")
Back to Journals » Biologics: Targets and Therapy » Volume 13
Remicade® (infliximab): 20 years of contributions to science and medicine
Authors Melsheimer R , Geldhof A , Apaolaza I, Schaible T
Received 1 March 2019
Accepted for publication 16 May 2019
Published 30 July 2019 Volume 2019:13 Pages 139—178
DOI https://doi.org/10.2147/BTT.S207246
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Doris Benbrook
Richard Melsheimer,1 Anja Geldhof,1 Isabel Apaolaza,1 Thomas Schaible2
1Medical Affairs, Janssen Biologics BV, Leiden, the Netherlands; 2Medical Affairs, Janssen Pharmaceuticals, Horsham, PA, USA
Abstract: On August 24, 1998, Remicade® (infliximab), the first tumor necrosis factor-α (TNF) inhibitor, received its initial marketing approval from the US Food and Drug Administration for the treatment of Crohn’s disease. Subsequently, Remicade was approved in another five adult and two pediatric indications both in the USA and across the globe. In the 20 years since this first approval, Remicade has made several important contributions to the advancement of science and medicine: 1) clinical trials with Remicade established the proof of concept that targeted therapy can be effective in immune-mediated inflammatory diseases; 2) as the first monoclonal antibody approved for use in a chronic condition, Remicade helped in identifying methods of administering large, foreign proteins repeatedly while limiting the body’s immune response to them; 3) the need to establish Remicade’s safety profile required developing new methods and setting new standards for postmarketing safety studies, specifically in the real-world setting, in terms of approach, size, and duration of follow-up; 4) the study of Remicade has improved our understanding of TNF’s role in the immune system, as well as our understanding of the pathophysiology of a range of diseases characterized by chronic inflammation; and 5) Remicade and other TNF inhibitors have transformed treatment practices in these chronic inflammatory diseases: remission has become a realistic goal of therapy and long-term disability resulting from structural damage can be prevented. This paper reviews how, over the course of its development and 20 years of use in clinical practice, Remicade was able to make these contributions.
Keywords: Remicade, infliximab, monoclonal antibody, immune-mediated inflammatory disease, TNF inhibition, Crohn’s disease, rheumatoid arthritis
Plain language summary
Prompted by the recent twentieth anniversary of the first approval of Remicade® (infliximab; Janssen Biotech, Inc., Horsham, PA, USA), a first-in-class monoclonal antibody tumor necrosis factor α (TNF) inhibitor, the authors have written this review in order to recognize the drug’s contributions to science and medicine. Remicade’s first therapeutic indication, Crohn’s disease, was followed by another five indications, all of which are immune-mediated inflammatory diseases (IMIDs). A common factor in these diseases is increased expression of the cytokine TNF, which drives the underlying inflammation causing them. Through inhibition of TNF, this chronic inflammation can be suppressed and the disease successfully treated. In the course of Remicade’s development and its use in clinical practice, several important firsts were achieved. Remicade established the proof of concept that targeted therapy (ie, blockade of a single inflammatory mediator) can be a successful treatment approach for IMIDs. It demonstrated that there is a role for monoclonal antibodies in the treatment of chronic diseases. The need to establish its safety profile led to the development of new methods and standards for postmarketing commitment studies. Studies in current indications, in indications where it was not efficacious, and of its safety profile have taught us much about the immune system and greatly improved our understanding of the pathophysiology of several IMIDs. Lastly, the availability of Remicade and other TNF inhibitors has transformed the practice of medicine in these diseases, where more ambitious goals of therapy are now possible. Each of these advancements has helped to bring about a revolution in medicine that is still ongoing today.
Introduction
On August 24, 1998, the monoclonal antibody (mAb) Remicade® (infliximab; Janssen Biotech, Horsham, PA, USA) received approval from the US Food and Drug Administration (FDA) for the treatment of Crohn’s disease (CD), thereby becoming the first tumor necrosis factor-α (TNF) inhibitor available for use in clinical practice. In the ensuing years, this initial indication was followed by approval in another five adult and two pediatric chronic inflammatory conditions both in the USA1 and around the globe (Figure 1). Beyond offering a welcome new treatment option for patients, it has contributed to several important advances in science and medicine, which will be the focus of this review.
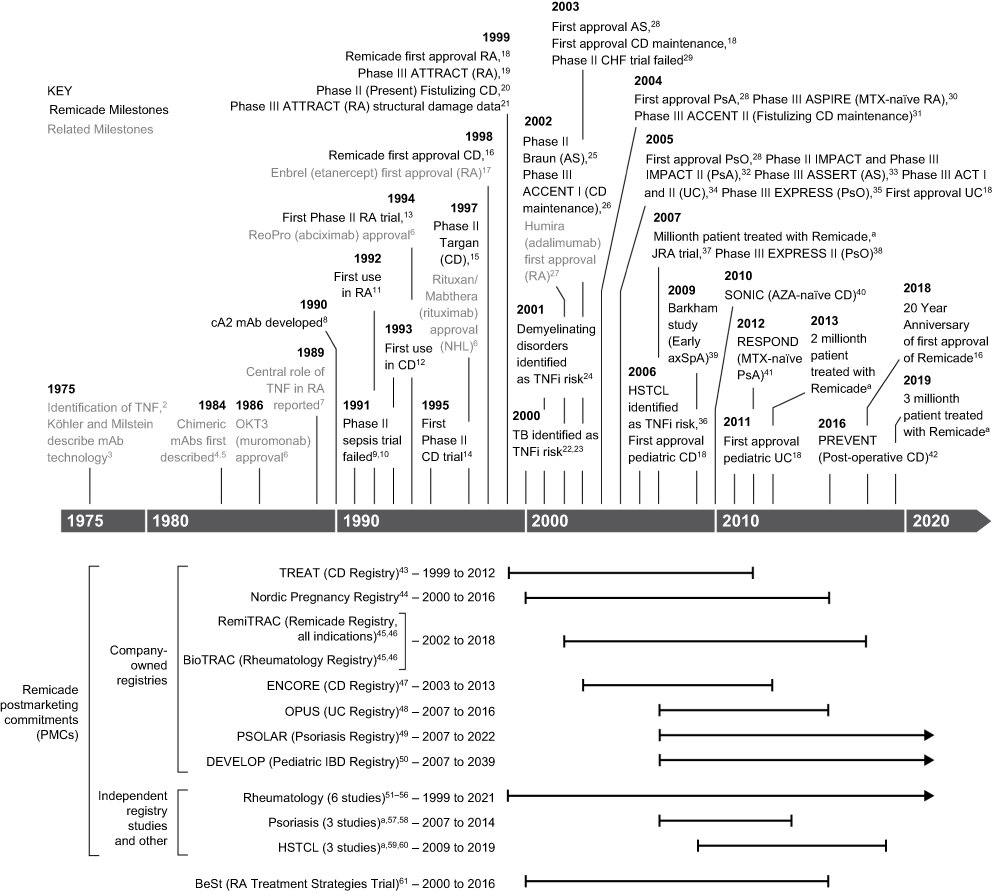 |
Figure 1 Key milestones in the development of Remicade.Note: aJanssen, data on file. Abbreviations: AS, ankylosing spondylitis; axSpA, axial spondyloarthritis; CD, Crohn’s disease; CHF, congestive heart failure; HSTCL, hepatosplenic T-cell lymphoma; IBD, inflammatory bowel disease; JRA, juvenile rheumatoid arthritis; MTX, methotrexate; mAb, monoclonal antibody; NHL, non-Hodgkin’s lymphoma; PBO, placebo; PsA, psoriatic arthritis; PsO, psoriasis; RA, rheumatoid arthritis; TB, tuberculosis; TNF, tumor necrosis factor; TNFi, TNF inhibition; UC, ulcerative colitis. |
Remicade’s approval represented the culmination of two independent sets of research that began in 1975 when both TNF and hybridoma technology, the method for producing monoclonal antibodies, were first described in the literature.2,3 Subsequent research on TNF identified its role not only in host response, but also in the pathophysiology of a number of immune-mediated inflammatory diseases (IMIDs), such as CD and rheumatoid arthritis (RA). This observation led to the question of whether blockade of TNF could serve as a treatment for these diseases. At the time, conventional treatments for IMIDs, such as immunosuppressants and corticosteroids, treated symptoms of disease, but not the underlying pathophysiology, and were associated with both limited efficacy and side effects.
However, the feasibility of targeted therapy in this setting was unknown. In a complex network of inflammatory mediators with pleiotropic, sometimes overlapping functions, could inhibiting a single cytokine such as TNF suppress inflammation in a clinically meaningful way? How could such a cytokine be inhibited? Because of their high ligand specificity and affinity, mAbs were obvious candidates, but had not met expectations as therapeutics. Could they achieve their potential? These questions were first answered in 1992 when Remicade, then known simply as cA2, was used to confirm the hypothesis that the inflammation driving RA is mediated by TNF and can be suppressed by its blockade.11
The development of Remicade up to that point and since then has been a classic example of how an improvement in understanding of pathophysiology can lead to a therapeutic breakthrough, which in turn leads to a deeper understanding of pathophysiology. Each new learning led to another question to be answered, which has resulted in the development of a significant body of scientific and medical research: a literature search for the word “infliximab” today reveals more than 13,000 publications, a number that approaches 50,000 when including other TNF inhibitors such as etanercept (Enbrel®; Immunex Corporation, Thousand Oaks, CA, USA) and adalimumab (Humira®; AbbVie, North Chicago, IL, USA). Many of the learnings are now common knowledge, but in 1992 were hypotheses. It is only with hindsight that we can put them into perspective (Figure 1).
This article reviews these learnings as they developed over time: the discovery of TNF; Remicade’s clinical development, primarily in RA and CD, which are its earliest and most prominent indications; its evaluation in other diseases; the establishment of its safety profile; and its role in changing clinical practice. It is important to note that the participants in this scientific journey were not only Janssen and its commercial partners and local distributors (Merck, Sharp and Dohme [MSD] in Europe, Turkey, and Russia, and Mitsubishi Tanabe Pharmaceutical Corporation in Japan, Taiwan, and Indonesia), but also a large number of independent researchers. Their collective work demonstrates that the learnings from a drug do not stop at the end of its formal development, but can continue for years afterward.
With the recent 20-year anniversary of Remicade’s first approval in 1998, it is fitting to reflect on its contributions to science and medicine.
TNF is a key driver of inflammation
In 1975, Carswell et al described an experiment in which tumor regression was observed in mice injected with endotoxin from the pathogenic bacterium Serratia marcescens. They isolated a substance in the serum of these mice that led to this regression and named it “tumor necrosis factor” (TNF).2 In 1985, Beutler et al studied a factor which caused cachexia, a wasting syndrome, by acting on lipoprotein lipase and other metabolic pathways.62 This factor, which they called “cachectin”, was later found to be TNF. Simultaneously, Dayer et al, while searching for a factor that mediated shock, isolated a substance from cells of monocytic lineage which was also found to be TNF.63
These three independent discoveries of TNF in separate fields of research display vividly the central and complex role that it plays in the immune system. It is a key driver and regulator of the body’s inflammatory response64 and is involved in immune surveillance and homeostasis.65,66
TNF is not usually detectable in healthy populations, but is increased in both serum and tissue under inflammatory and infectious conditions, and after tissue injury.67 It is one of the first cytokines to appear in the blood after injury or stress and does so within minutes,65 secreted primarily by macrophages and monocytes, but also by other immune cells, eg, neutrophils, T cells, and natural killer (NK) cells, as well as non-immune cells.67 The concentration of TNF in serum correlates with the severity of infection. Other pro-inflammatory cytokines, such as interleukin (IL)-6 and IL-1, appear later and are at least in part dependent on prior release of TNF.65 The 24-kDa membrane-bound form of TNF (tmTNF) is cleaved by a metalloproteinase enzyme, TNF-α-converting enzyme (TACE), to release a 17-kDa soluble form (sTNF). Both forms are biologically active.67 They mediate their effects through binding to either of two receptors, TNFR1 (p55) or TNFR2 (p75). TNFR1 is expressed on most cell types and is generally activated by sTNF, while TNFR2 is expressed primarily on immune, specifically T-regulatory, and endothelial cells and is preferentially activated by tmTNF.65,68,69 TNFR1 seems to be primarily involved in the inflammatory response and mediating apoptosis while TNFR2 appears to be important for tissue repair, immune modulation, and homeostasis.65,69
TNF exerts its pleiotropic effects via a number of mechanisms, such as macrophage activation, differentiation and phagosome formation,64 activation of neutrophils and NK cells, and promotion of cell adhesion, apoptosis, and cellular proliferation.70 These mechanisms contribute to the body’s common manifestations of inflammation, eg, fever, vasodilation/edema, sleep disruption, regulation of coagulation and tissue degeneration, as well as to the manifestations of downregulation of the inflammatory response, eg, promotion of immune modulation, tissue regeneration, formation and maintenance of granulomas, immune surveillance, and homeostasis.66,67 TNF’s functions are seemingly contradictory: it plays a role in both tissue degeneration and tissue repair, cellular proliferation and apoptosis, and has anti-tumor and tumor pro-growth properties.65 These functions are, in fact, complementary. Succinctly stated, in a time- and context-dependent manner, TNF drives a rapid and vigorous inflammatory response triggered by infection or injury (primarily via sTNF/TNFR1), and also functions to limit the extent and duration of this inflammatory response when the trigger has been resolved (primarily via tmTNF/TNFR2).69 These dual roles will, in part, explain later observations in patients treated with TNF inhibitors.
Studies in the 1980s and 1990s showed that levels of TNF were increased in a number of pathophysiological conditions. In patients with sepsis, elevated levels of TNF appeared to correlate with mortality. Moreover, peak elevation in monocyte TNF expression correlated with septic episodes.71 Overexpression was also observed across seemingly unrelated conditions, often in the absence of infection. Elevated levels were present in the mucosa and stools of patients with inflammatory bowel disease (IBD), and mucosal cells expressing TNF had been detected in patients with CD.72 Pro-inflammatory cytokines such as TNF and IL-1 were detected in the synovial fluid of patients with RA, and TNF surface receptors were found to be upregulated in active RA tissues.73
These findings led to the question: if TNF is a driver of inflammation and these conditions are primarily diseases of chronic inflammation, could blockade of TNF be a successful treatment strategy in these conditions?
Two key proofs of concept are established with Remicade
Would blocking a single cytokine be effective in these complex immune-mediated diseases? How could such a cytokine be blocked?
The advent of monoclonal antibodies
Traditional development of pharmaceuticals in the past had been empirical, where potential agents were not targeted to a specific mediator of disease and their mechanism of action was not precisely understood.65 In IMIDs, while conventional drugs such as steroids and immunosuppressants are beneficial, they have broad unspecific effects and are characterized by limited efficacy in itself or efficacy constrained by unacceptable toxicity. A deeper understanding of the pathophysiology of disease would have offered the possibility of testing a new approach to pharmaceutical development, where targeting specific mediators of disease provides benefit with fewer toxicities.
In the 1980s and 1990s, the most readily available approach to targeted therapy was through the use of mAbs, the largest class of therapeutic proteins derived from recombinant DNA techniques, known as biologics. However, 10–20 years after the publication of Köhler and Milstein first describing them,3 they had not met expectations as therapeutics.74 The mAbs available at that time were fully murine and were associated with a number of limitations. As foreign proteins, they were immunogenic and associated with high anti-drug antibody (ADA) rates, which resulted in both safety implications and a negative effect on pharmacokinetics (PK). In addition, murine mAbs were poor at inducing antibody effector function in humans. As late as 1993, only one mAb had received regulatory approval for clinical use, Janssen’s muromonab, also known as OKT3, for short-term use in transplant rejection with concomitant immunosuppression, a setting in which ADA risk was limited.6
Yet, advances in mAb technology were coming to fruition at that time. Under the assumption that replacing the murine content of mAbs with human equivalents would both reduce the risk of ADA and improve effector function, researchers used new molecular biology techniques to develop chimeric mAbs, which are produced from genes whose DNA sequences are approximately 75% human, where only the DNA sequence for the variable domain within the antigen-binding fragment (Fab) remains murine.4–6 The assumption proved correct and in 1994, Janssen’s abciximab (ReoPro®; Janssen Biotech, Horsham, PA, USA), a Fab fragment, became the first chimeric mAb to receive regulatory approval, for the prevention of platelet-mediated thrombosis during angioplasty.6 The first whole chimeric mAb to receive regulatory approval was rituximab (Rituxan®; Genentech, South San Francisco, CA, USA; MabThera®; Roche Registration GmbH, Grenzach-Wyhlen Germany) in 1997 for use in lymphoma. Both were approved for single or short-term use, leaving questions about long-term use of mAbs unanswered.6
In the early 1990s, Janssen’s Immunology unit (then known as Centocor) developed the chimeric anti-TNF mAb, cA2.8 Based on data showing that TNF blockade prevented septic shock in animals given a lethal dose of endotoxin,71,75 Janssen selected sepsis as the initial focus for the clinical development of cA2. Sepsis was an obvious candidate for targeted therapy with mAbs because of its high rate of mortality, which increased the acceptability of possible adverse drug reactions (ADRs), and the need for short-term therapy, limiting the possibility of immunogenicity as a concern. However, in the preliminary clinical study, there were no differences in patterns of cytokine activation or mortality with cA2 relative to placebo,9,10 and the program was discontinued. Most researchers in the field concluded that TNF inhibition failed in sepsis because blocking a single cytokine could not work in diseases characterized by a complex mixture of redundant inflammatory mediators.76 Based on these results, the expectations for targeted therapy in other inflammatory conditions were low.
Proof of concept in RA and CD
However, in parallel to the research in sepsis, Ravinder (Tiny) Maini and Marc Feldmann at the Kennedy Institute in London had completed research suggesting that blockade of a single cytokine could reap therapeutic benefit in an IMID. When evaluating a batch of synovium samples from patients with active RA, they observed elevated levels of pro-inflammatory cytokines in all of them.65 This was striking, since such cytokines are usually produced for short periods only (hours to 2 days), and suggested that in RA their production was continuous. The key breakthrough came in 1989, when Brennan et al suggested for the first time that TNF may be a pivotal cytokine in the pathophysiology of RA.7 Their study, in which several pro-inflammatory cytokines (TNF, IL-1, IL-6, granulocyte-macrophage colony-stimulating factor) were neutralized one by one, revealed that blocking TNF in synovial culture led to the inhibition of the others. This was consistent with the observation that TNF is one of the first cytokines to appear after injury or stress and led to the concept of a pro-inflammatory cascade at work in RA, with TNF at its beginning.
Based on these observations, in 1993, Elliott et al of the same group in London conducted a proof-of-concept study in humans, where 20 patients with severe RA were treated with either two 10 mg/kg or four 5 mg/kg infusions of cA2, given 2 weeks and 4 days apart, respectively.11 Positive clinical improvements, as measured by swollen and tender joint counts and pain, and biochemical responses, as measured by reduced levels of inflammatory mediators, were observed in all patients, providing the first evidence that TNF blockade reduced inflammation and improved symptoms in RA. The effect faded after a few weeks.
Elliott et al proceeded to conduct a 73-patient randomized, placebo-controlled trial of a single infusion of two doses (1 mg/kg and 10 mg/kg) of cA2 in RA.13 Both doses proved highly effective (combined dose groups with a 61% clinical response [Paulus 20%] vs 8% for placebo at week 4, P<0.001), but the maximal improvement and its duration were dose dependent. The rate of adverse events was similar across the cA2 arms and the placebo group. As found in the first study, the benefits disappeared within a few weeks. In a first exploration of retreatment, several initial responders from the first study were given cA2 for up to three additional cycles administered upon relapse.77 The patients regained their responses, but they were again temporary, and the time to relapse generally shortened with each successive cycle, raising concerns about ADAs developing over time (four of seven patients were ADA positive).
Gastroenterologists, who had observed increased levels of TNF in patients with CD, also explored its blockade as a possible treatment. In 1993, the group of Sander van Deventer at the Academic Medical Centre in Amsterdam reported on a female patient, age 12 years, with CD who was non-responsive to conventional therapies and received cA2 as a compassionate-use treatment.12 She received two infusions of cA2 10 mg/kg 2 weeks apart and responded immediately after the first dose. Clinical and endoscopic remission was observed, but, as with RA, this was temporary, with symptoms returning after 3 months. Van Dullemen et al from the same group in Amsterdam then conducted an open-label proof-of-concept study of a single dose of cA2 (10 mg/kg or 20 mg/kg) in 10 patients with steroid-non-responsive CD.14 Within 4 weeks, eight patients showed normalization of their Crohn's Disease Activity Index (CDAI) scores and near-complete healing of mucosal ulcerations, images of which merited display on the cover of the journal in which the data were published (Figure 2). The average duration of response after a single infusion was 4 months, and cA2 was well tolerated among all patients.
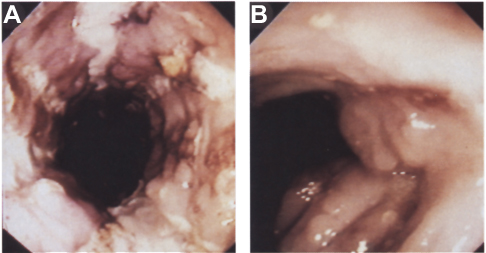 |
Figure 2 Healing of colonic ulcerations in a Crohn's disease patient (A) before treatment and (B) 4 weeks after a single infusion of Remicade 10 mg/kg.Notes: Gastroenterology by American Gastroenterological Association. Reproduced with permission of W.B./Saunders Co., from Treatment of Crohn’s disease with anti-tumor necrosis factor chimeric monoclonal antibody (cA2). van Dulleman HM, van Deventer SJ, Hommes DW, et al. Gastroenterology, volume 109, issue 1, 1995.14 |
This small, uncontrolled study supported the hypothesis that TNF was a major contributor to the pathophysiology of CD and paved the way for Targan et al to conduct a 12-week multicenter, double-blind, placebo-controlled trial in 108 patients with treatment-resistant CD.15 In 1997, they reported that 65% of patients receiving a single dose of cA2 (5, 10, or 20 mg/kg) had a clinical response by week 4 compared with 17% of placebo-treated patients (P<0.001). The rates of adverse events were similar among treatment groups. Rutgeerts et al explored retreatment in this trial, where patients with an initial response to cA2 were given an additional four open-label infusions of 10 mg/kg every 8 weeks (q8w) beginning 12 weeks after the initial infusion.78 Similarly to the previous experience in RA, they found that the initial benefit of cA2 could be regained and, owing to the regular q8w retreatment schedule rather than waiting for relapse, it was also sustained for the duration of the study. cA2 was well tolerated and the rate of immunogenicity was low (10%).
These studies showed that blocking one cytokine, TNF, could have profound, if temporary, clinical benefits in both RA and CD. The benefit observed in both conditions was profound and rapid. Moreover, the lack of significant safety issues despite these benefits was encouraging. While the cause of the TNF-driven inflammation was still unknown, and its blockade was not a cure, a possible method to suppress it had been found, suggesting potential for clinical use. However, for this therapeutic strategy to work, it was clear that sustained TNF blockade would be necessary. Initial experience with retreatment was positive, but would it work in the long term?
How can dose and concomitant medications influence the rate of efficacy and immunogenicity? Phase II trial in RA
The possibility of long-term TNF blockade with cA2 as a treatment strategy for chronic inflammatory diseases raised three fundamental questions: 1) Could mAbs be administered repeatedly as long-term therapy? Immunogenicity was the primary concern as it could be associated with ADRs (eg, allergic or hypersensitivity reactions) or limits on efficacy (neutralization and clearance of the mAb); 2) Would long-term TNF blockade succeed, or would the disease circumvent this blockade and restore the chronic inflammation via another pathway? 3) Would long-term TNF blockade be associated with an unacceptable safety risk? Given the function of TNF, infections and malignancies were of particular concern. Clinical development in RA and CD proceeded, starting with the first of these questions, immunogenicity.
The relationship between dose, PK, efficacy, safety, and immunogenicity of cA2 was first studied in a phase II, double-blind, placebo-controlled RA trial, conducted in 1995–1996, evaluating cA2 alone or in combination with methotrexate (MTX), an immunomodulator and the gold-standard conventional synthetic disease-modifying anti-rheumatic drug (csDMARD) in RA.79 In the trial, 101 patients with clinically active disease despite receiving MTX were randomized to receive cA2 at 1, 3, or 10 mg/kg, with or without MTX, or placebo plus MTX, at weeks 0, 2, 6, 10, and 14, and were followed through week 26. The rationale for an induction regimen at weeks 0 and 2 followed by 4-week intervals thereafter was two-fold: 1) that high-dose induction would suppress inflammation rapidly and profoundly, and 2) that early, high systemic exposure of the immune system to an antigen, in this case cA2, could result in increased tolerance, thereby reducing immunogenicity.
Approximately 60% of patients in all dose groups had responded by week 2, but the degree and duration of response varied by dose and MTX use. Patients receiving cA2 1 mg/kg without MTX experienced a rapid decline in response, while patients receiving 3 or 10 mg/kg without MTX showed sustained responses. In all three dose groups, the response was longer in duration when combined with MTX. Serum cA2 concentrations were stable in patients receiving 3 or 10 mg/kg alone or in combination, though consistently higher in those receiving MTX. In contrast, patients receiving 1 mg/kg without MTX showed an elimination of cA2 in the serum by the end of the 4 weeks, whereas stable, albeit low, levels were maintained in patients receiving MTX. On the important question of immunogenicity, there were two key observations: rates of ADA were lower for all doses when combined with MTX and were inversely related to cA2 dose administered (Figure 3).
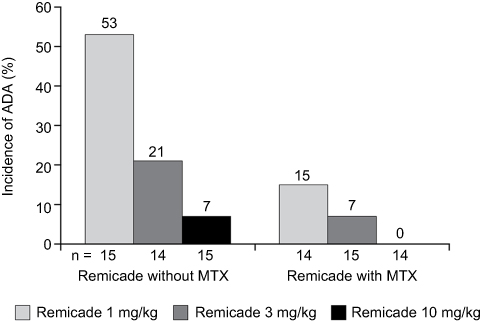 |
Figure 3 Incidence of ADAs in a phase II trial of MTX-refractory RA patients, by Remicade dose and use of concomitant MTX. ADAs were measured by a drug-sensitive immunoassay.Note: Data from Maini et al.79 Abbreviations: ADA, anti-drug antibody; MTX, methotrexate; RA, rheumatoid arthritis. |
These findings demonstrated not only that retreatment with TNF blockade could be effective in sustaining the initial treatment benefit, but also how immunogenicity of mAbs could be reduced: a high-dose induction regimen was associated with tolerance to cA2, levels of immunogenicity were inversely proportional to dose, and co-administration with MTX both reduced immunogenicity and improved the PK of cA2.
First approval of Remicade: CD
Despite the initial proof-of-concept study of targeted TNF blockade with cA2 being conducted in RA, CD was selected as the first indication for commercial development because it was believed to have a more expedited path to regulatory approval owing to its severity in patients not responding to conventional therapies.
Having completed the Targan study in luminal CD,15 the next trial was conducted in fistulizing CD, a debilitating complication observed in as many as 20% of CD patients where no therapy had been shown to be effective. Present et al evaluated the efficacy of cA2 to close draining fistulas in 94 patients randomized to receive an induction dose at 0, 2, and 6 weeks of placebo, or 5 or 10 mg/kg of cA2, and then followed through week 18.20 The primary endpoint was closure of ≥50% of fistulas for at least two consecutive visits 4 weeks apart. The response rate was 26% in placebo patients compared with 68% (P=0.002) and 56% (P=0.02) in the 5 and 10 mg/kg groups, respectively. The safety results were consistent with those observed in the initial studies.
The Targan and Present studies thus demonstrated the safe and effective use of cA2 in treatment-resistant, moderate-to-severe CD, and were the basis for its first regulatory application. The FDA granted accelerated review and approved cA2, renamed Remicade (infliximab), on August 24, 1998.16 Approval by the European Medicines Agency (EMA) in Europe was granted a year later, followed by approvals in Brazil (2000), Canada (2001), Japan (2002), and over 100 other countries (Janssen, data on file).
Importantly, the FDA and other health authorities granted approval for single treatment with Remicade, but recognized the need for maintenance treatment in CD and required that Janssen study it in the phase III setting as a condition of approval.
Can long-term blockade of TNF lead to sustained suppression of inflammation? Phase III trials in RA and CD
Maintenance treatment was first studied in the phase III ATTRACT trial, which evaluated Remicade over a 2-year period in moderate-to-severe RA despite treatment with MTX.19 Patients, all of whom remained on stable doses of MTX, were treated with an induction regimen (infusions at weeks 0, 2, and 6) of either placebo or Remicade 3 or 10 mg/kg, followed by infusions of the same dose administered every 4 weeks (q4w) or q8w thereafter (with placebo infusions as needed to maintain the blind). The clinical effects of all four Remicade groups were superior to placebo and similar to each other at the primary endpoint, the proportion of patients who achieved ≥20% improvement in the American College of Rheumatology response criteria (ACR20) at 6 months (50.0% in the 3 mg/kg q8w group [standard approved dose] vs 20.0% in the placebo group, P<0.001), and were sustained through the 2-year duration of the trial with a mild dose response.80 This dose response, combined with another trial evaluating dose escalation for Remicade in RA, START,81 led to the approval of dose increase above 3 mg/kg in RA for lack of initial response or later loss of response.
In CD, maintenance treatment with Remicade was studied in the phase III ACCENT I and II trials for luminal and fistulizing disease, respectively.26,31 Both trials had a randomized withdrawal design, where all patients were initially treated with Remicade, after which responders were randomized either to Remicade maintenance therapy (5 or 10 mg/kg in ACCENT I or 5 mg/kg in ACCENT II) or placebo maintenance q8w for 1 year. Patients who lost response to treatment crossed over to a dose of Remicade 5 mg/kg higher than their randomized dose. In ACCENT I, at crossover, the dose was administered upon symptom return, allowing the evaluation of episodic use of Remicade. In ACCENT II, the regular q8w maintenance schedule was continued after crossover. Both studies met their primary endpoints, which were clinical remission and time to loss of response at week 30 in ACCENT I and median time to loss of response (≥50 reduction in number of draining fistulas) in ACCENT II. In both studies, the initial Remicade induction response waned in patients randomized to placebo maintenance, while the benefits were largely sustained in patients randomized to continue Remicade (with a dose response in ACCENT I). Moreover, dose escalation resulted in regaining response in patients who lost their initial response.82 Episodic retreatment with Remicade in ACCENT I revealed important lessons. While response was regained, outcomes were generally worse and led to higher levels of ADA than in patients who maintained a regular q8w maintenance schedule.83 In both ACCENT studies (as in ATTRACT), ADA development was associated with an increased risk of infusion reactions and subsequent loss of response.26 These observations, confirmed independently,84 provided support to the then novel, now accepted, hypothesis of an important dynamic of immunogenicity and serum drug levels: not only do ADAs lead to low serum levels of drug, but low serum levels lead to increased ADAs.85 It is now well understood that maintaining target levels of the drug is important to minimize the development of ADAs.
Thus, the phase II/III program had answered all three initial questions about long-term TNF blockade with Remicade: 1) mAbs could be administered repeatedly as maintenance therapy, and several strategies were identified to reduce the risk of ADA; 2) sustained TNF blockade could result in long-term suppression of disease; 3) to the degree that the safety profile of a drug could be assessed with fewer than 2,000 patients treated for ≤2 years, and considering the efficacy observed, the overall benefit–risk profile was considered acceptable (see “Establishing the safety profile of Remicade”, later in this review). On the basis of the results of the ATTRACT and ACCENT I/II trials, Remicade was approved as induction and maintenance therapy for RA in 1999 and as maintenance therapy for CD in 2003.
Is TNF blockade disease-modifying?
At the time of the initial approvals in CD and RA, it was not known whether TNF blockade would be disease-modifying and thereby inhibit the progressive, irreversible structural damage caused by these diseases. This question was first answered in RA, where joint destruction is a hallmark of the disease and a predictor of poor functional outcome and disability. The ATTRACT study was the first to show that progression of joint damage could be inhibited with TNF blockade. The mean increase in radiographic progression score at 1 year (using the Sharp/van der Heijde score [SHS]), the co-primary endpoint of the trial, was 0.6 for Remicade across all doses versus 7.0 for the MTX group (P=0.001), indicating inhibition of joint damage progression in the majority of Remicade-treated patients, which was unprecedented for any therapy at that time (Figure 4).21 This effect was sustained through to the end of the 2-year trial.80 Importantly, the inhibition of progression was observed regardless of whether patients had a clinical response to Remicade or not. Further subgroup analysis showed that inhibition of joint progression occurred in patients with early disease as well as those with established disease, another important finding.
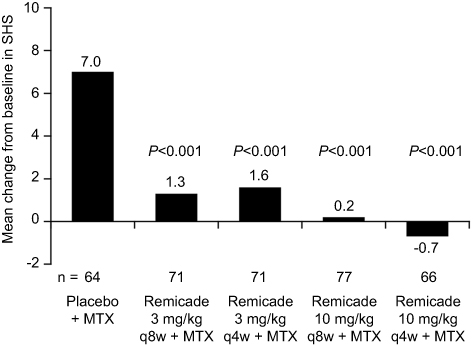 |
Figure 4 Progression of structural damage in RA at week 54 in the ATTRACT trial.Notes: Data from Lipsky.21 P-values vs placebo + MTX arm. Abbreviations: MTX, methotrexate; q4w, every 4 weeks; q8w, every 8 weeks; SHS, Sharp/van der Hejde score. |
Analogous work was done in the CD clinical trials, where the endpoints studied included the effect of Remicade on mucosal healing and the need for surgery. The original observation by Derkx et al12 and van Dullemen et al,14 that Remicade healed the mucosa in CD patients, was confirmed in ACCENT I, where the healing (defined as the absence of mucosal ulcerations in all segments where they had been observed on endoscopy at baseline) was observed as early as the end of induction and was sustained through to the end of the trial: 50% of initial responders receiving q8w maintenance therapy had complete mucosal healing at 1 year compared with only 7% of initial responders receiving episodic maintenance therapy (P=0.007).82 In addition, in both ACCENT I and ACCENT II, Remicade maintenance therapy was associated with a reduced rate of hospitalizations and surgeries.89,90
The inhibition of joint destruction and the mucosal healing effects with Remicade in patients with RA and CD demonstrated that TNF blockade was not just an effective agent on clinical symptoms, but was also disease-modifying, implying an impact on the course of the disease. Research into the exact mechanism of Remicade’s effect followed.
What is Remicade’s mechanism of action? Analyses from RA and CD clinical trials
It was of interest to determine whether the TNF-dependent inflammatory cascade observed in the initial synovial cell culture experiments77 occurred in vivo. In RA patients receiving Remicade, rapid decline in serum IL-6 levels on the day of treatment confirmed that a TNF-dependent cytokine cascade was indeed occurring.91 Reductions in other pro-inflammatory mediators and chemokines were also observed.10 Similarly, C-reactive protein (CRP, a serum marker of systemic inflammation) was observed to decline rapidly after administration of Remicade.19 Detailed mechanistic studies were performed, and multiple aspects of the disease were found to improve, including immune function, joint function and hematological parameters.65 Immunohistological studies were conducted to investigate changes in the synovium, where reductions in the expression of adhesion molecules and in cell infiltration were observed, as were reductions in angiogenic factors and angiogenesis.10 Taylor et al demonstrated in a neutrophil radiolabeling study that the influx of granulocytes was reduced by approximately 50% in the joints within 2 weeks of a single dose of Remicade, indicating that reduced recruitment of these and other leukocytes to the joints is an important aspect of the mechanism of anti-TNF therapy.92
Similar mechanistic studies were performed in CD. As with RA, Remicade treatment of CD patients was associated with a rapid reduction of CRP.15 Histological evaluation of colonic biopsies revealed a reduction in detectable TNF after treatment and provided evidence of reduced infiltration of inflammatory cells and other inflammatory markers at these sites. Analysis of lamina propria mononuclear cells of the intestinal mucosa showed that Remicade treatment caused a reduction in the number of cells capable of expressing TNF and interferon-γ.93,94
As an antibody, Remicade functions in two ways: it binds directly to s/tmTNF via its Fab (antigen-binding) region and has a functional Fc (constant) region. Through both, its possible mechanisms of action of TNF inhibitors generally fall into two categories: 1) blockade of TNF-receptor-mediated signaling through neutralization of sTNF and tmTNF; and 2) removal of TNF-expressing cells by induction of Fc- or tmTNF-mediated effector mechanisms, such as antibody-dependent cellular cytotoxicity (ADCC) and apoptosis (Table 1). The relative contribution of these mechanisms to Remicade’s efficacy remains uncertain and possibly differs by disease. For example, the role of anti-TNF-induced apoptosis of immune cells in reducing inflammation in RA synovial tissue is unclear, while evidence exists for such a process in the bowel mucosa in IBD.
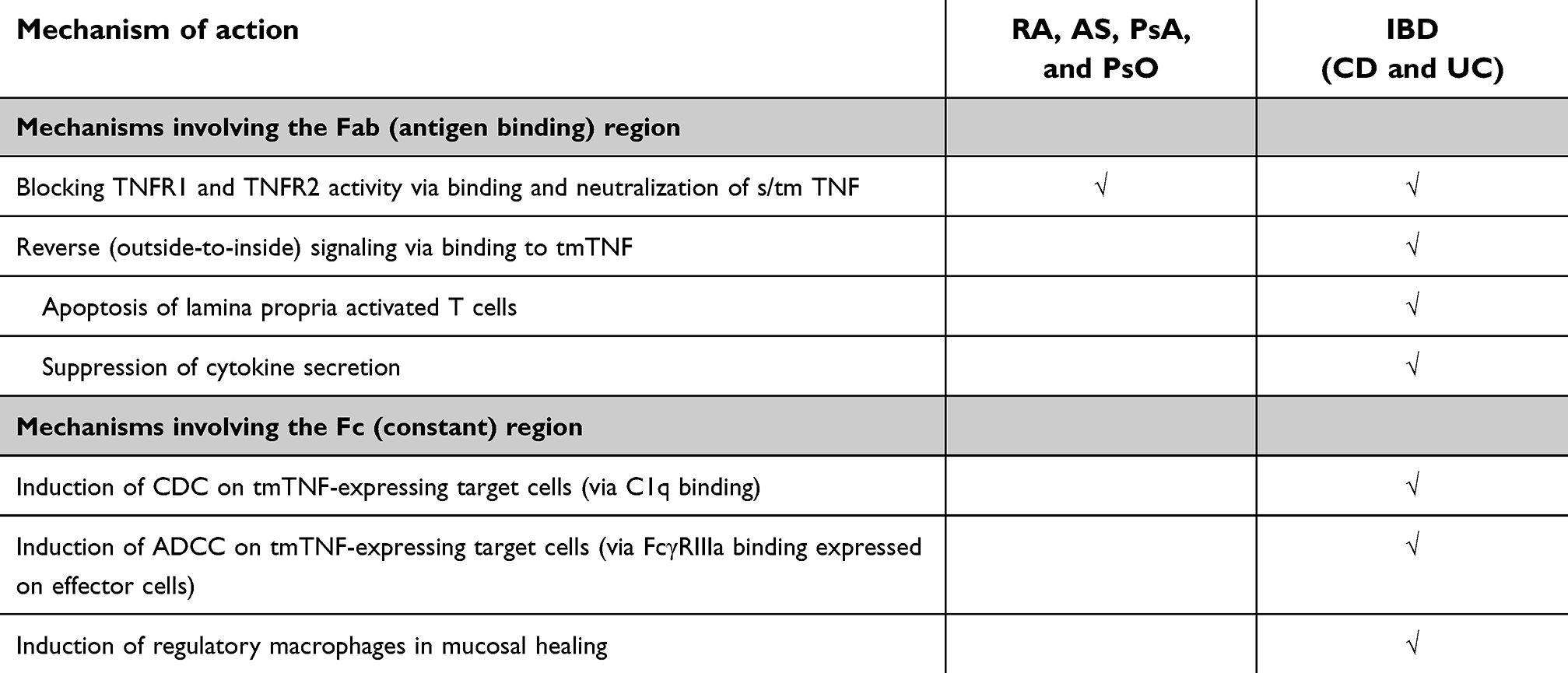 |
Table 1 Possible mechanisms of clinical efficacy of TNF blockade with Remicade |
Early clinical evidence that the mechanism of action of anti-TNF therapy in CD differs from RA was the observation that etanercept, a p75(TNFR2)-IgG Fc receptor fusion protein, did not show efficacy in CD.95 Certolizumab (Cimzia®; UCB, Smyrna, GA, USA), a PEGylated IgG Fab fragment, demonstrated efficacy in CD, but seemingly less so than Remicade and adalimumab (both of which are full antibodies), especially in induction of clinical response.96 A comparison of the differing characteristics of the TNF inhibitors tested in CD provides insight into possible explanations for these differences. All TNF inhibitors bind to both sTNF and tmTNF, but the nature of this binding differs between them. Remicade and adalimumab, as full antibodies, are bivalent; ie, capable of binding two molecules of TNF simultaneously, enabling them to form complexes.97 Certolizumab and etanercept are both monovalent. Remicade, adalimumab, and certolizumab have high affinity for tmTNF, while etanercept has lower affinity for tmTNF than the antibodies.94 Remicade and adalimumab have fully functional Fc fragments, while etanercept has lower Fc activity than the full mAbs and certolizumab has no Fc fragment at all.94 Together, this evidence suggests that in CD, in addition to the neutralization of TNF, one or more effector mechanisms are involved in the resolution of inflammation and mucosal healing (Table 1). Research continues today to determine exactly which mechanisms contribute to the efficacy of these agents.
The results of mechanistic studies of TNF inhibitors combined with their demonstrated efficacy confirmed the role of TNF in the pathogenesis of both RA and CD. The next question was whether TNF inhibition would be an effective therapeutic strategy in other conditions characterized by TNF elevation.
TNF elevation does not always mean TNF mediation
Is TNF blockade effective in diseases beyond CD and RA?
Remicade approval in additional IMIDs
The first evidence of efficacy of TNF blockade beyond RA and CD came from anecdotal reports in clinical practice, in which patients treated with Remicade for CD experienced improvements in extraintestinal manifestations of their disease, specifically ankylosing spondylitis (AS)98 and psoriasis (PsO).99 These findings led to clinical development in these indications. Psoriatic arthritis (PsA) and ulcerative colitis (UC) were also studied owing to their related pathogenesis to RA and CD. Health authority approvals for AS, PsA, PsO, and UC were received between 2003 and 2006, followed by pediatric CD (2006) and pediatric UC (2011).18
The efficacy of Remicade in each of these conditions was comparable with that seen in RA and CD (Figure 5), and these results brought new insights to the understanding of the pathogenesis of each. For instance, effective treatment of UC, previously viewed to be a Th2-mediated disease, with the blockade of TNF, a cytokine associated with Th1-mediated diseases such as RA, CD, and psoriasis, contributed to a reassessment of the Th1/Th2 paradigm in the pathogenesis of IBD.100 Treatment of PsA with Remicade demonstrated that TNF drives the inflammation not just in joint-related signs and symptoms and structural damage,101 but also in its other major clinical manifestations, such as PsO, enthesitis, and dactylitis.32 In AS, TNF inhibition demonstrated efficacy on signs and symptoms of disease, but, in contrast to RA and PsA, it had no apparent effect on structural damage in clinical trials, despite a reduction in bone/spinal inflammation as measured by magnetic resonance imaging (MRI). Later non-randomized studies suggested that TNF blockade reduces progression of structural damage in the long term (≥2 years).102,103 In psoriasis, two targeted therapies (alefacept, an anti-CD2 agent, and efalizumab, an anti-CD11 agent) were approved by the FDA and the latter by the EMA, but had modest efficacy, and in the case of efalizumab, emerging safety issues in the postmarketing setting. They were later removed from the market.104–106 In contrast, TNF inhibitors provided evidence that targeted systemic therapy could have both proven efficacy and acceptable safety in the setting of psoriasis.
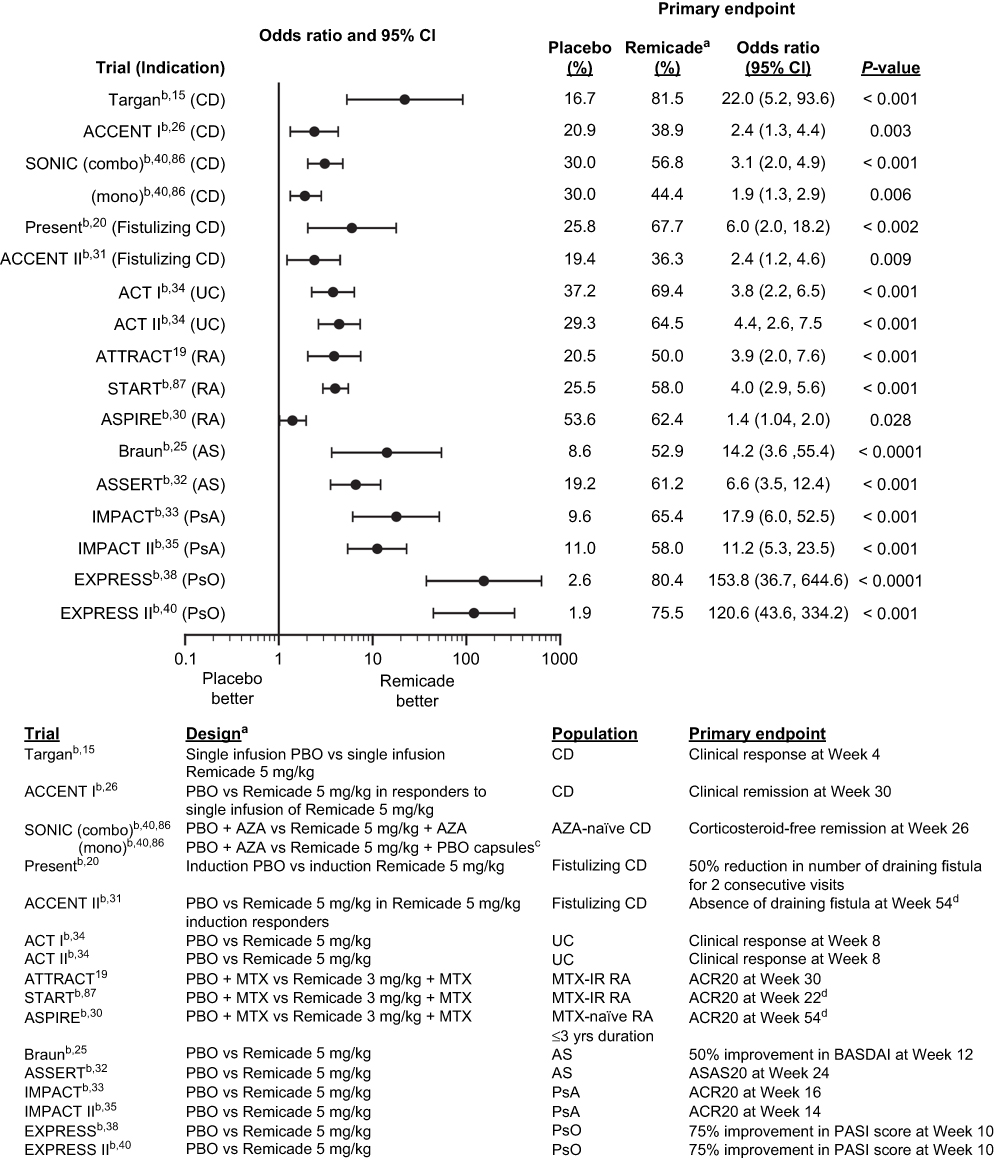 |
Figure 5 Efficacy of Remicade in pivotal phase II/III clinical trials in adult indications: odds ratio of primary endpoint.Notes: aMost trials evaluated multiple doses of Remicade. In the treatment arms shown (the generally approved doses), the Remicade dosing regimen tested was induction (infusions at weeks 0, 2, and 6) followed by q8w maintenance, with the exceptions of the Targan and Present studies, where single infusion and induction only, respectively, were tested, and of the AS trials (Braun and ASSERT), where induction followed by q6w maintenance was tested. bJanssen, data on file. cPBO of AZA. dMajor secondary endpoint. The primary endpoint was a continuous variable for which an odds ratio was not calculated. Abbreviations: ACR20, American College of Rheumatology 20% response; AS, ankylosing spondylitis; ASAS20, Ankylosing Spondylitis Activity Score 20% response; AZA, azathioprine; BASDAI, Bath Ankylosing Spondylitis Disease Activity Index; CD, Crohn’s disease; MTX, methotrexate; MTX-IR, methotrexate inadequate responders; PASI 75, Psoriasis Area and Severity Index 75% reduction; PBO, placebo; PsA, psoriatic arthritis; PsO, psoriasis; q6w, every 6 weeks; q8w, every 8 weeks; RA, rheumatoid arthritis; UC, ulcerative colitis. |
Unsuccessful clinical indications of TNF blockade
In addition to the approved indications, both Janssen and independent investigators have evaluated Remicade in other disorders associated with elevated TNF, including both other IMIDs, such as asthma and multiple sclerosis (MS), and diseases not generally characterized as IMIDs, such as infectious diseases, cancer, and cardiovascular conditions (Table 2). In each of these disorders, there were mechanistic, in vitro and/or animal data supporting the hypothesis that TNF blockade could be beneficial. In a few, such as systemic lupus erythematosus (SLE) and cancer, there was reason to believe that TNF blockade could either exacerbate the disease or treat it, reflective of the multiple functions of TNF. However, even in these conditions, the unmet medical need and the potential benefit justified testing TNF blockade as a treatment.
 |
Table 2 Diseases beyond approved indications where Remicade was studied as treatment |
In several of the IMIDs, efficacy was observed in initial clinical studies, but for various reasons full development did not follow. Based on small studies, Remicade received regulatory approval in Japan for two rare diseases prevalent in the Japanese population, Behҫet’s and Kawasaki disease.110,111,117,118 In hidradenitis suppurativa and uveitis, initial studies suggesting efficacy with Remicade led to the development and approval of other TNF inhibitors in these indications.116,131,148 In autoimmune hepatitis and SLE, efficacy was observed in proof-of-concept studies, but was outweighed by the negative safety profile,109,127–129 and in sarcoidosis and asthma, only marginal benefit was observed in formal phase II trials and development was discontinued.123 In the remaining IMIDs evaluated (Table 2), Remicade showed no or only short-term clinical benefit. In all non-IMIDs studied, TNF blockade with Remicade proved ineffective or insufficiently effective to justify further clinical development.
In two instances, unexpected safety signals arose in clinical studies with Remicade. Despite preclinical evidence that TNF blockade improved an animal model of MS,119 two patients with rapidly progressive MS treated with Remicade in a phase I safety trial experienced a transient increase in the number of gadolinium-enhancing lesions and other signs suggestive of immune activation and increased disease activity.119 A similar worsening of symptoms was found in a double-blind placebo-controlled study in MS with another TNF inhibitor, lenercept,149 and in clinical practice TNF inhibitors have been associated with cases of new onset and exacerbation of central and peripheral nervous system demyelinating disorders, such as optic neuritis and Guillain–Barré syndrome.150
In the second situation, preclinical and early clinical data suggested that TNF played an important role in the pathogenesis and progression of congestive heart failure (CHF). Yet in the phase II ATTACH study of 150 patients with stable class III or IV CHF, short-term TNF blockade with Remicade showed no clinical benefit, and high doses (10 mg/kg) were associated with an elevated risk of death or hospitalization.29 Poor outcomes were also observed with etanercept in clinical trials of patients with CHF, confirming that TNF blockade is not an effective strategy in the treatment of moderate-to-severe CHF.151
What are the possible reasons for failure of TNF blockade?
Two possible explanations for the lack of demonstrated benefit of Remicade in these indications, both related to trial design, are that the dose or duration of treatment was not sufficient or that, as proof-of-concept studies, the trials were underpowered and unable to detect a benefit. However, given the general understanding of Remicade dosing, and the consistency of results across multiple trials with other TNF inhibitors, it seems more likely that TNF blockade is simply not the right treatment strategy in these diseases. But why not?
In the non-IMID indications, investigators identified two main possible explanations for lack of effect of TNF blockade. First, the inflammation in these diseases is possibly not driven by TNF, but rather the high production of TNF could be a downstream effect. Alternatively, the inflammation, even if TNF driven, is just one of several ongoing pathologies such that suppressing it does not have an impact on the disease as a whole.
It is less clear why TNF blockade works so effectively in some IMIDs with elevated TNF, but is ineffective or leads to worsening in others. It is possible that the timing of treatment with respect to disease course or the selected patient population was not optimal. Sarcoidosis, similar to CD, is both granulomatous in nature and mediated by Th1. Theoretically, TNF blockade should have been effective, yet the phase II trial showed only marginal benefit with Remicade. The investigators noted that one possible explanation for the trial’s results is that it included patients with stable disease, which may have diminished a possible response to Remicade, owing to a lack of inflammation and/or high levels of fibrosis, which TNF blockade would not treat. An exploratory subgroup analysis revealed that patients with severe disease were more likely to benefit. Janssen subsequently conducted another phase II trial with a follow-up TNF inhibitor, golimumab (Simponi®; Janssen Biotech, Horsham, PA, USA), enrolling specifically this severe population, and yet again only marginal benefit was observed.152 The sarcoidosis experience emphasizes the challenge of identifying a suitable population for a targeted treatment (eg, patients with TNF-driven disease) even when a sound understanding of the disease pathophysiology is present.
Another possible explanation for the lack of benefit in IMIDs treated with TNF blockade is that the ongoing inflammation is not TNF-driven, not solely TNF-driven, or not TNF-driven at each stage of disease, ie, where drivers of inflammation change over time or in different circumstances. It is known that the inflammation in IMIDs has different etiologies, mediated by Th1, Th2, and/or the more recently discovered Th17 responses. Elevated levels of TNF are associated with diseases thought to be driven by Th1 and/or Th17 responses, such as RA and CD. A number of the diseases where Remicade failed to work are thought to be Th2-mediated, including asthma (Janssen, data on file) and scleroderma,125 where TNF, although elevated, is possibly not central to the underlying inflammation. In others, multiple pathways could be active. For instance, in view of the negative results with Remicade in Sjögren’s syndrome126 and atopic dermatitis,108 investigators speculated that TNF blockade could have shifted the balance of Th1/Th2-mediated inflammation in favor of Th2. Whereas recent research has shown that some diseases, such as PsO and CD, can be effectively treated by blockade of cytokines elevated in either the Th1 or Th17 pathway,35,40,153,154 others perhaps require blockade of both. One analysis, for instance, suggested that the inflammation in giant cell arteritis (GCA) is driven by cytokines from both the Th1 and Th17 pathways and successful treatment will require blockade of both.155
Several hypotheses have been proposed for worsening disease with TNF blockade. In the MS trials, investigators identified possible mechanisms by which TNF blockade could lead to further immune activation contributing to the disease’s pathogenesis or to interruption of TNF-mediated tissue repair via TNFR2.150,156 In CHF, disease worsening occurred despite decreases in both CRP and IL-6 with Remicade treatment. This led the investigators to propose that cytokine activation, including TNF, was beneficial, serving as part of the body’s adaptive response to CHF, and that blocking it disrupted this response.29
It is clear that despite an improved understanding of the immune system, there are still many unknowns. The learnings from these trials have demonstrated that TNF’s effect and, by extension, those of TNF blockade, are context driven and are difficult to predict. Remicade is effective in a number of IMIDs with a TNF-driven inflammatory component. In the other diseases where Remicade was studied, the results have provided useful insights into their pathophysiology and helped researchers in the search for new therapeutic targets.
Establishing the safety profile of Remicade
As Remicade was both the first TNF inhibitor and the first mAb to be used in chronic diseases, there was little precedent to draw upon, leaving researchers to hypothesize on its safety profile. Given the physiological role of TNF in host defense and immune surveillance, Remicade was expected to be associated with an increase in infections and, over the long term, malignancy, specifically lymphoma. As a foreign protein, Remicade also brought concerns of immunogenicity and consequent allergic and hypersensitivity reactions. Finally, as with any new drug, but in particular with this new form of targeted therapy, there were concerns about unforeseen risks.
The process of fully defining Remicade’s safety profile took years and is still ongoing (Table 3). It has required Janssen to use a combination of data sources: phase II/III clinical trials, routine pharmacovigilance, postmarketing studies, large clinical databases, and independent research. Depending on the frequency of a given ADR, eg, common, uncommon, rare, and unexpected, different data sources and methods of analysis have been utilized. Several examples are described in this section to demonstrate how this was done.
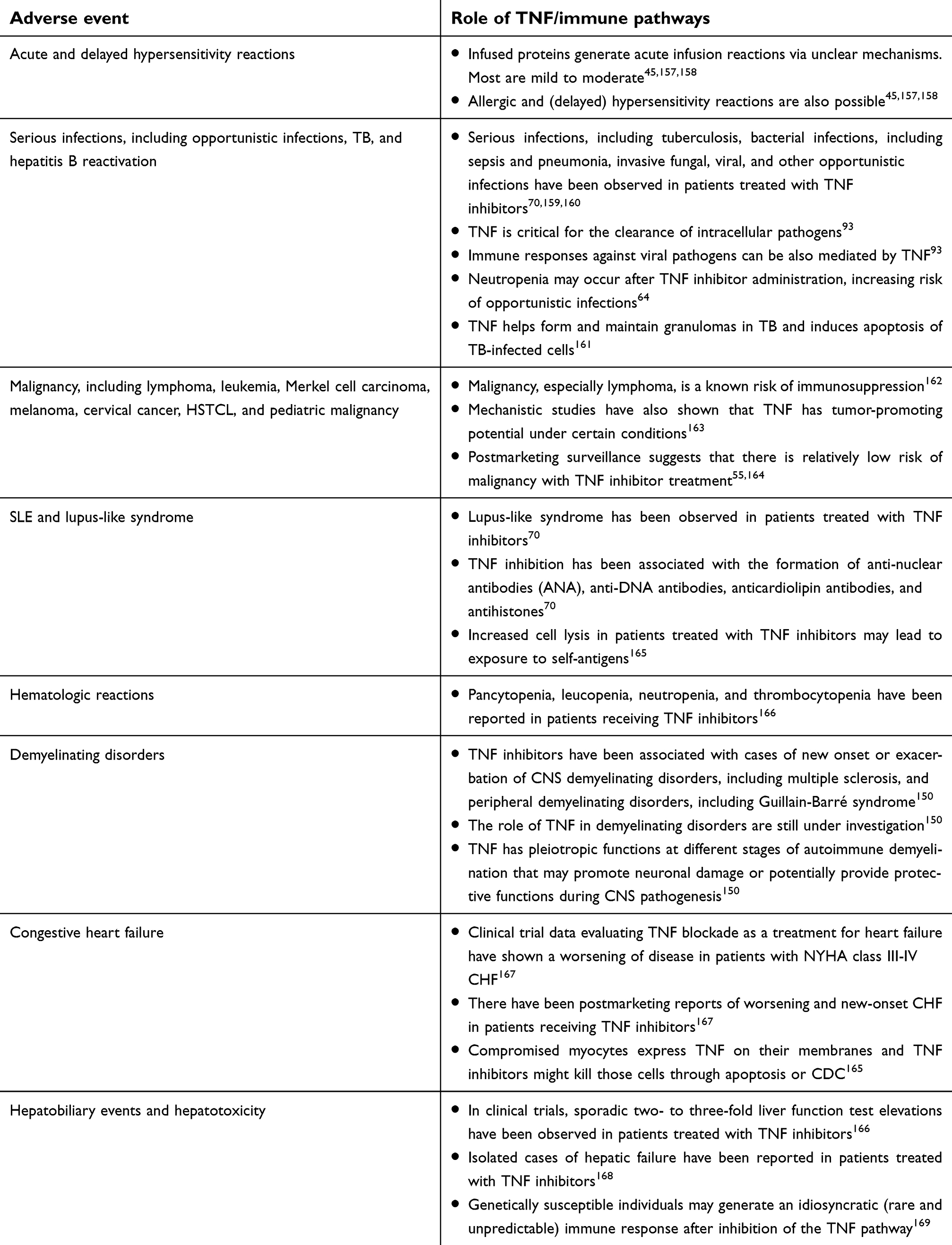 |
Table 3 Overview of Remicade’s safety profile |
What could the phase II/III program tell us?
The number of patients exposed to Remicade across the trials completed at the time of initial approval was not large (<200 patients in CD, approximately 500 across all clinical trials), yet based on this limited experience, the observed profile was aligned with expectations of TNF blockade. In Remicade-treated patients across all trials, ADAs were observed in 28.3% of patients. One or more infusion reactions occurred in 15.9% of patients (in 7.6% of infusions) (Janssen, data on file), most of which were mild to moderate in severity. Other than infusion reactions, allergic/hypersensitivity reactions were infrequent. Infections, including serious infections, occurred at a higher rate in patients treated with Remicade than with placebo (21.0% vs 11.0% for infections and 2.4% vs 1.8% for serious infections, respectively) (Janssen, data on file). Malignancies, including lymphomas, were observed, but were too infrequent to assess any possible association with Remicade (seven malignancies, of which five were lymphoid). One unexpected phenomenon, an increase in the risk of autoimmunity, was identified. In Remicade-treated patients, there was a net increase in new anti-nuclear antibodies (ANA) of 12% (from 24% to 36% of Remicade-treated patients), and anti-dsDNA antibodies developed in 9% of patients (from 0% to 9%). Isolated cases of (reversible) lupus-like syndrome were observed (Janssen, data on file). Viewed in the context of moderate-to-severe CD unresponsive to conventional therapies, this benefit–risk profile was viewed as positive, and Remicade received approval for CD.
As clinical development in additional indications proceeded, the phase II/III/IIIb trial database increased in size and had, by the end of development in 2016, grown to over 10,000 patients in more than 50 trials conducted across the globe in the six approved adult indications, and included pediatric populations in RA, CD, and UC (Janssen, data on file). While this was a much larger data set than was available at the time of initial approval, it was still not sufficient to fully establish Remicade’s safety profile, a situation typical of clinical development programs. Clinical trials enroll a highly selected patient population and are not designed to detect rare safety events or events with long latency periods. To remedy this, manufacturers employ routine pharmacovigilance activities to monitor a drug’s safety profile in clinical practice, the key part of which includes analysis of safety events reported to the company and in the medical literature.
What was the scale of the challenge of defining Remicade’s safety profile? Common, uncommon, rare, and unexpected adverse events
Considering that a number of foreseeable risks needed further quantification and qualification, routine pharmacovigilance activities were not sufficient to define Remicade’s emerging safety profile. For this purpose, postmarketing commitment (PMC) safety studies were agreed with or required by the health authorities for each new indication as it was granted. (In this manuscript, the term PMCs will be used collectively to refer to all studies agreed with or required by health authorities as a follow-up measure to provide additional data on safety or efficacy in the post-approval setting, known as postmarketing requirements [PMRs] and postmarketing commitments [PMCs] for the FDA, and postapproval measures [PAMs] for the EMA.)
The primary goals of the PMC program were to evaluate Remicade’s long-term safety, specifically infections and malignancies, and its safety in vulnerable populations where it was expected to be used, specifically, pediatric patients and women exposed during pregnancy. The program was also to serve as a data source and hypothesis-generating tool for other possible adverse events.
A major consideration for the health authorities when determining the scope of the PMC program was the estimation of how broadly Remicade was to be used. It was indicated for six different diseases, which were serious but not generally regarded as life-threatening in nature. Moreover, their collective prevalence consisted of millions of patients. Given that the indicated diseases themselves differed in demographics, background safety risks, comorbidities, and conventional therapies, key safety questions would sometimes need to be evaluated separately by therapeutic area, ie, rheumatology, gastroenterology, and dermatology, and occasionally for each individual indication, ie, CD and UC. With the need to study Remicade in multiple diseases, as well as in demographically and geographically diverse populations, the PMC program needed to include tens of thousands of patients and would take years to execute.
The majority of the Remicade PMCs have sourced data from prospective, observational registries of specific diseases. Registries, while neither randomized nor containing the level of detail collected in controlled clinical trials, have the advantages of large size, long duration of patient treatment and follow-up, and inclusion of a broad population reflecting real-world use. Where registries already existed, Janssen initiated collaborations to meet its PMC requirements. When such independent initiatives were not sufficiently available, de novo disease registries were set up by Janssen and its commercial partners.
In total, the Remicade PMC program in approved indications consisted of seven company registries, 12 registries studies (ie, those based on analyses from independent registries), and three additional studies addressing specific safety topics (Table 4). Of the registries/registry studies, one included patients across all indications, seven included patients with rheumatic diseases, five were in IBD, including two pediatric registries, four were in PsO, and two evaluated Remicade in pregnancy across multiple indications. All seven company registries were designed and recruited by Janssen or its partners specifically to address Remicade PMCs. In general, the registry-based PMCs had two key design features: 1) they followed Remicade patients as well as comparator cohorts, including those receiving conventional therapies, and later, when they became available, those exposed to other biologics; and 2) Janssen committed to long-term patient follow-up, ranging from 5 to 20 years. The three additional studies were designed specifically to evaluate the risk of hepatosplenic T-cell lymphoma (HSTCL) with Remicade.
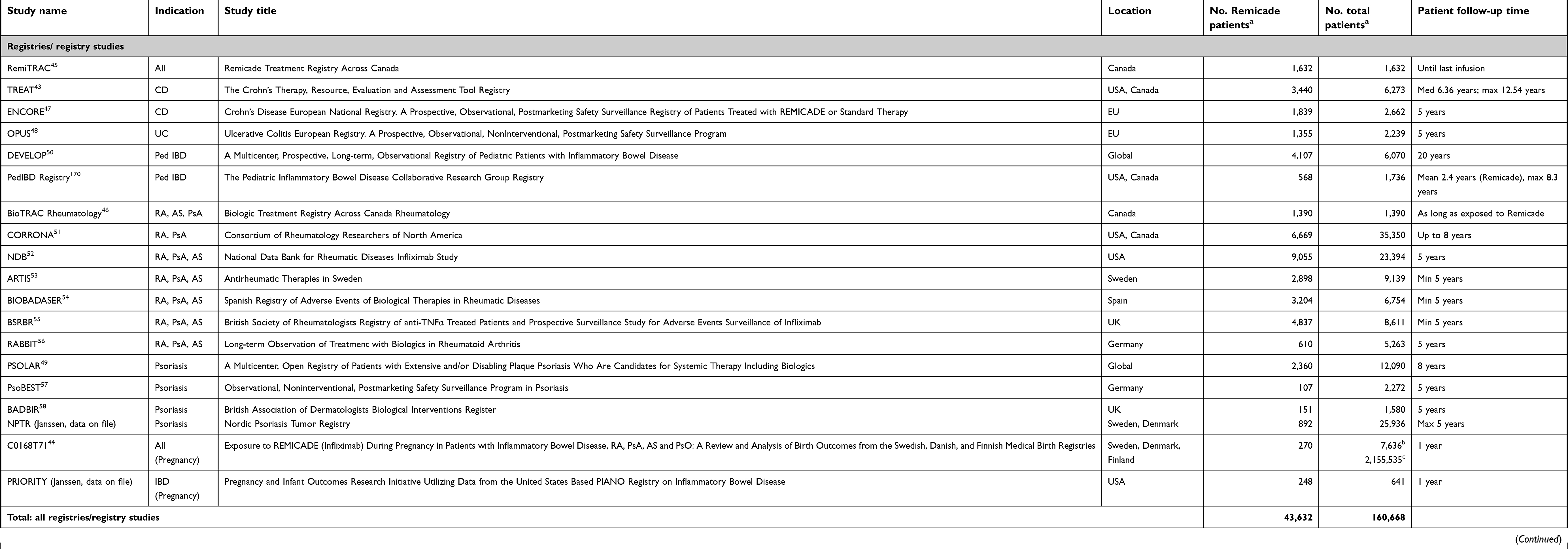 |
Table 4 Remicade postmarketing commitment program |
To date, approximately 44,000 patients exposed to Remicade and 117,000 patients in the comparator cohorts have been included in the PMC program, a figure which does not include patients followed in local post-approval safety monitoring programs, such as those required in Japan. From the beginning of each PMC through study closure, health authorities worldwide receive comprehensive reports on a regular basis providing updated analyses of key safety risks to support the prescribing information. Several of these PMCs are still ongoing today.
How can registries inform us about common adverse events?
The major common adverse event to be studied in the PMC program was infection, including the subgroup of serious infections. TREAT, a US-based Janssen registry in CD started in 1999, is a good example of the scale and design needed for this purpose. Over 5 years it enrolled two cohorts, those receiving treatment with Remicade and those receiving conventional therapies, and followed them until it was closed in 2012. It was the largest registry in IBD at the time, enrolling more than 6,000 patients with a median follow-up time of 6.36 years.43
Importantly, TREAT served as a data source also for studying the risks in conventional treatments. At the time of initiation of the program, thorough understanding of the risk of serious infection with conventional medications (specifically corticosteroids, analgesics, and the immunosuppressants thiopurines and MTX) was lacking. It was necessary in the Remicade PMC registries to gain an understanding of the background risks of these conventional agents as Remicade itself, used mostly after or in combination with these therapies, could not be judged in the absence of such knowledge. Similar understanding on the interactions between certain disease characteristics, such as severity and the risk of infection, was also required to understand the risks of Remicade and was also obtained from TREAT.
Analyses in TREAT confirmed the increased risk of serious infections with Remicade observed in the phase II/III program (unadjusted rates: 2.04 and 1.00 per 100 patient-years for Remicade and conventional therapies, respectively; adjusted HR=1.43, 95% CI 1.11 to 1.84, P=0.006).171 Further analysis revealed that other significant predictors of serious infection were older age, use of prednisone, narcotic analgesics, moderate/severe disease, colonic disease, and disease duration at enrollment. The ENCORE and OPUS PMC registries in Europe for CD and UC, respectively, yielded confirmatory results to these findings and showed that results were generalizable for patients treated in clinical practice across geographic borders.47,48
In rheumatology, data from several biologics registries in the USA and Europe were published indicating a similar association between TNF inhibitors and serious infection risk to that found in CD,172,173 with similar additional risk factors for serious infection such as disease severity. It was also established that there is a time-dependent increase in serious infection risk, where the maximum risk is reached within the first 6 months of therapy, with a gradual decline thereafter.159,174,175
Can registries inform us about uncommon events?
TREAT also demonstrated how registries can be used to better characterize the risk of uncommon events, for example lymphoma, for which they are often not adequately powered despite their size and duration. During the 13 active years of TREAT, just 15 cases of lymphoma were reported, evenly distributed between the Remicade and conventional therapy cohorts.43 Owing to this limited number of cases, predictors associated with the risk of lymphoma could not be further estimated in regression models. This low rate of lymphoma was also an occurrence in registries across the other indications.48,55,175–178 Although an increased risk in patients treated with Remicade could not be excluded with these limited numbers, they suggested that there was a relatively low rate of lymphoma with TNF blockade, and that this rate did not differ from that observed with conventional therapies or from the background rate in these diseases. Additional studies, especially meta-analyses including population-wide data, suggested that an increased risk is associated with profound persistent immunosuppression, eg, combination therapy with thiopurines and TNF inhibitors, and with cumulative disease activity.162,179–181
Can big data play a role in evaluating uncommon safety risks?
To establish an association between treatment and an uncommon event such as lymphoma, much larger sample sizes are needed than can be found in most registries. After 20 years on the market, the number of patients treated with TNF inhibitors as a class is large enough and technology has improved enough to enable analyses with the power necessary for low-frequency events. Lemaitre et al published a study from the French National Health Insurance Database (SNIIRAM) evaluating the incidence of lymphoma among 189,000 IBD patients with median follow-up of 6.7 years.182 In this study, 336 patients had claims for lymphoma, which was sufficient for statistical analysis. The lymphoma incidence rate was found to be 0.26 per 1,000 person-years in IBD patients exposed to neither TNF inhibitors nor thiopurines, 0.54 in patients exposed to thiopurines, 0.41 in patients exposed to TNF inhibitors, and 0.95 in patients exposed to combination therapy. Adjusted HRs were 2.6 for thiopurines, 2.41 for TNF inhibitors, and 6.11 for the combination, relative to those unexposed to these drugs, all of which were statistically significant. As this study was based on administrative databases, clinical information, such as disease activity, smoking history, disease phenotype, and information on inflammatory burden, was not available to the authors, and the relative role of these risk factors could not be determined.
An analysis of similar size was also conducted in RA. Eleven biologics registries participated in a collaborative project to investigate the risk of malignancy, including lymphoma, associated with use of TNF inhibitors in RA.55 Among 124,997 patients, 533 lymphomas were reported. Consistent with smaller analyses that did not suggest an increased risk of lymphoma with TNF inhibition or other treatments over and above the elevated risk already present in patients with active RA,175–177 this large, multiregistry study confirmed comparable crude lymphoma rates across treatment cohorts and showed further that lymphoma subtype distribution was similar in biologic-naïve patients with RA and those treated with TNF inhibitors.55
How have PMCs continued to evolve?
Janssen’s registries initiated later in Remicade’s life cycle benefited from learnings from the early experience. An example of this is PSOLAR, begun in 2007 as a PMC for Remicade in psoriasis. Enrollment in the Remicade cohort was ended in 2013 and by which time this registry had enrolled over 12,000 patients with psoriasis. Going beyond TREAT, PSOLAR was a disease-based registry, allowing all patients with comparable psoriatic disease to enter, irrespective of treatment status.49 Where TREAT could not include patients receiving other biologics because none was available at the time, PSOLAR included other TNF inhibitors and, later, other new biologic classes. Further, PSOLAR provided external validity and generalizability as the participating sites represented a mix of community-based, academic, and/or hospital-affiliated practices from around the globe. It also included formal sample size calculations permitting signal detection of adverse events, whereby hypotheses could be generated for later analyses. Lastly, Janssen took measures to ensure the objectivity and transparency of PSOLAR. In contrast to registries of academic or professional organizations, whose independence was implied, possible bias introduced by Janssen’s conflicts of interest was a concern with its company-owned registries. To address this, PSOLAR enlisted from inception a formal steering committee with leaders in their respective fields, whose role was to ensure that the integrity of the registry would be maintained by reviewing, approving, and monitoring research projects, and by driving the interpretation and dissemination of results. Strict publication rules were stipulated in advance, chief among them having independent experts as leading authors.
PMCs evaluating long-term safety in special (vulnerable) populations require unique design considerations. To evaluate the risk of malignancy in pediatric patients with IBD, Janssen designed the global DEVELOP registry, where children treated with Remicade or other therapies were enrolled and are being followed for 20 years.50 Achieving this length of follow-up requires extensive patient retention efforts owing to administrative and healthcare system challenges. For instance, at the age of 18, registry participants transition from pediatric care under the supervision of DEVELOP investigators to adult care by clinicians not involved in the registry. Similarly, in some countries, a lack of a personal identifier makes tracking patients across different healthcare providers over time difficult. Yet these efforts have served their purpose. An interim analysis from DEVELOP published in 2017, based on 5,766 patients with a median follow-up time of 4.7 years, indicated that Remicade therapy was not associated with an increased risk of malignancy compared with a matched general population.50 The data further demonstrated a trend toward an increased risk of malignancy in thiopurine-exposed patients, irrespective of biologic exposure, reinforcing findings from other independent research efforts in adults.162,179
Setting up a pregnancy registry with sufficient power to detect possible safety signals posed particular challenges and required an innovative approach to address. After review of existing databases and discussion with experts, Janssen learned that in the Nordic countries, government-held nationwide healthcare databases, including complete birth registries, could be linked to the local biologics registries. Working with data access providers in Sweden, Denmark, and Finland, Janssen was able to design a study which accrued patients starting from 2000. After 10 years, it had finally accrued enough women exposed to Remicade during pregnancy for a meaningful analysis. The national databases included a total of 2,155,535 births collected between 2000 and 2013, from which the outcomes of 270 births in women exposed to Remicade during pregnancy could be compared with 906 births in women exposed to other TNF inhibitors, 6,460 births in women with similar diseases exposed to non-biologic conventional treatments, and births in the general population.44 Additional information on the infants born to these women was collected during their first year of life.
The study found that exposure to Remicade was not associated with increased rates of congenital anomalies or infant death, the most relevant medical outcomes of interest to health authorities. Remicade in combination with conventional immunosuppressive treatments was associated with other adverse birth outcomes (eg, low birth weight). However, this was not observed with Remicade monotherapy. The potential contribution of exposure versus severity of underlying disease in these outcomes remains unclear.
Janssen’s first experience with combining and linking population-based databases established a proof of principle and led to other, similar collaborations for Remicade (Table 4) (NPTR, HSTCL-Kaiser Permanente) and other drugs in its portfolio (Janssen, data on file).183
How can rare and unexpected events be detected?
Perhaps the biggest challenge to establishing the safety profile of a drug is the detection of rare and idiosyncratic safety events. Even large databases are not of sufficient size to detect, analyze, or quantify such events, so special methods are needed. Janssen, like all pharmaceutical companies, relies on healthcare professionals to report to the company any ADRs occurring in patients treated with their drugs (known as spontaneous reporting). When the company identifies a possible safety signal, a full investigation is triggered. This includes trending analyses of events reported to the company over time (itself a complex process requiring accurate estimates of patients exposed to the drug by indication, age, geography, etc, for which Remicade demanded special methods), disproportionality analyses comparing rates with Remicade with rates observed across all other drugs in the company safety database, detailed clinical follow-up on each case report, a literature review of the adverse event, analyses from the clinical trial database and PMC registries, and consultation with experts. If necessary, mechanistic and preclinical studies to help understand the observation are also conducted. Finally, regular communication with health authorities is required.
A well-known example of signal detection from spontaneous reports is that of tuberculosis (TB) in patients treated with Remicade. Both the phase II/III trials and the PMCs included sites located primarily in North America and Western Europe, and enrolled a population of patients at relatively low risk of being exposed to TB. In 2000, once use of Remicade in clinical practice started to grow and included patients from a broad geographic area, Janssen began receiving spontaneous reports of disseminated TB in patients treated with Remicade.22,23,160 The company’s ensuing investigation found independent research ongoing at that time, which revealed a role of TNF in maintaining granulomas, which are responsible for sequestration of Mycobacterium tuberculosis and sustaining latency of disease, and in the induction of apoptosis of TB-infected cells. Inhibition of TNF disrupts these immune responses and likely leads to breakdown of granulomas and reactivation of latent TB infections.184
Perhaps the most prominent example of detecting a rare adverse event with Remicade is that of HSTCL, a very rare and usually fatal form of lymphoma. It occurs predominantly in adolescent and young males, in whom widespread use of Remicade began only with its approval in pediatric CD in 2006. After several cases were reported,36 Janssen committed to monitoring its various data sources, including the PMC registries, for additional cases. However, given the rarity of HSTCL and its occurrence in a specific, small subgroup of patients, no cases were found in the existing registries and it was unlikely that many would be found in the future. The company then agreed with health authorities to conduct three PMCs specifically for the study of HSTCL: 1) an analysis to calculate the incidence of HSTCL in IBD using the claims databases of Kaiser Permanente, a US health insurance company60 2) a study of the incidence and prevalence of HSTCL in the general population and in IMIDs using PALGA, a nationwide network and registry of histopathology and cytopathology centers in the Netherlands59 and 3) a Janssen study to collect samples from IBD patients diagnosed with HSTCL to identify biomarkers that may allow earlier identification of a patient’s risk of developing HSTCL. After extensive review of each case of HSTCL in IBD patients both with and without treatment with Remicade identified in Janssen’s global safety database, the FDA’s MedWatch system, and the medical literature, analysis showed that nearly all cases occurred in patients treated either with thiopurines only or with a combination of TNF blockade and thiopurines, with few cases in patients receiving TNF inhibitor monotherapy.36,60 The Remicade prescribing information was updated accordingly to warn prescribers of the possible risk.
As with the examples of lymphoma and pregnancy, the HSTCL experience demonstrated the challenges of identifying sufficient cases of rare and even low-frequency events despite routine pharmacovigilance activities and a PMC program comprised of studies with both large populations and long-term follow-up. Combined with a lack of information on possible confounding factors, there are limits to the ability to draw firm conclusions from these data sources on the quantitative (eg, incidence rates) and qualitative (eg, event subtypes, predictors, latency, severity) aspects of these risks. Nevertheless, the Remicade postmarketing safety program has provided and continues to provide essential information needed to assess the risk profile of Remicade, and has confirmed the overall positive benefit–risk balance originally observed in the clinical development program.
What has Remicade’s safety profile taught us about the physiological role of TNF and the effects of TNF blockade?
The profile of safety events associated with TNF blockade has been of great interest to researchers from an immunological perspective. Commonly compared with conventional immunosuppression, TNF blockade differs from it mechanistically. Where immunosuppressants prevent activation and proliferation of lymphocytes, TNF inhibitors, by virtue of blocking a single cytokine, are a targeted approach to modulating immune responses and therefore are not broadly immunosuppressive.93,94 While its safety profile is similar to that of conventional immunosuppressants, specific blockade of TNF does not have the off-target effects of many immunosuppressants or steroids, nor is there evidence of cumulative toxicity with TNF blockade.66,185 It is noteworthy that blockade of a cytokine so central to host defense can be blocked without a greater and broader risk of adverse effects.
The study of Remicade has confirmed and refined much of what was known about TNF. It plays a complex role in innate immunity, particularly against mycobacterial, invasive fungal, and (other) intracellular infections, and, not surprisingly, its blockade is associated with a small increase in these and other opportunistic infections.93 Similarly, reports of reactivation of hepatitis B infections confirm the role of TNF in the immune response against certain viral pathogens.
Less clear is the extent of effect that TNF inhibitors have on immune surveillance against malignancies, including lymphomas. Whereas data from large registries of patients with RA (see Table 4) indicate that disease activity, rather than TNF antagonism, is likely to be responsible for the observed increased risk of lymphoma, data from large IBD studies182 suggest a possible increase, albeit small, associated with TNF blockade. Specific malignancies, such as Merkel cell carcinoma, melanoma, and leukemia, have also been associated with TNF blockade. Investigation on these and other malignancies continues.
Use of TNF blockade has also led to an unexpected finding, paradoxical adverse events, defined as the occurrence of a pathological condition that usually responds to a drug. For example, TNF inhibitors are effective in psoriasis, yet psoriatic skin lesions have been reported as an adverse event following TNF inhibitor treatment for other conditions.186 The mechanism of this is unclear, but one hypothesis posits that inhibiting TNF may, in certain settings, increase the production of other cytokines, such as interferons and IL-17s, underlying the role of alternative inflammatory pathways in the pathogenesis of these diseases.186 Another line of investigation suggests that TNF blockade could lead to the downregulation of T-regulatory cells which would normally modulate TNF-driven inflammation.187,188 It is a fascinating observation and yet another example of the use of TNF blockade bringing new insights to our understanding of the immune system.
Once identified, how are safety risks to be mitigated?
The final step in the risk-management process consists of the actions a company should take to mitigate safety risks once they have been identified. Routine measures include updating the product’s prescribing information with a warning and description of the event, and inclusion in the company’s standard medical information, educational, and promotional materials. When these measures are insufficient, a manufacturer can implement additional risk mitigation activities. In the case of Remicade, several risks have led to the need for such extra measures. An example is the risk of mycobacterial and invasive fungal infections, where cases of delayed or missed diagnoses were still occurring in TNF inhibitor-treated patients despite routine risk-mitigation measures. In response, Janssen implemented a global educational program on the risk of these infections. In the USA, this took the form of a formal Risk Evaluation and Mitigation Strategy, as per FDA requirements, where communication and education tools for physicians were developed and deployed emphasizing the importance of maintaining a high index of suspicion for opportunistic infections in Remicade-treated patients, the need for screening measures and possible pretreatment, and how these infections should be managed. In Europe, Janssen and its partners were required not only to implement educational activities, but also to demonstrate their effectiveness by evaluating levels of awareness among physicians of the risks and how to reduce them. There were also educational efforts aimed at patients, the most important of which was a reminder card to be given at the beginning of therapy notifying them of specific risks and the need to inform all healthcare providers responsible for their care that they are on Remicade. After review of the outcomes of these programs and consultation with the health authorities, the formal requirements of the additional measures have been fulfilled and Janssen continues routine activities to this day.
Changes in treatment paradigms due to the advent of TNF blockade
Therapeutic advances often lead to changes in treatment paradigms. In the case of TNF inhibitors, the impact was extensive as it occurred across several relatively common diseases which are chronic, progressive, and often accompanied by disability and severe comorbidities, and where standard treatments had remained unchanged for years. The breakthrough of Remicade and other TNF inhibitors set off a revolution, leading to a reassessment of conventional therapies, changes in treatment goals and new treatment strategies. These paradigm shifts occurred in parallel across indications, as the learnings from one informed the others.
When Remicade first became available, its use was limited to patients who had failed conventional therapies. Clinical experience confirmed the efficacy observed in the phase II/III trials and clinicians soon learned how to optimize the results further. Better patient management methods mitigated safety risks such as infusion reactions and infections.157,158,189,190 By treating the underlying pathophysiology of disease, TNF blockade offered the possibility of treating multiple manifestations with a single therapy. Many patients could be treated for years at a time, with both sustained response and an acceptable tolerability,66,185 a welcome departure from standard treatments.191,192
Within a few years, the positive experience with Remicade in clinical practice led researchers to ask several questions about expanding its use. If treatment stops structural damage and avoids long-term, irreversible sequelae of disease, why wait until the structural damage is manifest before using it? Would treating early, perhaps even as an alternative to conventional treatments, avoid the damage in the first place?
Could TNF blockade be used earlier in the disease course?
IBD
In CD, initial studies exploring the use of Remicade in patients early in the course of disease were conducted by independent investigators. GETAID, a French IBD study group, investigated Remicade in steroid-refractory patients and D’Haens et al conducted the Step-up-Top-down study evaluating Remicade in early CD.193,194 Both studies suggested that early use may be beneficial, and led to SONIC, a Janssen-sponsored phase III study, which compared Remicade with the standard maintenance treatment in CD, azathioprine (AZA, a thiopurine), rather than testing its effect where this treatment had failed.40
In SONIC, CD patients naïve to AZA and early in the course of disease (median duration of disease 2.4 years) were randomized to one of three treatment arms: AZA alone, Remicade alone, and the combination of AZA and Remicade. The primary endpoint was the new gold standard of efficacy, corticosteroid-free clinical remission at week 26, and was achieved in 30.0% of patients receiving AZA alone, 44.4% of patients receiving Remicade alone (P=0.006), and 56.8% of patients receiving the combination (P<0.001) (Figure 6). Importantly, endoscopic assessment of the healing of mucosal lesions demonstrated a similar pattern of results across the three groups. The incidence of ADA was lower in the combination group (0.9%) compared with Remicade alone (14.6%), consistent with that observed with the combination of Remicade and MTX in RA. Safety was comparable in all three groups. These results showing the superiority of both Remicade regimens versus AZA triggered a reassessment of the use and timing of not only TNF blockade but also AZA in CD.
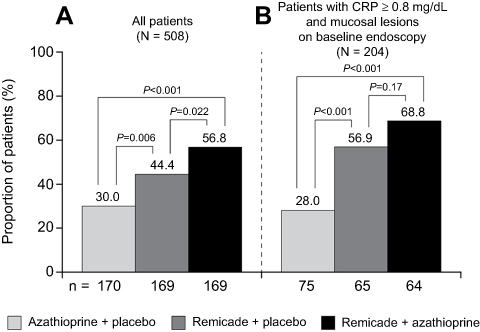 |
Figure 6 SONIC trial: primary endpoint (steroid-free remission) at week 26 in (A) all patients and (B) patients with elevated CRP (≥0.8 mg/dL) and mucosal lesions at baseline. (A) From Colombel JF, Sandborn WJ, Reinisch W, et al. Infliximab, azathioprine, or combination therapy for Crohn’s disease. N Engl J Med. 2010;362:1383–1395. Copyright © 2010 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society;40 and (B) from Colombel JF, Sandborn WJ, Reinisch W, et al. Infliximab, azathioprine, or combination therapy for Crohn’s disease. N Engl J Med. 2010;362(15):1383–1395. Supplementary Material. Copyright © 2010 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.84 Abbreviation: CRP, C-reactive protein. |
A study of similar design in UC, UC-SUCCESS, showed comparable results.195 The researchers explored whether Remicade, either as monotherapy or in combination with AZA, was superior to AZA alone in patients who were failing corticosteroids and naïve to AZA. The combination was superior to either agent alone for induction of steroid-free remission at week 16, the primary endpoint, and resulted in greater improvement in quality of life of UC patients. Remicade, both as monotherapy and in combination with AZA, was more likely to achieve a clinical response and mucosal healing than in those treated with AZA alone.195 Remicade was also assessed as an alternative to standard therapies, intravenous steroids and cyclosporine, in the management of severe, acute UC, an emergent, life-threatening form of the disease usually resulting in colectomy. Remicade proved to be better than intravenous steroids196 and equal to cyclosporine197 at avoiding colectomy, but with the benefits of improved tolerability, ease of use, and the possibility of use as a maintenance therapy.
Rheumatology indications
In RA, the exploration of earlier use followed a similar trajectory. The first signal that Remicade could be used early in the course of disease was from an ATTRACT subgroup analysis of patients with shorter duration of disease (<3 years) and therefore lower baseline levels of joint damage than the trial’s full population. Results suggested that Remicade provided long-term benefits in this subgroup by preventing radiographic progression and preserving joint integrity,198 and supported the hypothesis of a window of opportunity to avoid joint damage early in the disease, after which it would appear and begin to impair normal function.199–201
This analysis led to ASPIRE, a phase III trial to assess formally the use of Remicade in patients with severely active RA naïve to MTX and disease duration <3 years, in which patients were randomized to either MTX alone or MTX in combination with Remicade for 1 year.30,202 This trial demonstrated that the combination provided both clinical and structural benefits early in the course of disease compared to MTX alone. A similar trial, RESPOND, was performed with Remicade in patients with PsA and who were naïve to MTX, helping to establish the efficacy of early use of TNF blockade in that disease as well.41
Earlier use of TNF blockade in AS was explored in another independent study from Barkham et al, where patients with very early inflammatory back pain (indicative of future AS) were randomized to Remicade or placebo.39 Compared with placebo at week 20, patients receiving Remicade had a greater reduction in inflammatory lesions, as measured by total MRI score, and better scores on clinical and patient-reported outcomes. This was the first study to show that TNF blockade was effective for early sacroiliitis or “preradiographic” axial spondyloarthritis (axSpA), ie, without structural damage on X-ray and therefore not (yet) AS.
Although the results from these trials were positive, there were several reasons not to implement earlier use of TNF blockade. While generally well tolerated, it is not risk free and its use in larger populations with earlier, possibly milder forms of disease raised questions about the balance of benefits versus risks. Moreover, less costly conventional therapies, while perhaps inferior to TNF blockade, were still effective drugs, particularly in RA and psoriasis, where the benefits of MTX, for example, are substantial. Clinicians then asked two questions, the answers to which might guide decisions on early use of TNF blockade. Were there predictors of response which might help to identify the subgroups of patients most likely to need and to benefit from early TNF blockade (eg, patients at high risk of rapid progression) or not likely to respond to TNF blockade at all? Had conventional therapies and treatment strategies been optimized or was there still room to improve them?
Are there predictors of response which can identify patients who will benefit from (early) treatment with TNF inhibition? Are there predictors of lack or loss of response?
Predictors of response
SONIC was relevant not only because it identified the optimal maintenance therapy for early symptomatic control, but also because of its implications in the context of CD as a progressive disease, where early suppression of the underlying inflammation could lead to avoidance of permanent structural bowel damage characteristic of its later stages. Subgroup analyses from SONIC revealed that patients with objective signs of inflammation at baseline, specifically mucosal lesions and/or elevated levels of CRP, had relatively greater benefit of therapy compared with the trial population as a whole. In the subgroup with both characteristics, 69% of patients receiving combination therapy were in steroid-free remission at week 26 (Figure 6).86 This confirmed that it was now possible not only to identify but also to treat patients at early risk of progression due to inflammation.
In early RA, an analysis from ASPIRE evaluated a range of baseline patient characteristics and found that swollen joint counts, rheumatoid factor positivity, and increased serum markers of inflammation (erythrocyte sedimentation rate [ESR] and/or CRP) were all predictors of both disease progression and response to TNF blockade. Visual matrices based on these parameters were then developed to identify populations at high risk of rapid radiographic progression (Figure 7). In the trial, these patients were unlikely to respond to MTX alone, but benefited from its use combined with Remicade.203 ASPIRE was helpful in understanding when MTX alone was sufficient and when early combination therapy was warranted.
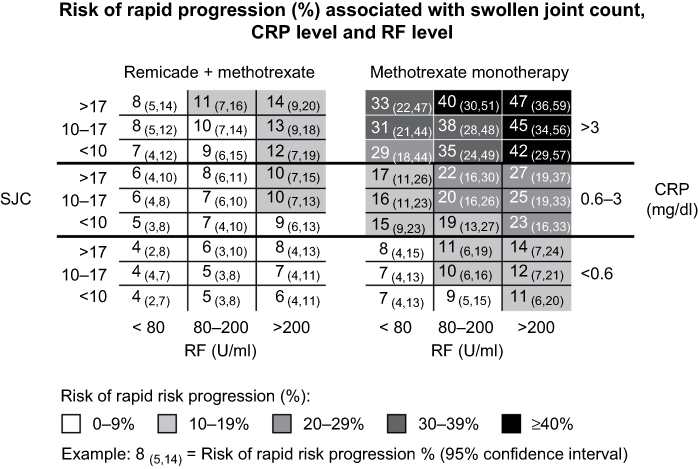 |
Figure 7 Prediction model of rapid radiographic progression in RA: matrix model from the ASPIRE trial in MTX-naïve, early RA. Rapid progression was defined as a threshold change in modified Sharp/van der Heijde (SHS) score of ≥5 units/year. From Vastesaeger N, Xu S, Aletaha D, et al. A pilot risk model for the prediction of rapid radiographic progression in rheumatoid arthritis. Rheumatology. 2009;48(9):1114–1121. doi:10.1093/rheumatology/kep155. Reproduced by permission of Oxford University Press on behalf of the British Society for Rheumatology.203 Abbreviations: CRP, C-reactive protein; MTX, methotrexate; RA, rheumatoid arthritis; RF, rheumatoid factor; SJC, swollen joint count, 28 joints assessed. |
The Barkham study in axSpA had shown that early sacroiliitis could be treated before substantial damage to the spine had occurred, providing an impetus for early use of TNF blockade.39 However, for this early use to be practical, there was a need for better differential diagnosis of axSpA from other causes of back pain. Research found that patients with back pain >3 months beginning before the age of 45 years in the presence of human leukocyte antigen-B27 positivity or sacroiliitis combined with typical features of SpA (eg, extraspinal manifestations, response to NSAIDs, family history) were likely to develop axSpA. This led to the formal definition of a new disease entity, non-radiographic axial spondyloarthritis (nr-axSpA), now recognized for clinical trial purposes and by health authorities as a new indication.204,205 In the case of AS, therefore, the option to treat early with TNF inhibitors had led to a fundamental reassessment of disease.
Possible reasons for lack or loss of response
As efficacious as TNF blockade is, not all patients respond and many patients who do will lose their response over time. In Remicade clinical trials across all indications, as well as in trials of other TNF inhibitors, 20–40% of patients did not respond to induction therapy (primary non-response) (Figure 4), and in clinical use another 5–15% have been observed to lose response annually thereafter (secondary non-response).206,207 Considerable effort has gone into understanding and predicting non-response in the hope of further optimizing therapy with Remicade and other TNF inhibitors.
Causes for this treatment failure generally fall into two categories.208 The first is insufficient dosing. This can be due to several reasons, such as fast drug clearance, high inflammatory burden, and ADAs, and can frequently be resolved with dose escalation.81,82,209 To identify possible underdosing, both monitoring of serum drug levels and ADA testing have become common in the management of patients on TNF inhibitors, particularly in IBD.210–212
The second cause of lack or loss of response is more interesting from a pathophysiological perspective, and more difficult to address: the inflammation behind the disease in these patients is perhaps not (or not consistently) driven by TNF. In IBD, for example, the European Crohn's and Colitis Organisation (ECCO), a leading professional organization in the field, held a workshop on reasons for failure of TNF inhibition and hypothesized that TNF-independent (or not fully dependent) pathways may be involved in inflammation or that TNF blockade even induces or promotes pro-inflammatory pathways, reminiscent of the reasons for the failure of TNF blockade in other diseases or for paradoxical adverse events.213 New agents targeting different inflammatory mediators have already proven to be effective, including specifically in patients who do not respond to or have lost response to TNF inhibition, such as inhibitors of IL-12/23 and integrin-α4β7 in IBD,212,214 of IL-6, T-cells, and B-cells in RA,215,216 and of IL-17 and IL-23 in psoriasis.153,217 Independent studies aimed at identifying predictors of response to TNF inhibition have evaluated a wide range of genetic, serum protein, or transcriptomic markers. Several potential predictors of response have been identified and may be helpful for guiding clinical decision in the future.218,219
Had conventional therapies been fully optimized? Treatment strategy trials in RA and CD
The availability of highly efficacious TNF inhibitors for RA renewed interest among rheumatologists in optimizing csDMARDs, alone or in combination, and the use of accelerated step-up therapy when they were not working. Confident in the knowledge that treatment alternatives with biologics existed if conventional therapies failed, physicians now monitored patients frequently in the expectation of high levels of efficacy, a practice known as “tight control”, and moved on to the next line of therapy if their treatment goal had not been met (and reduced treatment if it had), a practice now known as “treat-to-target”.220–222 The question at hand was which of the various treatment strategies available, now including TNF inhibitors, was most likely to treat RA patients successfully?
This question was answered with the BeSt trial, an independent study conducted in the Netherlands, whose elegant design compared the safety and efficacy of the key therapeutic strategies in RA available to rheumatologists.223 BeSt was a 10-year, multicenter clinical trial of csDMARD- and TNF inhibitor-naïve, early (≤2 years) RA patients randomized to one of four treatment strategies: sequential csDMARD monotherapy (group 1), step-up csDMARD combination therapy (group 2), initial csDMARD combination therapy with tapered high-dose prednisone (group 3), and initial combination therapy of Remicade plus MTX (group 4). Patients were assessed every 3 months, “tight control”, and treatment was adjusted according to a predefined treatment progression in each arm based on the ability to achieve low disease activity (LDA), a score ≤2.4 on the RA Disease Activity Score (DAS), “treat-to-target”. The overall objective of the study was to compare a step-up approach (groups 1 and 2) with an aggressive approach (groups 3 and 4). Of note, groups 1, 2, and 3 could also receive Remicade plus MTX at some point during follow-up via progression to later steps of treatment if LDA was not achieved. Patients in group 4 could stop Remicade and even MTX if certain LDA and remission targets were achieved and sustained.223,224
At the end of year 1, mean scores on the Health Assessment Questionnaire (HAQ) were better with early aggressive treatment compared to the early step-up approach (P=0.009). Median increases in total radiographic joint score were higher in groups 1 and 2 than in groups 3 and 4 (P<0.001) and lowest of all in group 4, with no significant differences in adverse event or withdrawal rates among groups.223 Annual assessments during the trial revealed how graduation to the next steps in the treatment algorithm based on achieving LDA or not led to smaller amounts of joint progression each successive year, highlighting the possibility of avoiding such damage by the use of an aggressive strategy early in the treatment timeline (Figure 8).225
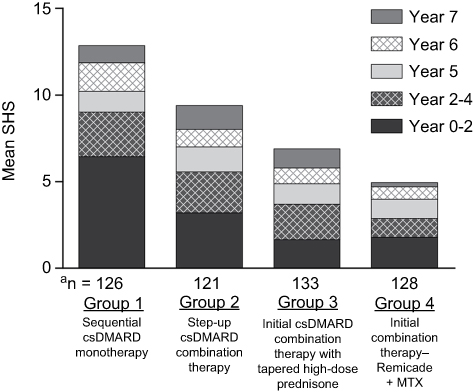 |
Figure 8 Cumulative progression of joint damage through year 7 in BeSt. Reproduced with permission from Van den Broek M, Lems WF, Allaart CF. BeSt practice: the success of early-targeted treatment in rheumatoid arthritis. Clin Exp Rheumatol. 30(4 Suppl 73):S35–S38. Copyright © 2012 Clinical and Experimental Rheumatology SAS.225 aSample size represents the population originally randomized to each arm. Numbers of patients at the end of each year of follow-up differed. Abbreviations: csDMARD, conventional synthetic disease-modifying anti-rheumatic drug; MTX, methotrexate; SHS, Sharp/van der Heijde score. |
After a decade of a treat-to-target approach in the BeSt study, radiographic progression remained low in all arms (mean increase in SHS at year 10 was estimated to be 14.2, 14.1, 14.6, and 8.9 in groups 1 to 4, respectively, P=0.046 for group 1 vs group 4) and functional ability approached normality for all groups (mean HAQ 0.6). Patients originally randomized to the combination of Remicade plus MTX (group 4) had the lowest structural damage and disability rates61 and the lowest need to progress in the assigned treatment algorithm. Early aggressive treatment in groups 3 and 4 was not associated with worse safety outcomes, probably due to the overall improvement of symptoms of disease. The rate of mortality in the trial across all four arms was not increased compared with the normal population, which was unexpected for this disease and possibly related to effective suppression of inflammation. This unique study of treatment strategies in patients with early RA showed that a tight-control, treat-to-target approach prevents long-term progression of structural damage and disability, and that an early aggressive approach leads to better results than an early step-up approach.
In IBD, gastroenterologists asked questions similar to those asked in BeSt. Beyond SONIC and UC-SUCCESS, several trials of TNF inhibitors have investigated a variety of treatment strategies, including accelerated step-up therapy versus conventional treatment strategies (Step Up, Top Down, REACT),194,226 managing patients based on clinical symptoms alone or serum levels of drug (TAXIT, TAILORIX),227,228 basing treatment decisions on symptoms alone or in combination with objective markers of disease, such as fecal calprotectin (FCal) and CRP (CALM),229 and how best to prevent recurrence post-surgery (PREVENT, POCER).42,230 The results of these trials and others have led to conclusions parallel to those drawn from BeSt.
How has TNF blockade influenced treatment guidelines and health authority requirements for next-generation drugs?
Many of the changes in practice patterns made as a result of the use of TNF inhibitors have become standard and can be found in the treatment guidelines for each of their indicated diseases (Table 5). Across several leading professional organizations in IBD, such as ECCO and the American College of Gastroenterology (ACG),212,214,252 CD and UC are recognized to be progressive diseases leading to irreversible damage in a substantial number of patients, driven in large part by inflammation that can be suppressed with TNF blockade. Goals of therapy are now steroid-free remission, rather than clinical response, and should include reductions in objective measures of inflammation, such as CRP and FCal, healing of the inflamed mucosa,253 and ultimately, improved quality of life. Moreover, physicians should evaluate patients individually and frequently, stratify by risk, and take a proactive approach to achieving treatment goals. Should these goals not be met within designated times, eg, 6 months, then the treatment should be optimized.232,254 Use of steroids should be limited and monotherapy with azathioprine is now discouraged.255
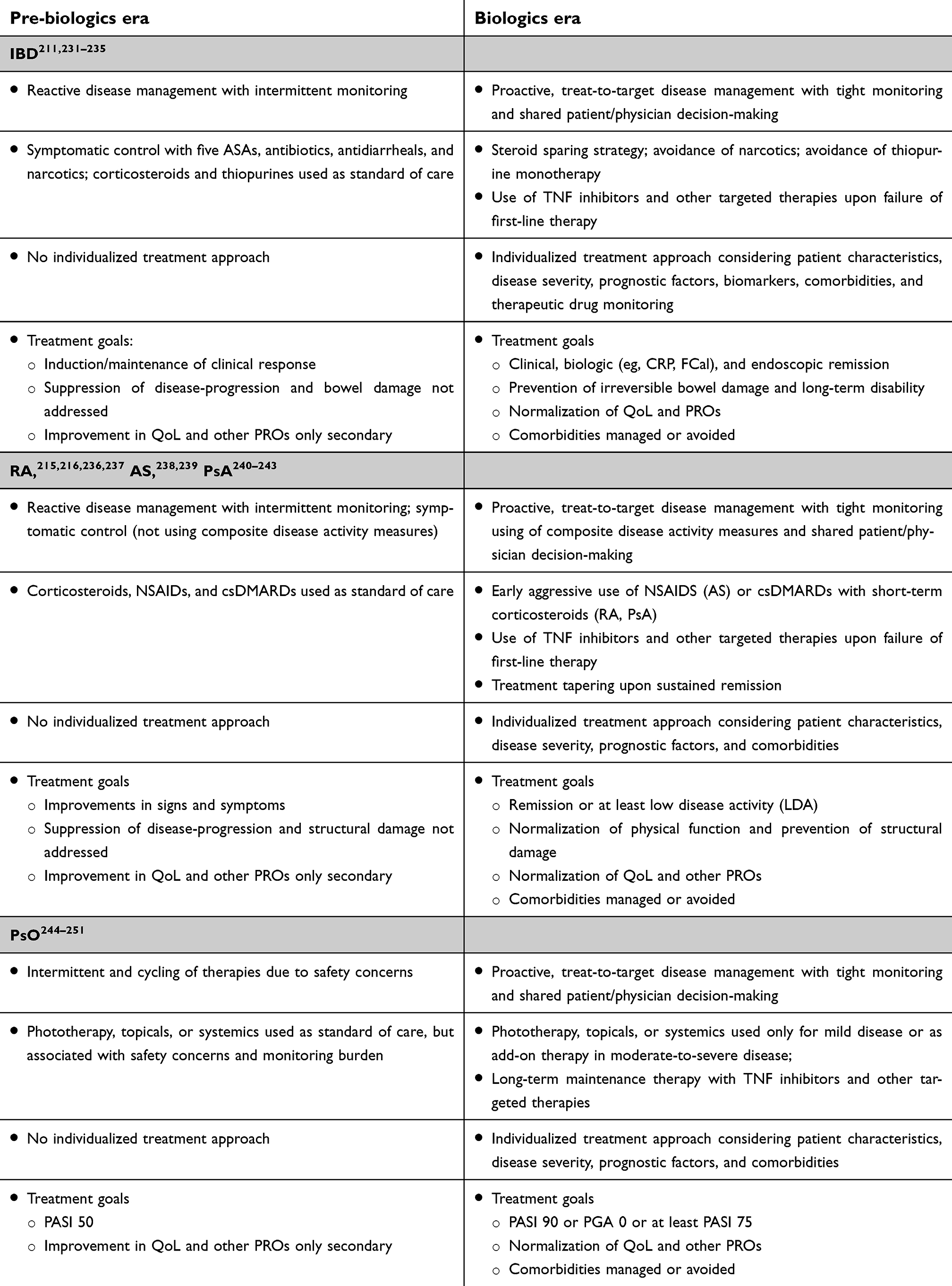 |
Table 5 Changes in treatment practices in the TNF era |
In RA, leading professional organizations, such as the European League Against Rheumatism (EULAR) and ACR,215,216 now incorporate the general principles of tight control and treat-to-target in their treatment guidelines: early assessment of prognostic factors to weigh risk of disease progression; immediate use of MTX in combination with short-term steroids as a preferred first-line therapy; assessment of response to therapy every 3–6 months, including objective markers of inflammation, with LDA, if not clinical remission, and inhibition of joint progression as treatment goals; and continued and quick progression to the next line of therapy when treatment goals are not met.
Even in psoriasis, where Remicade is often overlooked because of the preference for subcutaneous TNF inhibitors and next-generation biologics, it was recognized early on for its quick onset of action and high degree of efficacy.106 The advent of biologics has led to more stringent treatment goals and earlier treatment.246 Prior to their availability, a 50% reduction in a patient’s Psoriasis Area and Severity Index (PASI) score, measuring the proportion of skin covered with psoriatic plaques, was the goal of therapy. With today’s new therapies, an improvement of this limited magnitude would likely trigger a change in therapy, with the goal being at least a 75% reduction in PASI and an improved quality of life (Dermatology Life Quality Index ≤5). With the next-generation biologics, IL-23 and IL-17 inhibitors, 90% PASI reductions and even full clearance of disease are realistic goals. While this disease lacks a long-term structural damage component found in the other indications of TNF blockade, it is associated with compromised psychosocial status, which the evidence suggests might be limited or even reversed with successful treatment.256 Treatment guidelines now reflect tight-control and treat-to-target approaches with use of TNF inhibitors and other biologics recommended if phototherapy and conventional systemic agents fail to provide an adequate response.241 Some experts go further and consider that with the long-term experience gathered with the use of biologics, no reason supports reserving them for second-line use.185,217,257
The improvement in outcomes offered by TNF inhibitors has also contributed to changes in the requirements of health authorities for the evaluation of new treatments. This is most evident in RA, where, for example, both the EMA and the FDA require greater improvement in symptoms than before TNF inhibitors were available. Where both had previously required evidence of improvement in single or composite measures of disease activity (eg, tender and swollen joint counts, ACR20), remission or measures of LDA should now be considered as evidence of efficacy.258–260 The availability of improved therapies has provided a rationale for limiting the exposure of patients to placebo or ineffective therapies for a prolonged period of time; treatment longer than 12 weeks should include either an active comparator as the control or provisions for escape to rescue treatment for patients with active disease. Both health authorities recognize that demonstrating a benefit on structural damage is increasingly difficult because joint erosions are now unlikely to develop in this setting.
In both CD and UC the pattern is similar. Major health authorities today consider clinical remission, as opposed to response, to be an appropriate endpoint on which to evaluate a drug’s efficacy. Moreover, while necessary, symptomatic response is no longer adequate. Both the FDA and the EMA now also require evidence of endoscopic improvement of disease, in recognition of mucosal inflammation as the underlying cause.261–265
Summary and conclusion
In 1992, when Janssen first provided the cA2 antibody to the Kennedy Institute to test the hypothesis that inflammation in RA is driven by TNF and could be suppressed with TNF blockade, no one could have foreseen the contributions the drug would make to science and medicine over the subsequent 20+ years. As described in this review, the learnings from Remicade can be divided into five categories, ranging from principles of drug development to practice of medicine to our understanding of the immune system.
First, Remicade was one of the first mAbs to establish the proof of principle of their use as the highly targeted therapies imagined at the time of their introduction in 1975, and was the first to establish a role for them in chronic diseases. Early studies with Remicade demonstrated how to administer mAbs while limiting their immunogenicity, enabling them to be used long term. Combined with the success of other early mAbs, eg, rituximab, trastuzumab, and related constructs, eg, etanercept, they have come of age, with more than 60 approved for use targeting a variety of specific mediators, over 30 of which are in chronic diseases.266,267 At least 250 mAbs are in development today.268 Research has already moved from chimeric to fully human mAbs and continues to advance to next-generation agents, such as bispecific antibodies and antibody–drug conjugates.266
Second, the proof of principle that targeted therapy could be an effective strategy to treat IMIDs was first demonstrated with Remicade. Despite the complexity of the immune system, initial studies confirmed that, at least in some diseases, the inflammatory response works in cascades; and inhibition of a single cytokine initiating a cascade can have a profound effect on disease. Targeted blockade of a single cytokine can yield a safety profile generally consistent with that of conventional immunosuppressants, yet without their cumulative and off-target toxicity risks and with a degree of efficacy well beyond what they provide. Today, most new drugs in development, biologics and small molecules alike, target specific mediators of disease.
Third, the Remicade postmarketing safety surveillance program, including both PMCs and routine pharmacovigilance, has been pioneering in establishing methodological and operational requirements for safety studies and in setting standards for design, scale, rigor, and duration of follow-up, specifically in the real-world setting. The program provided new understanding of the risks of conventional therapies and of the diseases themselves, knowledge that is essential when judging the relative risks and benefits of new therapies. Innovative solutions, such as use of big data, claims databases, and combining various data sources, have been established as feasible and acceptable methods of safety monitoring. Strong working relationships with a large network across academia are required to access existing data sources and to seek independent expertise in study design, conduct, and interpretation of results. Similarly, cooperation and transparency with health authorities are imperative, as is a commitment to publish results. Above all, adequate resources need to be allocated by the company, often for years, to handle the volume of work and to provide the expertise required across multiple disciplines.
Fourth, Remicade, surely one of the most rigorously studied biologics in medicine today, has taught us much about the role of TNF in the immune system and in the pathophysiology of several diseases. TNF drives the inflammation found in several IMIDs and does so in early as well as late disease. Conversely, the lack of benefit of TNF blockade in a number of diseases where it is elevated indicates that TNF elevation does not always mean that inflammation is mediated by or dependent on TNF. The studies where TNF blockade was not successful were not without scientific benefit, as they provided insights into the pathophysiology of these diseases. The effective use of TNF blockade in some diseases has given us hope that targeted therapy might work in others, and has fueled research that has already led to the approval of drugs targeting other components of the immune system (cytokines, receptors, integrins). Study of TNF blockade has also confirmed much of what was suspected about TNF’s role in the immune system, in particular regarding infection, and broadened it further, as seen in demyelinating disorders, CHF, and paradoxical adverse events. Importantly, studies with Remicade and subsequent TNF inhibitors demonstrated that certain cytokines with a broad function in the immune system can be blocked without triggering an unacceptable level of safety risk.
The final category of Remicade’s contributions is its influence on the practice of medicine. Across six different diseases, the improved efficacy of Remicade and other TNF inhibitors compared with conventional therapies has led to more ambitious treatment goals. Rather than just reducing symptoms, TNF blockade was able to address the underlying pathophysiology driving these diseases, thereby modifying their course, such that irreversible damage to the joints and the bowel could be avoided. This led to earlier treatment, use of predictors of disease progression and response, tighter monitoring and the use of treat-to-target approaches, and a reassessment of conventional therapies to optimize their safety and efficacy. In the case of rheumatology, csDMARDs, in particular MTX, experienced new life as therapies. In the case of IBD, the limitations of steroids and thiopurines became apparent. Clinical research on TNF inhibitors has led to different ways of assessing disease, the most vivid example being AS, where use early in the course of the disease led to the definition of a new disease entity, non-radiographic axial SpA. Lastly, the availability of TNF blockade has led to higher standards for assessing new therapies.
Even today, the list of learnings is still growing. Several studies in the Remicade PMC program are still ongoing (Table 4). Other studies continue to refine the use of TNF inhibitors in such areas as combinations with conventional therapies, predictors of response, dose titration, and possible discontinuation after successful, sustained response. Independent investigators are conducting proof-of-concept studies with Remicade as treatment for various conditions not yet evaluated, including bipolar disorder, hypertension, and type 1 diabetes (Table 2). The study of the effect of TNF blockade on long-term comorbidities continues, prompted by the hypothesis that the inflammation driving IMIDs is also a contributor to these comorbidities, ie, cardiovascular complications or colorectal cancer in UC.
Perhaps Remicade’s most important legacy is the impact it has had on the lives of many of the estimated 3 million patients it has been used to treat (Janssen, data on file). Recent studies across its indications have found that patients treated today with Remicade have lower levels of disease activity and shorter duration of disease at the time of initiation of treatment than in the past, resulting in lower rates of disability and improved quality of life. When combined with the patients receiving other TNF inhibitors, this amounts to a substantial decrease in the burden of some of the most common IMIDs in our society.269–273 Amid Remicade’s other considerable contributions to science and medicine, on which this manuscript has focused, this contribution to public health is perhaps the most important of all.
Abbreviation list
ACR, American College of Rheumatology; ACR20, 20% reduction in ACR response criteria; ADA, anti-drug antibody; ADCC, antibody-dependent cellular cytotoxicity; ADR, adverse drug reaction; AS, ankylosing spondylitis; axSpA, axial spondyloarthritis; AZA, azathioprine; BASDAI, Bath Ankylosing Spondylitis Disease Activity Index; CD, Crohn’s disease; CDAI, Crohn's Disease Activity Index; CHF, congestive heart failure; CNS, central nervous system; CRP, C-reactive protein; csDMARD, conventional synthetic disease-modifying anti-rheumatic drug; DAS, disease activity score; DLQI, Dermatology Life Quality Index; ECCO, European Crohn's and Colitis Organisation; EMA, European Medicines Agency; Fab, (antibody) fragment antigen-binding; Fc, (antibody) fragment constant; FCal, fecal calprotectin; FDA, US Food and Drug Administration; GM-CSF, granulocyte-macrophage colony-stimulating factor; HAQ, Health Assessment Questionnaire; HSTCL, hepatosplenic T-cell lymphoma; IBD, inflammatory bowel disease; IgG, immunoglobulin G, antibody type G; IL, interleukin; IMID, immune-mediated inflammatory disease; JRA, juvenile rheumatoid arthritis; LDA, low disease activity; mAb, monoclonal antibody; MRI, magnetic resonance imaging; MS, multiple sclerosis; MTX, methotrexate; NHL, non-Hodgkin’s lymphoma; NK, natural killer; nr-axSpA, non-radiographic axial spondyloarthritis; NSAID, non-steroidal anti-inflammatory drug; NYHA, New York Heart Association; p75, another name for TNFR2; PASI, Psoriasis area and severity index; PBO, placebo; PGA, Physician Global Assessment; PK, pharmacokinetics; PMC, postmarketing commitment; PRO, patient-reported outcome; PsA, psoriatic arthritis; PsO, psoriasis; QoL, quality of life; q4w, every 4 weeks; q8w, every 8 weeks; RA, rheumatoid arthritis; SHS, Sharp/van der Heijde score; SJC, swollen joint count; SLE, systemic lupus erythematosus; SpA, spondyloarthritis; sTNF, soluble TNF; TB, tuberculosis; tmTNF, transmembrane TNF; TNF, tumor necrosis factor; TNFR1, TNF receptor 1, also known as p55; TNFR2, TNF receptor 2, also known as p75; UC, ulcerative colitis; VEGF, vascular endothelial growth factor.
Acknowledgments
Editorial assistance was provided by Ashfield Healthcare Communications and funded by Janssen Biologics. The authors acknowledge the following contributors for their subject matter expertise: Daniel Baker, Sandra Belzner, Angels Bermejo Colom, Freddy Cornillie, Merle Elloso, Suzan Esslinger, Long Long Gao, Andrew Greenspan, Jens Heile, Soteroulla Ioannou, James Izanec, Russell Ludowyke, Tony (Tianshu) Ma, Flore-Anne Mayne, Salvatore Morello, Francois Nantel, Alessandra Oortwijn, Marijo Otero Lobato, Bethany Paxson, Antonio Pires, Michael Plotnick, Sharon Popik, Alla Rhoge, Pawel Tulinksi, Roger Van Kruchten, and Rob Vermeulen; Brenda Loux provided assistance with graphic design. The authors thank the patients, clinical practitioners, and staff over the past 20 years for their participation in clinical trials involving Remicade. Janssen provided financial support for the development of this article.
Disclosure
RM, AG, and IA are current employees of Janssen Pharmaceuticals and stockholders in Johnson & Johnson, Janssen’s parent company. TS is a former Janssen employee and a stockholder in, and receives a pension from Johnson & Johnson. The authors report no other conflicts of interest in this work.
References
1. REMICADE® [Package Insert]. Horsham, PA: Janssen Pharmaceuticals Inc; 2013.
2. Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci U S A. 1975;72(9):3666–3670. doi:10.1073/pnas.72.6.2091
3. Köhler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256(5517):495–497. doi:10.1038/256495a0
4. Morrison SL, Johnson MJ, Herzenberg LA, Oi VT. Chimeric human antibody molecules: mouse antigen-binding domains with human constant region domains. Pro Natl Acad Sci U S A. 1984;81(21):6851–6855. doi:10.1073/pnas.81.21.6851
5. Boulianne GL, Hozumi N, Shulman MJ. Production of functional chimeric mouse/human antibody. Nature. 1984;312(5995):643–646. doi:10.1038/312643a0
6. Reichert J. Monoclonal antibodies in the clinic. Nat Biotechnol. 2001;19(9):819–822. doi:10.1038/nbt0901-819
7. Brennan FM, Chantry D, Jackson A, et al. Inhibitory effect of TNF alpha antibodies on synovial cell interleukin-1 production in rheumatoid arthritis. Lancet. 1989;2(8657):244–247.
8. Knight D, Trinh H, Le J, et al. Construction and initial characterization of a mouse-human chimeric anti-TNF antibody. Mol Immunol. 1993;30(16):1443–1453. doi:10.1016/0161-5890(93)90106-L
9. Zimmerman J, Dillon K, Campbell W, et al. Phase I/II1trial of cA2, a chimeric anti-TNF antibody in patients with sepsis [abstract]. Intensive Care Med. 1994;20(Suppl 1):S151.
10. Vilcek J, Feldmann M. Historical review: cytokines as therapeutics and targets of therapeutics. Trends Pharmacol Sci. 2004;25(4):201–209. doi:10.1016/j.tips.2004.09.009
11. Elliott MJ, Maini RN, Feldmann M, et al. Treatment of rheumatoid arthritis with chimeric monoclonal antibodies to tumor necrosis factor alpha. Arthritis Rheum. 1993;36(12):1681–1690.
12. Derkx B, Taminiau J, Radema S, et al. Tumour-necrosis-factor antibody treatment in crohn’s disease. Lancet. 1993;342(8864):173–174. doi:10.1016/0140-6736(93)91375-V
13. Elliott MJ, Maini RN, Feldmann M, et al. Randomised double-blind comparison of chimeric monoclonal antibody to tumour necrosis factor alpha (cA2) versus placebo in rheumatoid arthritis. Lancet. 1994;344(8930):1105–1110.
14. van Dullemen HM, van Deventer SJ, Hommes DW, et al. Treatment of crohn’s disease with anti-tumor necrosis factor chimeric monoclonal antibody (cA2). Gastroenterology. 1995;109(1):129–135. doi:10.1016/0016-5085(95)90277-5
15. Targan SR, Hanauer SB, van Deventer SJ, et al. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for crohn’s disease. Crohn’s disease cA2 study group. N Engl J Med. 1997;337(15):1029–1035. doi:10.1056/NEJM199710093371502
16. Food and Drug Administration [FDA]. Product approval for infliximab [Crohn’s disease], August 24, 1998. https://www.accessdata.fda.gov/drugsatfda_docs/appletter/1998/inflcen082498L.htm.
17. Food and Drug Administration [FDA]. Product approval for Enbrel. https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=103795. Accessed February 13, 2019.
18. Food and Drug Administration [FDA]. Product approval for Infliximab [several indications] https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=103772. Accessed February 13, 2019.
19. Maini R, St Clair EW, Breedveld F, et al. Infliximab (chimeric anti-tumour necrosis factor alpha monoclonal antibody) versus placebo in rheumatoid arthritis patients receiving concomitant methotrexate: a randomised phase III trial. ATTRACT study group. Lancet. 1999;354(9194):1932–1939. doi:10.1016/S0140-6736(99)00434-1
20. Present DH, Rutgeerts P, Targan S, et al. Infliximab for the treatment of fistulas in patients with Crohn’s disease. N Engl J Med. 1999;340(18):1398–1405. doi:10.1056/NEJM199905133401909
21. Lipsky PE, van der Heijde D, St. Clair EW, et al. Infliximab and methotrexate in the treatment of rheumatoid arthritis. N Engl J Med. 2000;343(22):1594–1602. doi:10.1056/NEJM200011303432202
22. Keane J, Gershon S, Wise RP, et al. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N Engl J Med. 2001;345(15):1098–1104. doi:10.1056/NEJMoa011110
23. Gomez-Reino J, Carmona L, Valverde V, Mola EM, Montero MD. Treatment of rheumatoid arthritis with tumor necrosis factor inhibitors may predispose to significant increase in tuberculosis risk: a multicenter active-surveillance report. Arthritis Rheum. 2003;48(8):2122–2127. doi:10.1002/art.11137
24. Mohan N, Edwards ET, Cupps TR, et al. Demyelination occurring during anti-tumor necrosis factor alpha therapy for inflammatory arthritides. Arthritis Rheum. 2001;44(12):2862–2869.
25. Braun J, Brandt J, Listing J, et al. Treatment of active ankylosing spondylitis with infliximab: a randomised controlled multicentre trial. Lancet. 2002;359(9313):1187–1193.
26. Hanauer SB, Feagan BG, Lichtenstein GR, et al. Maintenance infliximab for crohn’s disease: the ACCENT I randomised trial. Lancet. 2002;359(9317):1541–1549. doi:10.1016/S0140-6736(02)07571-2
27. Food and Drug Administration [FDA]. Product approval for Humira. https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=125057.
28. European Medicines Agency. https://www.ema.europa.eu/.
29. Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT. Anti-TNF therapy against congestive heart failure investigators. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: results of the anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation. 2003;107(25):3133–3140.
30. St Clair EW, van der Heijde DM, Smolen JS, et al. Combination of infliximab and methotrexate therapy for early rheumatoid arthritis: a randomized, controlled trial. Arthritis Rheum. 2004;50(11):3432–3443. doi:10.1002/art.20568
31. Sands BE, Anderson FH, Bernstein CN, et al. Infliximab maintenance therapy for fistulizing crohn’s disease. N Engl J Med. 2004;350(9):876–885. doi:10.1056/NEJMoa030815
32. Antoni C, Krueger GG, de Vlam K, et al. Infliximab improves signs and symptoms of psoriatic arthritis: results of the IMPACT 2 trial. Ann Rheum Dis. 2005;64(8):1150–1157. doi:10.1136/ard.2004.030221
33. Van der Heijde D, Dijkmans B, Geusens P, et al. Efficacy and safety of infliximab in patients with ankylosing spondylitis: results of a randomized, placebo-controlled trial (ASSERT). Arthritis Rheum. 2005;52(2):582–591. doi:10.1002/art.20852
34. Rutgeerts P, Sandborn WJ, Feagan BG, et al. Infliximab for induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2005;353(23):2462–2476. doi:10.1056/NEJMoa050516
35. Reich K, Nestle FO, Papp K, et al. Infliximab induction and maintenance therapy for moderate-to-severe psoriasis: a phase III, multicentre, double-blind trial. Lancet. 2005;366(9494):1367–1374. doi:10.1016/S0140-6736(05)67528-9
36. Kotlyar DS, Blonski W, Diamond RH, Wasik M, Lichtenstein GR. Hepatosplenic T-cell lymphoma in inflammatory bowel disease: a possible thiopurine-induced chromosomal abnormality. Am J Gastroenterol. 2010;105(10):2299–2301. doi:10.1038/ajg.2010.213
37. Ruperto N, Lovell DJ, Cuttica R, et al. A randomized, placebo-controlled trial of infliximab plus methotrexate for the treatment of polyarticular-course juvenile rheumatoid arthritis. Arthritis Rheum. 2007;56(9):3096–3106. doi:10.1002/art.22862
38. Menter A, Feldman SR, Weinstein GD, et al. A randomized comparison of continuous vs. intermittent infliximab maintenance regimens over 1 year in the treatment of moderate-to-severe plaque psoriasis. J Am Acad Dermatol. 2007;56(1):
39. Barkham N, Keen HI, Coates LC, et al. Clinical and imaging efficacy of infliximab in HLA-B27-positive patients with magnetic resonance imaging-determined early sacroiliitis. Arthritis Rheum. 2009;60(4):946–954. doi:10.1002/art.24408
40. Colombel JF, Sandborn WJ, Reinisch W, et al. Infliximab, azathioprine, or combination therapy for Crohn’s disease. N Engl J Med. 2010;362(15):1383–1395. doi:10.1056/NEJMoa0904492
41. Baranauskaite A, Raffayová H, Kungurov NV, et al. Infliximab plus methotrexate is superior to methotrexate alone in the treatment of psoriatic arthritis in methotrexate-naive patients: the RESPOND study. Ann Rheum Dis. 2011;71(4):541–548. doi:10.1136/ard.2011.152223
42. Regueiro M, Feagan BG, Zou B, et al. Infliximab reduces endoscopic, but not clinical, recurrence of Crohn’s disease after ileocolonic resection. Gastroenterology. 2016;150(7):1568–1578. doi:10.1053/j.gastro.2016.02.072
43. Lichtenstein GR, Feagan BG, Cohen RD, et al. Infliximab for crohn’s disease: more than 13 years of real-world experience. Inflamm Bowel Dis. 2018;24(3):490–501. doi:10.1093/ibd/izx072
44. Broms G, Granath F, Ekbom A, et al. Low risk of birth defects for infants whose mothers are treated with anti-tumor necrosis factor agents during pregnancy. Clin Gastroenterol Hepatol. 2016;14(2):
45. Choquette D, Faraawi R, Chow A, Rodrigues J, Bensen WJ, Nantel F. Incidence and management of infusion reactions to infliximab in a prospective real-world community registry. J Rheumatol. 2015;42(7):1105–1111. doi:10.3899/jrheum.150470
46. Choquette D, Rahman P, Chow A, et al. Consolidated long-term safety of infliximab in inflammatory arthritis from a prospective, observational registry. Ann Rheum Dis. 2018;77:A1380.
47. D’Haens G, Reinisch W, Colombel JF, et al. Five-year safety data From ENCORE, a European observational safety registry for adults with Crohn’s disease treated with infliximab [Remicade®] or conventional therapy. J Crohns Colitis. 2017;11(6):680–689.
48. Panes J, Lindsay J, Teich N, et al. Five-year safety data from OPUS, a European observational safety registry for adults with ulcerative colitis treated with originator infliximab [Remicade®] or conventional therapy. J Crohns Colitis. 2019. doi:10.1093/ecco-jcc/jjz048 [Epub ahead of print].
49. Strober B, Gooderham M, de Jong E, et al. Depressive symptoms, depression, and the effect of biologic therapy among patients in Psoriasis Longitudinal Assessment and Registry (PSOLAR). J Am Acad Dermatol. 2018;78(1):70–80. doi:10.1016/j.jaad.2017.08.051
50. Hyams JS, Dubinsky MC, Baldassano RN, et al. Infliximab is not associated with increased risk of malignancy or hemophagocytic lymphohistiocytosis in pediatric patients with inflammatory bowel disease. Gastroenterology. 2017;152(8):1901–1914.e3. doi:10.1053/j.gastro.2017.02.004
51. Greenberg JD, Kremer JM, Curtis JR, et al. Tumour necrosis factor antagonist use and associated risk reduction of cardiovascular events among patients with rheumatoid arthritis. Ann Rheum Dis. 2011;70(4):576–582. doi:10.1136/ard.2010.139832
52. Wolfe F, Michaud K. Biologic treatment of rheumatoid arthritis and the risk of malignancy: analyses from a large US observational study. Arthritis Rheum. 2007;56(9):2886–2895. doi:10.1002/art.22862
53. Neovius M, Arkema EV, Olsson H, et al. Drug survival on TNF inhibitors in patients with rheumatoid arthritis comparison of adalimumab, etanercept and infliximab. Ann Rheum Dis. 2015;74(2):354–360. doi:10.1136/annrheumdis-2013-204128
54. Hernandez MV, Sanmarti R, Canete JD, et al. Cutaneous adverse events during treatment of chronic inflammatory rheumatic conditions with tumor necrosis factor antagonists: study using the Spanish registry of adverse events of biological therapies in rheumatic diseases. Arthritis Care Res (Hoboken). 2013;65(12):2024–2031. doi:10.1002/acr.22096
55. Mercer LK, Galloway JB, Lunt M, et al. Risk of lymphoma in patients exposed to antitumour necrosis factor therapy: results from the British society for rheumatology biologics register for rheumatoid arthritis. Ann Rheum Dis. 2017;76(3):497–503. doi:10.1136/annrheumdis-2016-209389
56. Strangfeld A, Hierse F, Rau R, et al. Risk of incident or recurrent malignancies among patients with rheumatoid arthritis exposed to biologic therapy in the German biologics register RABBIT. Arthritis Res Ther. 2010;12(1):R5. doi:10.1186/ar2904
57. Augustin M, Spehr C, Radtke MA, et al. German psoriasis registry PsoBest: objectives, methodology and baseline data. J Dtsch Dermatol Ges. 2014;12(1):48–57.
58. Warren RB, Smith CH, Yiu ZZN, et al. Differential drug survival of biologic therapies for the treatment of psoriasis: a prospective observational cohort study from the British Association of Dermatologists Biologic Interventions Register (BADBIR). J Invest Dermatol. 2015;135(11):2632–2640. doi:10.1038/jid.2015.208
59. Montgomery M, van Santen MM, Biemond BJ, Diamond RH, Pals ST. Hepatosplenic T-cell lymphoma: a population-based study assessing incidence and association with immune-mediated disease. Gastroenterol Hepatol (N Y). 2015;11(3):160–163.
60. Herrinton L, Liu L, Weng X, et al. Role of thiopurine and anti-TNF therapy in lymphoma in inflammatory bowel disease. Am J Gastroenterol. 2011;106:2146–2153. doi:10.1038/ajg.2011.283
61. Markusse IM, Akdemir G, Dirven L, et al. Long-term outcomes of patients with recent-onset rheumatoid arthritis after 10 years of tight controlled treatment: a randomized trial. Ann Intern Med. 2016;164(8):523–531. doi:10.7326/M15-0919
62. Beutler B, Greenwald D, Hulmes JD, et al. Identity of tumour necrosis factor and the macrophage-secreted factor cachectin. Nature. 1985;316(6028):552–554. doi:10.1038/316552a0
63. Dayer JM, Beutler B, Cerami A. Cachectin/tumor necrosis factor stimulates collagenase and prostaglandin E2 production by human synovial cells and dermal fibroblasts. J Exp Med. 1985;162(6):2163–2168. doi:10.1084/jem.162.3.781
64. Ali T, Kaitha S, Mahmood S, Ftesi A, Stone J, Bronze MS. Clinical use of anti-TNF therapy and increased risk of infections. Drug Healthc Patient Saf. 2013;5:79–99. doi:10.2147/DHPS.S28801
65. Monaco C, Nanchahal J, Taylor P, et al. Anti-TNF therapy: past, present and future. Int Immunol. 2015;27(1):55–62. doi:10.1093/intimm/dxv003
66. Calabrese L. The yin and yang of tumor necrosis factor inhibitors. Cleve Clin J Med. 2006;73(3):251–256.
67. Zelova H, Hosek J. TNF-α signalling and inflammation: interactions between old acquaintances. Inflamm Res. 2013;62(7):641–651. doi:10.1007/s00011-012-0548-1
68. Kim D, Cheon J. Pathogenesis of inflammatory bowel disease and recent advances in biologic therapies. Immune Netw. 2017;17(1):25–40. doi:10.4110/in.2017.17.1.25
69. Yang S, Wang J, Brand DD, Zheng SG. Role of TNF-TNF receptor 2 signal in regulatory T cells and its therapeutic implications. Front Immunol. 2018;9:784. doi:10.3389/fimmu.2018.00784
70. Connor V. Anti-TNF therapies: a comprehensive analysis of adverse effects associated with immunosuppression. Rheumatol Int. 2009;31(3):327–337. doi:10.1007/s00296-009-1292-x
71. Spooner CE, Markowitz NP, Saravolatz LD. The role of tumor necrosis factor in sepsis. Clin Immunol Immunopathol. 1992;62(1 Pt 2):S11–S17.
72. Van Deventer SJ. Tumour necrosis factor and crohn’s disease. Gut. 1997;40(4):443–448. doi:10.1136/gut.40.6.761
73. Feldmann M, Brennan FM, Maini RN. Role of cytokines in rheumatoid arthritis. Annu Rev Immunol. 1996;14:397–440. doi:10.1146/annurev.immunol.14.1.397
74. Hall S. Monoclonal antibodies at age 20: promise at last? Science. 1995;270(5238):915–916.
75. Cerami A. The value of failure: the discovery of TNF and its natural inhibitor erythropoietin. J Intern Med. 2011;269(1):8–15. doi:10.1111/j.1365-2796.2010.02319.x
76. Clark MA, Plank LD, Connolly AB, et al. Effect of a chimeric antibody to tumor necrosis factor-alpha on cytokine and physiologic responses in patients with severe sepsis–a randomized, clinical trial. Crit Care Med. 1998;26(10):1650–1659. doi:10.1097/00003246-199810000-00016
77. Elliott MJ, Maini RN, Feldmann M, et al. Repeated therapy with monoclonal antibody to tumour necrosis factor alpha (cA2) in patients with rheumatoid arthritis. Lancet. 1994;344(8930):1125–1127.
78. Rutgeerts P, D’Haens G, Targan S, et al. Efficacy and safety of retreatment with anti-tumor necrosis factor antibody (infliximab) to maintain remission in Crohn’s disease. Gastroenterology. 1999;117(4):761–769. doi:10.1016/S0016-5085(99)70332-X
79. Maini RN, Breedveld FC, Kalden JR, et al. Therapeutic efficacy of multiple intravenous infusions of anti-tumor necrosis factor alpha monoclonal antibody combined with low-dose weekly methotrexate in rheumatoid arthritis. Arthritis Rheum. 1998;41(9):1552–1563. doi:10.1002/1529-0131(199809)41:9<1552::AID-ART5>3.0.CO;2-W
80. Maini RN, Breedveld FC, Kalden JR, et al. Sustained improvement over two years in physical function, structural damage, and signs and symptoms among patients with rheumatoid arthritis treated with infliximab and methotrexate. Arthritis Rheum. 2004;50(4):1051–1065. doi:10.1002/art.20159
81. Rahman MU, Strusberg I, Geusens P, et al. Double-blinded infliximab dose escalation in patients with rheumatoid arthritis. Ann Rheum Dis. 2007;66(9):1233–1238. doi:10.1136/ard.2006.065995
82. Rutgeerts P, Diamond RH, Bala M, et al. Scheduled maintenance treatment with infliximab is superior to episodic treatment for the healing of mucosal ulceration associated with Crohn’s disease. Gastrointest Endosc. 2006;63(3):433–442. doi:10.1016/j.gie.2005.08.011
83. Rutgeerts P, Feagan BG, Lichtenstein GR, et al. Comparison of scheduled and episodic treatment strategies of infliximab in Crohn’s disease. Gastroenterology. 2004;126(2):402–413. doi:10.1053/j.gastro.2003.11.014
84. Baert F, Noman M, Vermeire S, et al. Influence of immunogenicity on the long-term efficacy of infliximab in Crohn’s disease. N Engl J Med. 2003;348(7):601–608. doi:10.1056/NEJMicm020037
85. Hanauer SB, Wagner CL, Bala M, et al. Incidence and importance of antibody responses to infliximab after maintenance or episodic treatment in Crohn’s disease. Clin Gastroenterol Hepatol. 2004;2(7):542–553. doi:10.1016/S1542-3565(04)00238-1
86. Colombel JF, Sandborn WJ, Reinisch W, et al. Infliximab, azathioprine, or combination therapy for Crohn’s disease [supplement]. N Engl J Med. 2010;362(15):1383–1395. doi:10.1056/NEJMoa0904492
87. Westhovens R, Yocum D, Han J, et al. The safety of infliximab, combined with background treatments, among patients with rheumatoid arthritis and various comorbidities: a large, randomized, placebo-controlled trial. Arthritis Rheum. 2006;54(4):1075–1086. doi:10.1002/art.21734
88. Antoni CE, Kavanaugh A, Kirkham B, et al. Sustained benefits of infliximab therapy for dermatologic and articular manifestations of psoriatic arthritis: results from the infliximab multinational psoriatic arthritis controlled trial (IMPACT). Arthritis Rheum. 2005;52(4):1227–1236. doi:10.1002/art.20967
89. Lichtenstein GR, Yan S, Bala M, et al. Remission in patients with Crohn’s disease is associated with improvement in employment and quality of life and a decrease in hospitalizations and surgeries. Am J Gastroenterol. 2004;99(1):91–96. doi:10.1111/j.1572-0241.2004.10370.x
90. Lichtenstein GR, Yan S, Bala M, Blank M, Sands BE. Infliximab maintenance treatment reduces hospitalizations, surgeries, and procedures in fistulizing Crohn’s disease. Gastroenterology. 2005;128(4):862–869. doi:10.1053/j.gastro.2005.01.048
91. Charles P, Elliott MJ, Davis D, et al. Regulation of cytokines, cytokine inhibitors, and acute-phase proteins following anti-TNF-alpha therapy in rheumatoid arthritis. J Immunol. 1999;163(3):1521–1528.
92. Taylor PC, Peters AM, Paleolog E, et al. Reduction of chemokine levels and leukocyte traffic to joints by tumor necrosis factor alpha blockade in patients with rheumatoid arthritis. Arthritis Rheum. 2000;43(1):38–47. doi:10.1002/1529-0131(200001)43:1<38::AID-ANR6>3.0.CO;2-L
93. Tracey D, Klareskog L, Sasso EH, Salfeld JG, Tak PP. Reduction of chemokine levels and leukocyte traffic to joints by tumor necrosis factor: a comprehensive review. Pharmacol Ther. 2008;117(2):244–279. doi:10.1016/j.pharmthera.2007.10.001
94. Oikonomopoulos A, van Deen WK, Hommes D. Anti-TNF antibodies in inflammatory bowel disease: do we finally know how it works? Curr Drug Targets. 2013;14(12):1421–1432. doi:10.2174/13894501113149990164
95. Sandborn WJ, Hanauer SB, Katz S, et al. Etanercept for active crohn’s disease: a randomized, double-blind, placebo-controlled trial. Gastroenterology. 2001;121(5):1088–1094. doi:10.1053/gast.2001.28674
96. Sandborn WJ, Feagan BC, Stoinov S, et al. Certolizumab pegol for the treatment of Crohn’s disease. N Eng J Med. 2007;357(3):228–238. doi:10.1056/NEJMoa067594
97. Rigby WFC. Drug insight: different mechanisms of action of tumor necrosis factor antagonists–passive-aggressive behavior? Nat Clin Pract Rheumatol. 2007;3(4):227–233. doi:10.1038/ncprheum0438
98. Van Den Bosch F, Kruithof E, De Vos M, De Keyser F, Mielants H. Crohn’s disease associated with spondyloarthropathy: effect of TNF-alpha blockade with infliximab on articular symptoms. Lancet. 2000;356(9244):1821–1822. doi:10.1016/S0140-6736(00)03239-6
99. Oh CJ, Das KM, Gottlieb AB. Treatment with anti-tumor necrosis factor alpha (TNF-alpha) monoclonal antibody dramatically decreases the clinical activity of psoriasis lesions. J Am Acad Dermatol. 2000;42(5 Pt 1):829–830. doi:10.1067/mjd.2000.105948
100. Mayer L. Evolving paradigms in the pathogenesis of IBD. J Gastroenterol. 2010;45(1):9–16. doi:10.1007/s00535-009-0138-3
101. Van der Heijde D, Kavanaugh A, Gladman DD, et al. Infliximab inhibits progression of radiographic damage in patients with active psoriatic arthritis through one year of treatment: results from the induction and maintenance psoriatic arthritis clinical trial 2. Arthritis Rheum. 2007;56(8):2698–2707. doi:10.1002/art.22862
102. Baraliakos X, Haibel H, Listing J, Sieper J, Braun J. Continuous long-term anti-TNF therapy does not lead to an increase in the rate of new bone formation over 8 years in patients with ankylosing spondylitis. Ann Rheum Dis. 2014;73(4):710–715. doi:10.1136/annrheumdis-2014-205310
103. Haroon N, Inman RD, Learch TJ, et al. The impact of tumor necrosis factor α inhibitors on radiographic progression in ankylosing spondylitis. Arthritis Rheum. 2013;65(10):2645–2654.
104. Krueger GG, Papp KA, Stough DB, et al. A randomized double-blind, placebo-controlled phase III study evaluating efficacy and tolerability of 2 courses of alefacept in patients with chronic plaque psoriasis. J Am Acad Dermatol. 2002;47(6):821–833. doi:10.1067/mjd.2002.127247
105. Gordon KB, Papp KA, Hamilton TK, et al. Efalizumab for patients with moderate to severe plaque psoriasis: a randomized control trial. JAMA. 2003;290(23):3073–3080. doi:10.1001/jama.290.23.3073
106. Reich K, Burden AD, Eaton JN, Hawkins NS. Efficacy of biologics in the treatment of moderate to severe psoriasis: a network meta-analysis of randomized controlled trials. Br J Dermatol. 2012;166(1):179–188. doi:10.1111/j.1365-2133.2011.10583.x
107. Naveau S, Chollet-Martin S, Dharancy S, et al. A double-blind randomized controlled trial of infliximab associated with prednisolone in acute alcoholic hepatitis. Hepatology. 2004;39(5):1390–1397. doi:10.1002/hep.20206
108. Jacobi A, Antoni C, Manger B, Schuler G, Hertl M. Infliximab in the treatment of moderate to severe atopic dermatitis. J Am Acad Dermatol. 2005;52(3 Pt 1):522–526. doi:10.1016/j.jaad.2004.11.022
109. Weiler-Normann C, Schramm C, Quaas A, et al. Infliximab as a rescue treatment in difficult-to-treat autoimmune hepatitis. J Hepatol. 2013;58(3):529–534. doi:10.1016/j.jhep.2012.11.010
110. Hibi T, Hirohata S, Kikuchi H, et al. Infliximab therapy for intestinal, neurological, and vascular involvement in Behcet disease: efficacy, safety, and pharmacokinetics in a multicenter, prospective, open-label, single-arm phase 3 study. Medicine (Baltimore). 2016;95(24):e3863. doi:10.1097/MD.0000000000004864
111. Sfikakis PP, Theodossiadis PG, Katsiari CG, et al. Effect of infliximab on sight-threatening panuveitis in Behçet’s disease. Lancet. 2001;358(9278):295–296. doi:10.1016/S0140-6736(01)05497-6
112. Hoffman GS, Cid MC, Rendt-Zagar KE, et al. Infliximab for maintenance of glucocorticosteroid-induced remission of giant cell arteritis: a randomized trial. Ann Intern Med. 2007;146(9):621–630. doi:10.7326/0003-4819-146-9-200705010-00004
113. de Menthon M, Cohen P, Pagnoux C, et al. Infliximab or rituximab for refractory Wegener’s granulomatosis: long-term follow up. A prospective randomised multicentre study on 17 patients. Clin Exp Rheumatol. 2011;29(1 Suppl 64):S63–S71.
114. Salvarani C, Macchioni P, Manzini C, et al. Infliximab plus prednisone or placebo plus prednisone for the initial treatment of polymyalgia rheumatic: a randomized trial. Ann Intern Med. 2007;146(9):631–639. doi:10.7326/0003-4819-146-9-200705010-00005
115. Couriel DR, Saliba R, de Lima M, et al. A phase III study of infliximab and corticosteroids for the initial treatment of acute graft-versus-host disease. Biol Blood Marrow Transplant. 2009;15(12):1555–1562. doi:10.1016/j.bbmt.2009.08.003
116. Grant A, Gonzalez T, Montgomery MO, Cardenas V, Kerdel FA. Infliximab therapy for patients with moderate to severe hidradenitis suppurativa: a randomized, double-blind, placebo-controlled crossover trial. J Am Acad Dermatol. 2010;62(2):205–217. doi:10.1016/j.jaad.2009.06.050
117. Burns JC, Best BM, Mejias A, et al. Infliximab treatment of intravenous immunoglobulin-resistant kawasaki disease. J Pediatr. 2008;153(6):833–838. doi:10.1016/j.jpeds.2008.06.011
118. Masuda H, Kobayashi T, Hachiya A, et al. Infliximab for the treatment of refractory kawasaki disease: a nationwide survey in Japan. J Pediatr. 2018;195:115–120. doi:10.1016/j.jpeds.2017.10.013
119. van Oosten BW, Barkhof F, Truyen L, et al. Increased MRI activity and immune activation in two multiple sclerosis patients treated with the monoclonal anti-tumor necrosis factor antibody cA2. Neurology. 1996;47(6):1531–1534. doi:10.1212/WNL.47.6.1531
120. Hall RP, Fairley J, Woodley D, et al. A multicentre randomized trial of the treatment of patients with pemphigus vulgaris with infliximab and prednisone compared with prednisone alone. Br J Dermatol. 2015;172(3):760–768. doi:10.1111/bjd.13350
121. Schiffenbauer A, Garg M, Castro C, et al. A randomized, double-blind, placebo-controlled trial of infliximab in refractory polymyositis and dermatomyositis. Semin Arthritis Rheum. 2018;47(6):858–864. doi:10.1016/j.semarthrit.2017.10.010
122. Hommes DW, Erkelens W, Ponsioen C, et al. A double-blind, placebo-controlled, randomized study of infliximab in primary sclerosing cholangitis. J Clin Gastroenterol. 2008;42(5):522–526. doi:10.1097/MCG.0b013e3181662426
123. Baughman RP, Drent M, Kavuru M, et al. Infliximab therapy in patients with chronic sarcoidosis and pulmonary involvement. Am J Respir Crit Care Med. 2006;174(7):795–802. doi:10.1164/rccm.200510-1659PP
124. Vorselaars AD, Crommelin HA, Deneer VH. Effectiveness of infliximab in refractory FDG PET-positive sarcoidosis. Eur Respir J. 2015;46(1):175–185. doi:10.1183/09031936.00227014
125. Denton CP, Engelhart M, Tvede N, et al. An open-label pilot study of infliximab therapy in diffuse cutaneous systemic sclerosis. Ann Rheum Dis. 2009;68(9):1433–1439. doi:10.1136/ard.2008.096123
126. Mariette X, Ravaud P, Steinfeld S, et al. Inefficacy of infliximab in primary Sjogren’s syndrome: results of the randomized, controlled Trial of Remicade in Primary Sjogren’s Syndrome (TRIPSS). Arthritis Rheum. 2004;50(4):1270–1276. doi:10.1002/art.20146
127. Aringer M, Graninger WB, Steiner G, Smolen JS. Safety and efficacy of tumor necrosis factor alpha blockade in systemic lupus erythematosus: an open-label study. Arthritis Rheum. 2004;50(10):3161–3169. doi:10.1002/art.20576
128. Aringer M, Houssiau F, Graninger WB, et al. TNF blocade in SLE: long term efficacy and safety aspects of open-label infliximab [abstract]. Ann Rheum Dis. 2008;67(Suppl II):211.
129. Uppal SS, Hayat SJ, Raghupathy R. Efficacy and safety of infliximab in active SLE: a pilot study. Lupus. 2009;18(8):690–697. doi:10.1177/0961203309102557
130. University of Chicago. Islet transplantation in patients with “Brittle” type I diabetes. Available from:https://www.clinicaltrials.gov/ct2/show/NCT01630850?term=infliximab&cond=Diabetes&rank=5.NLMidentifierNCT01630850.
131. Suhler EB, Smit JR, Wertheim MS, et al. A prospective trial of infliximab therapy for refractory uveitis. Arch Ophthalmol. 2005;123(7):903–912. doi:10.1001/archopht.123.3.321
132. Cooper C, Shafran S, Greenbloom S, et al. Single-dose infliximab in hepatitis C genotype 1 treatment-naive patients with high serum tumour necrosis factor-alpha does not influence the efficacy of pegylated interferon alpha-2b/ribavirin therapy. Can J Gastroenterol Hepatol. 2014;28(1):35–40.
133. Walker RE, Spooner KM, Kelly G, et al. Inhibition of immunoreactive tumor necrosis factor-α by a chimeric antibody in patients infected with human immunodeficiency virus type I. J Infect Dis. 1996;174(1):63–68. doi:10.1093/infdis/174.1.63
134. Charles KA, Kulbe H, Soper R, et al. The tumor-promoting actions of TNF-alpha involve TNFR1 and IL-17 in ovarian cancer in mice and humans. J Clin Invest. 2009;119(10):3011–3023. doi:10.1172/JCI39065
135. Harrison ML, Obermueller E, Maisey NR, et al. Tumor necrosis factor alpha as a new target for renal cell carcinoma: two sequential phase II trials of infliximab at standard and high dose. J Clin Oncol. 2007;25(29):4542–4549. doi:10.1200/JCO.2007.11.2136
136. Baron F, Suciu S, Amadori S, et al. Value of infliximab (Remicade®) in patients with low-risk myelodysplastic syndrome: final results of a randomized phase II trial (EORTC trial 06023) of the EORTC leukemia group. Haematologica. 2012;97(4):529–533. doi:10.3324/haematol.2011.044347
137. Jatoi A, Ritter HL, Dueck A, et al. A placebo-controlled, double-blind trial of infliximab for cancer-associated weight loss in elderly and/or poor performance non-small cell lung cancer patients (N01C9). Lung Cancer. 2010;68(2):234–239. doi:10.1016/j.lungcan.2009.06.020
138. Wiedenmann B, Malfertheiner P, Friess H, et al. A multicenter, phase II study of infliximab plus gemcitabine in pancreatic cancer cachexia. J Support Oncol. 2008;6(1):18–25.
139. Diaz LA
140. De Faria A, Ritter A, Santa Catharina A, et al. Effects of TNF-alpha inhibition on hemodynamic parameters and biomarkers in resistant hypertension: background, rationale, and design [abstract]. J Hypertension. 2016;34(e–Suppl 2):e176. doi:10.1097/01.hjh.0000491827.30246.46
141. Raison CL, Rutherford RE, Woolwine BJ, et al. A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: the role of baseline inflammatory biomarkers. JAMA Psychiatry. 2013;70(1):31–41. doi:10.1001/2013.jamapsychiatry.4
142. Lee Y, McIntyre R Anhedonia and inflammation in bipolar I/II depression: preliminary results from a 12-week, phase II, double-blind, placebo-controlled clinical trial investigating the efficacy of infliximab on anhedonia.
143. Wascher TC, Lindeman J, Sourij H, Pacini G, Roden M. Chronic treatment with infliximab does not affect insulin resistance or endothelial function in men with the metabolic syndrome - a placebo-controlled pilot trial [abstract]. J Diabetes. 2009;1(s1):A33–A82.
144. Nussenblatt RB, Byrnes G, Sen HN, et al. A randomized pilot study of systemic immunosuppression in the treatment of age-related macular degeneration with choroidal neovascularization. Retina. 2010;30(10):1579–1587. doi:10.1097/IAE.0b013e3181e7978e
145. Rennard SI, Fogarty C, Kelsen S, et al. The safety and efficacy of infliximab in moderate to severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2007;175(9):926–934. doi:10.1164/rccm.200701-094ED
146. Koninckx PR, Craessaerts M, Timmerman D, et al. Anti-TNF-alpha treatment for deep endometriosis-associated pain: a randomized placebo-controlled trial. Hum Reprod. 2008;23(9):2017–2023. doi:10.1093/humrep/dem367
147. Korhonen T, Karppinen J, Paimela L, et al. The treatment of disc-herniation-induced sciatica with infliximab: one-year follow-up results of FIRST II, a randomized controlled trial. Spine (Phila Pa 1976). 2006;31(24):2759–2766. doi:10.1097/01.brs.0000245873.23876.1e
148. Humira Injection [Package Insert]. North Chicago, IL: AbbVie Inc; 2013.
149. Lenercept Study Group. TNF neutralization in MS: results of a randomized, placebo-controlled multicenter study. Neurology. 1999;53(3):457–465. doi:10.1212/WNL.53.3.457
150. Kemanetzoglou E, Andreadou E. CNS demyelination with TNF-alpha blockers. Curr Neurol Neurosci Rep. 2017;17(4):36. doi:10.1007/s11910-017-0742-1
151. Mann DL, McMurray JJ, Packer M, et al. Targeted anticytokine therapy in patients with chronic heart failure: results of the Randomized Etanercept Worldwide Evaluation (RENEWAL). Circulation. 2004;109(13):1594–1602. doi:10.1161/01.CIR.0000124490.27666.B2
152. Judson M, Baughman RP, Costabel U, et al. Safety and efficacy of ustekinumab or golimumab in patients with chronic sarcoidosis. Eur Respir J. 2014;44(5):1296–1307. doi:10.1183/09031936.00003814
153. Blauvelt A, Papp KA, Griffiths CE, et al. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the continuous treatment of patients with moderate to severe psoriasis: results from the phase III, double-blinded, placebo- and active comparator-controlled VOYAGE 1 trial. J Am Acad Dermatol. 2017;76(3):405–417.
154. Feagan BG, Sandborn WJ, D’Haens G, et al. Induction therapy with the selective interleukin-23 inhibitor risankizumab in patients with moderate-to-severe crohn’s disease: a randomised, double-blind, placebo-controlled phase 2 study. Lancet. 2017;389(10080):1699–1709. doi:10.1016/S0140-6736(17)30570-6
155. Deng J, Younge BR, Olshen RA, Goronzy JJ, Weyand CM. Th17 and Th1 T-cell responses in giant cell arteritis. Circulation. 2010;121(7):906–915. doi:10.1161/CIRCULATIONAHA.109.872903
156. Pegoretti V, Baron W, Laman JD, Eisel ULM. Selective modulation of TNF-TNFRs signaling: insights for multiple sclerosis treatment. Front Immunol. 2018;9:925. doi:10.3389/fimmu.2018.00925
157. Cheifetz A, Mayer L. Monoclonal antibodies, immunogenicity, and associated infusion reactions. Mt Sinai J Med. 2005;72(4):250–256.
158. Lequerré T, Vittecoq O, Klemmer N, et al. Management of infusion reactions to infliximab in patients with rheumatoid arthritis or spondyloarthritis: experience from an immunotherapy unit of rheumatology. J Rheumatol. 2006;33(7):1307–1314.
159. Lahiri M, Dixon WG. Risk of infection with biologic antirheumatic therapies in patients with rheumatoid arthritis. Best Pract Res Clin Rheumatol. 2015;29(2):290–305. doi:10.1016/j.berh.2015.05.009
160. Solovic I, Sester M, Gomez-Reino JJ, et al. The risk of tuberculosis related to tumour necrosis factor antagonist therapies: a TBNET consensus statement. Eur Respir J. 2010;36(5):1185–1206. doi:10.1183/09031936.00028510
161. Yasui K. Immunity against mycobacterium tuberculosis and the risk of biologic anti-TNF-alpha reagents. Pediatr Rheumatol Online J. 2014;12:45. doi:10.1186/1546-0096-12-45
162. Kotlyar DS Lewis JD
163. Lebrec H, Ponce R, Preston BD, Iles J, Born TL, Hooper M. Tumor necrosis factor, tumor necrosis factor inhibition, and cancer risk. Curr Med Res Opin. 2015;31(3):557–574. doi:10.1185/03007995.2015.1011778
164. Lichtenstein GR, Feagan BG, Cohen RD, et al. Drug therapies and the risk of malignancy in Crohn’s disease: results from the TREAT Registry. Am J Gastroenterol. 2014;109(2):212–223. doi:10.1038/ajg.2013.441
165. Van Assche G, Vermeire S, Rutgeerts P. Safety issues with biological therapies for inflammatory bowel disease. Curr Opin Gastroenterol. 2006;22(4):370–376. doi:10.1097/01.mog.0000231810.87901.e8
166. Cush J. Safety overview of new disease-modifying antirheumatic drugs. Rheum Dis Clin North Am. 2004;30(2):237–255. doi:10.1016/j.rdc.2004.02.003
167. Sinagra E, Perricone G, Romano C, Cottone M. Heart failure and anti tumor necrosis factor-alpha in systemic chronic inflammatory diseases. Eur J Intern Med. 2013;24(5):385–392. doi:10.1016/j.ejim.2012.12.015
168. Menghini V, Arora AS. Infliximab-associated reversible cholestatic liver disease. Mayo Clin Proc. 2001;76(1):84–86. doi:10.4065/76.1.84
169. Bonacini M, Ghabril M, Bonkovsky HL. Hepatotoxicity of anti-TNF agents. Dig Dis Sci. 2014;59(5):1070–1071. doi:10.1007/s10620-014-3109-3
170. Hyams JS, Lerer T, Griffiths A, et al. Outcome following infliximab therapy in children with ulcerative colitis. Am J Gastroenterol. 2010;105(6):1430–1436. doi:10.1038/ajg.2009.759
171. Lichtenstein GR, Feagan BG, Cohen RD, et al. Serious infection and mortality in patients with crohn’s disease: more than 5 years of follow-up in the TREAT™ registry. Am J Gastroenterol. 2012;107(9):1409–1422. doi:10.1038/ajg.2012.218
172. Askling J, Fored CM, Brandt L, et al. Time-dependent increase in risk of hospitalisation with infection among Swedish RA patients treated with TNF antagonists. Ann Rheum Dis. 2007;66(10):1339–1344. doi:10.1136/ard.2006.062760
173. Listing J, Gerhold K, Zink A. The risk of infections associated with rheumatoid arthritis, with its comorbidity and treatment. Rheumatology (Oxford). 2013;52(1):53–61. doi:10.1093/rheumatology/kes305
174. Galloway JB, Hyrich KL, Mercer LK, et al. Anti-TNF therapy is associated with an increased risk of serious infections in patients with rheumatoid arthritis especially in the first 6 months of treatment: updated results from the British society for rheumatology biologics register with special emphasis on risks in the elderly. Rheumatology (Oxford). 2010;50(1):124–131. doi:10.1093/rheumatology/keq242
175. Askling J, Fored CM, Baecklund E, et al. Haematopoietic malignancies in rheumatoid arthritis: lymphoma risk and characteristics after exposure to tumour necrosis factor antagonists. Ann Rheum Dis. 2005;64(10):1414–1420. doi:10.1136/ard.2004.030221
176. Askling J, Baecklund E, Granath F, et al. Anti-tumour necrosis factor therapy in rheumatoid arthritis and risk of malignant lymphomas: relative risks and time trends in the Swedish biologics register. Ann Rheum Dis. 2009;68(5):648–653. doi:10.1136/ard.2007.085852
177. Baecklund E, Askling J, Rosenquist R, Ekbom A, Klareskog L. Rheumatoid arthritis and malignant lymphomas. Curr Opin Rheumatol. 2004;16(3):254–261. doi:10.1097/00002281-200405000-00014
178. Fiorentino D, Ho V, Lebwohl MG, et al. Risk of malignancy with systemic psoriasis treatment in the psoriasis longitudinal assessment registry. J Am Acad Dermatol. 2017;77(5):845–854.e5. doi:10.1016/j.jaad.2017.07.013
179. Siegel CA, Marden SM, Persing SM, Larson RJ, Sands BE. Risk of lymphoma associated with combination anti-tumor necrosis factor and immunomodulator therapy for the treatment of Crohn’s disease: a meta-analysis. Clin Gastroenterol Hepatol. 2009;7(8):874–881. doi:10.1016/j.cgh.2009.01.004
180. de La Forest Divonne M, Gottenberg JE, Salliot C. Safety of biologic DMARDs in RA patients in real life: a systematic literature review and meta-analyses of biologic registers. Joint Bone Spine. 2017;84(2):133–140. doi:10.1016/j.jbspin.2016.02.028
181. Baecklund E, Iliadou A, Askling J, et al. Association of chronic inflammation, not its treatment, with increased lymphoma risk in rheumatoid arthritis. Arthritis Rheum. 2006;54(3):692–701. doi:10.1002/art.21675
182. Lemaitre M, Kirchgesner J, Rudnichi A, et al. Association between use of thiopurines or tumor necrosis factor antagonists alone or in combination and risk of lymphoma in patients with inflammatory bowel disease. Jama. 2017;318(17):1679–1686. doi:10.1001/jama.2017.16071
183. European Network of Centres for Pharmacoepidemiology and pharmacovigilance. A non-interventional observational longitudinal Post-Authorization Safety Study (PASS) of SIMPONI® in treatment of ulcerative colitis using Nordic National Health Registries (MK-8259-013). http://www.encepp.eu/encepp/viewResource.htm?id=27697.
184. Raupach B, Kaufmann SH. Immune responses to intracellular bacteria. Curr Opin Immunol. 2001;13(4):417–428. doi:10.1016/S0952-7915(00)00236-3
185. Hsu S, Papp KA, Lebwohl MG, et al. Consensus guidelines for the management of plaque psoriasis. Arch Dermatol. 2012;148(1):95–102. doi:10.1001/archdermatol.2011.1410
186. Wendling D, Prati C. Paradoxical effects of anti-TNF-α agents in inflammatory diseases. Expert Rev Clin Immunol. 2014;10(1):159–169. doi:10.1586/1744666X.2014.866038
187. Fischer R, Kontermann R, Maier O. Targeting sTNF/TNFR1 signaling as a new therapeutic strategy. Antibodies. 2015;4(1):48–70. doi:10.3390/antib4010048
188. Chen X, Oppenheim JJ. Therapy: paradoxical effects of targeting TNF signalling in the treatment of autoimmunity. Nat Rev Rheumatol. 2016;12(11):625–626. doi:10.1038/nrrheum.2016.145
189. Fautrel B, Constantin A, Morel J, et al. Recommendations of the French society for rheumatology. TNFalpha antagonist therapy in rheumatoid arthritis. Joint Bone Spine. 2006;73(4):433–441. doi:10.1016/j.jbspin.2006.05.002
190. Han PD, Cohen RD. Managing immunogenic responses to infliximab: treatment implications for patients with crohn’s disease. Drugs. 2004;64(16):1767–1777. doi:10.2165/00003495-200464160-00004
191. Fidder H, Schnitzler F, Ferrante M, et al. Long-term safety of infliximab for the treatment of inflammatory bowel disease: a single-centre cohort study. Gut. 2009;58(4):501–508. doi:10.1136/gut.2008.163642
192. Vander Cruyssen B, Durez P, Westhovens R, De Keyser F. Seven-year follow-up of infliximab therapy in rheumatoid arthritis patients with severe long-standing refractory disease: attrition rate and evolution of disease activity. Arthritis Res Ther. 2010;12(3):R77. doi:10.1186/ar2997
193. Lémann M, Mary JY, Duclos B, et al. Infliximab plus azathioprine for steroid-dependent Crohn’s disease patients: a randomized placebo-controlled trial. Gastroenterology. 2006;130(4):1054–1061. doi:10.1053/j.gastro.2006.02.014
194. D’Haens G, Baert F, van Assche G, et al. Early combined immunosuppression or conventional management in patients with newly diagnosed Crohn’s disease: an open randomised trial. Lancet. 2008;371(9613):660–667. doi:10.1016/S0140-6736(08)60304-9
195. Panaccione R, Ghosh S, Middleton S, et al. Combination therapy with infliximab and azathioprine is superior to monotherapy with either agent in ulcerative colitis. Gastroenterology. 2014;146(2):392–400. doi:10.1053/j.gastro.2013.10.052
196. Järnerot G, Hertervig E, Friis-Liby I, et al. Infliximab as rescue therapy in severe to moderately severe ulcerative colitis: a randomized, placebo-controlled study. Gastroenterology. 2005;128(7):1805–1811. doi:10.1053/j.gastro.2005.03.003
197. Laharie D, Bourreille A, Branche J, et al. Ciclosporin versus infliximab in patients with severe ulcerative colitis refractory to intravenous steroids: a parallel, open-label randomised controlled trial. Lancet. 2012;380(9857):1909–1915. doi:10.1016/S0140-6736(12)61084-8
198. Breedveld FC, Emery P, Keystone E, et al. Infliximab in active early rheumatoid arthritis. Ann Rheum Dis. 2004;63(2):149–155. doi:10.1136/ard.2003.006312
199. Anderson JJ, Wells G, Verhoeven AC, Felson DT. Factors predicting response to treatment in rheumatoid arthritis: the importance of disease duration. Arthritis Rheum. 2000;43(1):22–29. doi:10.1002/(ISSN)1529-0131
200. Lard LR, Visser H, Speyer I, et al. Early versus delayed treatment in patients with recent-onset rheumatoid arthritis: comparison of two cohorts who received different treatment strategies. Am J Med. 2001;111(6):446–451. doi:10.1016/S0002-9343(01)00872-5
201. O’Dell JR. Treating rheumatoid arthritis early: a window of opportunity? Arthritis Rheum. 2002;46(2):283–285. doi:10.1002/art.10092
202. Smolen JS, Van Der Heijde DM, St Clair EW, et al. Predictors of joint damage in patients with early rheumatoid arthritis treated with high-dose methotrexate with or without concomitant infliximab: results from the ASPIRE trial. Arthritis Rheum. 2006;54(3):702–710. doi:10.1002/art.21678
203. Vastesaeger N, Xu S, Aletaha D, St Clair EW, Smolen JS. A pilot risk model for the prediction of rapid radiographic progression in rheumatoid arthritis. Rheumatology (Oxford). 2009;48(9):1114–1121. doi:10.1093/rheumatology/kep155
204. Rudwaleit M, Landewé R, van der Heijde D, et al. The development of assessment of spondyloarthritis international society classification criteria for axial spondyloarthritis (part I): classification of paper patients by expert opinion including uncertainty appraisal. Ann Rheum Dis. 2009;68(6):770–776. doi:10.1136/ard.2009.108217
205. Rudwaleit M, van der Heijde D, Landewé R, et al. The development of assessment of spondyloarthritis international society classification criteria for axial spondyloarthritis (part II): validation and final selection. Ann Rheum Dis. 2009;68(6):777–783. doi:10.1136/ard.2009.108233
206. Gisbert JP, Panés J. Loss of response and requirement of infliximab dose intensification in Crohn’s disease: a review. Am J Gastroenterol. 2009;104(3):760–767. doi:10.1038/ajg.2008.88
207. Souto A, Maneiro JR, Gómez-Reino JJ. Rate of discontinuation and drug survival of biologic therapies in rheumatoid arthritis: a systematic review and meta-analysis of drug registries and health care databases. Rheumatology (Oxford). 2016;55(3):523–534.
208. Desroches M, Louis G, Gleizes A, Krzysiek R, Emilie D. Treatment failure with antagonists of TNF-α: mechanisms and implications for the care of patients. Eur Cytokine Netw. 2010;21(4):226–231.
209. Schnitzler F, Fidder H, Ferrante M, et al. Long-term outcome of treatment with infliximab in 614 patients with Crohn’s disease: results from a single-centre cohort. Gut. 2009;58(4):492–500. doi:10.1136/gut.2008.155812
210. Ordás I, Mould DR, Feagan BG, Sandborn WJ. Anti-TNF monoclonal antibodies in inflammatory bowel disease: pharmacokinetics-based dosing paradigms. Clin Pharmacol Ther. 2012;91(4):635–646. doi:10.1038/clpt.2011.328
211. Steenholdt C, Bendtzen K, Brynskov J, Ainsworth MA. Optimizing treatment with TNF inhibitors in inflammatory bowel disease by monitoring drug levels and antidrug antibodies. Inflamm Bowel Dis. 2016;22(8):1999–2015. doi:10.1097/MIB.0000000000000772
212. Gomollón F, Dignass A, Annese V, et al. 3rd European evidence-based consensus on the diagnosis and management of crohn’s disease 2016: part 1: diagnosis and medical management. J Crohns Colitis. 2017;11(1):3–25. doi:10.1093/ecco-jcc/jjw168
213. Chowers Y, Sturm A, Sans M, et al. Report of the ECCO workshop on anti-TNF therapy failures in inflammatory bowel diseases: biological roles and effects of TNF and TNF antagonists. J Crohns Colitis. 2010;4(4):367–376. doi:10.1016/j.crohns.2010.05.011
214. Lichtenstein GR, Loftus EV, Isaacs KL, Regueiro MD, Gerson LB, Sands BE. ACG clinical guideline: management of crohn’s disease in adults. Am J Gastroenterol. 2018;113(4):481–517. doi:10.1038/s41395-018-0120-x
215. Smolen JS, Landewé R, Bijlsma J, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann Rheum Dis. 2017;76(6):960–977. doi:10.1136/annrheumdis-2016-210715
216. Singh JA, Saag KG, Bridges SL
217. Gisondi P, Del Giglio M, Girolomoni G. Treatment approaches to moderate to severe psoriasis. Int J Mol Sci. 2017;18(11):2427. doi:10.3390/ijms18112427
218. Gazouli M, Anagnostopoulos AK, Papadopoulou A, et al. Serum protein profile of crohn’s disease treated with infliximab. J Crohns Colitis. 2013;7(10):e461–e470. doi:10.1016/j.crohns.2012.10.011
219. Prieto-Pérez R, Almoguera B, Cabaleiro T, Hakonarson H, Abad-Santos F. Association between genetic polymorphisms and response to anti-TNFs in patients with inflammatory bowel disease. Int J Mol Sci. 2016;17(2):225. doi:10.3390/ijms17020225
220. Möttönen T, Hannonen P, Leirisalo-Repo M, et al. Comparison of combination therapy with single-drug therapy in early rheumatoid arthritis: a randomised trial. FIN-RACo trial group. Lancet. 1999;353(9164):1568–1573. doi:10.1016/S0140-6736(98)09449-5
221. Grigor C, Capell H, Stirling A, et al. Effect of a treatment strategy of tight control for rheumatoid arthritis (the TICORA study): a single-blind randomised controlled trial. Lancet. 2004;364(9430):263–269. doi:10.1016/S0140-6736(04)16676-2
222. Boers M, Verhoeven AC, Markusse HM, et al. Randomised comparison of combined step-down prednisolone, methotrexate and sulphasalazine with sulphasalazine alone in early rheumatoid arthritis. Lancet. 1997;350(9074):309–318.
223. Goekoop-Ruiterman YP, de Vries-Bouwstra JK, Allaart CF, et al. Clinical and radiographic outcomes of four different treatment strategies in patients with early rheumatoid arthritis (the BeSt study): a randomized, controlled trial. Arthritis Rheum. 2005;52(11):3381–3390. doi:10.1002/art.21211
224. van der Kooij SM, Goekoop-Ruiterman YP, de Vries-Bouwstra JK, et al. Drug-free remission, functioning and radiographic damage after 4 years of response-driven treatment in patients with recent-onset rheumatoid arthritis. Ann Rheum Dis. 2009;68(6):914–921. doi:10.1136/ard.2008.092254
225. Van Den Broek M, Lems WF, Allaart CF. BeSt practice: the success of early-targeted treatment in rheumatoid arthritis. Clin Exp Rheumatol. 2012;30(4 Suppl 73):S35–S38.
226. Khanna R, Bressler B, Levesque BG, et al. Early combined immunosuppression for the management of Crohn’s disease (REACT): a cluster randomised controlled trial. Lancet. 2015;386(10006):1825–1834. doi:10.1016/S0140-6736(15)61137-0
227. Vande Casteele N, Ferrante M, Van Assche G, et al. Trough concentrations of infliximab guide dosing for patients with inflammatory bowel disease. Gastroenterology. 2015;148(7):1320–1329. doi:10.1053/j.gastro.2015.02.031
228. D’Haens G, Vermeire S, Lambrecht G, et al. Increasing infliximab dose based on symptoms, biomarkers, and serum drug concentrations does not increase clinical, endoscopic, and corticosteroid-free remission in patients with active luminal crohn’s disease. Gastroenterology. 2018;154(5):1343–1351. doi:10.1053/j.gastro.2018.01.004
229. Colombel JF, Panaccione R, Bossuyt P, et al. Effect of tight control management on crohn’s disease (CALM): a multicentre, randomised, controlled phase 3 trial. Lancet. 2018;390:2779–2789. doi:10.1016/S0140-6736(17)32641-7
230. De Cruz P, Kamm MA, Hamilton AL, et al. Crohn’s disease management after intestinal resection: a randomised trial. Lancet. 2015;385(9976):1406–1417. doi:10.1016/S0140-6736(14)61908-5
231. Panés J, O’Connor M, Peyrin-Biroulet L, Irving P, Petersson J, Colombel JF. Improving quality of care in inflammatory bowel disease: what changes can be made today? J Crohns Colitis. 2014;8(9):919–926. doi:10.1016/j.crohns.2014.02.022
232. Peyrin-Biroulet L, Sandborn W, Sands BE, et al. Selecting therapeutic targets in inflammatory bowel disease (STRIDE): determining therapeutic goals for treat-to-target. Am J Gastroenterol. 2015;110(9):1324–1338. doi:10.1038/ajg.2015.233
233. Pineton de Chambrun G, Blanc P, Peyrin-Biroulet L. Current evidence supporting mucosal healing and deep remission as important treatment goals for inflammatory bowel disease. Expert Rev Gastroenterol Hepatol. 2016;10(8):915–927.
234. Serban ED. Treat-to-target in Crohn’s disease: will transmural healing become a therapeutic endpoint? World J Clin Cases. 2018;6(12):501–513. doi:10.12998/wjcc.v6.i12.501
235. Lega S, Dubinsky MC. What are the targets of inflammatory bowel disease management. Inflamm Bowel Dis. 2018;24(8):1670–1675. doi:10.1093/ibd/izy064
236. Abbasi M, Mousavi MJ, Jamalzehi S, et al. Strategies toward rheumatoid arthritis therapy; the old and the new. J Cell Physiol. 2019;234(7):10018–10031. doi:10.1002/jcp.27860
237. Smolen JS, Breedveid FC, Burmester GR, et al. Treating rheumatoid arthritis to target: 2014 update of the recommendations of an international task force. Ann Rheum Dis. 2016;75(1):3–15. doi:10.1136/annrheumdis-2015-207524
238. Marzo-Ortega H, Gaffney KM, Gaffney K. Defining the target: clinical aims in axial spondyloarthritis. Rheumatology (Oxford). 2018;57(Suppl6):vi18–vi22. doi:10.1093/rheumatology/key176
239. van der Heijde D, Ramiro S, Landewé R, et al. 2016 update of the ASAS-EULAR management recommendations for axial spondyloarthritis. Ann Rheum Dis. 2017;76(6):978–991. doi:10.1136/annrheumdis-2016-210770
240. Tucker LJ, Ye W, Coates LC. Novel concepts in psoriatic arthritis management: can we treat to target? Curr Rheumatol Rep. 2018;20(11):71. doi:10.1007/s11926-018-0781-x
241. Coates LC, Gossec L, Ramiro S, et al. New GRAPPA and EULAR recommendations for the management of psoriatic arthritis. Rheumatology (Oxford). 2017;56(8):1251–1253.
242. Singh JA, Guyatt G, Ogdie A, et al. Special article: 2018 American college of rheumatology/National psoriasis foundation guideline for the treatment of psoriatic arthritis. Arthritis Care Res (Hoboken). 2019;71(1):2–29. doi:10.1002/acr.23885
243. Ward MM, Deodhar A, Aki EA, et al. American college of rheumatology/Spondylitis association of America/Spondyloarthritis research and treatment network 2015 recommendations for the treatment of ankylosing spondylitis and nonradiographic axial spondyloarthritis. Arthritis Rheumatol. 2016;68:282–298. doi:10.1002/art.39298
244. Van de Kerkhof PCM. Therapeutic strategies: rotational therapy and combinations. Clin Exp Dermatol. 2001;26(4):356–361. doi:10.1046/j.1365-2230.2001.00829.x
245. Mrowietz U, Kragballe K, Reich K, et al. Definition of treatment goals for moderate to severe psoriasis: a European consensus. Arch Dermatol Res. 2011;303(1):1–10. doi:10.1007/s00403-010-1080-1
246. Augustin M, Alvaro-Garcia JM, Bagot M. A framework for improving the quality of care for people with psoriasis. J Eur Acad Dermatol Venereol. 2012;26(Suppl 4):1–16. doi:10.1111/j.1468-3083.2012.04576.x
247. Cather JC, Crowley JJ. Use of biologic agents in combination with other therapies for the treatment of psoriasis. Am J Clin Dermatol. 2014;15(6):467–478. doi:10.1007/s40257-014-0097-1
248. Puig L. PASI90 response: the new standard in therapeutic efficacy for psoriasis. J Eur Acad Dermatol Venereol. 2015;29(4):645–648. doi:10.1111/jdv.12669
249. Manalo IF Gilbert KE, Wu JJ. Time to raise the bar to psoriasis area severity index 90 and 100. J Drugs Dermatol. 2015;14(10):1086–1088.
250. Nast A, Gisondi P, Ormerod AD, et al. European S3-Guidelines on the systemic treatment of psoriasis vulgaris–update 2015–short version–EDF in cooperation with EADV and IPC. J Eur Acad Dermatol Venereol. 2015;29(12):2277–2294. doi:10.1111/jdv.12669
251. Blome C, Gosau R, Radtke MA. Patient-relevant treatment goals in psoriasis. Arch Dermatol Res. 2016;308(2):69–78. doi:10.1007/s00403-015-1613-8
252. Reinisch W, Van Assche G, Befrits R, et al. Recommendations for the treatment of ulcerative colitis with infliximab: a gastroenterology expert group consensus. J Crohns Colitis. 2012;6(2):248–258. doi:10.1016/j.crohns.2011.11.001
253. Im JP, Ye BD, Kim YS, Kim JS. Changing treatment paradigms for the management of inflammatory bowel disease. Korean J Intern Med. 2017;33(1):28–35. doi:10.3904/kjim.2017.400
254. Danese S Colombel J, Reinisch W, et al. Review article: infliximab for Crohn’s disease treatment–shifting therapeutic strategies after 10 years of clinical experience. Aliment Pharmacol Ther. 2011;33(8):857–869. doi:10.1111/j.1365-2036.2011.04598.x
255. Louis E, Baumgart DC, Ghosh S, et al. What changes in inflammatory bowel disease management can be implemented today? J Crohns Colitis. 2012;6(Suppl 2):S260–S267. doi:10.1016/S1873-9946(12)60506-6
256. Sampogna F, Puig L, Spuls P, et al. Reversibility of alexithymia with effective treatment of moderate-to-severe psoriasis: longitudinal data from EPIDEPSO. Br J Dermatol. 2019;180(2):397–403. doi:10.1111/bjd.17259.
257. Daudén E, Puig L, Ferrándiz C et al. Consensus document on the evaluation and treatment of moderate-to-severe psoriasis: psoriasis group of the spanish academy of dermatology and venereology. J Eur Acad Dermatol Venereol. 2016;30(Suppl 2):1–18. doi:10.1111/jdv.13652
258. European Medicines Agency [EMA]. Points to consider on clinical investigation of medicinal products other than NSAIDS for treatment of rheumatoid arthritis. https://www.ema.europa.eu/documents/scientific-guideline/points-consider-clinical-investigation-medicinal-products-other-nsaids-treatment-rheumatoid_en.pdf. Adopted on December 2003. Accessed on February 13, 2019.
259. European Medicines Agency [EMA]. Guideline on clinical investigation of medicinal products for the treatment of rheumatoid arthritis. https://www.ema.europa.eu/documents/scientific-guideline/guideline-clinical-investigation-medicinal-products-treatment-rheumatoid-arthritis_en.pdf. Adopted by CHMP, December 14, 2017.
260. Food and Drug Administration [FDA]. Guidance for industry rheumatoid arthritis: developing drug products for treatment. https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm354468.pdf. Published May 2013.
261. European Medicines Agency [EMA]. Guideline on the development of new medicinal products for the treatment of crohn’s disease. https://ukapps.kp360group.com/eRoomReq/Files/AHC4/CodonMedicalJanssenRemicade/0_53fd2/EMA%20CHMP%20Guideline%20Drug%20Development%20in%20CD%202008.pdf.
262. European Medicines Agency [EMA]. Guideline on the development of new medicinal products for the treatment of crohn’s disease. https://www.ema.europa.eu/documents/scientific-guideline/guideline-development-new-medicinal-products-treatment-crohns-disease-revision-2_en.pdf. Adopted on June 28, 2018.
263. European Medicines Agency [EMA].Guideline on the development of new medicinal products for the treatment of ulcerative colitis. https://www.ema.europa.eu/documents/scientific-guideline/guideline-development-new-medicinal-products-treatment-ulcerative-colitis-revision-1_en.pdf. Adopted June 28, 2018.
264. Food and Drug Administration [FDA]. Ulcerative colitis: clinical trial endpoints – guidance for the industry. https://www.fda.gov/downloads/Drugs/Guidances/UCM515143.pdf. Published August 2016.
265. Food and Drug Administration [FDA]. Gastroenterology regulatory endpoints and the advancements of therapeutics, public workshop. https://www.federalregister.gov/documents/2015/01/29/2015-01625/gastroenterology-regulatory-endpoints-and-the-advancement-of-therapeutics-public-workshop. Published January 2015.
266. Carter PJ, Lazar GA. Next generation antibody drugs: pursuit of the ‘high-hanging fruit’. Nat Rev Drug Discov. 2018;17(3):197–223. doi:10.1038/nrd.2017.227
267. Beck A, Wurch T, Bailly C, Corvaia N. Strategies and challenges for the next generation of therapeutic antibodies. Nat Rev Immunol. 2010;10(5):345–352. doi:10.1038/nri2747
268. Ribatti D. From the discovery of monoclonal antibodies to their therapeutic application: an historical reappraisal. Immunol Lett. 2014;161(1):96–99. doi:10.1016/j.imlet.2014.05.010
269. Thorne C, Bensen WG, Choquette D, et al. Effectiveness and safety of infliximab in rheumatoid arthritis: analysis from a Canadian multicenter prospective observational registry. Arthritis Care Res. 2014;66(8):1142–1151. doi:10.1002/acr.22290
270. Rahman P, Choquette D, Bensen WG, et al. Biologic Treatment Registry Across Canada (BioTRAC): a multicentre, prospective, observational study of patients treated with infliximab for ankylosing spondylitis. BMJ Open. 2016;6(4):e009661. doi:10.1136/bmjopen-2015-009661
271. van Den Reek JMPA, Seyger MMB, van Lümig PPM, et al. The journey of adult psoriasis patients towards biologics: past and present - results from the BioCAPTURE registry. J Eur Acad Dermatol Venereol. 2018;32(4):615–623. doi:10.1111/jdv.14282
272. Jeuring SF, Bours PH, Zeegers MP, et al. Disease outcome of ulcerative colitis in an era of changing treatment strategies: results from the Dutch population-based IBDSL cohort. J Crohns Colitis. 2015;9(10):837–845. doi:10.1093/ecco-jcc/jjv129
273. Jeuring SF, van Den Heuvel TR, Liu LY, et al. Improvements in the long-term outcome of crohn’s disease over the past two decades and the relation to changes in medical management: results from the population-based IBDSL cohort. Am J Gastroenterol. 2017;112(2):325–336. doi:10.1038/ajg.2016.524
274. Food and Drug Administration. FDA Briefing Document Arthritis Advisory Committee Meeting February 09, 2016. Available from: https://www.fda.gov/media/95987/download. Accessed June 28, 2019.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.