")
Back to Journals » Journal of Inflammation Research » Volume 14
Regulation of the Immune Microenvironment by an NLRP3 Inhibitor Contributes to Attenuation of Acute Right Ventricular Failure in Rats with Pulmonary Arterial Hypertension
Authors Guo L, Qin G, Cao Y, Yang Y, Dai S, Wang L, Wang E
Received 4 September 2021
Accepted for publication 21 October 2021
Published 2 November 2021 Volume 2021:14 Pages 5699—5711
DOI https://doi.org/10.2147/JIR.S336964
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Lizhe Guo,1 Gang Qin,1 Yanan Cao,1 Yue Yang,1 Sisi Dai,1 Lu Wang,1 E Wang1,2
1Department of Anesthesiology, Xiangya Hospital Central South University, Changsha, People’s Republic of China; 2National Clinical Research Center for Geriatric Disorders (Xiangya Hospital), Changsha, People’s Republic of China
Correspondence: E Wang
Department of Anesthesiology, Xiangya Hospital Central South University, Xiangya Road #87, Changsha, Hunan, 410008, People’s Republic of China
Tel/Fax +86-731-84327413
Email [email protected]
Background: Right heart failure is the terminal stage of PAH. When PAH patients suffer from pulmonary infection or puerperal infection heart failure often rapidly develops. Low dose of lipopolysaccharide induces rapid right ventricular failure in rats with pulmonary arterial hypertension.
Purpose: The objective of this study was to investigate whether the NLRP3 inflammasome mediates disturbance of the ventricular immune microenvironment of PAH rats and promotes right ventricular failure.
Methods: Intraperitoneal injection of monocrotaline was used to induce PAH in rats. Right ventricular function was measured via echocardiography before and after the rats were treated with lipopolysaccharide and MCC950. The degree of immune microenvironment disturbance in right ventricular tissue was measured with a rat chemokine and cytokine antibody array, Western blot, flow cytometry and quantitative real-time PCR analysis.
Results: After the rats were injected with LPS, they exhibited right ventricular dysfunction and a significant increase in right ventricular tissue inflammation with elevated M1 macrophage proportion. Administration of MCC950 suppressed inflammation and improved right ventricular function. The number of M1 macrophages was decreased after MCC950 treatment. NLRP3 inflammasome inhibition ameliorated LPS-induced changes in the immune microenvironment in the right heart and right ventricular dysfunction in rats with PAH.
Conclusion: Selective inhibition of NLRP3 pathway interfered the interaction between hypertrophic cardiomyocytes and macrophages in the initial stage of inflammation and maintained the immune microenvironment balance, eventually contributing to attenuation of LPS-induced acute heart failure in PAH rats.
Keywords: pulmonary arterial hypertension, right ventricular failure, NLRP3 inflammasome, inflammation, immune microenvironment
Introduction
Right ventricular failure (RVF) is the most important prognostic factor for both morbidity and mortality in patients with pulmonary arterial hypertension (PAH).1 When PAH patients suffer from pulmonary infection or puerperal infection heart failure often rapidly develops.2 To date, unlike research on left heart failure, research on RVF is insufficient.3
Inflammation may play an important role in left heart failure.4 NACHT, LRR and PYD domain-containing protein 3 (NLRP3) is an intracellular danger sensor that oligomerizes upon activation and initiates assembly of the inflammasome by recruiting apoptosis speck-like protein containing a caspase recruitment domain (ASC) and pro-casp1.5,6 Formation of the NLRP3 inflammasome leads to IL-1β maturation and release, which is a critical, finely regulated step in the process of amplification of the inflammatory response following cell or tissue injury.7 Many studies have highlighted the association of NLRP3 activation with hypertension and cardiovascular disease.8–10 A recent study demonstrated that through the NLRP3 inflammasome pathway, low-dose lipopolysaccharide (LPS; 2 mg/kg) is sufficient to induce cardiac dysfunction in NLRP3-A350V/CreT mutant mice.11 NLRP3 mediates IL-1β expression and release, but the mechanism by which NLRP3 participates in regulation of the myocardial immune microenvironment in the inflammatory state by mediating the interaction between cardiomyocytes and macrophages has not yet been reported, and whether the NLRP3 inflammasome pathway contributes to RVF and the underlying mechanisms are largely unknown. We verified that the NLRP3 inflammasome is involved in regulating macrophage polarization and promoting the occurrence and development of acute right heart failure by promoting myocardial pyroptosis and monocyte chemoattractant protein-1 (MCP-1) expression.
Materials and Methods
Animal Experimental Protocols
All experiments were carried out in accordance with the recommendations of national and international animal care and ethical guidelines and were approved by the Ethics Committee for Animal Research of Xiangya Hospital of Central South University (permit code: 2020sydw0350). Sprague-Dawley (SD) rats (weighing 220–250 g) were used for the present study. The rats were administered a single intraperitoneal injection of monocrotaline (MCT, 55 mg/kg; Sigma-Aldrich, St. Louis, MO) and then fed for 28 days. Then, transthoracic echocardiography was performed every two days to measure the maximum velocity of tricuspid regurgitation. A maximum tricuspid regurgitation velocity greater than or equal to 3 m/s was the criterion for successful establishment of the PAH model.
LPS and MCC950 Experiments
In experiment one, normal and PAH rats were randomly assigned to the saline injection group or low-dose LPS (1 mg/kg, serotype O55:B5, Sigma-Aldrich) injection group. Cardiac function data were obtained, and right ventricular tissues were harvested six hours after injection (Figure S1A).
In experiment two, PAH rats were randomly assigned to two groups. One hour before LPS injection, MCC950 (10 mg/kg; MedChem Express, Monmouth Junction, NJ) or the same volume of saline was intravenously injected through the caudal vein. Cardiac function data were obtained, and right ventricular tissues were harvested six hours after injection (Figure S1B).
Cell Culture and Treatment
H9C2 cells (Cell Bank of the Chinese Academy of Science, Shanghai, China) were cultured in Dulbecco’s modified Eagle’s medium with 10% foetal bovine serum (FBS) and 1% penicillin/streptomycin. H9C2 cells were exposed to arginine vasopressin (AVP, 1 µmol/mL, S80117; Shanghai Yuanye Bio-Technology Co., Ltd) for 72 h to induce hypertrophy.12
Rat primary macrophages from bone marrow (RMa-bm cells, cat. #1920; ScienCell, Carlsbad, CA, USA) were cultured in macrophage medium (MaM, cat. #1921; ScienCell) with 5% FBS (ScienCell) and 1% penicillin/streptomycin (cat. #0503; ScienCell).
To investigate the interaction between cardiomyocytes and macrophages, the supernatant of H9C2 cells was collected for stimulation of macrophages. MCC950 (100 nM; MedChem Express, Monmouth Junction, NJ) was added to H9C2 medium to inhibit NLRP3 inflammasome activation 1 hour before LPS administration.
The cells were grown at 37°C in a humidified atmosphere with 95% air and 5% CO2, and the medium was changed every 3 days. Eventually, the cells were collected for RNA and protein detection.
Echocardiographic Analysis of Cardiac Function
Invasive and noninvasive detection methods were used to verify the right heart function measurements in rats. All rats were anaesthetized by inhalation of 6% to 8% (v/v) sevoflurane, and anaesthesia was maintained with 2% (v/v) sevoflurane. All rats were placed on a heating pad to maintain a body temperature of 37°C. Echocardiography was performed with the Vivid E7 system (General Electric Vingmed, Horten, Norway). Right ventricular systolic pressure (RVSP) was equal to cross-valve pressure plus an estimated right atrial pressure (RAP) of 10 mmHg. Cross-valve pressure was estimated by calculating the maximum velocity of the tricuspid regurgitant (TRmax) jet using the modified Bernoulli equation. Right ventricular function was determined by measuring the fractional area change (FAC), the ratio between the difference in end-diastolic and end-systolic area and the end-diastolic area of the right ventricle.
Cardiac Positron Emission Tomography (PET) and Chest Computed Tomography (CT)
PET scans were performed with a Mediso NanoScan sequential PET/CT scanner (Mediso Ltd., Budapest, Hungary) to observe morphological changes in PAH rat hearts (Figure 1B). Briefly, anaesthesia was maintained with 2% (v/v) isoflurane, and the animals were visually monitored throughout the whole procedure. Body temperature was maintained with warm blankets. The rats were administered a bolus injection of 300–450 μCi 18F-fluorodeoxyglucose (FDG) via the tail vein at the beginning of the scan. The amount of tracer in the syringe before and after the injection was measured with a dose calibrator to determine the actual amount of the administered isotope; 20 min after FDG injection, a 10-minute CT scan followed by a 30-minute PET scan was performed.
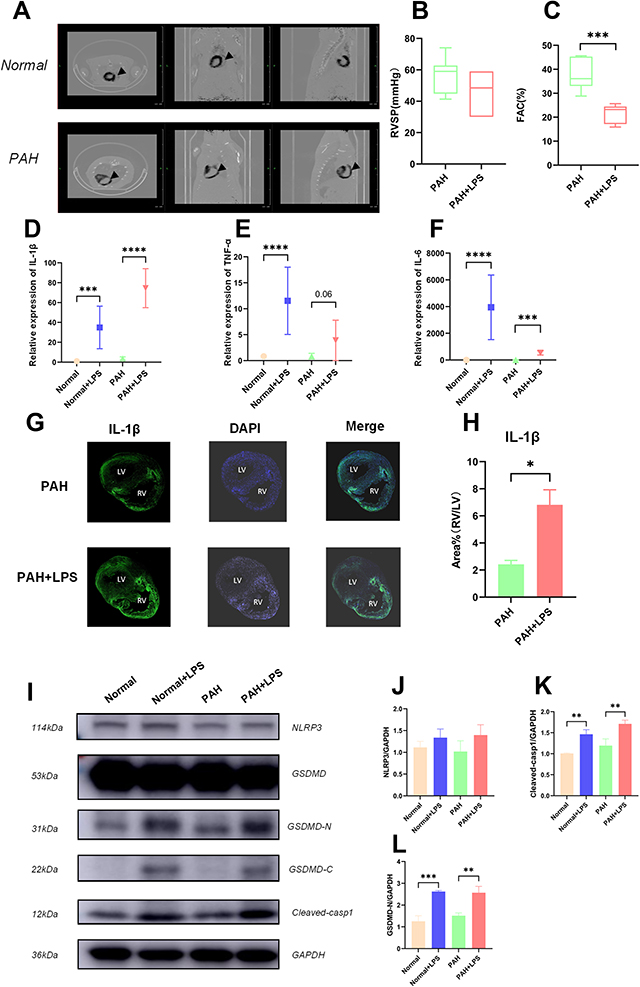 |
Figure 1 Activation of the NLRP3 pathway was associated with RVF in PAH rats. (A) PET-CT image of PAH rat hearts showing a thickened wall and dilated chamber of the right ventricle in a rat with pulmonary hypertension. (B) The RVSP of the PAH rats before and after LPS injection is shown. The significance of differences was determined with unpaired Welch’s t-test (n = 6). (C) The FAC [FAC = (end-diastolic area - end-systolic area)/end-diastolic area×100%] in the PAH rats before and after LPS injection is shown. The significance of differences was analysed with unpaired Welch’s t-test (n=6). (D) The IL-1β mRNA level in normal rats (SD rats were fed for 28 days after intraperitoneal injection of saline, and the samples were collected 6 hours after another intraperitoneal injection of saline), normal+LPS rats (SD rats were fed for 28 days after intraperitoneal injection of saline, and the samples were collected 6 hours after intraperitoneal injection of LPS), PAH rats (SD rats were fed for 28 days after intraperitoneal injection of MCT, and the samples were collected 6 hours after intraperitoneal injection of saline), and PAH+LPS rats (SD rats were fed for 28 days after intraperitoneal injection of MCT, and the samples were collected 6 hours after intraperitoneal injection of LPS). The significance of differences among groups was determined by one-way ANOVA (n = 5). (E) The TNF-α mRNA level in the right ventricles of rats in the abovementioned four groups is shown. The significance of differences among groups was determined by one-way ANOVA (n = 5). (F) The IL-6 mRNA level in the right ventricles of rats in the four abovementioned groups is shown. The significance of differences among the groups was determined by one-way ANOVA (n = 5). (G) IL-1β immunofluorescence in the right ventricle of PAH rats and PAH+LPS rats 6 h after LPS injection. (H) LPS significantly increased IL-1β expression in the right ventricle compared with the left ventricle in rats with pulmonary hypertension. The ratio of the IL-1β area% in the right ventricle to that in the left ventricle is shown. The significance of differences was determined with an unpaired Welch’s t-test (n = 3). (I) NLRP3, cleaved-casp1, GSDMD, GSDMD-N and GAPDH protein expression in the right ventricles of rats in the abovementioned four groups. (J) NLRP3 is an important upstream molecule of the pyroptosis pathway, which is involved in the formation of inflammasomes and cleavage of pro-casp1. The NLRP3 protein level in the right ventricle of rats in the abovementioned four groups is shown. The significance of the difference was analysed by one -way ANOVA (n = 3). (K) Cleaved casp1 cleaved the IL-1B precursor to generate mature IL-1B and cleaved GSDMD to expose GSMDM-N, thereby promoting cell lysis and pyroptosis. The cleaved casp1 protein level in the right ventricles of rats in the abovementioned four groups is shown. The significance of the difference was analysed by one -way ANOVA (n = 3). (L) GSDMD is an executor of pyroptosis, and the GSDMD N-terminal domain can assemble membrane pores to induce pyroptosis. The GSDMD-N protein level in the right ventricle of rats in the abovementioned four groups is shown. The significance of the difference was analysed by one -way ANOVA (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. |
Western Blot Analysis
Protein levels were measured with specific primary antibodies against NLRP3 (1:300, ab214185), pro casp1+p10+p12 (1:1000, ab179515), GSDMD (1:1000, ab219800; all from Abcam, San Francisco, CA), phospholamban (PLN 1:1000, #14562), ATP2A2/SERCA2 (1:1000, #4388), and phospho-PLN (1:1000, #8496; all obtained from Cell Signaling Technology, Danvers, MA, US). A GAPDH antibody (1:5000, A531; Bioworld Technology, St. Louis Park, MN, USA) was used to measure the level of GAPDH as an internal control.
Total proteins were separated on 10% or 12% SDS-polyacrylamide electrophoresis gels and transferred to a PVDF membrane (Millipore, Merck, Darmstadt, Germany). The membrane was blocked with 5% FBS in Tris-buffered saline with 0.1% Tween 20 (TBST) at room temperature for 2 hours and incubated with a specific primary antibody at 4°C overnight. The membrane was then incubated with a secondary horseradish peroxidase (HRP)-conjugated antibody (1:20000) at room temperature for 90 min. Finally, enhanced chemiluminescence (ECL) reagent was used to detect the bands. The obtained images were analysed with Image Lab software. The membrane was washed by stripping buffer (NCM biotech, Suzhou, China) for incubation with next primary antibody.
Quantitative Real-Time PCR
Total RNA was isolated from rat right ventricular tissue and cultured cells using a Total RNA Kit II (R6934-01; Omega Bio-tek, Norcross, GA, USA) according to the manufacturer’s instructions. An RT Reagent Kit with gDNA Eraser (No. RR047A; Takara, Tokyo, Japan) was used for cDNA synthesis. Real-time PCR was performed using All-in-OneTM qPCR Mix (No: QP001; GeneCopoeia, Germantown, MD, USA) according to the manufacturer’s protocol. Most primer sequences are listed in Table 1. Primers for TNF-α, ARG1, and PLN were purchased from GeneCopoeia (Germantown, MD, USA).
 |
Table 1 Sequences of Primers Used for Real-Time qPCR Experiments |
Immunofluorescence Analysis
Immunofluorescence staining was performed on paraffin-embedded tissue sections. The paraffin sections were dewaxed and placed in a box filled with EDTA antigen retrieval buffer (pH 8.0, G1206; Wuhan Servicebio Technology Co., Ltd., Wuhan, China) and heated in a microwave oven for antigen retrieval. The slides were placed in phosphate-buffered saline (PBS) (pH 7.4) and washed 3 times for 5 min each. After the sections were slightly dried, a circle was drawn around the tissue with a PAP pen. Blocking solution (5% BSA) was added to the tissue for 30 min and then gently shaken off. Primary antibody against IL-1β (1:200, GB11113; Wuhan Servicebio Technology Co., Ltd., Wuhan, China) was added to the sections, and the sections were placed flat in a wet box and incubated overnight at 4°C. Then, the sections were placed in PBS (pH 7.4) and washed 3 times for 5 min each. After the sections were slightly shaken and dried, secondary antibody (1:400, GB21301, Wuhan Servicebio Technology Co., Ltd., Wuhan, China) was added dropwise to the circle, and the sections were incubated at room temperature in the dark for 50 min. Then, the sections were placed in PBS (pH 7.4) and washed 3 times for 5 min each. Finally, the sections were slightly dried, sealed with an anti-fluorescence quenching agent (G1401; Wuhan Servicebio Technology Co., Ltd., Wuhan, China) and observed under a fluorescence microscope, and images were collected.
Analysis of the Rat Intracardiac Cytokine Protein Profile with a Rat Chemokine and Cytokine Antibody Array
Multiple ELISA antibody array detection was performed using a Rat Cytokine Array Q67 Kit (QAR-CAA-67; RayBiotech, Norcross GA) according to the manufacturer’s protocol. The Rat Cytokine Array Q67 Kit was used to simultaneously and quantitatively assay multiple (67) rat factors, including chemokines, inflammatory cytokines, growth factors, and receptors (commonly referred to in this report as soluble factors), in samples. We used rat right ventricular tissue homogenates to perform the assay. Each sample was tested individually in quintuplicate, and the values were averaged. The actual concentration of each cytokine was directly quantified according to a standard curve in parallel as part of the array.
Flow Cytometry Analysis
Single-cell suspensions of CD45-positive inflammatory cells extracted from myocardial tissue were prepared as described above.
To block non-specific binding of the antibody to the Fcγ receptor, the single-cell suspension was first incubated at 4°C with an anti-CD32 antibody (1:400 550273; BD Biosciences, Brøndby, Denmark) for 15 min. Subsequently, the cells were incubated at 4°C with Fixable Viability Stain 510 (1:400 564406; BD Biosciences, Brøndby, Denmark) for 30 min. Next, the cells were incubated with a variety of flow cytometry antibodies (shown in Table 2) at 4°C in the dark for 30 min and stained for cell identification markers. Flow cytometry analysis and cell sorting were performed on a DxP AthenaTM instrument (Cytekbio, Landing Parkway Fremont, USA), and the data were analysed using FlowJo software (Tree Star).
 |
Table 2 Flow Cytometry Antibody |
Separation of CD45+ Cells
Single-cell suspensions were prepared using a neonatal heart dissociation kit (no. 130-098-373; Miltenyi Biotec, Bergisch Gladbach, Germany) following the manufacturer’s instructions. The cell suspensions were then incubated with CD45-labelled magnetic beads (Miltenyi Biotec, Bergisch Gladbach, Germany) at 37°C for 30 min. Strongly magnetically labelled cells were passed through LS Columns (Miltenyi Biotec, Bergisch Gladbach, Germany) for magnetic separation to enrich CD45-positive cells in right ventricular tissues.
Statistical Analysis
All replicate data are expressed as the means ± SD. The distribution of the data was assessed with the Shapiro–Wilk normality test. The significance of differences between two independent experimental groups was assessed using unpaired Welch’s t-test or one-way ANOVA, whereas the data between groups with repeated measurements were compared using two-way ANOVA. The value of statistical significance was set at p < 0.05. Statistical analyses were performed using GraphPad Prism 8 software (GraphPad Software Inc., USA).
Results
NLRP3 Inflammasome Activation in the Right Ventricles of RVF Rats
MCT caused wall thickening and ventricular dilatation in the right ventricle of PAH rats 28 days after administration (Figure 1A). Echocardiography (Figure 1B, n = 6) and right heart catheterization (Figure S1C) showed that with the stably elevated right ventricular systolic pressure (RVSP) caused by MCT, intraperitoneal injection of LPS (1 mg/kg) caused rapid progression to RVF in PAH rats but not in normal rats within six hours (Figure 1C, n = 6, ***p < 0.001). Quantitative real-time PCR (qPCR) showed that the SERCA2a (Figure S1D, n = 6, ***p < 0.001) and type 2 ryanodine receptor (RyR2) mRNA levels (Figure S1E, n = 6, *p < 0.05) were significantly decreased in the right ventricle of RVF rats.
We then detected the mRNA levels of inflammatory factors that have already been reported to be associated with cardiac dysfunction (IL-1β, IL-6 and TNF-α) in tissue (Figure 1D–F, n = 6, **p < 0.01, ***p < 0.001). After LPS injection, the IL-1β, IL-6 and TNF-α mRNA levels were generally elevated in the right hearts of rats. Among them, the IL-1β mRNA level was the most elevated in rats in the PAH+LPS group, indicating that it was more relevant to heart dysfunction. Immunofluorescence showed asymmetry in IL-1β expression between the left and right hearts under inflammatory stimulation (Figure 1G), specifically, after intraperitoneal injection of LPS (1 mg/kg), the right ventricle expressed more IL-1β than the left ventricle in PAH rats (Figure 1H, n = 3, **p < 0.01).
The NLRP3 pathway is the direct upstream pathway for IL-1β maturation and release. Western blotting analysis showed that LPS did not significantly increased the NLRP3 expression (Figure 1I and J, n = 3) but significantly promoted cleaved-casp1 production (Figure 1I and K, n = 3, **p < 0.01) and exposure to the N-fragment of GSDMD (GSDMD-N) (Figure 1I and L, n = 3, **p < 0.01, ***p < 0.001) in right ventricular tissues.
Systemic Inhibition of NLRP3 Ameliorated the RVF Progression
We administered MCC950 to inhibit the NLRP3 pathway to explore the role of NLRP3 in the pathogenesis of acute RVF. Western blotting analysis demonstrated that MCC950 effectively inhibited cleaved-casp1 expression (Figure S2A and C, n = 5, **p < 0.01) and GSDMD-N production (Figure S2A and D, n = 5, ***p < 0.001) without affecting NLRP3 expression (Figure S2A and B, n = 3), which was consistent with previous research. Echocardiography showed that intravenous injection of MCC950 significantly prevented RVF caused by subsequent intraperitoneal LPS injection in PAH rats (Figure 2A, n = 6, ***p < 0.001). Survival analysis showed that nearly half of PAH rats died within 6 hours after LPS injection, and MCC950 application reduced the mortality of PAH rats within 6 hours after LPS injection (Figure S2E, n = 18–21). In addition, immunofluorescence results showed that MCC950 alleviated the asymmetrically increased IL-1β expression in myocardial tissue of the right ventricle of PAH rats (Figure 2B and C, n = 3, *p < 0.05, **p < 0.01), which was not seen in normal rats (Figure S2F and G n = 3). And qPCR showed application of MCC950 protected SERCA2a and RYR2 mRNA from declining (Figure S2H and I n = 6, *p < 0.05, **p < 0.01).
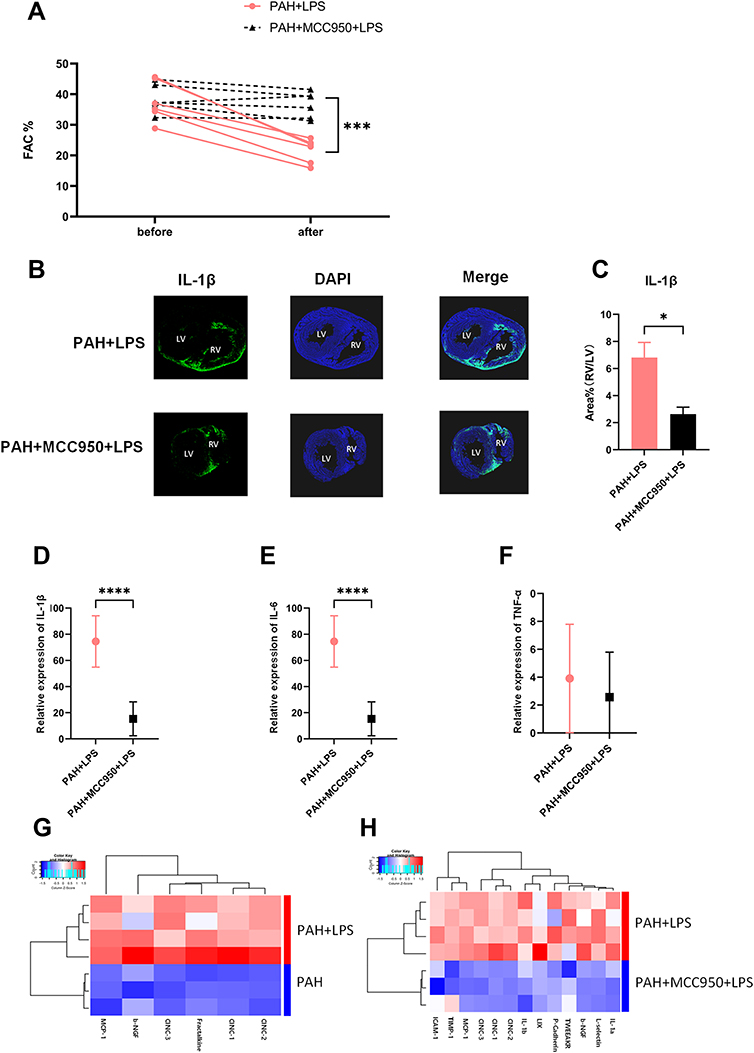 |
Figure 2 NLRP3 inhibition effectively protected right ventricular function, relieved the asymmetrical IL-1β elevation and erased the change in inflammation in the right ventricle. (A) The FAC in PAH+LPS rats (SD rats were fed for 28 days after intraperitoneal injection of MCT; intravenous injection of saline was performed 1 hour before LPS injection, and the samples were collected 6 hours after intraperitoneal injection of LPS) and PAH+MCC950+LPS rats (SD rats were fed for 28 days after intraperitoneal injection of MCT; intravenous injection of MCC950 was performed 1 hour before LPS injection, and the samples were collected 6 hours after intraperitoneal injection of LPS) before and after LPS injection is shown. The significance of differences was determined with two-way ANOVA (n = 6). (B) IL-1β immunofluorescence in the right ventricle of PAH+LPS rats and PAH+MCC950+LPS rats 6 h after LPS injection. (C) Pretreatment with MCC950 significantly alleviated the asymmetrically increased IL-1β expression in myocardial tissue of the right ventricle. The ratio of the IL-1β area% in the right ventricle to that in the left ventricle is shown. The significance of differences was determined with unpaired Welch’s t-test (n = 3). (D) The IL-1β mRNA level in the right ventricles of rats in the two abovementioned groups. The significance of differences was determined with unpaired Welch’s t-test (n = 6). (E) The TNF-α mRNA level in the right ventricles of rats in the two abovementioned groups. The significance of differences was determined with unpaired Welch’s t-test (n = 6). (F) The IL-6 mRNA level in the right ventricle of rats in the two abovementioned groups. The significance of differences was determined with unpaired Welch’s t-test (n = 6). (G) A total of 67 cytokines and chemokines in right ventricular tissue were tested with a cytokine array. The heat map shows that the expression of 6 factors (MCP-1, b-NGF, CINC-1, CINC-2, CINC3, and fractalkine) was increased in the right ventricular myocardium of PAH rats after LPS injection. (H) Heat map showing that 13 factors (MCP-1, b-NGF, CINC-1, CINC-2, CINC3, ICAM-1, TIMP-1, LIX, IL-1β, IL-1α, L-selectin, TWEAK R, and P-cadherin) remained at low levels in the right ventricular myocardium of PAH+MCC950+LPS rats after LPS injection. The significance of differences between two groups was determined with an unpaired Student’s t-test (n = 3–4). *p < 0.05, ***p < 0.001, ****p < 0.0001. |
Systemic Blockade of NLRP3 Ameliorated LPS-Induced Immune Microenvironment Fluctuations in Right Heart Tissue
We next examined changes in other classic injurious inflammatory cytokines by qPCR and found that after MCC950 administration, not only IL-1β (Figure 2D, n = 6, ***p < 0.001) but also TNF-α (Figure 2E, n = 6) and IL-6 (Figure 2F, n = 6, ***p < 0.001) remained at relatively low levels in RVF rat heart. To confirm the global changes in the immune microenvironment of the right ventricle regulated by NLRP3, we performed chemokine and cytokine antibody array and flow cytometry analyses. Heatmaps showed changes in the expression of inflammatory cytokines, chemokines, growth factors and adhesion molecules in the right ventricular tissues of PAH rats after LPS application and after MCC950 pretreatment (Figure 2G and H, n = 3–4). Elevated cytokines after LPS injection were compared with cytokines that were maintained at relatively lower levels in the MCC950 preconditioning group (Figure S3A), five chemokines and growth factors (MCP-1, CINC1, CINC2, CINC3, and b-NGF) were significantly elevated in the RVF rat hearts and remained at low levels after pharmacological inhibition of the NLRP3 pathway. We verified the array results for MCP-1, CINC1, CINC2, and CINC3 (Figure S3B–E, n = 6, *p < 0.05, **p < 0.01, ***p < 0.001) via q-PCR.
Flow cytometry was used to analyse the infiltrating inflammatory cell components in rat right ventricular tissue. CD45-positive cells and neutrophils were identified in single-cell suspensions obtained from intact fresh right ventricular free walls (Figure S3F). CD11b, CD86, and CD68 staining was performed on CD45-positive single-cell suspensions obtained with CD45-labelled magnetic beads (Figure S3G), and the purification efficiency of the magnetic beads was determined to be above 80% (Figure S3H and I, n = 16, ***p < 0.001). Flow cytometry showed that more CD45+ inflammatory cells (Figure 3A and D, n = 3–4) and CD45-positive/CD11b-positive mononuclear macrophages (Figure 3B and E, n = 3–4, **p < 0.01) accumulated in right heart tissue in the PAH group than in the normal group. After LPS injection, the proportion of CD68-positive/CD86-positive M1-type macrophages (Figure 3C and F, n = 3–4, **p < 0.01, ***p < 0.001) in the right heart tissue was significantly increased in the PAH group, and treatment with MCC950 prevented this effect.
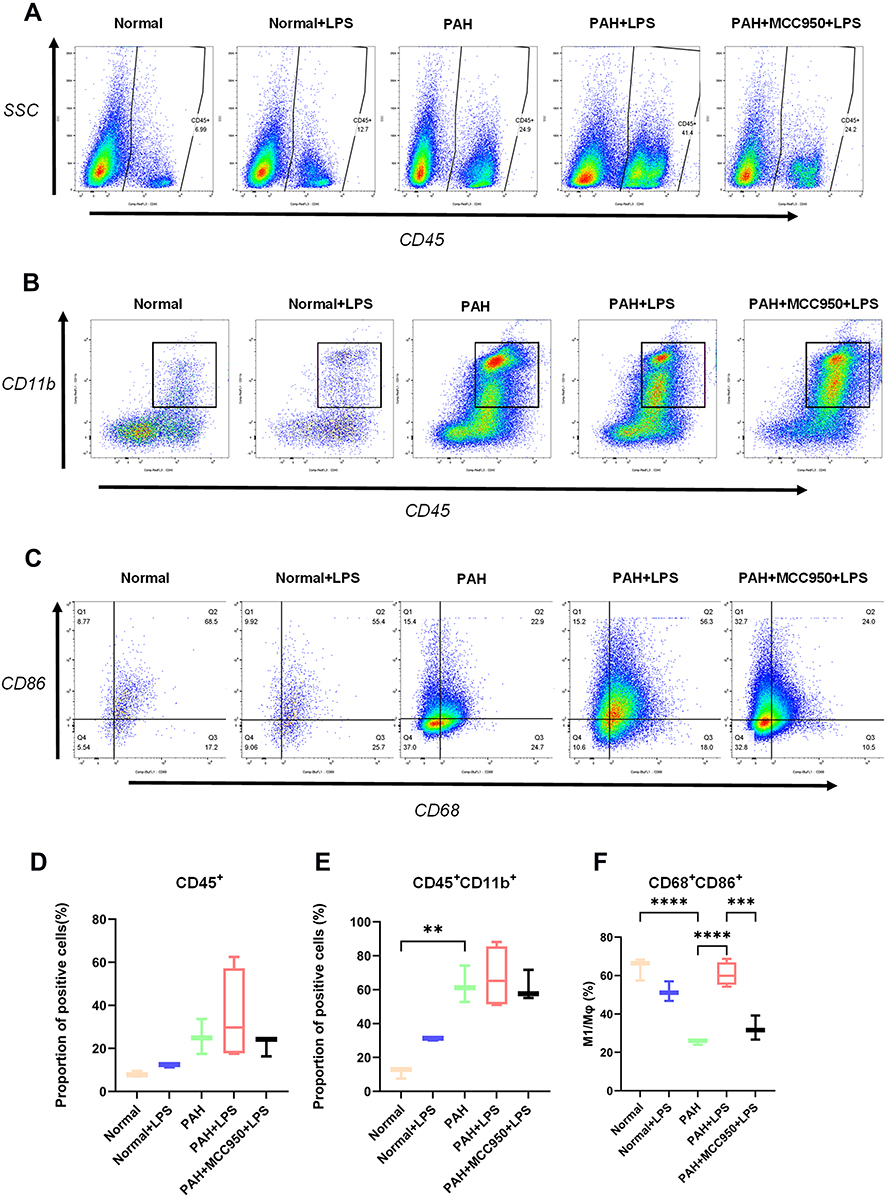 |
Figure 3 Changes in the proportion of immune cells in myocardial tissue were analysed via flow cytometry. (A) The proportion of CD45+ cells in each group was detected by flow cytometry. (B) The proportion of CD45+CD11b+ cells in each group was detected by flow cytometry. (C) The proportion of CD68+CD86+ cells in each group was detected by flow cytometry. (D) Pulmonary hypertension and LPS caused a significant increase in the proportion of CD45-positive inflammatory cells in right ventricular tissue (normal vs PAH, normal vs normal + LPS, n = 3). (E) CD45-positive/CD11b-positive mononuclear macrophage infiltration was significantly elevated in right ventricular tissues from rats subjected to PAH or LPS (normal vs PAH, normal vs normal + LPS, normal + LPS vs PAH + LPS, n = 3). (F) The proportion of CD68-positive/CD86-positive M1 macrophages was decreased in the hearts of PAH rats and significantly increased in the right ventricular tissues of PAH rats stimulated with LPS, but MCC950 application significantly inhibited the change in the proportion of CD68-positive/CD86-positive M1 macrophages (n = 3). The significance of the difference was analysed by one-way ANOVA, **p < 0.01, ***p < 0.001, ****p < 0.0001. |
NLRP3 Mediated the Interaction Between Cardiomyocytes and Macrophages and Contributed to Macrophage Polarization to the Proinflammatory M1 Phenotype
First, we confirmed that under LPS stimulation, the NLRP3 pathway was active in normal H9C2 cells and hypertrophic H9C2 cells, and the in vitro inhibitory effect of MCC950 was also verified by Western blotting (Figure S4A–D, n = 3, *p < 0.05, ***p < 0.001). Through q-PCR, we found that H9C2 cells expressed very little IL-1β, and IL-1β in activated macrophages was not affected by NLRP3 inhibition (Figure S4E, n = 3, *p < 0.05).
On the other hand, LPS induced elevated NLPR3 and MCP-1 mRNA levels in hypertrophic H9C2 cells (Figure 4A and B, n = 3, *p < 0.05, **p < 0.01, ***p < 0.001), and the NLRP3 and MCP-1 mRNA levels showed a strong correlation (Figure 4C, n = 14, r2=0.5840, ***p < 0.001). Intriguingly, the inhibitory effect of MCC950 on the NLRP3 pathway significantly downregulated MCP-1 mRNA without any influence on NLRP3 mRNA (Figure 4D and E, n = 6, **p < 0.01). We then collected the supernatant from different groups of myocardial cells and added it to the medium to stimulate macrophages (Figure 4F). We found that comparing to normal H9C2 cell with LPS stimulation, the supernatant of LPS-stimulated hypertrophic cardiomyocytes could cause higher iNOS (Figure 4G, n = 3, *p < 0.05) and TNF-α (Figure 4H, n = 3, *p < 0.05) transcription in macrophages as M1-type markers. This effect was abolished by MCC950 treatment (Figure 4J and K, n = 3, *p < 0.05, ***p < 0.001). As for M2-type markers, there was no significant difference in arg1 and il-10 expression between NLM (Macrophage+H9C2+LPS) and ALM (Macrophage+AVP conditioned H9C2+LPS) group (Figure S4F and G), and the application of NLRP3 inhibitor had no affection on M2-type markers transcription (Figure S4H and I).
 |
Figure 4 Through the NLRP3 pathway, cardiomyocytes participate in the macrophage polarization process through increased MCP-1 expression and cell pyroptosis under LPS stimulation. (A) We exposed H9C2 cells to AVP (1 µmol/mL) for 72 h to simulate hypertrophy of the right ventricular myocardium in rats with pulmonary hypertension, to MCC950 (100 mM) for 1 h to inhibit the NLRP3 pathway, and to LPS (100 µg/mL) for 6 h to mimic the in vivo inflammation state in the RVF model. The NLRP3 mRNA level in the H9C2, H9C2+LPS, H9C2+AVP, H9C2+AVP+LPS, and H9C2+AVP+MCC950+LPS cell groups is shown. The significance of the difference was analysed by one-way ANOVA (n = 3). (B) The MCP-1 mRNA level in the five abovementioned groups of cells is shown. The significance of the difference was analysed by one-way ANOVA (n = 3). (C) We analysed the correlation between NLRP3 and MCP-1 mRNA expression in H9C2 cell samples from different treatment groups. MCP-1 expression was highly correlated with NLRP3 expression (n = 14, r2 = 0.5840, p < 0.001). (D) The NLRP3 mRNA level in the H9C2+AVP+LPS and H9C2+AVP+MCC950+LPS cell groups is shown. The significance of differences was determined with an unpaired Welch’s t-test (n = 6). (E) NLRP3 inhibition decreased MCP-1 transcription, and the MCP-1 mRNA level in the H9C2+AVP+LPS and H9C2+AVP+MCC950+LPS cell groups. The significance of differences between two groups was determined with an unpaired Welch’s t-test (n = 6). (F) We exposed macrophages to supernatant collected from H9C2 cells in different groups and extracted RNA for subsequent analysis. (G) The supernatant collected from AVP+LPS-conditioned H9C2 cells enhanced iNOS transcription in macrophages. The iNOS mRNA level in the macrophage+H9C2+PBS, macrophage+H9C2+LPS, macrophage+AVP-conditioned H9C2+PBS and macrophage+AVP-conditioned H9C2+LPS cell groups is shown. The significance of differences between two groups was determined with unpaired Welch’s t-test (n = 3). (H) The supernatant collected from AVP+LPS-conditioned H9C2 cells enhanced TNF-α transcription in macrophages. The TNF-α mRNA level in the macrophage+H9C2+PBS, macrophage+H9C2+LPS, macrophage+AVP-conditioned H9C2+PBS and macrophage+AVP-conditioned H9C2+LPS cell groups is shown. The significance of differences between two groups was determined with an unpaired Welch’s t-test (n = 3). (I) The IL-1β mRNA level in the macrophage+H9C2+PBS, macrophage+ H9C2+LPS, macrophage+AVP-conditioned H9C2+PBS and macrophage+AVP-conditioned H9C2+LPS cell groups is shown. The significance of differences between two groups was determined with an unpaired Welch’s t-test (n = 3). (J) MCC950 administration cancelled the enhanced effect of AVP+LPS-conditioned H9C2 cell supernatant on iNOS transcription in macrophages. The iNOS mRNA level in the macrophage+AVP-conditioned H9C2+LPS and macrophage+AVP-conditioned H9C2+MCC950+LPS cell groups is shown. The significance of differences was determined with unpaired Welch’s t-test (n = 3). (K) MCC950 administration cancelled the enhanced effect of AVP+LPS-conditioned H9C2 cell supernatant on TNF-α transcription in macrophages. The TNF-α mRNA level in the macrophage+AVP-conditioned H9C2+LPS and macrophage+AVP-conditioned H9C2+MCC950+LPS cell groups is shown. The significance of differences was determined with unpaired Welch’s t-test (n = 3). (L) The IL-1β mRNA level in macrophage+AVP-conditioned H9C2+LPS and macrophage+AVP-conditioned H9C2+MCC950+LPS cell groups is shown. The significance of differences was analysed with unpaired Welch’s t-test (n = 3). *p < 0.05, ***p < 0.001, ****p < 0.0001. |
Discussion
In this study, we demonstrated that the NLRP3 inflammasome plays a significant role in mediating acute RVF in PAH rats. When PAH rats were subjected to LPS, the immune microenvironment dramatically changed, with increased inflammatory cytokine, chemokine, growth factor expression and an increased proportion of M1 macrophage polarization. Inhibition of NLRP3 considerably reduced cardiomyocyte inflammation levels and prevented M1 macrophage polarization, eventually ameliorating cardiac dysfunction and maintaining the expression of calcium homeostasis-related proteins. This study is the first to investigate the relationship between the NLRP3 pathway and the myocardial immune microenvironment in an RVF model.
Pyroptosis, which was first observed in macrophages,13 has now been reported in a variety of other cell types, including neutrophils, dendritic cells, endothelial cells, and cardiomyocytes, and participates in the occurrence and progression of many diseases.14–21 Both in vivo and in vitro, Lid et al found that LPS can activate the NLRP3/pyroptosis pathway in the myocardium to mimic a sepsis-induced cardiomyopathy, inducing cardiac dysfunction and inflammation.22 In our study, despite the large accumulation of CD45-positive inflammatory cells, the right ventricle of rats with pulmonary hypertension was in an immune microenvironment with anti-inflammatory factors dominant; in this microenvironment, the inflammation level, chemokines expression levels, and the proportion of M1 macrophages were low. After LPS activated the NLRP3 pathway in cardiomyocytes, the immune microenvironment balance was destroyed. Cardiomyocytes changed the polarization trend of macrophages by releasing a large amount of cell contents and MCP-1 through pyroptosis, and under LPS stimulation, M1 macrophages released more inflammatory factors that damaged cardiomyocytes.
Another possible explanation for the effect of intravenous MCC950 on the overall level of the NLRP3 pathway, including in cardiomyocytes and macrophages, is that it may prevent amplification of the inflammatory cascade by inhibiting IL-1β maturation and release, thus reducing the overall level of inflammatory factor transcription in right heart tissue. These two mechanisms may coexist, but more research is needed to confirm this speculation.
We also discovered that the NLRP3 inflammasome may regulate MCP-1 expression in a manner independent of IL-1β. Our experimental results show that LPS induces activation of the NLRP3 inflammasome in hypertrophic cardiomyocytes, thus increasing MCP-1 expression to help stimulate polarization of surrounding infiltrating macrophages to the M1 phenotype. The correlation between NLRP3 and MCP-1 expression has been demonstrated in many previous studies.23–26 However, the change in MCP-1 expression is generally considered part of the regulatory effect of NLRP3 on the overall inflammation level. Jung Hwa Ko noted in his research that the NLRP3 inflammasome promotes the IL-1β maturation and release and allows IL-1β to bind to the IL-1 receptor, activating the transcription of subsequent inflammatory factors, such as IL-6, IL-8 and MCP-1.23 In our study, the IL-1β mRNA level in H9C2 cells was low and stable. We found that the MCP-1 and NLRP3 transcription levels were significantly correlated without the presence of IL-1β, and the MCP-1 mRNA level was significantly reduced by MCC950-mediated NLRP3 inhibition. We believe that, the NLRP3 inflammasome has a direct regulatory effect on MCP-1 transcription that is independent of IL-1β, which is very important because many studies have demonstrated the important role of MCP-1 in the macrophage polarization towards the M1 phenotype and in impairment of cardiac function.27–29 Notably, the role of MCP-1 in driving the direction of macrophage polarization has been controversial in many nonheart disease studies.30–36 However, in studies related to cardiovascular disease and heart failure, the role of the MCP-1-CCR2 pathway in promoting M1 polarization and the proinflammatory effect of CCR2+ macrophages have been more widely reported,37–43 which is consistent with our experimental results. We speculate that cardiomyocytes may release certain secretory factors that can collaborate with the MCP-1-CCR2 pathway and participate in M1 macrophage polarization. Further work is needed to analyse the composition and function of the components released from cardiomyocytes in different disease models.
We conclude that LPS activates the NLRP3 pathway in myocardial cells, increases MCP-1 expression and induces release of other pyroptosis products, thus destroying the immune microenvironment which was formerly anti-inflammatory. The increase in the proportion of M1 macrophages is responsible for inducing the release of IL-1β and other inflammatory factors to further impair cardiac function. Selective suppression of the NLRP3 inflammasome by MCC950 interfered with the interaction between hypertrophic cardiomyocytes and macrophages in the initial stage of inflammation and maintained the immune microenvironment balance, eventually contributing to attenuation of LPS-induced acute heart failure in PAH rats.
Acknowledgments
This work was financially supported by the National Nature Science Foundation of China (grant number 81873508, 81800058, 81700275).
Author Contributions
Lizhe Guo was responsible for the conception, study design and execution.
Yanan Cao and Sisi Dai were responsible for the study design and acquisition of data.
Yue Yang was responsible for data analysis and interpretation.
Lu Wang, Gang Qin were responsible for the study execution.
Lizhe Guo was responsible for drafting and substantially revising the manuscript, Yanan Cao, Yue Yang, Sisi Dai, Lu Wang and Gang Qin have critically reviewed the article.
E Wang was responsible for all aspects of this study, including the study design, experimental execution, data analysis, data interpretation, and manuscript production.
All authors listed have agreed on submitting to this journal, and have reviewed and agreed on all versions of the article. All authors have agreed to take responsibility and be accountable for the contents of the article.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Samson N, Paulin R. Epigenetics, inflammation and metabolism in right heart failure associated with pulmonary hypertension. Pulm Circ. 2017;7(3):572–587. doi:10.1177/2045893217714463
2. Zhang J, Cao Y, Gao X, et al. Lipopolysaccharide acutely suppresses right-ventricular strain in rats with pulmonary artery hypertension. Pulm Circ. 2018;8(1):2045893217744504. doi:10.1177/2045893217744504
3. Mehra MR, Park MH, Landzberg MJ, Lala A, Waxman AB. Right heart failure: toward a common language. J Heart Lung Transplant. 2014;33(2):123–126. doi:10.1016/j.healun.2013.10.015
4. Sun XQ, Abbate A, Bogaard HJ. Role of cardiac inflammation in right ventricular failure. Cardiovasc Res. 2017;113(12):1441–1452. doi:10.1093/cvr/cvx159
5. Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13(6):397–411. doi:10.1038/nri3452
6. Wang L, Manji GA, Grenier JM, et al. PYPAF7, a novel PYRIN-containing Apaf1-like protein that regulates activation of NF-kappa B and caspase-1-dependent cytokine processing. J Biol Chem. 2002;277(33):29874–29880. doi:10.1074/jbc.M203915200
7. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10(2):417–426. doi:10.1016/s1097-2765(02)00599-3
8. De Miguel C, Pelegrín P, Baroja-Mazo A, Cuevas S. Emerging role of the inflammasome and pyroptosis in hypertension. Int J Mol Sci. 2021;22(3):1064. doi:10.3390/ijms22031064
9. Pellegrini C, Martelli A, Antonioli L, Fornai M, Blandizzi C, Calderone V. NLRP3 inflammasome in cardiovascular diseases: pathophysiological and pharmacological implications. Med Res Rev. 2021;41(4):1890–1926. doi:10.1002/med.21781
10. Sandanger Ø, Ranheim T, Vinge LE, et al. The NLRP3 inflammasome is up-regulated in cardiac fibroblasts and mediates myocardial ischaemia-reperfusion injury. Cardiovasc Res. 2013;99(1):164–174. doi:10.1093/cvr/cvt091
11. Toldo S, Mezzaroma E, McGeough MD, et al. Independent roles of the priming and the triggering of the NLRP3 inflammasome in the heart. Cardiovasc Res. 2015;105(2):203–212. doi:10.1093/cvr/cvu259
12. Zhao J, Lei Y, Yang Y, Gao H, Gai Z, Li X. Metoprolol alleviates arginine vasopressin-induced cardiomyocyte hypertrophy by upregulating the AKT1-SERCA2 cascade in H9C2 cells. Cell Biosci. 2020;10(1):72. doi:10.1186/s13578-020-00434-y
13. Brennan MA, Cookson BT. Salmonella induces macrophage death by caspase-1-dependent necrosis. Mol Microbiol. 2000;38(1):31–40. doi:10.1046/j.1365-2958.2000.02103.x
14. Edgeworth JD, Spencer J, Phalipon A, Griffin GE, Sansonetti PJ. Cytotoxicity and interleukin-1beta processing following Shigella flexneri infection of human monocyte-derived dendritic cells. Eur J Immunol. 2002;32(5):1464–1471. doi:10.1002/1521-4141(200205)32:5<1464::Aid-immu1464>3.0.Co;2-g
15. Zhang Y, Liu X, Bai X, et al. Melatonin prevents endothelial cell pyroptosis via regulation of long noncoding RNA MEG3/miR-223/NLRP3 axis. J Pineal Res. 2018;64(2):e12449. doi:10.1111/jpi.12449
16. Ryu JC, Kim MJ, Kwon Y, et al. Neutrophil pyroptosis mediates pathology of P. aeruginosa lung infection in the absence of the NADPH oxidase NOX2. Mucosal Immunol. 2017;10(3):757–774. doi:10.1038/mi.2016.73
17. Qiu Z, Lei S, Zhao B, et al. NLRP3 inflammasome activation-mediated pyroptosis aggravates myocardial ischemia/reperfusion injury in diabetic rats. Oxid Med Cell Longev. 2017;2017:9743280. doi:10.1155/2017/9743280
18. Pan J, Han L, Guo J, et al. AIM2 accelerates the atherosclerotic plaque progressions in ApoE-/- mice. Biochem Biophys Res Commun. 2018;498(3):487–494. doi:10.1016/j.bbrc.2018.03.005
19. Teng JF, Mei QB, Zhou XG, et al. Polyphyllin VI induces caspase-1-mediated pyroptosis via the induction of ROS/NF-κB/NLRP3/GSDMD signal axis in non-small cell lung cancer. Cancers. 2020;12(1):193. doi:10.3390/cancers12010193
20. Jiang C, Jiang L, Li Q, et al. Acrolein induces NLRP3 inflammasome-mediated pyroptosis and suppresses migration via ROS-dependent autophagy in vascular endothelial cells. Toxicology. 2018;410:26–40. doi:10.1016/j.tox.2018.09.002
21. Zhang H, Chen X, Zong B, et al. Gypenosides improve diabetic cardiomyopathy by inhibiting ROS-mediated NLRP3 inflammasome activation. J Cell Mol Med. 2018;22(9):4437–4448. doi:10.1111/jcmm.13743
22. Li N, Zhou H, Wu H, et al. STING-IRF3 contributes to lipopolysaccharide-induced cardiac dysfunction, inflammation, apoptosis and pyroptosis by activating NLRP3. Redox Biol. 2019;24:101215. doi:10.1016/j.redox.2019.101215
23. Ko JH, Yoon SO, Lee HJ, Oh JY. Rapamycin regulates macrophage activation by inhibiting NLRP3 inflammasome-p38 MAPK-NFκB pathways in autophagy- and p62-dependent manners. Oncotarget. 2017;8(25):40817–40831. doi:10.18632/oncotarget.17256
24. Verma V, Gupta S, Kumar P, et al. Involvement of NLRP3 and NLRC4 inflammasome in uropathogenic E. coli mediated urinary tract infections. Front Microbiol. 2019;10. doi:10.3389/fmicb.2019.02020
25. Xin JZ, Wu JM, Hu GM, et al. α(1)-AR overactivation induces cardiac inflammation through NLRP3 inflammasome activation. Acta Pharmacol Sin. 2020;41(3):311–318. doi:10.1038/s41401-019-0305-x
26. Mridha AR, Wree A, Robertson AAB, et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J Hepatol. 2017;66(5):1037–1046. doi:10.1038/s41401-019-0305-x
27. Bajpai G, Schneider C, Wong N, et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat Med. 2018;24(8):1234–1245. doi:10.1038/s41591-018-0059-x
28. Alkhatib K, Poseno TM, Diaz Perez A, Durdik JM, Stenken JA. Iloprost affects macrophage activation and CCL2 concentrations in a microdialysis model in rats. Pharm Res. 2018;35(1):20. doi:10.1007/s11095-017-2277-1
29. Rajasekaran M, Sul OJ, Choi EK, Kim JE, Suh JH, Choi HS. MCP-1 deficiency enhances browning of adipose tissue via increased M2 polarization. J Endocrinol. 2019;242(2):91–101. doi:10.1530/joe-19-0190
30. Deci MB, Ferguson SW, Scatigno SL, Nguyen J. Modulating macrophage polarization through CCR2 inhibition and multivalent engagement. Mol Pharm. 2018;15(7):2721–2731. doi:10.1021/acs.molpharmaceut.8b00237
31. Do DC, Mu J, Ke X, et al. miR-511-3p protects against cockroach allergen-induced lung inflammation by antagonizing CCL2. JCI Insight. 2019;4(20). doi:10.1172/jci.insight.126832
32. Du Q, Fu YX, Shu AM, et al. Loganin alleviates macrophage infiltration and activation by inhibiting the MCP-1/CCR2 axis in diabetic nephropathy. Life Sci. 2021;272:118808. doi:10.1016/j.lfs.2020.118808
33. Abid S, Marcos E, Parpaleix A, et al. CCR2/CCR5-mediated macrophage-smooth muscle cell crosstalk in pulmonary hypertension. Eur Respir J. 2019;54(4):1802308. doi:10.1183/13993003.02308-2018
34. Schmall A, Al-Tamari HM, Herold S, et al. Macrophage and cancer cell cross-talk via CCR2 and CX3CR1 is a fundamental mechanism driving lung cancer. Am J Respir Crit Care Med. 2015;191(4):437–447. doi:10.1164/rccm.201406-1137OC
35. Sierra-Filardi E, Nieto C, Domínguez-Soto A, et al. CCL2 shapes macrophage polarization by GM-CSF and M-CSF: identification of CCL2/CCR2-dependent gene expression profile. J Immunol. 2014;192(8):3858–3867. doi:10.4049/jimmunol.1302821
36. Liu T, Guo Z, Song X, et al. High-fat diet-induced dysbiosis mediates MCP-1/CCR2 axis-dependent M2 macrophage polarization and promotes intestinal adenoma-adenocarcinoma sequence. J Cell Mol Med. 2020;24(4):2648–2662. doi:10.1111/jcmm.14984
37. Bajpai G, Bredemeyer A, Li W, et al. Tissue resident CCR2- and CCR2+ cardiac macrophages differentially orchestrate monocyte recruitment and fate specification following myocardial injury. Circ Res. 2019;124(2):263–278. doi:10.1161/circresaha.118.314028
38. Lu H, Chen R, Barnie PA, et al. Fibroblast transdifferentiation promotes conversion of M1 macrophages and replenishment of cardiac resident macrophages following cardiac injury in mice. Eur J Immunol. 2020;50(6):795–808. doi:10.1002/eji.201948414
39. Tan X, Hu L, Shu Z, et al. Role of CCR2 in the development of streptozotocin-treated diabetic cardiomyopathy. Diabetes. 2019;68(11):2063–2073. doi:10.2337/db18-1231
40. Miyabe C, Miyabe Y, Bricio-Moreno L, et al. Dectin-2-induced CCL2 production in tissue-resident macrophages ignites cardiac arteritis. J Clin Invest. 2019;129(9):3610–3624. doi:10.1172/jci123778
41. Falkenham A, Myers T, Wong C, Legare JF. Implications for the role of macrophages in a model of myocardial fibrosis: CCR2(-/-) mice exhibit an M2 phenotypic shift in resident cardiac macrophages. Cardiovasc Pathol. 2016;25(5):390–398. doi:10.1016/j.carpath.2016.05.006
42. Watts JA, Gellar MA, Obraztsova M, Kline JA, Zagorski J. Role of inflammation in right ventricular damage and repair following experimental pulmonary embolism in rats. Int J Exp Pathol. 2008;89(5):389–399. doi:10.1111/j.1365-2613.2008.00610.x
43. Heo GS, Kopecky B, Sultan D, et al. Molecular imaging visualizes recruitment of inflammatory monocytes and macrophages to the injured heart. Circ Res. 2019;124(6):881–890. doi:10.1161/circresaha.118.314030
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.