")
Back to Journals » Drug Design, Development and Therapy » Volume 14
Remogliflozin Etabonate in the Treatment of Type 2 Diabetes: Design, Development, and Place in Therapy
Authors Mohan V , Mithal A, Joshi SR, Aravind SR , Chowdhury S
Received 4 January 2020
Accepted for publication 15 May 2020
Published 24 June 2020 Volume 2020:14 Pages 2487—2501
DOI https://doi.org/10.2147/DDDT.S221093
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jianbo Sun
Viswanathan Mohan,1 Ambrish Mithal,2 Shashank R Joshi,3 SR Aravind,4 Subhankar Chowdhury5
1Madras Diabetes Research Foundation & Dr. Mohan’s Diabetes Specialties Centre, Chennai, Tamil Nadu, India; 2Endocrinology and Diabetology, Max Healthcare Hospital, Gurgaon, India; 3Joshi Clinic, Lilavati Hospital, Apollo Sugar Clinic and Bhatia Hospital, Mumbai, India; 4Diacon Hospital, Bengaluru, India; 5Deptartment of Endocrinology, IPGME&R and SSKM Hospital, Kolkata, India
Correspondence: Viswanathan Mohan Tel +91 44 4396 8888
Fax +91 44 2835 0935
Email [email protected]
Abstract: Type 2 diabetes mellitus (T2DM) is an emerging epidemic in Asian countries, especially in India. With the advent of the SGLT2 inhibitor class of drugs demonstrating benefits beyond glycemic control, viz. weight loss, blood pressure reduction, and cardiovascular and renal protection, the management of T2DM has taken a quantum leap. Remogliflozin etabonate (RE) is the latest addition to the SGLT2 inhibitor class of drugs that have been recently approved in India for the management of T2DM. RE is a potent and selective inhibitor of SGLT2 with the unique distinction of being administered as a prodrug, existence of active metabolites, and short half-life necessitating twice-daily dosing. The Phase III study of RE demonstrated it to be an efficacious and safe agent and non-inferior to the currently available SGLT2 inhibitors. This paper reviews not only the pharmacokinetics, pharmacodynamics, clinical efficacy, and safety profile of RE but also its molecular and clinical development program. This review has taken into consideration all available published as well as unpublished literature on RE and discusses the individual studies performed during its development for characterization of pharmacological profile.
Keywords: remogliflozin, etabonate, SGLT2 inhibitor, design, development, place in therapy, T2DM
Introduction
The incidence and prevalence of type 2 diabetes mellitus (T2DM) are increasing globally, with about one in 11 adults having diabetes mellitus and 90% of them having T2DM. Asia is emerging as an epidemic area for diabetes, with China and India being epicenters.1 In India today, 8.9% of the total population, equivalent to 77 million people, have diabetes.2 The American Association of Clinical Endocrinologists (AACE) provides guidelines for T2DM management, which include lifestyle therapy, medically assisted weight loss, and individual goals of achieving hemoglobin A1C (HbA1C) level of ≤6.5%. The patient characteristics, like glycemic index and weight, lifestyle, co-morbidities, and undesirable side effects of pharmaco-therapeutic management, determine the choice of anti-diabetic agents. The commonly associated side effects with oral anti-diabetic agents are hypoglycemia, weight gain due to hyperinsulinemia, gastrointestinal symptoms, and hepato-renal toxicity.3,4 The increase in adverse effects demands a safer anti-diabetic agent. The critical effects under consideration are the drug’s potential for hypoglycemia, weight gain, and long term side effects.5
Modulation of renal glucose handling by inhibition of the sodium-glucose co-transporter 2 (SGLT2) in renal tubules provided a novel insulin-independent mechanism of plasma glucose reduction without intolerable adverse effects in type 2 diabetes mellitus (T2DM) patients, paving the way for the development of SGLT2 inhibitor class of drugs for controlling hyperglycemia. Insulin independent action can benefit patients with insulin resistance.6 SGLT2 inhibitors provide another advantage in reducing weight by increasing glucose excretion in urine and other mechanisms.6 Moreover, this class of drugs was reported to provide additional cardiovascular (CV) benefits in various clinical trials along with glycemic benefits.7–10 Also, these CV benefits were reported in non-diabetic animal models,11 and new trials are investigating cardiac benefits in non-diabetic heart failure patients.12
The AACE guidelines recommended SGLT2 inhibitors as one of the first-line anti-diabetic agents.5 The American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD) suggest using SGLT2 inhibitors for patients with diabetic co-morbidities like cardiovascular disease (including heart failure, and atherosclerotic cardiovascular disease) and, chronic kidney disease.13
There are two types of sodium-glucose cotransporter: SGLT1, high affinity, low capacity glucose cotransporter, and SGLT2, low affinity, and high capacity glucose cotransporter that were characterized and which demonstrated the renal (proximal tubule) glucose reabsorption.3 Phlorizin, the first natural SGLT inhibitor, came into existence in the year 1835. Phlorizin inhibits both SGLT1 and SGLT2, with low selectivity. SGLT1 inhibition, however, caused several gastrointestinal side effects. Phlorizin has a hydrolytic metabolite, phloretin, which inhibits glucose transporter 1 (GLUT1), obstructing glucose uptake in tissues.3
To overcome these limitations, researchers focused on orally-available SGLT2 selective, O-glucoside congeners of phlorizin namely (T-1095, sergliflozin, remogliflozin, and AVE2268).3 This article aims to review the literature on published as well as unpublished literature of remogliflozin about its mechanism of action, efficacy, side effects, drug interaction, contraindications, and potential place in the treatment of T2DM.
Materials and Methods
A Pubmed and Cochrane Advance search was carried for “remogliflozin,” using keywords like “Remogliflozin,” “Remogliflozin Etabonate,” “Remogliflozin AND Diabetes,” and “Remogliflozin Etabonate AND Diabetes.” Papers and abstracts on animal studies and those in non-English language were excluded, including any duplicates. Any useful references cited in these papers/abstracts were also reviewed. Clinical trial registries were reviewed for information on past and ongoing studies. The approved Summary of Product Characteristics (SmPC), briefings, and press releases was also referred. The unpublished data was acquired from the marketing authorization holder of remogliflozin etabonate. The overall process of selection has been depicted in the flow chart (Figure 1). A total of 26 clinical studies (published and unpublished) were considered for this review depicted.
 |
Figure 1 Diagrammatic representation of methods. Overall process of followed for selection of evidence considered for review. |
Scientific Summary
Pharmacokinetic (PK)/Pharmacodynamic (PD) Profile
Remogliflozin is administered in prodrug form viz. remogliflozin etabonate (RE) (Figure 2A) in an immediate release (IR) tablet formulation. The recommended dosage in T2DM patients is 100mg administered twice daily. After administration, RE is de-esterified by non-specific esterases present in mucosal cells of the gastrointestinal tract to get converted into its active form remogliflozin (Figure 2B). 14 RE is rapidly and almost completely absorbed and available in the plasma within 10 minutes with a Tmax of 0.5–1 hour.6 The administration with standard breakfast slightly delayed the Tmax by approximately 0.5–1.5 hours (hrs); however, there was no significant difference in the Cmax or Area under Curve (AUC) relative to the fasting state. Hence RE can be administered with or without food.15
 |
Figure 2 Chemical structure of remogliflozin etabonate and remogliflozin. (A) Chemical structure of remogliflozin etabonate. (B) Chemical structure of remogliflozin. (C) Portions of structure of remogliflozin showing glycone, aglycone portions and O-glycosidic bond. The O-glycosidic bond makes the drug susceptible to beta-glucosidase enzyme. (D) Remogliflozin is esterified with etabonic acid to overcome metabolism by beta-glucosidase enzyme. |
The plasma protein binding of remogliflozin was around 65%. Either RE or remogliflozin was not preferentially distributed to blood cells, and there was no selective association of RE or its metabolites with melanin-containing tissues.16
In the systemic circulation, remogliflozin is extensively metabolized, leading to N-dealkylation, O-dealkylation, oxidation, loss of glucose, and glucuronidation. In vitro studies have demonstrated that the primary enzyme involved in the CYP-based metabolism of remogliflozin is CYP34A, with a minor contribution from CYP2C19.16
Remogliflozin gets metabolized to two active metabolites, namely: GSK279782 and GSK333081.17 The major active metabolite GSK279782 has been shown to account for approximately 16–22% of the concentration of remogliflozin in circulation. The exposure of GSK333081 was found to be extremely low after single-dose studies and hence not considered clinically significant. Remogliflozin has multiple pathways of elimination, which are CYP as well as non-CYP pathways. The mean plasma elimination half-life of remogliflozin and GSK 279782 were around 1.5 to 1.9 hrs and 2.3 to 3.8 hrs, respectively, in healthy volunteers after a single dose of RE at 100 mg or 250 mg. In the same study, the mean plasma half-life of prodrug was mostly around 0.4 hrs to 0.7 hrs. Metabolic products of RE are eliminated from the body through renal excretion.14 In radiolabelled absorption, metabolic, and excretion (AME) studies, approximately 93% was excreted in the urine, of which about 11% of the dose was recovered as remogliflozin in urine; the majority of drug-related material is eliminated via the urine as inactive glucuronide metabolites.14
The inhibitory concentration of remogliflozin was evaluated, and Ki values of 12.4 and 4520 nmol/l for SGLT2 and SGLT1, respectively, were demonstrated. This shows that remogliflozin is a selective inhibitor of SGLT2.18,19
A single dose, dose-escalation study in healthy human volunteers, and T2DM patients observed 24-hour urine glucose excretion (UGE) to be 17.5–40.5g and 66.6 to 112.6g, respectively, in a dose-dependent manner. The UGE showed a dose-dependent increase in total urine glucose excretion (UGE) from 0 to 24 hrs in fasted and fed conditions. However, UGE increased less proportionally with an increase in dose from 150 mg to 500 mg, indicating a plateau effect, as observed with drugs of this class. Urinary glucose excretion was higher in patients with T2DM than in volunteers because of higher plasma glucose concentrations in patients. On correcting the UGE according to circulating plasma glucose concentrations and creatinine clearance, to estimate the percentage filtered glucose load, it was found to be similar in both healthy individuals as well as T2DM patients.6
Clinically significant increase in UGE and urine volumes were observed in 12-week dose-ranging (50–1000 mg) study in drug naïve T2DM patients. A dose-ordered increase at 12 weeks from baseline was observed in UGE over 24 hrs ranging from 61 to 96 g/ day. A similar dose-ordered increase at 12 weeks in Urine volume was observed (~0.5L/day).17
The key PK and PD studies that assisted the characterization of clinical profiles have been discussed in subsequent sections.
Clinical Efficacy and Safety
In a Phase III trial, T2DM patients with uncontrolled hyperglycemia (HbA1c 7% to 10%) on metformin monotherapy were administered RE 100mg twice daily for a period of 24 weeks. These participant patients observed early and consistent reduction in HbA1c with a reduction of 0.72% at 24 weeks. The observed reduction in fasting and postprandial plasma glucose levels was 17.86 mg/dL and 39.2 mg/dL, respectively, at 24 weeks. The reduction in body weight observed at the end of 24 weeks was 2.7 kgs. The systolic and diastolic pressure was observed to be reduced by −2.6 mmHg and 2 mmHg, respectively).17,20 The overall incidence of treatment-emergent adverse events was 8.5% with a low incidence of hypoglycemia (1.3%), genital mycotic infections (1.7%), and urinary tract infections (3.1%). Therefore remogliflozin therapy of 100mg BID was found to be safe, tolerable, and efficacious.20
Current Status
Based on the Phase III study, RE 100 mg tablets twice daily have been approved by health regulatory authority in India for adequate glycemic control, together with diet and exercise, in adults aged 18 years or older with T2DM.20,21
Developmental History
Commercial History
Remogliflozin etabonate was initially discovered and developed by Kissei Pharmaceutical, Japan, and later licensed to GlaxoSmithKline (GSK) in 2002. GSK executed a robust clinical development program from 2002 to 2009 for the treatment of type-2 diabetes mellitus in many countries worldwide and obesity in the United Kingdom. Eighteen Phase I and two Phase II studies were performed characterizing the pharmacokinetics (PK), pharmacodynamics (PD), and clinical dosage regimens for RE. In 2009, GSK discontinued the development of remogliflozin as well as sergliflozin, an additional SGLT2 inhibitor licensed to GSK, due to commercial reasons.17
From 2011–2016 Brighthaven Ventures LLC (BHV) Pharma, a subsidiary of Avolynt, took the development of remogliflozin further and conducted three studies. BHV signed an agreement with India-based Glenmark Pharmaceuticals Limited to develop and market remogliflozin. In 2017, Glenmark conducted a Phase I PK study in the Indian population and initiated a Phase III study with a sample size of 906. In 2019, all participants had completed the clinical phase of the trial.17,19 The summary of the clinical studies conducted during the developmental program of RE has been depicted in Figure 3.
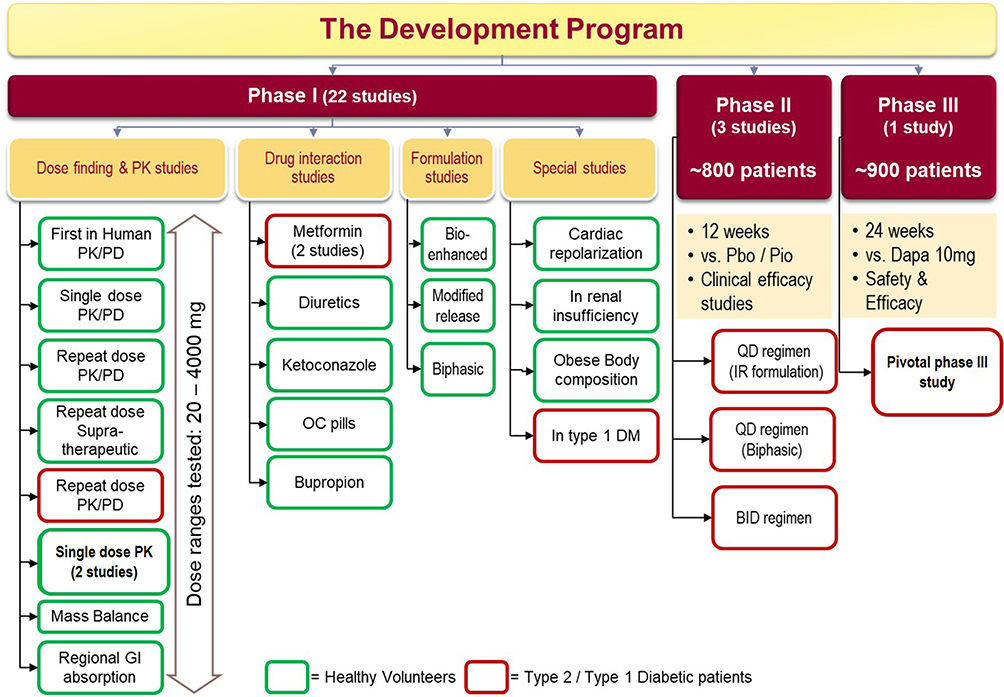 |
Figure 3 Clinical studies in development program of remogliflozin etabonate. Abbreviations: PK, pharmacokinetics; PD, pharmacodynamics; GI, gastrointestinal; OC, oral contraceptive pills; DM, diabetes mellitus; Pbo, placebo; Pio, pioglitazone; QD, once daily; IR, immediate release; BID, twice daily; Dapa, dapagliflozin. |
Molecular Developmental and Structural Activity Relationship
Remogliflozin provides high-affinity (Ki ~12.4 nmol/L), specific (SGLT1:SGLT2 = 1:365), competitive inhibition of SGLT2.19 For all SGLT2 inhibitors, phlorizin was the prototype molecule used for development. The remogliflozin molecule, like phlorizin, has aglycone portion and aglycone portion. Remogliflozin interacts with the SGLT2 protein due to the structural resemblance of the glycone portion to glucose. The bulky aglycone portion hinders SGLT2 activity and facilitates the inhibition. As these two portions are connected by O-glycosidic bond, remogliflozin is chemically classified as O-glucoside. The O-glycosidic bond is susceptible to enzymatic cleavage by β-glucosidases present in the gastrointestinal tract (GI tract) (Figure 2C).
Other currently available SGLT2 inhibitors in India are molecularly remodeled derivatives of phlorizin (an O-glucoside) obtained by the exclusion of an O-glycosidic bond to overcome this enzymatic cleavage. Hence the glycone-aglycone bridge in these agents is carbon to carbon bond and is referred to as C-glucosides.
Remogliflozin was esterified with etabonic acid to overcome the enzymatic cleavage, and administered in the form of a prodrug, namely remogliflozin etabonate.22 (Figure 2D)
Clinical Development Studies (Phase I, Phase II and Phase III Studies)
This section describes the key clinical studies conducted during the developmental program of remogliflozin.
Phase I Studies
Early phase I studies were designed to obtain PK, PD, safety, tolerability, and profile characterization.
This first-in-human study was conducted by Kapur et al, following single doses of RE, ranging from 20 to 1000 mg in healthy subjects (n=10) and 50 and 500 mg in subjects with T2DM (n=6).6 It was observed that RE gets rapidly absorbed and extensively converted to its active entity remogliflozin. Drug exposures were proportional to the dose administered, both in healthy volunteers and T2DM patients. The parent drug, RE, was present in plasma in low concentrations and was eliminated rapidly from the plasma with a mean t1/2 of about 15–40 minutes across dose levels. The active entity remogliflozin appeared in plasma within 10 minutes of dosing, at high concentrations (AUC: 81 to 105 times the AUC of RE across dose levels) with Cmax within 1.5 hours and was eliminated from plasma more slowly than RE with a mean t1/2 of 1.38 to 2.86 hours across dose levels. The active metabolite GSK279782 was also present in relatively high concentrations in plasma (AUC: 33 to 46 times the AUCs of RE across dose levels) with plasma profiles following similar time course to that of remogliflozin. Twenty-four-hour UGE showed a dose-dependent increase, but the increase was less than proportional with increasing exposure to remogliflozin. The largest effect was observed with a 500 mg dose in healthy subjects. The UGE was 17.5–40.5 g (Healthy volunteers) and 66.6 to 112.6 g (T2DM patients). In healthy subjects, AUC values for plasma glucose were very similar following placebo or RE administration. In subjects with T2DM, the increase in plasma glucose and plasma insulin levels following an oral glucose tolerance test (OGTT) was attenuated in a dose-dependent manner following RE dosing compared to placebo. The median baseline adjusted AUC (0–4) for plasma glucose, following an OGTT, was 11.00 mmol.h/L for placebo, 5.30 mmol.h/L for RE 50 mg and 2.65 mmol.h/L for RE 500 mg, whereas median baseline adjusted AUC (0–4) for plasma insulin was 36.29 μIU.h/mL for placebo, 24.67 μIU.h/mL for RE 50 mg and 18.82 μIU.h/mL for RE 500 mg.6
A Mass Balance Study with radio-labeled [14C]-RE was performed to investigate the metabolic disposition of a 400 mg single, oral dose in healthy male subjects. The study demonstrated that after oral administration, RE was rapidly and extensively absorbed (~93% of the dose), followed by rapid conversion to remogliflozin. Remogliflozin is further metabolized to GSK279782 and GSK333081. [14C]-RE-related material in blood had limited blood cell partitioning. The data from the study suggested that RE/remogliflozin was extensively metabolized, with other metabolites representing the majority (>90%) of the circulating plasma [14C]-radiocarbon, and hence eliminated rapidly from plasma with mean half-lives ranging from 0.39 to 2.84 hrs. Approximately 93% of the total radioactivity was excreted in the urine and 3% eliminated in the feces. Only about 11% of the dose was recovered in urine as the active moiety, remogliflozin.16 Similarly, a study was performed for characterizing the regional gastrointestinal absorption of RE in healthy volunteers by employing noninvasive remote-controlled InteliSite Companion Capsule for site-specific drug delivery in the GI tract. With various formulations tested, RE was found to be extensively absorbed and converted to remogliflozin primarily in the mid-small intestine, with minimal absorption and/or metabolic conversion in cecum/colon. Among all treatments, the 100 mg immediate release tablet of RE demonstrated the highest extent of bioavailability of remogliflozin in plasma, and the least inter-subject variability in the pharmacokinetics of remogliflozin and GSK279782.23
The single-dose PK in a fasted and fed state in healthy Indian volunteers showed a dose-proportional increase in Cmax, AUC, and similar half-lives at 100mg and 250mg,indicating linear pharmacokinetics. In general, under a fasted state, the maximum plasma concentrations were achieved rapidly for RE, remogliflozin, and GSK279782 with median Tmax ranging between 0.5 to 1.5 hours. With food, there was a slight delay in the Tmax (1.50 to 3.00 hours), but AUC and Cmax of RE, remogliflozin, and GSK279782 were generally comparable between fasted and fed states. The Cmax was similar in fed and fasted states (fed/fasted ratios 0.77–1.44 and 0.81–1.12) of the 100 and 250 mg doses, respectively. The AUC also demonstrated similar trends in fed and fasted conditions (fed/fasted ratio 1.22–1.35); however, RE and GSK279782 AUCs were slightly higher in the fed state. For both doses in the fed or fasted state, remogliflozin had the highest exposure, followed by GSK279782, and RE.15,17 The PK parameters, as observed in Indian patients, are depicted in Table 1.
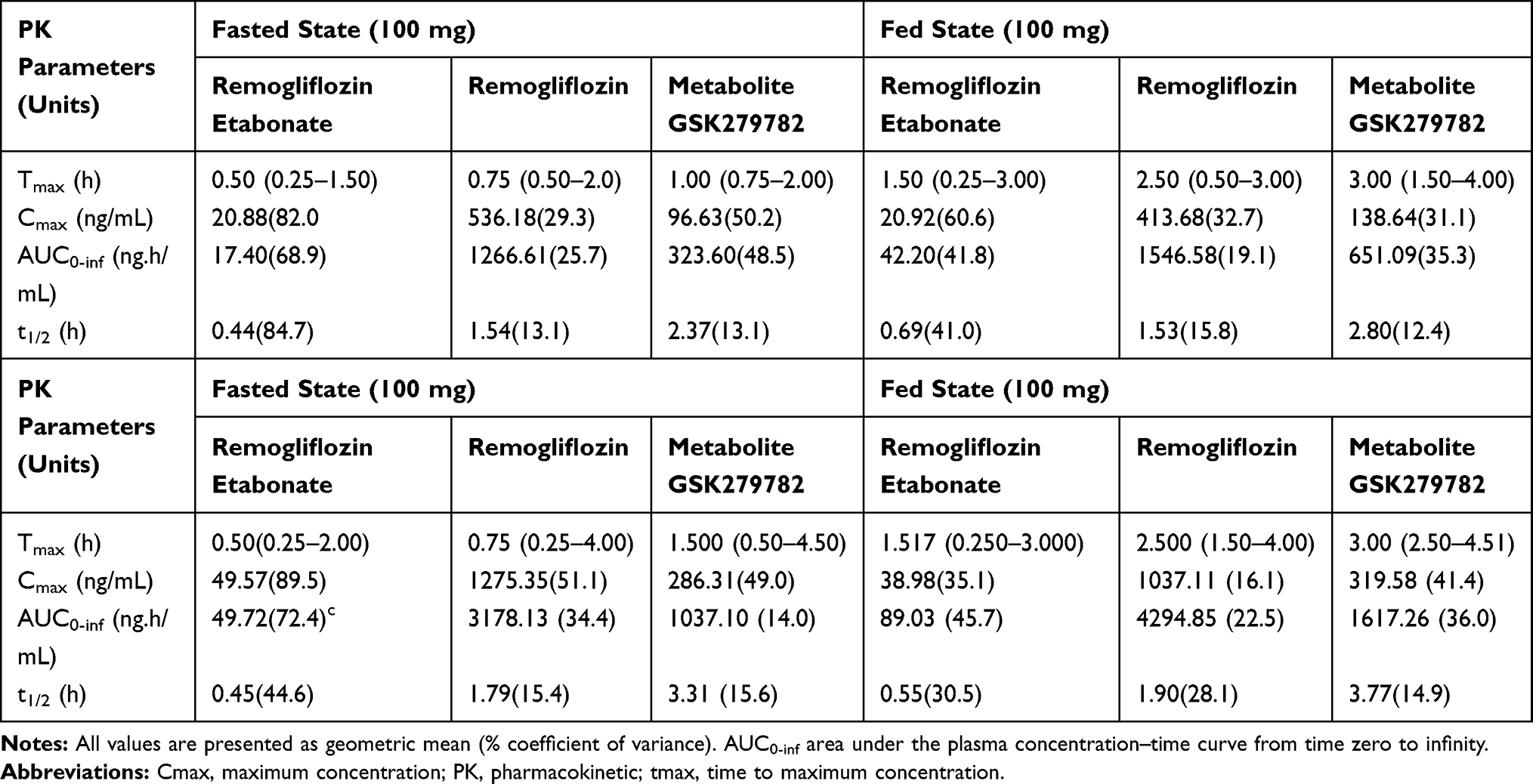 |
Table 1 Pharmacokinetic Profile of Single Dose of RE 100 mg and 250 mg in Indian Patients Under Fasting and Fed Conditions |
A single-dose PK Study of RE in healthy Japanese volunteers assessed the PK/PD in males as well as postmenopausal females. The effect of timing of meals (fasting condition or 15 minutes before meals) on the PK/PD parameter was evaluated in healthy males. Single RE dose of 100 mg or higher, in males or post-menopausal females (200 mg) under fasting conditions,suppressed the increase in postprandial plasma glucose and serum insulin levels after lunch compared to identical pre-dose lunch on the previous day. Likewise, a single RE dose of 200 mg, administered to healthy males 15 minutes before breakfast, suppressed abrupt increase in plasma glucose and serum insulin levels after breakfast, resulting in delayed peak time compared to identical pre-dose breakfast on the previous day. The 24-hour cumulative urinary glucose excretion increased in a relatively dose-dependent manner for the 50–1000 mg dose range. An increased UGE for 0–4 hours post-dose after pre-prandial administration as compared to fasting conditions and equivalent UGEs in the later period was demonstrated between the two modes of administration. All dose levels showed decreased AUC plasma glucose levels post-dose compared to pre-dose AUCs. The PK profile was found to be similar,as demonstrated in previous studies. No sex- or age-related difference was observed for UGE and PK parameters.24
The PK of repeat doses of RE was tested at three dose levels (100 mg BID, 1000mg BID, and 1000 mg QD) in subjects with T2DM. Pharmacokinetics of RE and its metabolites were similar to those following single-dose administration, with no apparent changes in the absorption or elimination characteristics across the three dosing regimens. No accumulation or time-dependent changes were observed, consistent with its short t1/2 estimates. The PD assessments showed a statistically significant decrease from baseline in pre-dose fasting plasma glucose concentration and AUC 0–24 weighted mean plasma glucose for BID dosing regimens of RE on Day 11 or Day 12. There was also a statistically significant decrease from baseline in pre-dose fasting plasma glucose (FPG) concentrations compared to placebo for the 100 mg and 1000 mg BID groups on Day 12 (–23.2 mg/dL and –41.2 mg/dL, respectively). An increase from baseline in the percentage of filtered glucose excreted in urine was observed for all active treatment groups relative to placebo. The magnitude of increase on Day 11 was greatest for the RE 1000 mg BID group (48%), followed by the 1000 mg QD (44%), and was slightly lower for the 100 mg BID group (37%).25
A multiple-dose study of safety, tolerability, and PK at supra-therapeutic doses of RE (2000 mg and 4000 mg QD) was conducted before formal evaluation of these doses on cardiac repolarization (QTc interval). The steady-state plasma exposure after 3-days administration of RE and its metabolites showed a dose-related increase in exposures (at least 2-fold higher than levels observed in previous studies and 4-fold higher than likely therapeutic dose); however, the increases were slightly less as compared to the dose-proportional increase. RE was found to be safe and well-tolerated after repeated supratherapeutic doses in healthy volunteers. The subsequent cardiac repolarization study (discussed later), high dose (500 mg QD), and supra-therapeutic dose (4000 mg) were evaluated, and PK was found to be similar to as observed in this study. Urine glucose excretion was consistent with saturation of the SGLT2 transporter at the doses administered in the study. The median amount of glucose excreted over zero (0) to 24 hours was 65.88 gm with RE 2000 mg and 71.8 gm with RE 4000 mg. The median percentage of filtered glucose excreted in the urine over 24 hours was 44% and 46%, respectively, following the RE 2000 mg and 4000 mg doses. There were no treatment-related trends compared to placebo in urine electrolyte excretion.26,27
Another repeat-dose study was conducted in healthy adult Japanese male subjects to evaluate the PK of RE on receiving, 200 mg or 600 mg of RE orally once or twice daily. It additionally evaluated the effect of timing relative to food. No significant effect of food or timing was observed for AUC, although Tmax was prolonged, and Cmax was decreased for all analytes when RE was administered 30 minutes after the start of the meal compared to when administered 15 minutes before a meal. The linearity index (LI) at Day 11 for RE, remogliflozin, and its active metabolite for 200 mg dose was1.53, 1.20, and 0.97, respectively, and for 600 mg dose was 0.97, 1.11 and 0.86 respectively. The results provided a linear PK profile, and there was no accumulation by repeat dose administration.28
All the Phase I studies reported RE to be well-tolerated at all dose levels tested without deaths, serious adverse events (SAEs), or clinically significant adverse events (AEs).6
Apart from these studies, two other PK/PD studies were performed evaluating varied bio-enhanced and modified-release formulations of RE (unpublished studies).
Two studies were conducted in subjects with T2DM to evaluate the combination of RE and metformin. One study23 was a randomized open-label, repeat dose study in which metformin and RE as co-administration were evaluated with 500 mg BID doses of each for three days, whereas another Two-Week Randomized Double-Blind, Repeat-Dose study with metformin ≥2000 mg was co-administered with RE at either a dose of 500 mg BID or 750 mg BID.29,30 There was no effect of RE on metformin PK parameters in either study. The 3-day study showed no effect of metformin on the PK parameters of RE and its metabolites, while a 2-week study demonstrated no accumulation of RE and no significant rise in serum lactic acid when RE was co-administered with metformin (≥2000 mg/day). The pharmacodynamics evaluation in terms of fasting glucose values, mean glucose and insulin following OGTT decreased in both RE treatment groups over the dosing period. The RE treatment groups observed more weight loss (1 kg) by Day 14 vs the placebo group. The percent of filtered glucose in the urine in subjects with T2DM observed with repeated doses of RE was not diminished by the co-administration of metformin.
A co-administration of RE with diuretics (furosemide/hydrochlorothiazide [HCTZ]) was evaluated for safety, tolerability, and PD effect on serum electrolytes. The 2-week study involved co-administration of RE (500 mg) BID with Furosemide (20 mg) QD or HCTZ (25 mg) QD. Except for small increases in serum magnesium, changes in other serum electrolytes were unremarkable. Serum sodium and potassium concentrations remained relatively stable in all treatment groups. The addition of RE to a diuretic increased the quantity of UGE (~72g/24 hours), increased urine osmolality and osmolar clearance,and brought about a decrease in free water clearance. The fluid balance showed an initial negative trend that resolved within six days of treatment with the combination of diuretic and RE. Decreased creatinine clearance (increase serum creatinine and urea) indicating possible volume depletion with the RE-diuretic combination. No meaningful differences from the placebo groups were observed in plasma renin activity (PRA) or aldosterone when RE was combined with diuretics. Modest reductions in systolic and diastolic BP (~3mmHg) and body weight (~1 kg) were observed in the RE treatment groups. Overall, the combined administration of RE with diuretics was generally well tolerated with potential safety issues of postural dizziness/orthostasis resulting from volume contraction.31,32
An open-label, drug–drug interaction study was conducted to investigate the effect of CYP3A4 inhibition (by six days repeat administration of Ketoconazole 400 mg OD) on the PK profile of a single dose of 250 mg RE in healthy adults. Though CYP inhibition by Ketoconazole caused only a modest change in plasma profiles of RE, measurable increases in remogliflozin (24%, 30%, and 33% increase in Cmax, AUC, and t1/2 estimates respectively) and reduced formation of oxidated active metabolites (including GSK279782) were observed. The PK results, therefore, show that Ketoconazole increases the remogliflozin AUC by 1.75-fold and prolongs the t½ by approximately 1 hour. The data also demonstrated that CYP3A4 is not the only important metabolic pathway for remogliflozin metabolism. Given the wide therapeutic index observed, no dose-adjustment would be clinically required when RE is administered with a CYP3A4 inhibitor.16
This drug–drug interaction study was conducted to investigate the effect of RE on the PK of bupropion. Concomitant administration of bupropion 150 mg with RE did not affect the plasma profiles and PK of RE, remogliflozin, and metabolites. The lack of PK interactions between RE and bupropion was expected based on the knowledge that RE and bupropion seem to be metabolized by multiple CYP and non-CYP pathways, show low inhibitory potential towards most CYP enzymes except for bupropion for CYP2D6, and have no known induction potential clinically. No treatment-related effects in PD endpoints were observed.33,34
An open-label, 21-day repeat-dose study investigated the effects of RE on PK of Oral contraceptive pills when co-administered to healthy female volunteers. The study observed low levels of ethinylestradiol and norethindrone, probably due to sporadic lack of absorption in women receiving oral contraceptive pills in combination with RE. As the effectiveness of the oral contraceptive may be negatively impacted, it is recommended that an appropriate alternative method for avoiding pregnancy should be utilized.35,36
As briefly discussed above, a placebo-controlled study evaluated the effect of 3-day repeat dose, high dose (500 mg OD) and supra-therapeutic dose (4000 mg) on cardiac repolarization (QTc interval) when compared to placebo and moxifloxacin single dose (400 mg) in healthy adults. Moxifloxacin was used as a positive control to determine the sensitivity to detect a mean difference in QTc. Since the moxifloxacin arm successfully revealed a positive QTc effect, the study was considered valid with respect to the active control. In contrast, there was no significant effect on cardiac repolarization for either of the RE treatment group.37,38
This study was an open-label, single-dose, and parallel-group study, conducted by O’Connor-Semmes et al14 that evaluated the effect of mild (estimated creatinine clearance (CLcr) of 50 to 80 mL/min) and moderate (estimated CLcr of 30 to 50 mL/min) renal impairment on PK and PD of RE. The mean renal clearance (CLr) of remogliflozin and GSK279782 was reduced by about 40% to 50% or 65% to 75% in subjects with mild to moderate renal impairment, respectively, and were highly correlated with CLcr. Mean plasma AUCs of active metabolites increased by 30% to 70%. Mild and moderate renal impairment did not result in statistically significant or clinically meaningful effects on the plasma exposure or plasma half-lives of RE and remogliflozin, consistent with the known extensive metabolism as the primary elimination pathway for these compounds. The amount of glucose excreted in the urine was lower than the respective matched groups. The results showed a decrease in urine glucose excretion to be positively correlated with renal clearance. However, the percent of filtered glucose excreted was similar between renal impairment (mild −21.9%; moderate-22.2%) and matched control groups. The PK parameters in renal impairment and matched groups are tabulated in Table 2. The PK, PD, and safety results suggested no dose adjustment of RE in mild or moderate renal impairment.14
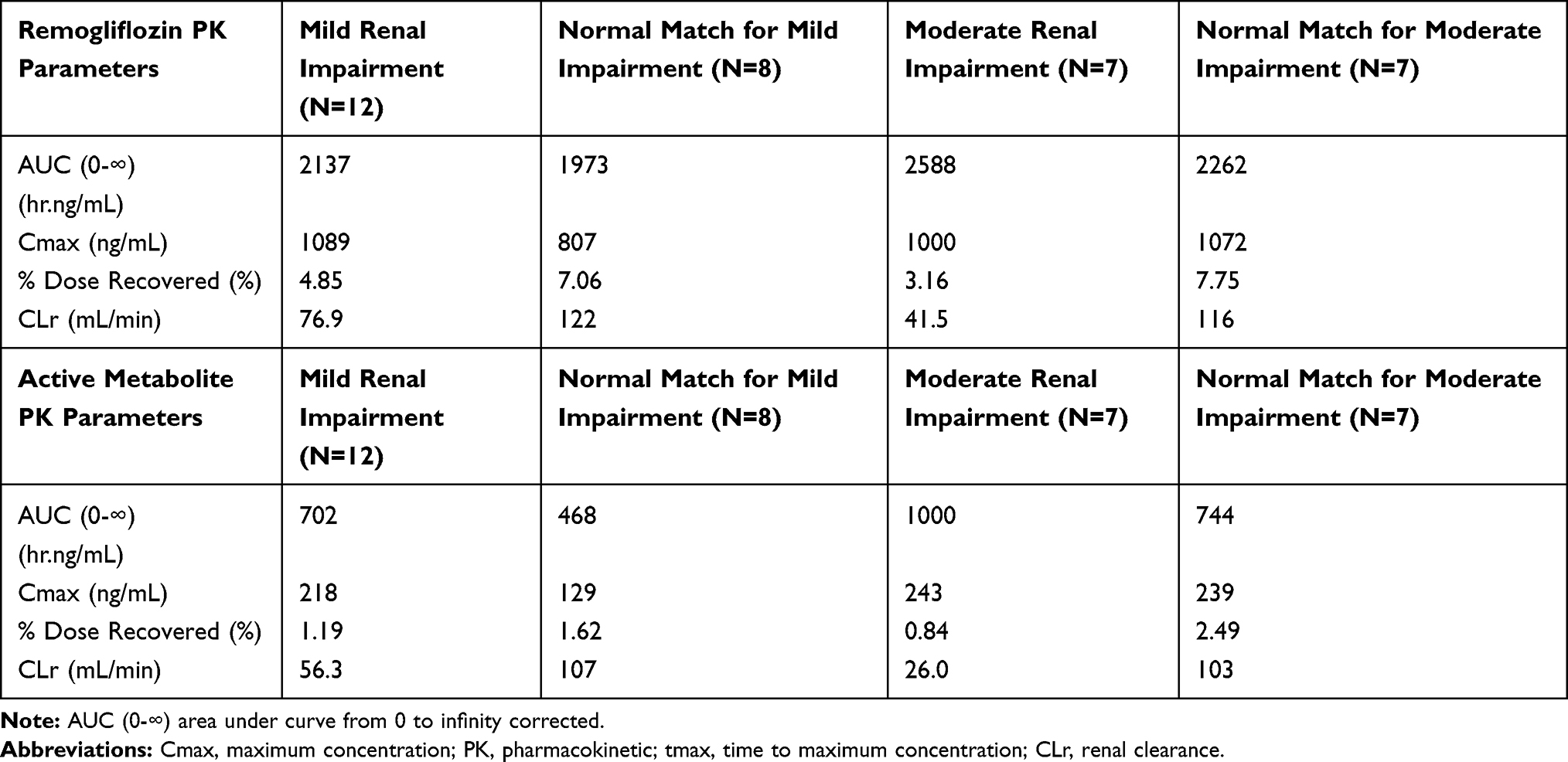 |
Table 2 Pharmacokinetic Parameters of Remogliflozin and Active Metabolite in Each Renal Functional Group |
An exploratory study of RE 250 mg TID administered for eight weeks in healthy obese subjects was carried out to evaluate the effect on body weight and body composition. With this dose regimen, the mean UGE was 72 g/24 hr (or 238 kcal/24h) at eight weeks. The treatment-related increase in calorie excretion was associated with a similar measurable treatment-related change in fat mass assessed by both QMR Echo-MRI (Quantitative magnetic resonance Echo-magnetic resonance imaging) and 4-compartment model modalities. Statistically significant (p<0.001) decreases from baseline values to Week 8 for body fat mass, and weight were observed (–4.8 kg and –7.6 kg, respectively for RE), but the difference from placebo did not achieve statistical significance in this small study. The decreases in fat mass were consistent with similar changes in clinical measures, including body mass index (BMI), and waist and hip circumference. The reported free water amount remained stable across the study period with GSK189075 treatment (mean of 0.60 kg at baseline and mean of 0.63 kg at Week 8).39
The exploratory study administered prandial insulin injections, or single oral doses of placebo, RE 50 mg, 150 mg and 500 mg administered in addition to basal insulin in 10 subjects with T1DM. With RE administration, the increase in plasma glucose concentrations following oral glucose (75 g) at breakfast, as well as a mixed meal at lunchtime, was blunted compared to placebo. No dose-response was apparent. The effect on mean glucose concentration was less with RE compared to bolus insulin. The AUC0-∞ and Cmax increased dose-proportionally from 50 mg to 500 mg for RE and the active metabolite. Glucose excreted in the urine was minimal with mealtime insulin injection+ placebo group (24.3 mg) and increased dose proportionally in RE groups (104.2 mg, 108.3 mg, and 138.6 mg for RE 50 mg, 150 mg, and 500 mg respectively) The administration of RE in T1DM patients and prandial/ basal insulin were generally well tolerated.40
Phase II Studies
The Phase IIa studies were designed to obtain safety, tolerability, and dose regimen to be utilized in pivotal studies in patients with T2DM. Three dose-ranging Phase II studies were conducted to evaluate the safety and efficacy of RE: once daily (QD) regimen of IR formulation, twice daily (BID) regimen of IR formulation and once daily (QD) regimen of Biphasic formulation in T2DM patients.
This was a multicenter, randomized, double-blind, placebo-controlled, parallel-group, dose-ranging study in 336 drug naïve type 2 diabetes mellitus patients to evaluate the efficacy, safety, and tolerability of a range of BID doses of RE (50, 100, 250, 500 and 1000 mg) vs 30 mg pioglitazone (PIO), compared with placebo, administered as monotherapy over 12 weeks. The primary efficacy endpoint was change in HbA1c at the end of study treatment (12 weeks) along with the secondary objectives to determine the change from baseline in fasting plasma glucose (FPG) and body weight at 12 weeks.
All RE doses produced a statistically and clinically significant decrease in HbA1c from baseline at 12 weeks (p<0.001) as well as vs placebo (p<0.001). The decrease in HbA1c was generally rapid, beginning as early as Week 4 and continuing through to Week 12.
Clinically and statistically significant dose-related decreases from baseline in FPG (15.6 to 37.3 mg/dL) were observed at Week 12 in all RE doses compared with placebo, with changes seen as early as Week 2. Similarly, 24-hour UGE increased from baseline in all RE treatment groups at Week 12, the mean increase ranging from 61.2 to 95.9 mg/24h.
There was a 1.36 to 3.51 kg of weight loss in the RE treatment groups. The most common reported AEs were headache, diarrhea, and constipation with ≥5% frequency. There was no increase in the occurrence of urinary tract infection (UTI) in RE compared with placebo and active group.41
This study was similar in design except for the evaluation of a range of QD doses of RE (100, 250, 500, and 1000 mg), compared with RE 250 mg BID, 30 mg pioglitazone (PIO) and placebo in 252 drug naïve T2DM patients. There was significant evidence of a trend in HbA1c reduction in the doses above 100 mg once daily, and significant reductions in HbA1c compared to placebo for the 250 mg once daily, 1000 mg once daily and 250 mg BID treatment groups. There were non-significant reductions in HbA1c for the 100 mg once daily, 500 mg once daily and pioglitazone groups. The decreases in HbA1c were rapid for the once-daily and BID dosing and were seen as early as Week 4 and continuing through to Week 12.
Clinically and statistically significant decreases from baseline in FPG were observed at Week 12 compared with placebo in all RE once-daily doses except for 500 mg once daily. Changes in FPG were seen as early as Week 2 and were maintained throughout till Week 12. Also, the 24-hour UGE level increased from baseline in all RE treatment groups (ranging from 19.44 to 69.8 mg/24h). There were no significant changes in lipid profile with RE administration. The reported AE of UTI was low in RE treatment groups.42
This was a Phase IIb, 12-week randomized, double-blind, parallel-group, placebo-controlled study to determine the efficacy and safety of biphasic RE when administered to subjects with type 2 diabetes mellitus. The primary objective of the study was to determine the efficacy of different doses of biphasic RE administered once daily compared to placebo based on the change from baseline in HbA1c at Week 12. The results observed that remogliflozin etabonate showed a significant decrease in placebo-adjusted HbA1c ranging from 0.44% to 0.6% (p < 0.02), a decrease in body weight and fasting glucose at all three doses tested. Additionally, there was a decrease in placebo-adjusted systolic (−2.4 to −5.6 mmHg) and diastolic (−1.7 to −3.7 mmHg) blood pressure. No significant safety or tolerability issues were identified with remogliflozin biphasic dose administration.43,44
Phase III Study
In Phase III, randomized, double-blind, double-dummy parallel-group, multicentre, active-controlled clinical trial, 906 subjects were enrolled. The efficacy and safety of RE 100 mg and RE 250 mg twice daily were compared with dapagliflozin 10 mg once daily. Of the 906 subjects, the first analysis of 612 subjects to test the non-inferiority of remogliflozin etabonate (any dose) vs Dapagliflozin was carried out.The 612 subjects were randomized in dapagliflozin 10 mg (n=146), RE 100 mg (n=225), and RE 250 mg (n=241) respectively. Treatment with RE 100 mg and RE 250 mg reduced HbA1c, fasting plasma glucose, and postprandial plasma glucose levels compared to control arm treatment at 24 weeks (Figure 4). No deaths were reported during the study period. The incidence of UTI was 6.6% in the RE 250 mg arm, 3.1% in the RE 100 mg arm, and 2.1% in the dapagliflozin 10 mg arm. The incidence of AEs was 34.4% in the RE 250 mg arm, 32.6% in RE 100 mg arm, and 29.5% in the dapagliflozin 10 mg arm.20
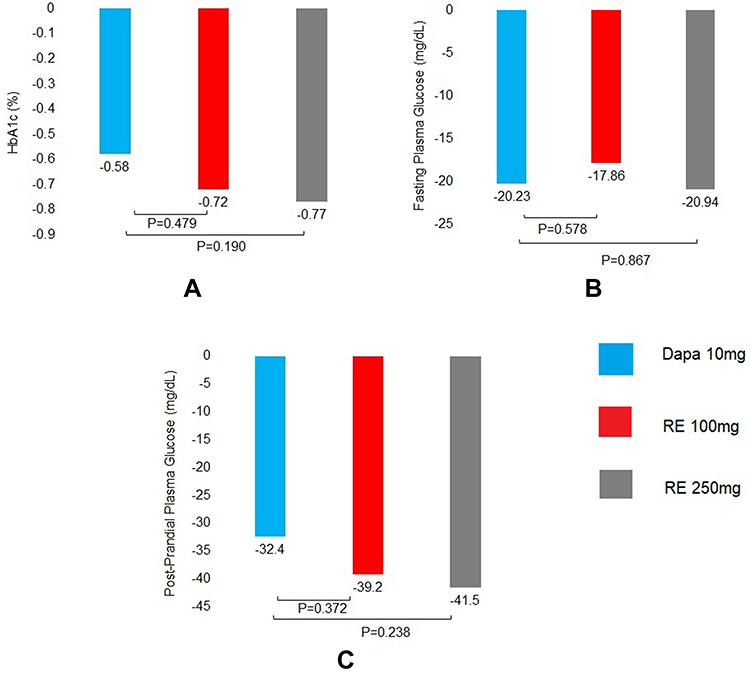 |
Figure 4 Mean change in HbA1c %, Fasting plasma glucose and post-prandial plasma glucose from baseline to week 24. (A) HbA1c at week 24. (B) Fasting blood glucose at week 24. (C) Post-prandial plasma glucose at week 24. Abbreviations: Dapa, dapagliflozin; RE, remogliflozin etabonate. |
The most frequent AEs reported were headache, genital fungal infections, hyperhidrosis, flatulence, dysuria, genital pruritus, nausea, toothache, dizziness, and blurred vision. All studies reported that RE was well tolerated as oral administration in healthy subjects and patients with T2DM.17,20,41,42
Discussion
Optimum glycemic control is a major challenge encountered while treating diabetic patients. Many times, it was noted that patients with Type II Diabetes Mellitus (T2DM) receiving intensive treatment fail to achieve the recommended glycated hemoglobin (HbA1c) targets. SGLT2 inhibitors can act effectively in individuals with long-standing diabetes with depleted β-cell reserve. This is due to their independent action, ie, mechanism non-dependent on insulin secretion and β-cell reserve.45 Among all treatment options available for T2DM, SGLT2 inhibition shows an increase in UGE, with corresponding reductions in plasma glucose and HbA1c levels, without causing hypoglycemia.
Remogliflozin is a recently launched candidate from the SGLT2 inhibitor class of drugs.17 The antidiabetic agent was shown to be effective, safe, and well-tolerated in a pivotal study.20 Remogliflozin demonstrated non-inferiority to existing SGLT2 inhibitor, namely dapagliflozin. The drug demonstrated comparable results in glycaemic, non-glycaemic, and safety parameters as compared to dapagliflozin.
The comparison of RE with the existing SGLT2 inhibitors showed the difference in the PK parameters. The noteworthy difference is a short half-life of the new molecule that necessitates a twice-daily dosing regimen.15,41 All other drugs in the same class have a longer half-life (around 12 hours).46–49 Hence, other approved SGLT2 inhibitors (empagliflozin, dapagliflozin, canagliflozin, ertugliflozin) are administered as once daily dose.50–53 The SGLT2 inhibitors are administered as active molecule except for RE; RE is a prodrug which gets converted to active metabolite in the GI tract and readily absorbed.14,17 However, like other drugs in the class, RE is also readily absorbed and attain peak plasma concentrations within 1–2 hrs.RE elimination shows high renal excretion on account of extensive metabolism by glucuronide conjugation of the drug as well as its metabolites. The PK/PD profile of existing SGLT2 inhibitors is presented with PK of remogliflozin. The comparative profile of PK of the currently approved SGLT2 inhibitors are presented in Table 3. 50–53
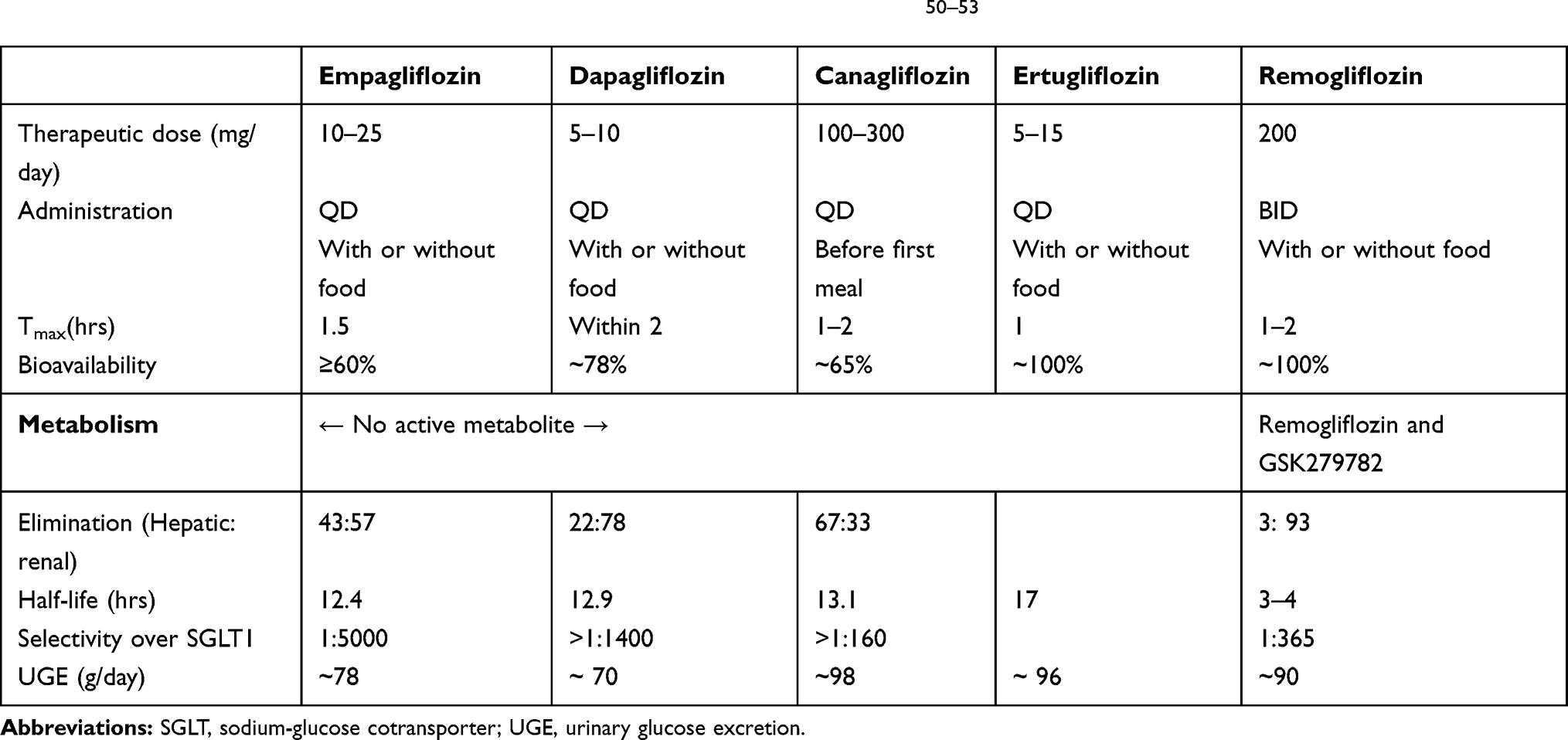 |
Table 3 Pharmacokinetic Properties of Currently Approved SGLT2 Inhibitors50–53 |
The PD in terms of UGE is observed to be similar across all available SGLT2 inhibitors compared to the approved regimen of RE in India (Table 3). Statistically significant and clinically meaningful reductions in HbA1C levels with RE 100 mg and 250 mg were reported within 24 weeks. Similarly, RE was observed to provide significant reductions in fasting and postprandial plasma glucose (FPG and PPG) levels in T2DM patients.20 The glycaemic control was similar to reduction observed in pivotal studies of other SGLT2 molecules in the class, ie, empagliflozin, dapagliflozin, and canagliflozin.54–56 Of note,Remogliflozin also elicited comparable pharmacodynamic profiles and effects to other molecules in the same class for extra glycaemic effects such as a reduction in blood pressure, and weight over a period of 24 weeks.7,20,54,55 Due to the twice-daily regimen of RE, it can be proposed that the drug would potentially offer the opportunity to truncate both postprandial peaks of Indian dietary patterns observed in T2DM patients viz. two large carbohydrate-rich meals daily. Moreover, the clinical benefits like a significant reduction in HbA1c and body weight are observed without any apparent increase in the incidence of hypoglycemia, as observed with SGLT2 inhibitor class of drugs.
Summary
In summary, data available from clinical trials of SGLT2 inhibitors reported the efficacy and safety of these drugs across a variety of patients with T2DM. Addition of SGLT2 inhibitors to the treatment algorithm, especially as an add-on therapy for achieving target HbA1c levels with an advantage of cardio-renal protection, bodyweight reduction, and reduced risk of hypoglycaemic events is a viable choice. Remogliflozin is candidate agent of SGLT2 inhibitor class of drugs that has been introduced in India after exhaustive clinical development. The pivotal study has showed it to provide clinically meaningful glycaemic control in T2DM patients along with non-glycaemic benefits as observed with other SGLT2 inhibitors. Apart from the control of FPG, RE showed a significant decrease in PPG levels of patients, which was considered to be a risk factor for microvascular and macrovascular complications. To conclude, remogliflozin, with its twice dosing regimen,appears to be a safe and effective therapeutic alternative for T2DM patients requiring SGLT2 inhibition for adequate glycaemic control.
Disclosure
Dr. Viswanathan Mohan and all other authors of this manuscript report personal fees as an Advisory Board Member of Glenmark Pharmaceuticals which markets Remogliflozin in India, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Zheng Y, Ley SH, Hu FB. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat Rev Endocrinol. 2017;14(2):88. doi:10.1038/nrendo.2017.151
2. International Diabetes Federation. IDF diabetes atlas 9th edition; 2019. Available from: https://www.diabetesatlas.org/en/resources/.
3. Choi CI. Sodium-glucose cotransporter 2 (SGLT2) inhibitors from natural products: discovery of next-generation antihyperglycemic agents. Molecules. 2016;21(9):1136. doi:10.3390/molecules21091136
4. Dey L, Attele AS, Yuan CS. Alternative therapies for type 2 diabetes. Altern Med Rev. 2002;7(1):45–58.
5. Tamez-Perez HE, Gonzalez-Guajardo EE, Tamez-Pena AL. Consensus statement by The American Association of Clinical Endocrinologists and American College of Endocrinology on the comprehensive type 2 diabetes management algorithm - 2019 executive summary. Endocr Pract. 2019;25(6):622.
6. Kapur A, O’Connor-Semmes R, Hussey EK, et al. First human dose-escalation study with remogliflozinetabonate, a selective inhibitor of the sodium-glucose transporter 2 (SGLT2), in healthy subjects and in subjects with type 2 diabetes mellitus. BMC Pharmacol Toxicol. 2013;14(1):26. doi:10.1186/2050-6511-14-26
7. Neal B, Perkovic V, Mahaffey KW, et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med. 2017;377(7):644–657. doi:10.1056/NEJMoa1611925
8. Zinman B, Wanner C, Lachin JM, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373(22):2117–2128. doi:10.1056/NEJMoa1504720
9. Wiviott SD, Raz I, Bonaca MP, et al. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2019;380(4):347–357. doi:10.1056/NEJMoa1812389
10. Zelniker TA, Wiviott SD, Raz I, et al. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials. Lancet. 2019;393(10166):31–39. doi:10.1016/S0140-6736(18)32590-X
11. Santos-Gallego CG, Requena-Ibanez JA, San Antonio R, et al. Empagliflozin ameliorates adverse left ventricular remodeling in nondiabetic heart failure by enhancing myocardial energetics. J Am Coll Cardiol. 2019;73(15):1931–1944. doi:10.1016/j.jacc.2019.01.056
12. Santos-Gallego CG, Garcia-Ropero A, Mancini D, et al. Rationale and design of the EMPA-TROPISM trial (ATRU-4): are the “cardiac benefits” of empagliflozin independent of its hypoglycemic activity? Cardiovasc Drugs Ther. 2019;33(1):87–95. doi:10.1007/s10557-018-06850-0
13. Davies MJ, D’Alessio DA, Fradkin J, et al. Management of hyperglycaemia in type 2 diabetes, 2018. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia. 2018;61(12):2461–2498. doi:10.1007/s00125-018-4729-5
14. O’Connor-Semmes R, Walker S, Kapur A, et al. Pharmacokinetics and pharmacodynamics of the SGLT2 inhibitor remogliflozinetabonate in subjects with mild and moderate renal impairment. Drug Metab Dispos. 2015;43(7):1077–1083. doi:10.1124/dmd.114.062828
15. Joshi S, Gudi G, Menon VC, et al. An open-label, single-period, two-stage, single oral dose pharmacokinetic study of RemogliflozinEtabonate tablet 100 and 250 mg in healthy asian indian male subjects under fasting and fed conditions. Clin Pharmacokinet. 2020;59(3):349–357. doi:10.1007/s40262-019-00819-4
16. Sigafoos JF, Bowers GD, Castellino S, et al. Assessment of the drug interaction risk for remogliflozinetabonate, a sodium-dependent glucose cotransporter-2 inhibitor: evidence from in vitro, human mass balance, and ketoconazole interaction studies. Drug Metab Dispos. 2012;40(11):2090–2101. doi:10.1124/dmd.112.047258
17. Markham A. RemogliflozinEtabonate: first global approval. Drugs. 2019;79(10):1157–1161. doi:10.1007/s40265-019-01150-9
18. Rosenstock J, Jelaska A, Frappin G, et al. Improved glucose control with weight loss, lower insulin doses, and no increased hypoglycemia with empagliflozin added to titrated multiple daily injections of insulin in obese inadequately controlled type 2 diabetes. Diabetes Care. 2014;37(7):1815–1823. doi:10.2337/dc13-3055
19. Fujimori Y, Katsuno K, Nakashima I, Ishikawa-Takemura Y, Fujikura H, Isaji M. Remogliflozinetabonate, in a novel category of selective low-affinity sodium glucose cotransporter (SGLT2) inhibitors, exhibits antidiabetic efficacy in rodent models. J Pharmacol Exp Ther. 2008;327(1):268–276. doi:10.1124/jpet.108.140210
20. Dharmalingam M, Aravind SR, Thacker H, et al. Efficacy and safety of RemogliflozinEtabonate, a new sodium glucose co-transporter-2 inhibitor, in patients with type 2 diabetes mellitus: a 24-week, randomized, double-blind, active-controlled trial. Drugs. 2020;11:1–4.
21. Central Drugs Standard Control Organization (CDSCO). List of new drugs approved in the year; 2019 till date. Available from: https://cdsco.gov.in/opencms/opencms/system/modules/CDSCO.WEB/elements/download_file_division.jsp?num_id=NDY2Nw==.
22. National Center for Biotechnology Information. PubChem database. Remogliflozinetabonate, CID=9871420. Available from: https://pubchem.ncbi.nlm.nih.gov/compound/Remogliflozin-etabonate.
23. O’Connor‐Semmes RL, Sandefer EP, Hussey EK, et al. Regional gastrointestinal delivery of remogliflozinetabonate in humans. Biopharm Drug Dispos. 2013;34(2):79–86. doi:10.1002/bdd.1824
24. GlaxoSmithKline. A phase I single-dose PK study in healthy subjects and postmenopausal females. Clinical study report (KGT-1101). 2006. (unpublished report)
25. Dobbins RL, O’Connor‐Semmes R, Kapur A, et al. Remogliflozinetabonate, a selective inhibitor of the sodium‐dependent transporter 2 reduces serum glucose in type 2 diabetes mellitus patients. Diabetes Obes Metab. 2012;14(1):15–22. doi:10.1111/j.1463-1326.2011.01462.x
26. GlaxoSmithKline. A randomized, placebo-controlled, double-blind, repeat dose, dose escalation trial to investigate the safety, tolerability and pharmacokinetics of supratherapeutic doses of GSK189075 administered for 3 days in healthy volunteers. clinical study report (KG2109799). 2007. (unpublished report)
27. GlaxoSmithKline. A study to evaluate and study drug levels in blood plasma when giving high doses of GSK189075 to healthy volunteers; 2007. Available from: https://clinicaltrials.gov/ct2/show/NCT00501930. NLM Identifier-NCT00501930.
28. GlaxoSmithKline.A phase I single-dose PK study in healthy subjects and postmenopausal females. Clinical study report (KGT-1102). 2007. (unpublished report)
29. GlaxoSmithKline. PK of concomitant administration of GSK189075 with metformin in subjects with type 2 diabetes mellitus. Clinical study report (KG2110243). 2008. (unpublished report)
30. GlaxoSmithKline. A study to assess the safety and tolerability of GSK189075 when given with a total daily dose of>/ 2000mg of metformin; 2007. Available from:https://clinicaltrials.gov/ct2/show/NCT00519480. NLM Identifier- NCT00519480.
31. GlaxoSmithKline. A study to compare effects of GSK189075 in combination with either furosemide or hydrochlorothiazide; 2008. Available from: https://clinicaltrials.gov/ct2/show/NCT00671424. NLM Identifier- NCT00671424.
32. GlaxoSmithKline. An evaluation of the safety, tolerability, and pharmacodynamics effects of GSK189075 when administered with furosemide or hydrochlorothiazide. Clinical study report (KG2105251). 2008. (unpublished report)
33. GlaxoSmithKline.A study to assess the safety of repeated doses of GSK189075 and WELLBUTRIN SR in healthy male subjects; 2008. Available from: https://clinicaltrials.gov/ct2/show/NCT00625859. NLM Identifier- NCT00625859.
34. GlaxoSmithKline. A double-blind, randomized, 6-sequence, 3-period, crossover drug-drug interaction study to evaluate the pharmacokinetics of WELLBUTRIN SR and GSK189075, when co-administered or administered alone in healthy male volunteers. Clinical study report (KGW111083). 2008. (unpublished report)
35. Remogliflozinetabonate [package insert]. Mumbai (India): Glenmark Pharmaceuticals Ltd; April 2019.
36. GlaxoSmithKline. A study of the effects of GSK189075 on PK profiles of an oral contraceptive pill when given to healthy female volunteers; 2009. Available from: https://clinicaltrials.gov/ct2/show/NCT00504816. NLM Identifier- NCT00504816.
37. GlaxoSmithKline. A randomized, double-blind, placebo-controlled, cross-over trial to investigate the effect of GSK189075 on cardiac repolarization as compared to placebo and a single dose of moxifloxacin in healthy adult subjects. Clinical study report (KG2107489). 2008. (unpublished report)
38. GlaxoSmithKline. A study to compare the effect on heart rhythm of 3 days of GSK189075, placebo, or moxifloxacin in healthy adults; 2007. Available from: https://clinicaltrials.gov/ct2/show/NCT00532610. NLM Identifier- NCT00532610.
39. Napolitano A, Miller S, Murgatroyd PR, et al. Exploring glycosuria as a mechanism for weight and fat mass reduction. A pilot study with remogliflozinetabonate and sergliflozinetabonate in healthy obese subjects. J Clin Transl Endocrinol. 2014;1(1):e3–8. doi:10.1016/j.jcte.2013.12.001
40. Mudaliar S, Armstrong DA, Mavian AA, et al. Remogliflozinetabonate, a selective inhibitor of the sodium-glucose transporter 2, improves serum glucose profiles in type 1 diabetes. Diabetes Care. 2012;35(11):2198–2200. doi:10.2337/dc12-0508
41. Sykes AP, O’Connor‐Semmes R, Dobbins R, et al. Randomized trial showing efficacy and safety of twice‐daily remogliflozinetabonate for the treatment of type 2 diabetes. Diabetes Obes Metab. 2015;17(1):94–97. doi:10.1111/dom.12391
42. Sykes AP, Kemp GL, Dobbins R, et al. Randomized efficacy and safety trial of once‐daily remogliflozinetabonate for the treatment of type 2 diabetes. Diabetes Obes Metab. 2015;17(1):98–101. doi:10.1111/dom.12393
43. BHV Pharma. Safety and efficacy of biphasic RemogliflozinEtabonate in the treatment of type 2 diabetes (BRID); 2015. Available from: https://clinicaltrials.gov/ct2/show/NCT02537470. NLM Identifier- NCT02537470.
44. Accesswire. Avolynt, Inc. announces positive topline data for phase 2b BRID study of SGLT2 remogliflozin [internet] North Carolina; Dec 2016 [
45. Singh AK, Singh R. Sodium-glucose co-transporter-2 inhibitors as add-on therapy to insulin: rationale and evidences. Expert Rev Clin Pharmacol. 2016;9(3):409–418. doi:10.1586/17512433.2016.1131121
46. Scheen AJ. Pharmacokinetic and pharmacodynamic profile of empagliflozin, a sodium glucose co-transporter 2 inhibitor. Clin Pharmacokinet. 2014;53(3):213–225. doi:10.1007/s40262-013-0126-x
47. Kasichayanula S, Liu X, LaCreta F, Griffen SC, Boulton DW. Clinical pharmacokinetics and pharmacodynamics of dapagliflozin, a selective inhibitor of sodium-glucose co-transporter type 2. Clin Pharmacokinet. 2014;53(1):17–27. doi:10.1007/s40262-013-0104-3
48. Devineni D, Curtin CR, Polidori D, et al. Pharmacokinetics and pharmacodynamics of canagliflozin, a sodium glucose co‐transporter 2 inhibitor, in subjects with type 2 diabetes mellitus. J Clin Pharmacol. 2013;53(6):601–610. doi:10.1002/jcph.88
49. Miao Z, Nucci G, Amin N, et al. Pharmacokinetics, metabolism, and excretion of the antidiabetic agent ertugliflozin (PF-04971729) in healthy male subjects. Drug Metab Dispos. 2013;41(2):445–456. doi:10.1124/dmd.112.049551
50. JARDIANCE® (empagliflozin) [package insert on the Internet]. U.S. Food and Drug Administration: BoehringerIngelheim Pharmaceuticals; January 2020. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/204629s023lbl.pdf.
51. INVOKANA (canagliflozin) [package insert on the Internet]. U.S. Food and Drug Administration: Janssen Pharmaceuticals; January 2020.Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/204042s036lbl.pdf.
52. FARXIGA® (dapagliflozin) [package insert on the Internet]. U.S. Food and Drug Administration: AstraZeneca Pharmaceuticals LP; January 2020. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/202293s022lbl.pdf.
53. STEGLATRO™ (ertugliflozin) [package insert on the Internet]. U.S. Food and Drug Administration: Merck & Co.; January 2020. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/209803s002lbl.pdf.
54. Fala L. Jardiance (empagliflozin), an SGLT2 inhibitor, receives FDA approval for the treatment of patients with type 2 diabetes. Am Health Drug Benefits. 2015;8(Spec Feature):92.
55. Ferrannini E, Ramos SJ, Salsali A, Tang W, List JF. Dapagliflozin monotherapy in type 2 diabetic patients with inadequate glycemic control by diet and exercise: a randomized, double-blind, placebo-controlled, phase 3 trial. Diabetes Care. 2010;33(10):2217–2224. doi:10.2337/dc10-0612
56. Stenlöf K, Cefalu WT, Kim KA, et al. Efficacy and safety of canagliflozin monotherapy in subjects with type 2 diabetes mellitus inadequately controlled with diet and exercise. Diabetes Obes Metab. 2013;15(4):372–382. doi:10.1111/dom.12054
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.