Y, Entelis N. Human mitochondrial tRNA quality control in health and disease: a channelling mechanism? RNA Biol. 2012;9:33–39. | |
Saada A, Shaag A, Arnon S, et al. Antenatal mitochondrial disease caused by mitochondrial ribosomal protein (MRPS22) mutation. J Med Genet. 2007;44:784–786. | |
Vedrenne V, Galmiche L, Chretien D, de Lonlay P, Munnich A, Rötig A. Mutation in the mitochondrial translation elongation factor EFTs results in severe infantile liver failure. J Hepatol. 2012;56:294–297. | |
O’Toole JF, Liu Y, Davis EE, et al. Individuals with mutations in XPNPEP3, which encodes a mitochondrial protein, develop a nephronophthisis-like nephropathy. J Clin Invest. 2010;120:791–802. | |
Vogtle FN, Wortelkamp S, Zahedi RP, et al. Global analysis of the mitochondrial N-proteome identifies a processing peptidase critical for protein stability. Cell. 2009;139:428–439. | |
Stames EM, O’Toole JF. Mitochondrial aminopeptidase deletion increases chronological lifespan and oxidative stress resistance while decreasing respiratory metabolism in S. cerevisiae. PLoS One. 2013;8:e77234. | |
de Lonlay P, Valnot I, Barrientos A, et al. A mutant mitochondrial respiratory chain assembly protein causes complex III deficiency in patients with tubulopathy, encephalopathy and liver failure. Nat Genet. 2001;29:57–60. | |
Tay SK, Sacconi S, Akman HO, et al. Unusual clinical presentations in four cases of Leigh disease, cytochrome C oxidase deficiency, and SURF1 gene mutations. J Child Neurol. 2005;20:670–674. | |
Honzik T, Tesarova M, Mayr JA, et al. Mitochondrial encephalocardio-myopathy with early neonatal onset due to TMEM70 mutation. Arch Dis Child. 2010;95:296–301. | |
Valnot I, von Kleist-Retzow JC, Barrientos A, et al. A mutation in the human heme A:farnesyltransferase gene (COX10) causes cytochrome c oxidase deficiency. Hum Mol Genet. 2000;9:1245–1249. | |
Prasad C, Melançon SB, Rupar CA, et al. Exome sequencing reveals a homozygous mutation in TWINKLE as the cause of multisystemic failure including renal tubulopathy in three siblings. Mol Genet Metab. 2013;108:190–194. | |
Ostergaard E, Hansen FJ, Sorensen N, et al. Mitochondrial encephalomyopathy with elevated methylmalonic acid is caused by SUCLA2 mutations. Brain. 2007;130:853–861. | |
Carrozzo R, Dionisi-Vici C, Steuerwald U, et al. SUCLA2 mutations are associated with mild methylmalonic aciduria, Leigh-like encephalomyopathy, dystonia and deafness. Brain. 2007;130:862–874. | |
Ostergaard E, Schwartz M, Batbayli M, et al. A novel missense mutation in SUCLG1 associated with mitochondrial DNA depletion, encephalomyopathic form, with methylmalonic aciduria. Eur J Pediatr. 2010;169:201–205. | |
Bourdon A, Minai L, Serre V, et al. Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat Genet. 2007;39:776–780. | |
Kollberg G, Darin N, Benan K, et al. A novel homozygous RRM2B missense mutation in association with severe mtDNA depletion. Neuromuscul Disord. 2009;19:147–150. | |
Tadiboyina VT, Rupar A, Atkison P, et al. Novel mutation in DGUOK in hepatocerebral mitochondrial DNA depletion syndrome associated with cystathioninuria. Am J Med Genet A. 2005;135:289–291. | |
Uusimaa J, Evans J, Smith C, et al. Clinical, biochemical, cellular and molecular characterization of mitochondrial DNA depletion syndrome due to novel mutations in the MPV17 gene. Eur J Hum Genet. Epub May 29, 2013. | |
Wortmann SB, Champion MP, van den Heuvel L, et al. Mitochondrial DNA m.3242G >A mutation, an under diagnosed cause of hypertrophic cardiomyopathy and renal tubular dysfunction? Eur J Med Genet. 2012;55:552–556. | |
Mimaki M, Hatakeyama H, Ichiyama T, et al. Different effects of novel mtDNA G3242A and G3244A base changes adjacent to a common A3243G mutation in patients with mitochondrial disorders. Mitochondrion. 2009;9:115–122. | |
Yorifuji T, Kawai M, Momoi T, et al. Nephropathy and growth hormone deficiency in a patient with mitochondrial tRNALeu(UUR) mutation. J Med Genet. 1996;33:621–622. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.