")
Back to Journals » Drug Design, Development and Therapy » Volume 14
Regulations of miR-183-5p and Snail-Mediated Shikonin-Reduced Epithelial-Mesenchymal Transition in Cervical Cancer Cells
Authors Tang Q, Liu L, Zhang H, Xiao J, Hann SS
Received 26 October 2019
Accepted for publication 29 January 2020
Published 11 February 2020 Volume 2020:14 Pages 577—589
DOI https://doi.org/10.2147/DDDT.S236216
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Qiongyu Guo
Qing Tang,1,* Lihua Liu,2,* Hongyan Zhang,2,* Jing Xiao,2 Swei Sunny Hann1,3
1Laboratory of Tumor Biology, The Second Clinical College of Guangzhou University of Chinese Medicine, Guangzhou, Guangdong Province 510120, People’s Republic of China; 2Department of Gynecology, The Second Clinical College of Guangzhou University of Chinese Medicine, Guangzhou, Guangdong Province 510120, People’s Republic of China; 3Guangdong Provincial Key Laboratory of Clinical Research on Traditional Chinese Medicine Syndrome, The Second Clinical College of Guangzhou University of Chinese Medicine, Guangzhou, Guangdong Province 510120, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Swei Sunny Hann; Jing Xiao
No. 111, Dade Road, Guangzhou, Guangdong Province 510120, People’s Republic of China
Tel +86 20-39318472
Email [email protected]; [email protected]
Background: Shikonin, the main ingredient of Lithospermum erythrorhizon, has been reported to have antitumor effects via multiple targets and signaling pathways. However, the detailed mechanism underlying the effects in cervical cancer still remained unknown.
Methods: MTT, wound-healing, transwell assays and flow cytometry experiments were used to measure cell growth, migration, invasion, and cell cycle analysis. Western blot was used to examine protein levels of Snail, Vimentin and E-cadherin. The expression level of miR-183-5p was measured via qRT-PCR. The E-cadherin promoter activity was detected via Secrete-PairTM Dual Luminescence Assay Kit. The transient transfection experiments were used for silencing of E-cadherin and overexpression of Snail genes. Tumor xenograft and bioluminescent imaging experiments were carried out to confirm the in vitro findings.
Results: We showed that shikonin inhibited cell viability, migration and invasion, and induced cell cycle arrest in a dose-dependent manner in cervical cancer Hela and C33a cells. Mechanistically, we found that shikonin increased miR-183-5p expression and inhibited expression of transcription factor Snail protein. The mimics of miR-183-5p reduced, while the inhibitors of miR-183-5p reversed shikonin-inhibited Snail protein expression. In addition, shikonin decreased Vimentin, increased E-cadherin protein expressions and E-cadherin promoter activity, the latter was reversed in cells transfected with exogenous Snail overexpression vectors. Moreover, silencing of E-cadherin significantly abolished shikonin-inhibited cervical cancer cell growth. Similar findings were also observed in vivo using one xenograft mouse model.
Conclusion: Our results show that shikonin inhibits EMT through inhibition of Snail and stimulation of miR-183-5p expressions, which resulted in induction of E-cadherin expression. Thus, blockade of EMT could be a novel mechanism underlying the anti-cervical cancer effects of shikonin.
Keywords: shikonin, EMT, cervical cancer, miR-183-5p, Snail, E-cadherin
Introduction
Cervical cancer is one of the most common cancers in women worldwide.1 Cervical cancer is amenable for early detection due to its long and relatively well-known natural history prior to its culmination as invasive disease.1 Genetic and epigenetic alterations, and mutations in host cell genes are crucial for development and progression of cervical precancerous lesions to invasive cancer. While concurrent chemoradiotherapy and subsequent chemotherapy are well tolerated and efficient treatment modalities, treatment outcome is rarely curative for advanced stage patients. Targeted therapies based on the knowledge of the molecular pathogenesis of the disease and immunotherapy targeting human papillomavirus (HPV) oncoproteins, such as E6 and E7, showed some promising.2,3 However, advanced-inoperable cervical cancer is still challenging due to increased percentage of regional and distant recurrences with dismal prognosis. Thus, better understanding of pathological changes and development of new treatment paradigm are strongly desirable.
Shikonin, a natural naphthoquinone component extracted from the root of Lithospermum erythrorhizon and Arnebia euchroma (Royle) Johnst native to China, hold promising potentials for antitumor effects via multiple-target mechanisms.4–8 It was reported that the anti-migration and anti-invasion activities of shikonin was through inhibition of c-Met followed by suppression of epithelial-mesenchymal transition (EMT) in lung cancer cells.9 Shikonin showed to exert anticancer effects on gallbladder cancer (GBC) cells by inducing apoptosis and regulating the cell cycle arrest via the c-Jun N-terminal kinase (JNK) signaling pathway.10 Preliminary clinical trials indicated the potential of shikonin for translation into application in clinical oncology. Shikonin was also found to have additive and synergistic interactions in combination with established chemotherapeutics and immunotherapeutic approaches, radiotherapy and other treatment modalities, which further strengthened the potential of this phytochemical to be integrated into standard treatment regimens in cancer.11,12 EMT is a fundamental process to regulate cell migration and invasion. Shikonin inhibited triple-negative breast cancer (TNBC) cell metastasis by targeting the EMT via glycogen synthase kinase-3β (GSK-3β), a serine/threonine protein kinase, mediated suppression of β-catenin signaling, which highlighted the importance of shikonin as a potential candidate for novel anticancer therapeutics against TNBC.13 Although the roles of shikonin in anti-cervical cancer were reported previously,14–16 its precise molecular antitumor mechanism still remained to be elucidated.
MiRNAs are small endogenous non-coding single-stranded RNAs that have been involved in the tumorigenesis, cell differentiation, tumor maintenance, distant metastasis and therapeutic resistance in cancer biology and played a critical role as potential biomarker and therapeutic target in cancer.17,18 Thus, identifying miRNAs and further inferring miRNA functions have become an important strategy in understanding physio-pathological processes, and their roles in cancer predictors and therapeutic targets.19,20 Expressions of miRNAs, such as miR-183-5p, have been shown to be associated with the growth and progression of cancer through multiple mechanisms.21–24 The inhibition of miR-183-5p significantly abolished the effects of tripartite motif-containing protein 65 (TRIM65), a critical regulator of a variety of cellular processes and tumor progression, on autophagy and cisplatin-induced apoptosis suggesting a critical role of miR-183-5p in mediating the TRIM65 – regulated autophagy and cisplatin resistance in human lung cancer A549/DDP cells.21 Another study showed that the expression of long non-coding RNA (lncRNA) taurine-upregulated gene 1 (TUG1) was increased in cervical cancer tissues, which was correlated with advanced clinical features and poor overall survival in patient with cervical cancer. Mechanistically, TUG1 could act as an endogenous sponge by directly binding to miR-183-5p thereby suppressing miR-183-5p expression via activating Wnt/β-catenin signaling pathway.25 Also, overexpression of miR-183-5p reduced proliferation, induced cell cycle arrests and apoptosis by suppressing silent information regulator-1 (SIRT1) expression in cervical cancer cells.26 Thus, miRNAs including miR-183-5p represent interesting strategies for diagnosis and prognosis in cervical cancer.27 Regardless, the detailed mechanisms underlying the anti-cervical cancer effect of miRNAs, such as miR-183-5p, still required to be determined.
In this study, we explored the potential molecular mechanism underlying the anti-cervical cancer effect. We showed that shikonin inhibited EMT through regulation of miR-183-5p and Snail expressions, and this result in induction of E-cadherin expression in vitro and in vivo.
Materials and Methods
Cell Culture and Reagents
Hela and C33a, both human cervical cancer cell lines, were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). All cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Gibco, Grand Island, NY, USA), supplemented with 10% heat-inactivated fetal bovine serum (FBS), 100 U/mL penicillin and 100 mg/mL streptomycin (Gibco, Grand Island, NY, USA) at 37°C in humidification environment encompassing 5% carbon dioxide (CO2). Shikonin was purchased from Meilun Biotechnology Co. (Dalian, China) and dissolved with dimethyl sulfoxide (DMSO) (Sigma-Aldrich, St. Louis, MO, USA). 3-(4, 5-Dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) was obtained from Promega (Madison, WI, USA). Cell Cycle staining kit was obtained from MultiSciences (Lianke) Biotechnology Co. (Hangzhou, China). Lipofectamine 3000 reagent was ordered from Life Technologies (AB &invitrogen, Carlsbad, CA, USA). Geneticin (G-418 Sulfate) was obtained from Life Technologies (Carlsbad, CA, USA). The D-luciferin was purchased from PerkinElmer (Waltham, MA, USA). Antibodies against Vimentin, E-cadherin, Snail and β-actin, and the secondary horseradish peroxidase (HRP)-labeled antibody were purchased from Cell Signaling Technology (CST; Beverly, MA, USA).
Cell Viability
MTT experiment was performed to measure cell viability of Hela and C33a cells. The cells seeded in 96-well plates (5×103 cells/well) were incubated at 37°C with 5% CO2 and treated with the increased dosages of shikonin for up to 72 h. Subsequently, cells were incubated with MTT tetrazolium salt at a working concentration of 5 mg/mL for 4 h. The absorbance of the dissolved formazan crystals in each well was measured using a Microplate Reader (Bio-Rad, Hercules, CA, USA) at 490 nm. Cell viability was calculated by the following equation: (absorbance of the experiment samples/absorbance of the control) × 100%.
Cell Cycle Analysis
Hela and c33a cells were cultured in 6-well plates and treated with increased doses of shikonin for 24 h. Cell cycle experiment was carried out using the cell cycle staining kit based on the manufacture’s instruction. In brief, the cells were washed and resuspended in PBS and then incubated with 0.05 mg 0.1% sodium citrate containing propidium iodide and 50 µg RNase for 1 h at room temperature. The cells were washed and subjected to FACSCalibur flow cytometric analysis (FC500, Beckman Coulter, FL, USA). The proportion (percentage) of cells within the G0/G1, S, and G2/M phases of the cell cycle was analyzed using the MultiCycle AV DNA Analysis software (Phoenix Flow Systems, Inc. San Diego, CA, USA).
Wound-Healing Assay
Cells were seeded at 3x105 cells per well and cultured for 24 h in 6 well plates. After scraping the cell monolayer with a sterile micropipette tip, the wells were washed with PBS and treated with indicated concentrations of shikonin for 24 h. The first image of each scratch was obtained at zero time. After 24 h, the image of each scratch was captured at the same location and the healed area was measured. Images of the scratch were taken at 100 × magnification and the extent of migration in each sample was photographed under Axiovert 200 microscope (Nikon, TI2-E, Tokyo, Japan) and measured by Image J software (National Institutes of Health, Bethesda, MD, USA) and expressed as percentage of control (0 h).
Invasion Assay
Cells were cultured in transwell plates with chambers that were equipped with 8 µm pore size and 6.5-mm diameter polyvinylpyrrolidone (PVP)-free polycarbonated membranes (Corning Costar Inc., NY, USA) according to instructions. The cells were seeded onto the upper chamber at a concentration of 5x104 cells/well and were cultured in the presence or absence of shikonin in the serum-free medium for 24 h. The lower chambers of the transwell were filled with complete medium containing 10% FBS. After incubation for 24 h, the cells were fixed with 4% paraformaldehyde and stained with 0.1% crystal violet. Pictures were taken under 100 x magnifications. Five fields on each membrane were randomly selected and the number of invaded cells was counted as percentage of control under microscope (Nikon, TI2-E, Tokyo, Japan).
Cell Transfection Assay
The miR-183-5p mimics and negative control were obtained from RIBO BIO Inc. (Guangzhou, China). Hela and c33a cells were cultured in 6-well plates before transfection. MiR-183-5p mimics were transiently transfected into cells by Lipofectamine RNAiMAX transfection reagent (AB & invitrogen, Carlsbad, CA, USA) complying with the manufacturer’s instructions. In separate experiments, the cells were transfected with the Snail overexpression vector or control vector purchased from Genechem Co (Shanghai, China) using Lipofectamine 3000 reagent (Invitrogen) according to the manufacturer’s protocol.
Western Blot Analysis
Hela and c33a cells were harvested, washed and lysed with radioimmunoprecipitation assay (RIPA) buffer (Beyotime, Shanghai, China). Protein concentration was determined by the Thermo bicinchoninic acid (BCA) protein assay Kit. Equal amounts of proteins from whole cell lysates were separated on 10% sodium dodecyl sulfate (SDS) polyacrylamide gels and then the proteins were transferred onto polyvinylidene fluoride (PVDF) membranes and incubated for immunoblotting with the relevant primary antibodies against Snail, Vimentin and E-cadherin (1:1000 dilutions) at 4°C overnight. The membranes were washed and incubated with a secondary antibody raised against rabbit IgG conjugated to horseradish peroxidase (Cell Signaling, Beverly, MA, USA). The membranes were washed again and transferred to freshly made enhanced chemiluminescence (ECL) solution (Immobilon Western; Millipore, Billerica, MA, USA), followed by observing signals using the Gel Imagine System (Bio-Rad, Hercules, CA, USA) and documenting the results. Image J software (National Institutes of Health, Bethesda, MD, USA) was applied to quantify and compare the intensity of single band between the control and proteins of interest.
Quantitative Reverse Transcription-Polymerase Chain Reaction (qRT-PCR)
RNA was prepared from either cells or tissue samples using Trizol (Invitrogen, Carlsbad, CA, USA). Reverse transcription reaction was conducted to synthesize complementary DNAs (cDNAs) by wielding Reverse Transcription Kit (Takara, Japan). The TaqMan Universal Master Mix II (Takara, Japan) were used for the analysis of miR-183-5p expression, and U6 was used for internal control for normalization purpose. These outcomes were assessed by using the 2−ΔΔCt method. The sequences of the miR-183-5p and U6 primers were provided as below: miR-183-5p forward: GCCGCTATGGCACTGCTA; miR-183-5p reverse: CAGAGCAGGGTCCGAGGTA; U6 forward: CGCTTCGGCAGCACATATAC; U6 reverse: TTCACGAATTTGCGTGTCATC. QRT-PCR was performed in a 20 μL mixture containing 2 μL of the cDNA preparation and using ChamQTM Universal SYBR Green qPCR Master Mix (Vazyme Biotech Co., Ltd. Nanjing, China) on an ABI 7500 Real-Time PCR System (Applied Biosystems, Grand Island, NY, USA). The PCR conditions were as follows: 30 sec. at 95°C, followed by 40 cycles of 10 sec. at 95°C and 30 sec. at 60°C. Each sample was tested in triplicate. Threshold values were determined for each sample/primer pair, the average and standard errors were calculated.
Dul-Luciferase Reporter Assay
The E-cadherin promoter plasmids were purchased from GeneCopoeia, Inc. Hela and C33a cells were transfected with the E-cadherin promoter plasmids (0.5 μg/well) in 24-well plates by using Lipofectamine 3000 reagent, and then transfected with Snail overexpression plasmid and the control vector purchased from GeneCopoeia for 24 h, followed by treating with shikonin for an additional 24 h. The preparation of cell extracts and measurement of luciferase reporter activities were determined using the Secrete-PairTM Dual Luminescence Assay Kit (GeneCopoeia, Inc., Rockville, MD, USA). Luciferase activity (actual luminescence units) was normalized with SEAP activity within each sample.
Establishment of Xenografts
Animal studies were performed according to the protocols approved by the Institutional Animal Care and Use Committee of Guangdong Provincial Hospital of Chinese Medicine (the Ethics Approval Number 2018067) and the National Institutes of Health guide for the care and use of Laboratory animals (NIH Publications No. 8023, revised 1978). The special pathogen-free grade BALB/c nude mice (4–6 weeks) were ordered from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). These selected mice were randomly divided into three groups (control group, shikonin low dose group and shikonin high dose group), The administrated Hela-Luc cells, which was carrying luciferase reporter gene (obtained from the Guangzhou Land Biological Technology Co., Guangzhou, China), were gathered and injected subcutaneously in the oxter and forelimb of mice. Xenografts were allowed to grow until the initial measurement was made with calipers. After this, the mice were treated with shikonin via intraperitoneal injection at the low dose (1.25 mg/kg) and high dose (2.5 mg/kg), respectively. The diameter of tumors was measured by calipers. Next, for bioluminescence imaging (BLI) procedure, mice were anesthetized by inhalation of 2% isoflurane. The substrate D-luciferin (Caliper Life Sciences, Hopkinton, MA, USA) was injected into the peritoneal cavity with a dose of 150 mg/kg in approximately 100 μL. The intensity of BLI signal was determined using the IVIS-200 imaging system (Xenogen/Caliper, Alameda, CA, USA). Quantification of bioluminescence was reported as photons/s. Tumor volume measurements were calculated using the formula for an oblong sphere: volume = (width2 × length) x 0.5. After 30 days, these mice were sacrificed, and the isolated tumor tissues were weighed, and prepared for the detection of the expressions of Snail, Vimentin and E-cadherin proteins, miR-183-5p by Western blot and qRT-PCR analysis, respectively.
Statistical Analysis
All data were expressed as mean ± SD of three independent experiments. Differences between groups were assessed by one-way ANOVA and significance of difference between particular groups was analyzed by Turkey’s Multiple Comparison Test for multiple comparison involved (GraphPad Prism 5.0 software, LaJolla, CA, USA). Difference was considered significant if p< 0.05.
Results
Shikonin Inhibited Cell Growth and Induced Cell Cycle in Hela and C33a Cells
We first detected the effect of shikonin on cell growth in Hela and C33a cells by MTT assay. We found that shikonin inhibited the viability of both cells in a time- and concentration-dependent manner (Figure 1A). To further assess the effects of shikonin on cell proliferation, we performed the flow cytometry experiment for cell cycle analysis. Compared with the untreated control cells, shikonin significantly increased the proportion of cells at G0/G1 phases, while the proportion of cells at S phases was reduced in Hela and C33a cells suggesting that cell cycle was arrested at G0/G1 phase by shikonin (Figure 1B). Together, these results indicated that shikonin inhibited cell growth and induced cell cycle arrest in cervical cancer Hela and C33a cells.
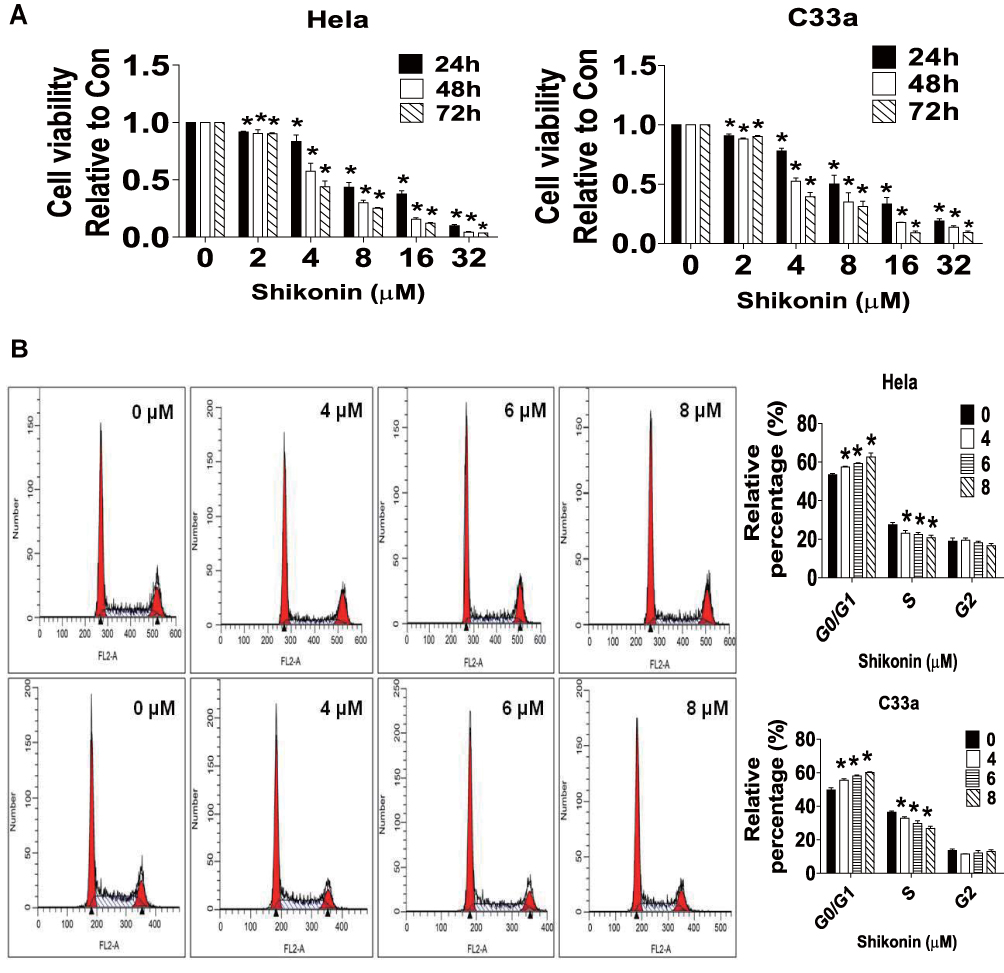 |
Figure 1 Shikonin inhibited proliferation and induced cell cycle arrest in Hela and C33a cells. (A) Hela and C33a cells were treated with an increasing concentration of shikonin for 24, 48 and 72 hrs. MTT assay was performed to detect the cell growth as described in the Materials and Methods section. (B) Hela and C33a cells were treated with indicated doses of shikonin for 24 hrs. Afterwards, the cells were collected and processed for analysis of cell cycle distribution by flow cytometry. The percentages of the cell population in each phase (G0/G1, S and G2/M) were assessed by Multicycle AV DNA Analysis Software. Values are given as the mean ± SD from 3 independent experiments performed in triplicate. *Indicates a significant difference from the control group (P<0.05). |
Shikonin Reduced Cell Migration and Invasion in Hela and C33a Cells
Next, the function of shikonin in migration and invasion of Hela and C33a cells were determined via wound-healing assays and transwell experiment, respectively. Hela and C33a cells were treated with shikonin for 24 h then performed the wound-healing and invasion experiments as described in the Materials and Methods section. The results showed that shikonin significantly restrained the cell migration and invasive abilities of Hela and C33a cells at the doses of 4, 6 and 8 μM, respectively (Figure 2A and B). These results indicated that shikonin inhibited migration and invasion of Hela and C33a cells.
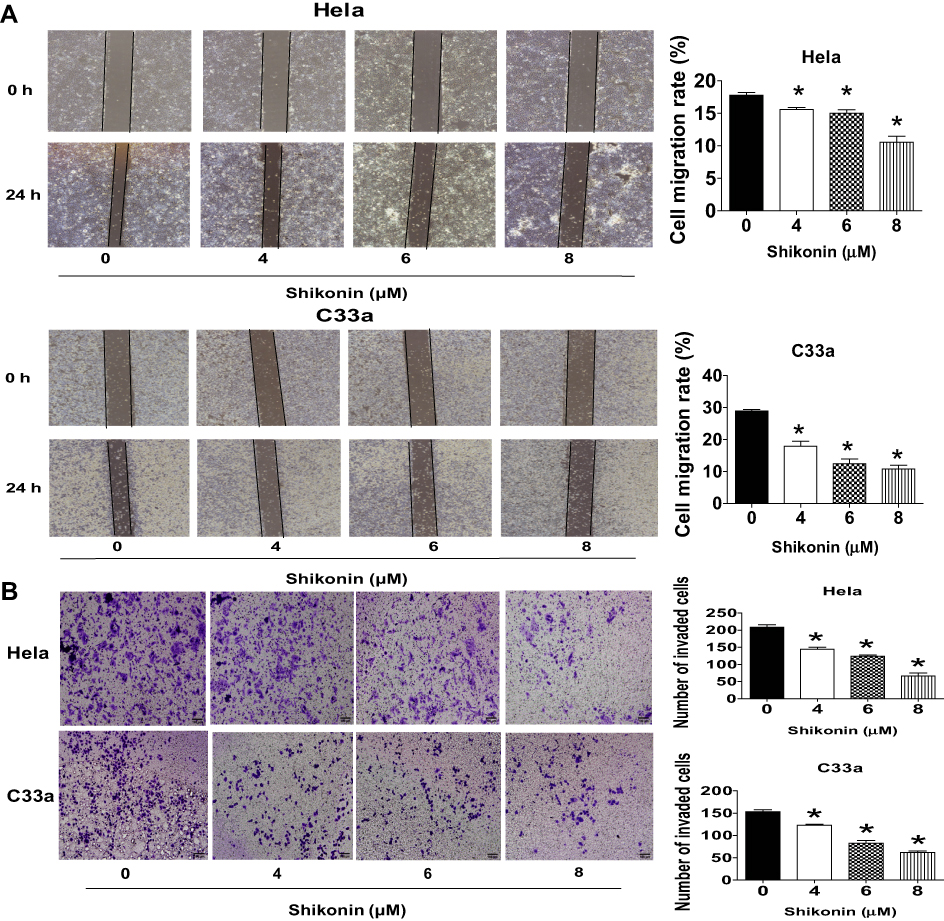 |
Figure 2 Shikonin inhibited cell migration and invasion in Hela and C33a cells. (A) Wound-healing assays were performed to detect cell migration with/without shikonin as described in the Materials and Methods section. Hela and C33a cells were scratched with a pipette tip, and then treated with indicated doses of shikonin for up to 24 h, Images of the scratch were photographed under Axiovert 200 microscope (magnification, ×100) and expressed as percentage of control (0 h). (B) Invasion ability was determined by transwell assay as described in the Materials and Methods section. Hela and C33a were incubated with indicated doses of shikonin for 24 hrs, and then fixed with 4% paraformaldehyde for 10 mins at room temperature, stained with 0.1% crystal violet, Images were photographed and counted under Axiovert 200 light microscope, Scale bar = 100 μm. *Indicates a significant difference from the control group (P<0.05). |
Shikonin Increased the Level of miR-183-5p and Decreased the Snail Protein Expression in Hela and C33a Cells
We then started to explore the potential functions and mechanisms underlying the inhibitory effect of shikonin in this process. MiRNA has been shown to play an important role in cancer predictors and therapeutic targets.19,20 Expression of miR-183-5p has been shown to be associated with the growth and progression of cancer through multiple mechanisms.21–24 EMT is a major process to regulate cell migration and invasion. Shikonin reversed the EMT in breast cancer cells via upregulating E-cadherin and downregulating Snail expressions.13 In this study, qRT-PCR analysis showed that miR-183-5p expression was clearly increased by shikonin in both Hela and C33a cells (Figure 3A). Western blot results revealed that shikonin significantly decreased the expression of EMT factor Snail protein in a dose-dependent manner (Figure 3B). To further explore the potential interaction between miR-183-5p and Snail, Hela and C33a cells were transfected with the miR-183-5p mimics or negative control for 48 h. Western blot analysis found that the mimics of miR-183-5p significantly repressed the expression of Snail protein (Figure 3C). Conversely, the inhibitors of miR-183-5p significantly reversed the shikonin-reduced Snail protein expression (Figure 3D). Of note, overexpression of Snail had no significant effect on miR-183-5p expression in Hela and C33a cells (Figure 3E). All these observations indicated that miR-183-5p acted as upstream factor and regulated Snail expression in this process.
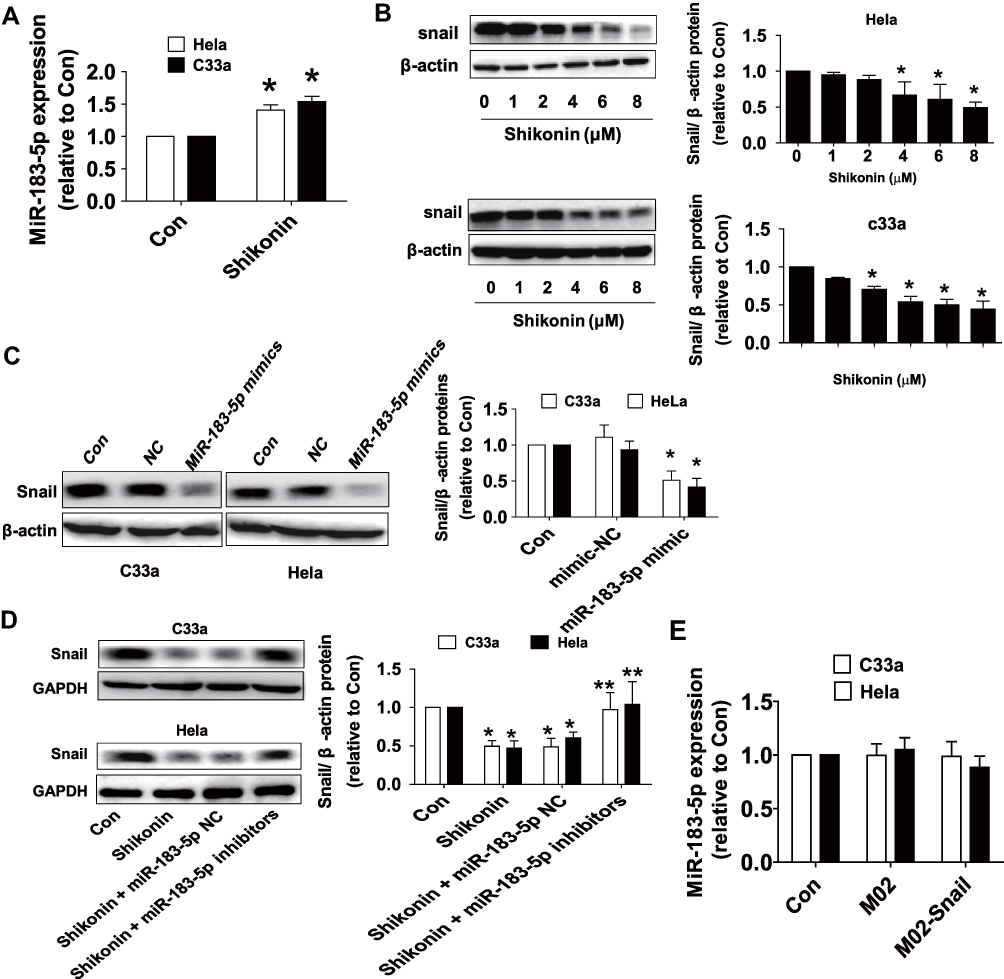 |
Figure 3 Shikonin induced the level of miR-183-5p and decreased the Snail protein expression in Hela and C33a cells. (A) Hela and C33a cells were treated with shikonin (6 μM) for 24 h, qRT-PCR analysis was used to measure the expression of miR-183-5p. (B) Cells were treated with increased doses of shikonin for 24 h, and then cells were harvested and lysed. The expression of Snail protein was detected via Western blot analysis. (C) MiR-183-5p mimics or negative control were transfected into Hela and C33a cells for up to 48 h, the expression of Snail protein was detected via Western blot analysis. (D) Hela and C33a cells were transfected with miR-183-5p inhibitors or negative control for 48 h; the protein expression of Snail was evaluated via Western blot analysis. The figures are representative cropped gels/blots that have been run under the same experimental conditions. (E) Hela and C33a cells were transfected with the control or Snail expression vectors for 24 h, followed by measuring the expression of miR-183-5p via qRT-PCR. *Indicates a significant difference from the control (P<0.05). **Represents significant difference compared to shikonin-treated alone group (P<0.05). |
Shikonin Inhibited Vimentin, Increased E-Cadherin Protein Expressions and Promoter Activity; Silencing of E-Cadherin Reversed Shikonin Inhibited Induced Cell Growth in Hela and C33a Cells
To further delineate the anti-tumor mechanism underlying shikonin-regulated miR-183-5p and Snail, we decipher the role of EMT markers, such as Vimentin and E-cadherin. Western blot results revealed that shikonin significantly inhibited Vimentin and increased E-cadherin protein expressions in a dose-dependent manner (Figure 4A). Moreover, dual-luciferase reporter assay showed that shikonin increased the activity of E-cadherin gene promoter. Interestingly, exogenously expression of Snail reversed shikonin-induced E-cadherin promoter activity (Figure 4B) and E-cadherin protein expression (Figure 4C). More importantly, silence of E-cadherin dramatically overcame the effect of shikonin-inhibited cell growth in Hela and C33a cells (Figure 4D). These results unveiled an important role of E-cadherin and indicated that shikonin-inhibited cell growth inhibition and EMT process via inhibition of Vimentin and repressed Snail-mediated induction of E-cadherin.
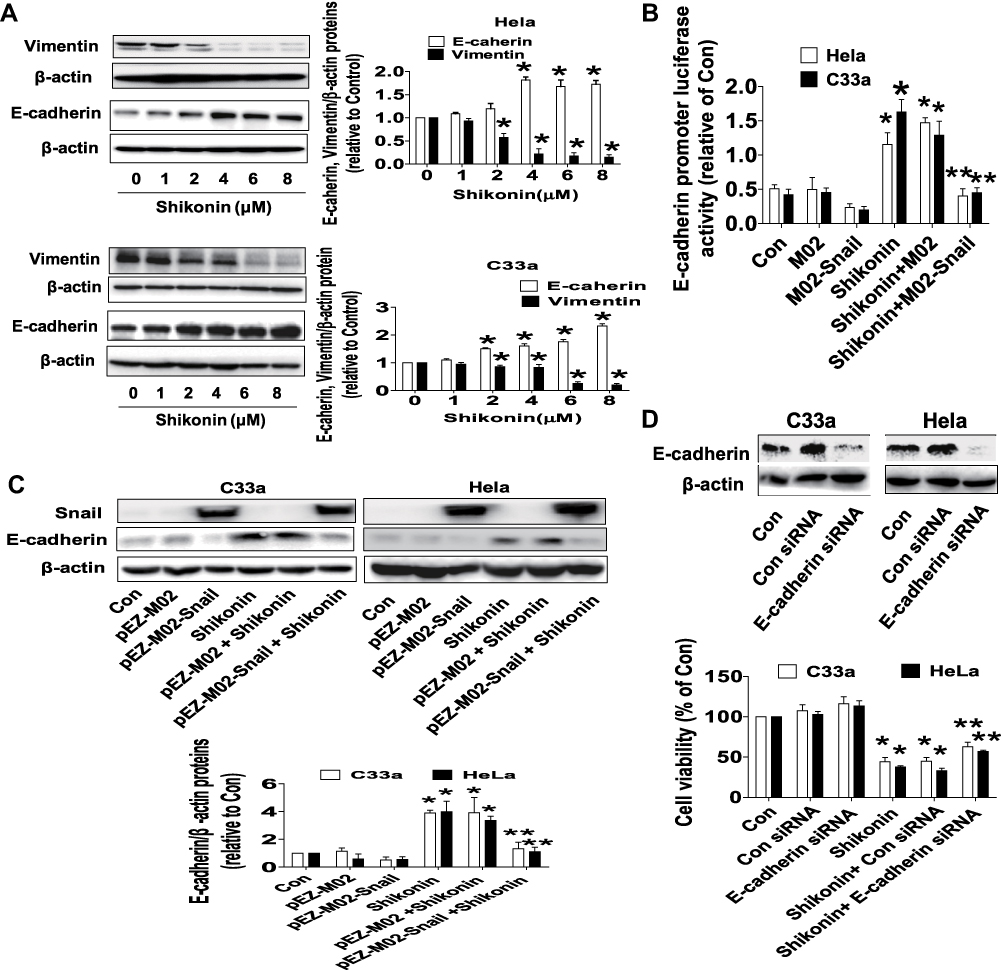 |
Figure 4 Shikonin inhibited Vimentin, increased E-cadherin protein expressions and promoter activity, silencing of E-cadherin reversed shikonin-inhibited cell growth in Hela and C33a cells. (A) Hela and C33a cells were treated with increasing doses of shikonin for 24 h. The expressions of Vimentin and E-cadherin proteins were measured via Western blot analysis. (B) Cells were transfected with E-cadherin promoter for 24 h, and then transfected with Snail overexpression vector or the control vector, and treated with shikonin (6 μM) for up to 48 h. Afterwards, the E-cadherin promoter activity was detected via Secrete-Pair Dual Luminescence Assay Kit as described in the Materials and Methods section. (C) Hela and C33a cells were transfected with Snail overexpression vector or negative control vector for 24 h, and then treated with shikonin (6 μM) for an additional 24 h. Western blot analysis was performed to measure the protein levels of E-cadherin protein expression. The figures are representative cropped gels/blots that have been run under the same experimental conditions. (D) E-cadherin siRNA or control siRNA were transfected into Hela and C33a cells for 24 h, and then treated with shikonin for an additional 24 h, MTT assay was used to measure cell viability of Hela and C33a cells. *Indicates a significant difference from the control (P<0.05). **Represents significant difference compared to shikonin-treated alone group (P<0.05). |
Shikonin Inhibited Tumor Growth and Regulated the Expressions of miR-183-5p, Snail, Vimentin and E-Cadherin in vivo
We used Hela-Luc cells to examine whether shikonin-inhibited cervical cancer growth in vivo. We found that shikonin significantly inhibited tumor growth (Figure 5A). In addition, shikonin caused significant decrease in tumor weight and sizes in the established Hela-Luc cells xenografted tumors compared with that of the vehicle-treated control animals (Figure 5B–D). Furthermore, the protein levels of Snail and Vimentin were significantly inhibited by shikonin, while miR-183-5p expression and E-cadherin protein were increased in shikonin-treated tumor tissues (Figure 5E and F). Together, the relevant discoveries revealed that shikonin suppressed tumor growth in vivo via inhibiting EMT by regulation of miR-183-5p/Snail/Vimentin/E-cadherin regulatory axis in cervical cancer cells, which were in line with those observation in vitro data.
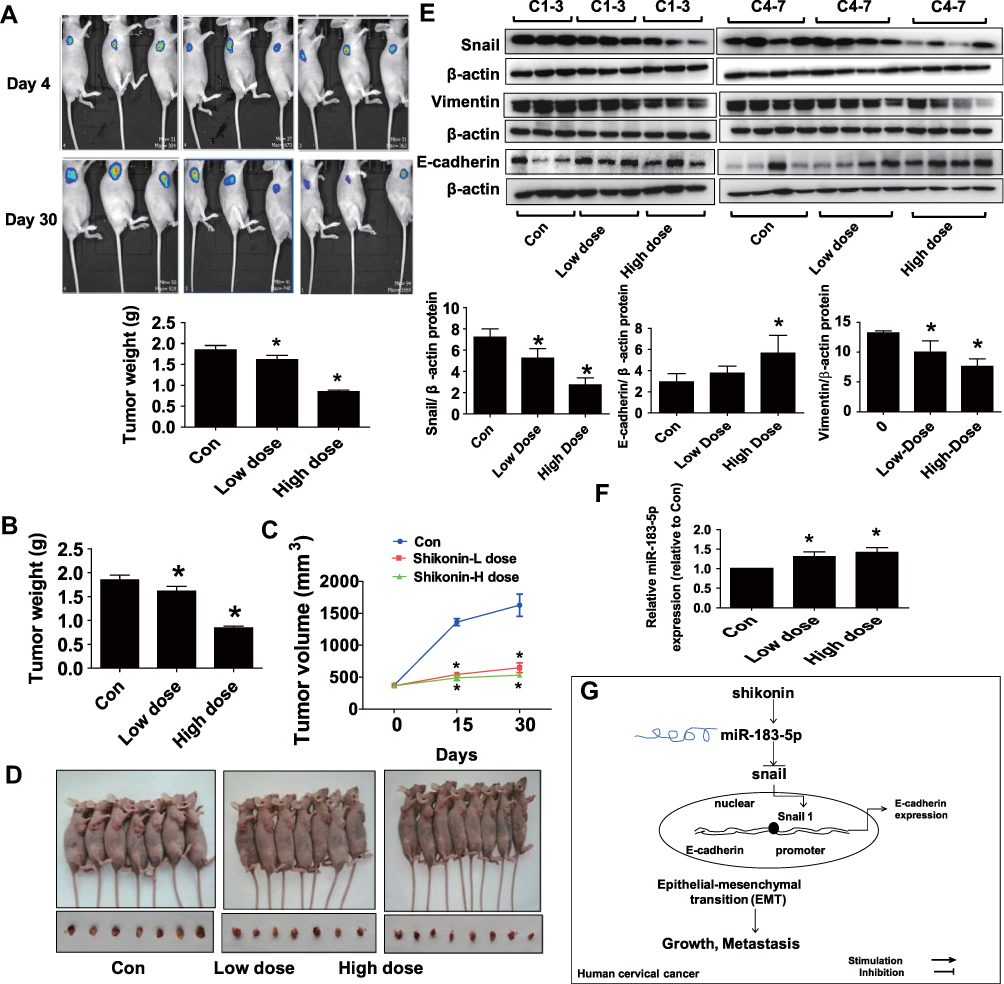 |
Figure 5 Shikonin inhibited tumor growth and regulated the expressions of miR-183-5p, Snail, Vimentin and E-cadherin expressions in vivo. (A) Two different concentrations of shikonin (low dose 1.25 mg/kg and high dose 2.5 mg/kg) were used to treat BALB/c nude mice. Tumor growth was monitored by injecting luciferin substrate, followed by measuring bioluminescence signals. Representative images are shown. (B, C) The xenografts were harvested on day 30, the tumor weight (B) and tumor volume (C) were measured. (D) The photographs of mice and xenografts that treated with low and high doses of shikonin, and the vehicle-treated were shown. (E) The tumor tissues in all mice were isolated and processed for detecting Snail, Vimentin and E-cadherin protein expressions via Western blot. (F) MiR-183-5p expression was measured via qRT-PCR analysis in tumor tissues in all mice. *Indicates a significant difference from the control (P<0.05). (G) Our results show that shikonin inhibits cell viability, migration and invasion, and induces cell cycle arrest through regulations of miR-138-5p and Snail, this results in induction of E-cadherin expression thereby resulting in inhibition of EMT process in cervical cancer cells. |
Discussion
Cervical cancer cells, seriously affect the health of women. The primary causes of mortality associated with cervical cancer are unsuccessful surgical treatment, tumor recurrence and metastasis.28 EMT is a biological process that involves the polarization of epithelial cells, which normally interact with the basement membrane via their basal surfaces. This polarization induces multiple biochemical changes that enable the cells to assume a mesenchymal cell phenotype.29 These changes impart several properties, such as migratory capacity and invasiveness. Currently, the EMT program is divided into three types: embryogenesis, fibrosis, and tumorigenesis. Type 1 and 2 EMT contribute to organ development and tissue regeneration.30 Type 3 EMT is involved in carcinogenesis and has been reported to be significantly associated with local invasion and distant metastasis.31 Shikonin, one major bioactive component extracted from the roots of Lithospermum erythrorhizon, was found to be capable of inhibiting cervical cancer via inducing cell apoptosis and inhibiting cell proliferation.14,15,32 Despite the inhibitory effect of shikonin in cell proliferation have been reported, the action of shikonin in EMT process in cervical cancer cells still remained unknown. Based on the results from the current study, we showed that shikonin inhibited cell proliferation, migration and invasion in both Hela and C33a cells. Western blot assay revealed the regulation effect of shikonin on EMT-related molecules (up-regulation of E-cadherin, down-regulation of vimentin and Snail). Cell-cell adhesion was known to be mediated by E-cadherin repression, which was lost during primary tumor formation, resulting in tumor cells breakthrough the basement membrane with increased invasive properties. Snail directly targeted and suppressed E-cadherin transcription to promote tumor EMT.33,34 Our results indicated that shikonin prevented proliferation, migration and invasion of cervical cancer cells through regulating EMT-associated pathway.
We demonstrated an important role of miR-183-5p in mediating the effect of shikonin in this process. Recent studies have shown that miR-183-3p regulated cell phenotypes and tumor growth in various cancers.35,36 MiR-183, acted as a tumor suppressor, suppressed metastasis-associated protein1 (MTA1) thereby inhibiting the proliferation, EMT, migration and invasion of human lung cancer cells.37 In the current study, we observed a role of miR-183-5p induction in mediating the inhibitory effect of shikonin in cervical cancer cell growth. In line with this, overexpression of miR-183-5p showed to reduce proliferation, induce cell cycle arrests and apoptosis by suppressing SIRT1 expression in cervical cancer cells.26 MiR-183-5p expression level has been found low in clinical tissues of cervical cancer and cells as compared to the normal specimens and normal cervical epithelial cells, suggesting that miR-183-5p significantly inhibited migration and invasion in cervical cancer cells.38 It was also shown that increased expression of miR-183-5p promoted apoptosis and suppressed the EMT, proliferation, invasion and migration of human endometrial cancer cells by downregulating Ezrin, a membrane-cytoskeleton linker protein.39 We also showed that induction of miR-183-5p has been involved in the shikonin-inhibited EMT process via inhibition of Snail and induction of E-cadherin expressions. As a zinc finger transcriptional repressor and a key regulator of EMT, the expression and regulation of Snail have been reported to be involved in EMT process in cancer cells. Snail is a master regulator of cellular identity and a strong repressor of specific target genes, such as the E-cadherin, in carcinogenesis leading to controlling EMT process during tumor progression.40 Our results suggested that miR-183-5p, acted as upstream factor, interacted and regulated Snail signaling, thereby increasing E-cadherin expression both at transcriptional and translational levels; this led to inhibition of proliferation, migration, invasion, and EMT process in cervical cancer cells. Consistent with these findings, similar results were also observed in other cancer cell types.41,42 Of note, whether there was a direct physical interaction between miR-183-5p and Snail still needs to be determined with more experimental approaches, such as RNA immunoprecipitation (RIP) and/or RNA pull-down, or 3ʹ-untranslated region (3ʹ UTR) luciferase reporter assays, in the future. Together, these results indicated the critical tumor suppressor role of miR-183-5p in repression of EMT process. However, opposite findings were also observed in other studies.43,44 Thus, the true role of miR-183 still required to be determined.
A critical molecular characteristic in the EMT is the loss of E-cadherin expression, as well as the EMT factors, such as Snail. Our results indicated transcriptional and translational regulation of E-cadherin by Snail. In consistent with this, metformin, an AMP-activated protein kinase (AMPK) activator, diminished the extracellular signal-regulated kinase (ERK) signaling by activation of AMPK pathway, this led to suppression of Snail resulting in upregulation of a tumor suppressor and critical EMT marker E-cadherin by binding to the E-cadherin promoter. Metformin, an effective agent widely used as a first-line treatment for type 2 diabetes, promoted EMT through an inverse interaction of AMPK and ERK signaling regulatory axis via regulation of E-cadherin expression, which provide a potential for reducing cancer occurrence in metformin-treated population.45 Functional E-cadherin loss is important for metastasis. H3K9 methylation was upregulated and H3K4 and H3K56 acetylation were downregulated at the E-cadherin promoter in Snail2-overexpressing cancer cells. Furthermore, Snail2 interacted with G9a and histone deacetylases (HDACs) to form a complex to suppress E-cadherin transcription suggesting epigenetic regulation of E-cadherin by Snail. Snail is a repressor of E-cadherin during carcinogenesis; demonstrated the importance of G9a- and HDACs-mediated regulation during Snail-induced E-cadherin repression and metastasis in lung cancer cells.46 Another study found that homeobox C8 (HOXC8) promoted EMT in lung cancer cells by binding to the E-cadherin promoter and acting as a transcriptional repressor to regulate E-cadherin transcription in lung cancer cells.47
More importantly, to further confirm the anti-tumor impacts of shikonin on cervical cancer; we established one xenografted tumor mouse model. The results showed that shikonin remarkably suppressed tumor formation in vivo. Furthermore, Vimentin and Snail protein levels in tumor tissues were significantly declined, while E-cadherin protein and miR-183-5p expression levels were increased by shikonin. These data further clarified the anti-tumor activity of shikonin in cervical cancer. The doses used in this were based on other studies,10,48–50 which showed substantial inhibitory effects without noticeable toxicity. Our findings suggested that shikonin suppressed growth of human cervical cancer via regulating miR-183-5p/Snail/Vimentin/E-cadherin signaling regulatory axis. Whether shikonin had potential in prolonging the survival and inhibiting metastasis in cervical cancer xenografted tumors required to be elucidated.
In conclusion, these findings indicated that shikonin-inhibited cervical cancer cells proliferation, migration and invasion. Moreover, shikonin inactivated EMT by regulating miR-183-5p and Snail, thereby induction of E-cadherin expression at transcriptional and translational levels (Figure 5G). Thus, blockade of EMT by shikonin could be a novel mechanism underlying the anti-cervical cancer effect of shikonin. These findings may unveil new insight for prevention of cervical cancer growth, EMT and perhaps metastasis by shikonin.
Acknowledgments
This work was supported in part by the grants from the National Natural Scientific Foundation of China (Nos. 81473716, 81703551, and 81871863) and the Major Program of National Natural Science Foundation of Guangdong (No. 2018B030311061).
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that they have no competing interests.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34. doi:10.3322/caac.v69.1
2. Menderes G, Black J, Schwab CL, Santin AD. Immunotherapy and targeted therapy for cervical cancer: an update. Expert Rev Anticancer Ther. 2016;16(1):83–98. doi:10.1586/14737140.2016.1121108
3. Shibata T, Lieblong BJ, Sasagawa T, Nakagawa M. The promise of combining cancer vaccine and checkpoint blockade for treating HPV-related cancer. Cancer Treat Rev. 2019;78:8–16. doi:10.1016/j.ctrv.2019.07.001
4. Zhang X, Cui JH, Meng QQ, Li SS, Zhou W, Xiao S. Advance in anti-tumor mechanisms of shikonin, alkannin and their derivatives. Mini Rev Med Chem. 2018;18(2):164–172. doi:10.2174/1389557517666170228114809
5. Guo N, Miao R, Gao X, et al. Shikonin inhibits proliferation and induces apoptosis in glioma cells via downregulation of CD147. Mol Med Rep. 2019;19(5):4335–4343. doi:10.3892/mmr.2019.10101
6. Zhang S, Gao Q, Li W, et al. Shikonin inhibits cancer cell cycling by targeting Cdc25s. BMC Cancer. 2019;19(1):20. doi:10.1186/s12885-018-5220-x
7. Xu J, Koizumi K, Liu M, et al. Shikonin induces an antitumor effect on murine mammary cancer via p38dependent apoptosis. Oncol Rep. 2019;41(3):2020–2026. doi:10.3892/or.2019.6966
8. Liu Y, Kang X, Niu G, et al. Shikonin induces apoptosis and prosurvival autophagy in human melanoma A375 cells via ROS-mediated ER stress and p38 pathways. Artif Cells Nanomed Biotechnol. 2019;47(1):626–635. doi:10.1080/21691401.2019.1575229
9. Hsieh YS, Liao CH, Chen WS, Pai JT, Weng MS. Shikonin inhibited migration and invasion of human lung cancer cells via Suppression of c-Met-Mediated Epithelial-to-Mesenchymal transition. J Cell Biochem. 2017;118(12):4639–4651. doi:10.1002/jcb.26128
10. Zhai T, Hei Z, Ma Q, et al. Shikonin induces apoptosis and G0/G1 phase arrest of gallbladder cancer cells via the JNK signaling pathway. Oncol Rep. 2017;38(6):3473–3480. doi:10.3892/or.2017.6038
11. Boulos JC, Rahama M, Hegazy MF, Efferth T. Shikonin derivatives for cancer prevention and therapy. Cancer Lett. 2019;459:248–267. doi:10.1016/j.canlet.2019.04.033
12. Wang Z, Yin J, Li M, et al. Combination of shikonin with paclitaxel overcomes multidrug resistance in human ovarian carcinoma cells in a P-gp-independent manner through enhanced ROS generation. Chin Med. 2019;14:7. doi:10.1186/s13020-019-0231-3
13. Chen Y, Chen ZY, Chen L, et al. Shikonin inhibits triple-negative breast cancer-cell metastasis by reversing the epithelial-to-mesenchymal transition via glycogen synthase kinase 3beta-regulated suppression of beta-catenin signaling. Biochem Pharmacol. 2019;166:33–45. doi:10.1016/j.bcp.2019.05.001
14. Wu Z, Wu LJ, Li LH, Tashiro S, Onodera S, Ikejima T. Shikonin regulates HeLa cell death via caspase-3 activation and blockage of DNA synthesis. J Asian Nat Prod Res. 2004;6(3):155–166. doi:10.1080/1028602032000169622
15. Lu D, Qian J, Li W, Feng Q, Pan S, Zhang S. beta-hydroxyisovaleryl-shikonin induces human cervical cancer cell apoptosis via PI3K/AKT/mTOR signaling. Oncol Lett. 2015;10(6):3434–3442. doi:10.3892/ol.2015.3769
16. Han HW, Zheng CS, Chu SJ, et al. The evaluation of potent antitumor activities of shikonin coumarin-carboxylic acid, PMMB232 through HIF-1alpha-mediated apoptosis. Biomed Pharmacother. 2018;97:656–666. doi:10.1016/j.biopha.2017.10.159
17. Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 2017;16(3):203–222. doi:10.1038/nrd.2016.246
18. Li X, Xu M, Ding L, Tang J. MiR-27a: a novel biomarker and potential therapeutic target in tumors. J Cancer. 2019;10(12):2836–2848. doi:10.7150/jca.31361
19. Long L, Zhang X, Bai J, Li Y, Wang X, Zhou Y. Tissue-specific and exosomal miRNAs in lung cancer radiotherapy: from regulatory mechanisms to clinical implications. Cancer Manag Res. 2019;11:4413–4424. doi:10.2147/CMAR.S198966
20. Ding L, Lan Z, Xiong X, et al. The dual role of MicroRNAs in colorectal cancer progression. Int J Mol Sci. 2018;19(9):2791. doi:10.3390/ijms19092791
21. Pan X, Chen Y, Shen Y, Tantai J. Knockdown of TRIM65 inhibits autophagy and cisplatin resistance in A549/DDP cells by regulating miR-138-5p/ATG7. Cell Death Dis. 2019;10(6):429. doi:10.1038/s41419-019-1660-8
22. Lu Z, He Q, Liang J, et al. miR-31-5p is a potential circulating biomarker and therapeutic target for oral cancer. Mol Ther Nucleic Acids. 2019;16:471–480. doi:10.1016/j.omtn.2019.03.012
23. He Z, Ruan X, Liu X, et al. FUS/circ_002136/miR-138-5p/SOX13 feedback loop regulates angiogenesis in Glioma. J Exp Clin Cancer Res. 2019;38(1):65. doi:10.1186/s13046-019-1065-7
24. Xiong Y, Zhang J, Song C. CircRNA ZNF609 functions as a competitive endogenous RNA to regulate FOXP4 expression by sponging miR-138-5p in renal carcinoma. J Cell Physiol. 2019;234(7):10646–10654. doi:10.1002/jcp.v234.7
25. Zhu J, Shi H, Liu H, Wang X, Li F. Long non-coding RNA TUG1 promotes cervical cancer progression by regulating the miR-138-5p-SIRT1 axis. Oncotarget. 2017;8(39):65253–65264. doi:10.18632/oncotarget.18224
26. Ou L, Wang D, Zhang H, Yu Q, Hua F. Decreased expression of miR-138-5p by lncRNA H19 in cervical cancer promotes tumor proliferation. Oncol Res. 2018;26(3):401–410. doi:10.3727/096504017X15017209042610
27. Berti FCB, Salviano-Silva A, Beckert HC, de Oliveira KB, Cipolla GA, Malheiros D. From squamous intraepithelial lesions to cervical cancer: circulating microRNAs as potential biomarkers in cervical carcinogenesis. Biochim Biophys Acta Rev Cancer. 2019;1872(2):188306. doi:10.1016/j.bbcan.2019.08.001
28. Qureshi R, Arora H, Rizvi MA. EMT in cervical cancer: its role in tumour progression and response to therapy. Cancer Lett. 2015;356(2Pt B):321–331. doi:10.1016/j.canlet.2014.09.021
29. Samatov TR, Tonevitsky AG, Schumacher U. Epithelial-mesenchymal transition: focus on metastatic cascade, alternative splicing, non-coding RNAs and modulating compounds. Mol Cancer. 2013;12(1):107. doi:10.1186/1476-4598-12-107
30. Kent CN, Guttilla Reed IK. Regulation of epithelial-mesenchymal transition in endometrial cancer: connecting PI3K, estrogen signaling, and microRNAs. Clin Transl Oncol. 2016;18(11):1056–1061. doi:10.1007/s12094-016-1492-2
31. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420–1428. doi:10.1172/JCI39104
32. Wu Z, Wu LJ, Tashiro S, Onodera S, Ikejima T. Phosphorylated extracellular signal-regulated kinase up-regulated p53 expression in shikonin-induced HeLa cell apoptosis. Chin Med J (Engl). 2005;118(8):671–677.
33. Galvan JA, Zlobec I, Wartenberg M, et al. Expression of E-cadherin repressors SNAIL, ZEB1 and ZEB2 by tumour and stromal cells influences tumour-budding phenotype and suggests heterogeneity of stromal cells in pancreatic cancer. Br J Cancer. 2015;112(12):1944–1950. doi:10.1038/bjc.2015.177
34. Lin Y, Dong C, Zhou BP. Epigenetic regulation of EMT: the Snail story. Curr Pharm Des. 2014;20(11):1698–1705. doi:10.2174/13816128113199990512
35. Zheng S, Zhong YF, Tan DM, Xu Y, Chen HX, Wang D. miR-183-5p enhances the radioresistance of colorectal cancer by directly targeting ATG5. J Biosci. 2019;44(4). doi:10.1007/s12038-019-9918-y
36. Li Y, He S, Zhan Y, et al. microRNA-183-3p inhibits progression of human prostate cancer by downregulating high-mobility group nucleosome binding domain 5. DNA Cell Biol. 2019;38(8):840–848. doi:10.1089/dna.2019.4642
37. Yang CL, Zheng XL, Ye K, et al. MicroRNA-183 acts as a tumor suppressor in human non-small cell lung cancer by down-regulating MTA1. Cell Physiol Biochem. 2018;46(1):93–106. doi:10.1159/000488412
38. Zhang W, Zhang M, Liu L, Jin D, Wang P, Hu J. MicroRNA-183-5p inhibits aggressiveness of cervical cancer cells by targeting integrin subunit beta 1 (ITGB1). Med Sci Monit. 2018;24:7137–7145. doi:10.12659/MSM.910295
39. Yan H, Sun BM, Zhang YY, et al. Upregulation of miR-183-5p is responsible for the promotion of apoptosis and inhibition of the epithelial-mesenchymal transition, proliferation, invasion and migration of human endometrial cancer cells by downregulating Ezrin. Int J Mol Med. 2018;42(5):2469–2480. doi:10.3892/ijmm.2018.3853
40. Al Khatib AM, Stepan AE, Margaritescu C, Simionescu C, Ciurea RN. E-cadherin and snail immunoexpression in colorectal adenocarcinomas. Curr Health Sci J. 2019;45(2):204–209. doi:10.12865/CHSJ.45.02.12
41. Lin X, Zheng L, Song H, et al. Effects of microRNA-183 on epithelial-mesenchymal transition, proliferation, migration, invasion and apoptosis in human pancreatic cancer SW1900 cells by targeting MTA1. Exp Mol Pathol. 2017;102(3):522–532. doi:10.1016/j.yexmp.2017.05.009
42. Meng F, Zhang L. miR-183-5p functions as a tumor suppressor in lung cancer through PIK3CA inhibition. Exp Cell Res. 2019;374(2):315–322. doi:10.1016/j.yexcr.2018.12.003
43. Wang H, Ma Z, Liu X, et al. MiR-183-5p is required for non-small cell lung cancer progression by repressing PTEN. Biomed Pharmacother. 2019;111:1103–1111. doi:10.1016/j.biopha.2018.12.115
44. Li H, Pan X, Gui Y, et al. Upregulation of miR-183-5p predicts worse survival in patients with renal cell cancer after surgery. Cancer Biomark. 2019;24(2):153–158. doi:10.3233/CBM-182047
45. Banerjee P, Surendran H, Chowdhury DR, Prabhakar K, Pal R. Metformin mediated reversal of epithelial to mesenchymal transition is triggered by epigenetic changes in E-cadherin promoter. J Mol Med (Berl). 2016;94(12):1397–1409. doi:10.1007/s00109-016-1455-7
46. Hu Y, Zheng Y, Dai M, et al. Snail2 induced E-cadherin suppression and metastasis in lung carcinoma facilitated by G9a and HDACs. Cell Adh Migr. 2019;13(1):285–292. doi:10.1080/19336918.2019.1638689
47. Zhang J, Yang M, Li D, et al. Homeobox C8 is a transcriptional repressor of E-cadherin gene expression in non-small cell lung cancer. Int J Biochem Cell Biol. 2019;114:105557. doi:10.1016/j.biocel.2019.06.005
48. Zhao X, Zhu Y, Hu J, et al. Shikonin inhibits tumor growth in mice by suppressing pyruvate kinase M2-mediated aerobic glycolysis. Sci Rep. 2018;8(1):14517. doi:10.1038/s41598-018-31615-y
49. Kim HJ, Hwang KE, Park DS, et al. Shikonin-induced necroptosis is enhanced by the inhibition of autophagy in non-small cell lung cancer cells. J Transl Med. 2017;15(1):123. doi:10.1186/s12967-017-1223-7
50. Ni F, Huang X, Chen Z, Qian W, Tong X. Shikonin exerts antitumor activity in Burkitt’s lymphoma by inhibiting C-MYC and PI3K/AKT/mTOR pathway and acts synergistically with doxorubicin. Sci Rep. 2018;8(1):3317. doi:10.1038/s41598-018-21570-z
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.