")
Back to Journals » Journal of Inflammation Research » Volume 14
Regulation of Pancreatic Fibrosis by Acinar Cell-Derived Exosomal miR-130a-3p via Targeting of Stellate Cell PPAR-γ
Authors Wang Q, Wang H, Jing Q, Yang Y, Xue D , Hao C, Zhang W
Received 31 December 2020
Accepted for publication 10 February 2021
Published 24 February 2021 Volume 2021:14 Pages 461—477
DOI https://doi.org/10.2147/JIR.S299298
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Qiang Wang,* Hao Wang,* Qingxu Jing, Yang Yang, Dongbo Xue, Chenjun Hao, Weihui Zhang
Department of General Surgery, Laboratory of Hepatosplenic Surgery, Ministry of Education, The First Affiliated Hospital of Harbin Medical University, Harbin, Heilongjiang Province, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Dongbo Xue; Chenjun Hao
Department of General Surgery, The First Affiliated Hospital of Harbin Medical University, Youzheng Street 23, Harbin, 150001, People’s Republic of China
Email [email protected]; [email protected]
Introduction: As endogenous miRNA carriers, exosomes play a role in the pathophysiological processes of various diseases. However, their functions and regulation mechanisms in pancreatic fibrosis remain unclear.
Methods: In this study, an RNA microarray was used to detect differentially expressed exosomal miR-130a-3p in AR42J cells before and after taurolithocholate (TLC) treatment. mRNA-seq was used to screen differentially expressed genes before and after pancreatic stellate cell (PSC) activation. We used the STRING database to construct a protein-protein interaction (PPI) network for differentially expressed genes, used CytoNCA to analyze the centrality of the PPI network, and identified 10 essential proteins in the biological network. Then, the TargetScan and miRanda databases were used to predict the target genes of miR-130a-3p. The intersections of the target genes and the mRNAs encoding the 10 essential proteins were identified to construct miR-130a-3p/peroxisome proliferator-activated receptor gamma (PPAR-γ) pairs. Fluorescence labeling of exosomes and dynamic tracing showed that exosomes can fuse with the cell membranes of PSCs and transport miR-130a-3p into PSCs. A luciferase reporter gene assay was used to confirm that miR-130a-3p can bind to PPAR-γ to inhibit PPAR-γ expression. In vitro and in vivo functional experiments were performed for gain-of-function studies and loss-of-function studies, respectively.
Results: The studies showed that acinar cell-derived exosomal miR-130a-3p promotes PSC activation and collagen formation through targeting of stellate cellular PPAR-γ. Knockdown of miR-130a-3p significantly improved pancreatic fibrosis. Notably, miR-130a-3p knockdown reduced serum levels of hyaluronic acid (HA) and β-amylase and increased the C-peptide level to protect endocrine and exocrine pancreatic functions and the function of endothelial cells.
Conclusion: This study revealed that the exosomal miR-130a-3p/PPAR-γ axis participates in PSC activation and the mechanism of chronic pancreatitis (CP) with fibrosis, thus providing a potential new target for the treatment of chronic pancreatic fibrosis.
Keywords: pancreatitis, exosomes, miR-130a-3p, pancreatic fibrosis, PPAR-γ
Introduction
Chronic pancreatitis (CP) is a common digestive disease in clinical practice. Typical CP pathological manifestations include pancreatic parenchymal fibrosis, acinar cell atrophy, pancreatic duct deformation, inflammatory cell infiltration, and extracellular matrix (ECM) deposition. The essence of fibrosis is an increase in ECM synthesis and a relative decrease in ECM degradation. The compromised dynamic balance between synthesis and degradation results in excessive ECM deposition. Activation of pancreatic stellate cells (PSCs) is the most important factor in the process of pancreatic fibrosis. PSC activation can produce type I and type III collagen, fibronectin, and laminin.1 In normal pancreatic tissues, PSCs remain in a quiescent state and do not promote pancreatic fibrosis. However, various stimulating factors in pancreatic injury can activate transformation of PSCs to myofibroblasts, which express α-SMA (α-smooth muscle actin) and secrete a large amount of ECM.2 Many studies have shown an increased number of PSCs in pancreatic fibrosis areas in human chronic pancreatitis specimens and animal pancreatic fibrosis models and positive α-SMA staining (PSC activation state) and collagen staining in double-stained sections, indicating that activated PSCs in the pancreas are an important source of fibrotic collagen production and play an important role in the development of pancreatic fibrosis.3
Pancreatic fibrosis is an important factor in the occurrence of pancreatic cancer. In most cases, CP is associated with pancreatic fibrosis development. Thus, understanding the mechanism of CP fibrosis is of great significance to pancreatic cancer prevention. Siech et al4 demonstrated that acinar cells can promote PSC activation in an acute pancreatitis (AP) model induced by alcohol and fat. Patel et al5 found that damaged acinar cells in CP accelerated the process of pancreatic fibrosis by promoting PSC activation. Therefore, we investigated the mechanism by which pancreatic acinar cells activate PSCs. In recent years, studies have shown that communication between cells is dependent on extracellular vesicles. Therefore, we must consider the effect of extracellular vesicles on CP fibrosis. Our studies indicate that during CP development, exosomes secreted by acinar cells have a high concentration of miRNAs.6,7 Among these mRNAs, miR-130a-3p shows significant expression, and the uptake of exosomes carrying miR-130a-3p activates PSCs to release large amounts of collagen fibers, causing fibrosis of the pancreas.
Methods
Cell Culture
Rat pancreatic acinar cells (AR42J, The Cell Bank of Type Culture Collection of Chinese Academy of Sciences) were cultured in Ham’s F12K medium containing 20% fetal bovine serum, 100 kU/L penicillin, and 100 mg/L streptomycin in a 37 °C and 5% CO2 incubator. PSCs were obtained from clean-grade healthy male Sprague-Dawley (SD) rats weighing 200–300 g. The SD rats were anesthetized by intraperitoneal injection of 10% chloral hydrate, followed by skin preparation and disinfection. After opening the abdomen along the midline, the pancreas was removed aseptically along the duodenum. The pancreas was washed with PBS twice and then cut into 1–2-mm3 pieces. The pancreas pieces were digested with GBSS digestion solution (containing 0.02% pronase, 0.05% collagenase P, and 0.1% DNase I) in a water bath at 37 °C for 20 min. The digested cell suspension was filtered through a 100-µm filter and washed with Dulbecco’s modified Eagle’s medium (DMEM), and the cells were resuspended in 9.5 mL of GBSS containing 5% FBS. The cells were resuspended in 9.5 mL of GBSS containing 5% FBS and mixed with 8 mL of 28.7% Nycodenz solution. Subsequently, 6 mL of GBSS containing 5% FBS was gently added to the mixture to provide a Nycodenz gradient, followed by centrifugation at 1400×g for 20 min at 4 °C. The resulting white flocculent layer between the GBSS liquid surface and the Nycodenz contained the PSCs. The PSCs were aspirated, washed twice with DMEM, and resuspended with medium containing 20% FBS. The viability of the cells was determined by Trypan blue staining. The cells were inoculated in culture flasks, and the medium was changed to 10% FBS on the second day and then every other day. Morphological observation: The morphological structure of PSCs, including lipid droplets in the cytoplasm, pseudopods, and nuclei, was observed under an Olympus inverted biomicroscope once a day. The cells were inoculated in culture flasks for further culture. PSC identification was performed every day.
Treatment of rat pancreatic acinar cells (Figure 1A): The cells in the control group were routinely cultured in DMEM, while the cells in the activation group were cultured in DMEM containing 200 μg taurolithocholate TLC-S for 48 h, and then the cells were separated and washed with PBS. Then, the culture medium was replaced with fresh medium, and the cell culture continued for another 48 h. The culture medium was collected and centrifuged. The supernatant was obtained and retained as the supernatant group. The white precipitate in the bottom of the vial was resuspended in DMEM and saved as the exosome group (a part of this suspension was saved as conditioned medium for future use, and the other part was used for extraction of exosomes and their miRNAs). Cell culture medium that was not centrifuged was used as the complete culture medium group. In total, six groups were examined in this experiment: the control exosome group, control supernatant group, control whole culture medium group, activated exosome group, activated supernatant group, and activated whole-culture medium group.
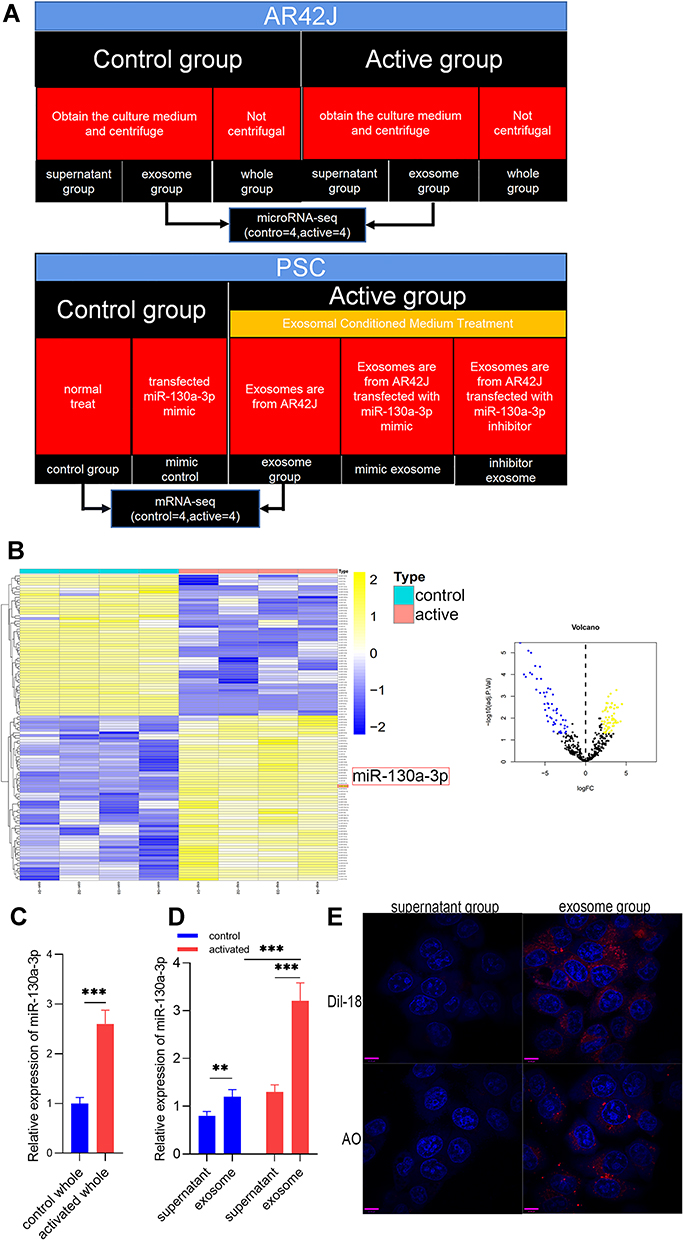 |
Figure 1 The expression, distribution, and trajectory of exosomal miR-130a-3p. (A) Cell experiment grouping. (B) The expression of exosomal miRNA in the control group and in the activation group in which miR-130a-3p expression is significantly high. (C and D) The distribution of exosomal miR-130a-3p. (E) Dynamic tracing of exosomes and exosomal miRNAs. DilC-18 was used to label exosome phospholipid bilayers, and AO was used to label exosomal miRNAs. Red fluorescence represents exosomes and exosomal miRNAs, and blue fluorescence represents the PSC nucleus (***p <0.001, **p <0.01). |
Treatment of rat PSCs (Figure 1A): Normal acinar cells and activated acinar cells were incubated with rat PSCs for 14 days, and then mRNA sequencing (mRNA-seq) was performed on the PSCs. The PSCs were divided into five groups. In the control group, PSCs were treated with normal culture medium for 14 days. In the exosome group, rat PSCs were treated with conditioned medium for 14 days. In the mimic control group, PSCs were transfected with miR-130a-3p mimic on the 11th day. In the mimic exosome group, PSCs were incubated for 14 days with exosomes extracted from AR42J cells transfected with miR-130a mimic. In the inhibitor exosome group, PSCs were incubated for 14 days with exosomes extracted from AR42J cells transfected with miR-130a inhibitor. Transfection was performed using LipofectamineTM 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. The miR-130a-3p mimic sequence was CAGUGCAAUGUUAAAAGGGCAU, and the miR-130a-3p inhibitor sequence was AUGCCCUUUUAACAUUGCACUG.
Animals
The Animal Research Center of the First Clinical College of Harbin Medical University provided 20 healthy male SD rats (250±20 g). The rats were randomly divided into four groups and allowed to acclimate for one week. The rats were fasted with free access to water for 12 h before surgery. A midline incision was made along the linea alba, and the pancreatic duct was exposed at the junction between the stomach and the duodenum. In the sham operation group, ligation of the pancreatic duct was not performed, and an equal volume of sterile saline was injected intraperitoneally. In the CP group, ligation of the pancreatic duct was performed with intraperitoneal injection of 50 μg/kg cerulenin two days later.8 In the negative interference lentivirus group, 100 μL of lentivirus with random interference sequences was injected into the biliary and pancreatic ducts in animals in the model group. In the inhibitor group, 100 μL of miR-130a inhibitor lentivirus (1×108 pfu/mL) was injected into the biliary and pancreatic ducts in animals in the model group.9 The skin incision was closed in layers. One month later, the animals were euthanized after blood was sampled from the inferior vena cava. Parts of the pancreatic tissues were collected and fixed in 10% neutral formaldehyde solution and 2.5% glutaraldehyde.
Extraction of exosomes and exosome miRNAs from rat pancreatic acinar cell culture medium
We used ExoQuickTM exosome precipitation solution (SBI, USA) for exosome extraction. TRIzol reagent was used to directly extract total RNA, including miRNA, from exosomes.
Microarray-Based Gene Expression Profiling
miRNAs: After qualified quality control of total RNA was confirmed, a microarray analysis of miRNAs was performed, including labeling, hybridization, signal amplification, and image acquisition. Then, data were extracted from the obtained images and analyzed. mRNA: The Agilent SurePrint G3 Rat Gene Expression v2 8x60K Microarray (design ID: 074036) was used in this experiment, and data analyses of the six samples were conducted by OE Biotechnology Co., Ltd. (Shanghai, China). Total RNA was quantified by the NanoDrop ND-2000 (Thermo Scientific), and RNA integrity was assessed using the Agilent Bioanalyzer 2100 (Agilent Technologies). Sample labeling, microarray hybridization, and washing were performed based on the manufacturer’s standard protocols. Briefly, total RNA was transcribed to double-stranded cDNA and then synthesized into cRNA and labeled with Cyanine-3-CTP. The labeled cRNAs were hybridized onto the microarray. After washing, the arrays were scanned by the Agilent Scanner G2505C (Agilent Technologies).
RT-PCR Quantitation
According to the instructions of an RNA extraction kit (BioTeke, RP1201, China), total RNA was extracted, cDNA was synthesized by reverse transcription, and real-time fluorescence quantitative analysis was performed on an ExicyclerTM 96 fluorescence detector. All data were analyzed via relative quantitative analysis using the 2-ΔΔCt method to detect the levels of peroxisome proliferator-activated receptor gamma (PPAR-γ), α-SMA, collagen I, and collagen III mRNAs. The experiment was repeated three times.
Dynamic Tracing of Exosomes and Exosomal RNAs
AR42J cells (7×106) were plated. pPACKH1 (45 μL) and CD9 Cyto-TraceTM plasmid (4.5 μg) were added into fresh culture medium with PureFectionTM (5.5 μL). The medium was mixed for 10 sec, maintained at room temperature for 15 min, and then added into the culture medium of the AR42J cells. The cells were cultured in a 37 °C and 5% CO2 incubator for 24 h. The AR42J cells in the culture medium were treated with Dil-C18 fluorescent dye for 1 h and then washed with PBS three times to label the phospholipids on the exosome membrane, and the exosomes were identified with a laser confocal microscope.
The principle of Exo-RedTM staining is based on the properties of Acridine Orange (AO). AO can pass through the cell membrane and fluorescently label single-stranded RNAs inside exosomes. The detailed protocol was as follows: the obtained exosomes were resuspended in 500 μL 1×PBS in a 1.5-mL Eppendorf (EP) tube; 50 μL of 10×Exo-RedTM was added to the resuspended exosome solution; the mixture was incubated at 37 °C for 10 min; 100 μL of ExoQuick-TCTM reagent was added to stop the labeling reaction; the mixture was placed on ice for 30 min and then centrifuged at 14,000×g for 3 min; the supernatant was removed; the labeled exosomes were resuspended in 500 μL of 1×PBS; 100 μL of labeled exosomes was added to each well of a six-well plate (approximately 1×105 PSCs per well); and the cells were observed using a confocal laser microscope after 24 h of culture.
Western Blot Analysis
A kit (Wanleibio, WLA019) was used to detect PPAR-γ, α-SMA, collagen I, and collagen III levels with β-actin antibody as an internal control.
ELISA and DNS
Serum was obtained by centrifugation of the inferior vena cava blood of rats in the four groups. β-Amylase activity was calculated using a β-amylase detection kit (Nanjing Jianshe, C016-2) and the 3,5–dinitrosalicylic acid (DNS) method. A hyaluronic acid (HA) detection kit (Ulsan, CEA182Ge) and C-peptide detection kit (Ulsan, CEA447Ra) were used to calculate the concentrations of HA and CP based on the ELISA method according to the manufacturer’s instructions.
H&E Staining, Masson Staining, and Immunohistochemistry
Hematoxylin and eosin (H&E) staining and Masson staining were performed on the pancreatic tissues of rats in the four groups. After staining, the stained sections were photographed under a microscope at 200× magnification. Immunohistochemistry for α-SMA was performed, and the sections were photographed under a microscope at 400× magnification. HistoQuest software was used for quantitative analysis of Masson and immunohistochemical staining intensity.
Construction of miRNA-mRNA Pairs
Data processing of the exosomal miRNA expression profile chip and analysis of differentially expressed genes (control=4, active=4): The limma package in Bioconductor was used to analyze differentially expressed genes. A false discovery rate (FDR) <0.05 and |log2FC|≥0.58 (that is, the expression difference between groups was not less than 1.5 fold) were set. Data processing of the PSC mRNA expression profile chip and analysis of differentially expressed genes (control=4, active=4): the limma package in Bioconductor was used to analyze differentially expressed genes. An FDR <0.05 and |log2FC|≥0.58 (that is, the expression difference between groups was not less than 1.5 fold) were set. Based on the STRING database, a protein-protein interaction (PPI) network was constructed for significantly differentially expressed mRNAs (the threshold was a PPI confidence score=0.4). Then, we used cytoNCA10,11 to analyze the centrality of the PPI network. We used eight centrality measures (Betweenness, Closeness Centrality, Degree Centrality, Eigenvector Centrality, Local Average Connectivity-based Centrality, Network Centrality, Subgraph Centrality, and Information Centrality) to identify 10 essential proteins in the biological network. Then, the TargetScan and miRanda databases were used to predict the target genes of miR-130a-3p. The intersection of the target gene and the mRNAs encoding the 10 essential proteins was obtained to construct miRNA-mRNA pairs.
Determination of the Luciferase Reporter Gene
The miRDB12 database and DIANA TOOLS13 database were used to predict the binding site of miR-130a-3p to the PPAR-γ gene. In the putative binding sites, the 3ʹ untranslated region (UTR) sequence of the PPAR-γ gene containing a 3-bp mutation, the 3ʹUTR sequence of PPAR-γ, and its mutant were inserted separately into the luciferase reporter plasmid psiCHECK-2 to construct wild-type (PPAR-WT) and mutant (PPAR-MIT) recombinant plasmids. Next, PSCs were transfected with miR-130a-3p mimic or negative control (NC) and the recombinant plasmids described above. Forty-eight hours after transfection, a dual-luciferase detection kit was used to detect luciferase activity. The PSCs were divided into four groups: the WT+NC, MUT+NC, Wt+mimic, and MUT+mimic groups.
Statistical Analysis
SPSS 20.0 software was used for statistical analysis, and the experimental data are expressed as the mean±standard deviation (x±s). The t-test and the Wilcox test were used to compare the means of two samples. Data with a normal distribution were analyzed with the t-test, while data without a normal distribution were analyzed with the Wilcox test (***p <0.001, **p <0.01, *p <0.05).
Results
Differentially Expressed miRNAs in Exosomes
The microarray analysis of miRNAs showed that the exosomes released by AR42J cells contained multiple miRNAs, which is consistent with findings in previous reports (Figure 1). Compared with the cells in the control group, activated AR42J cells secreted exosomes containing 53 underexpressed miRNAs and 62 overexpressed miRNAs. These differentially expressed miRNAs may be communication messengers between cells. In the current study, the expression of miR-130a-3p was significantly increased (Figure 1B). Recent studies have shown that miR-130a-3p participates in the fibrogenesis of renal, pulmonary, liver, and cartilage tissues.14–16 In addition to miR-130a-3, we speculate that miR-124 may be associated with hepatic fibrosis similar to other miRNAs, including downregulated miR-15/16, miR-17-5p, and let-7d and upregulated miR-29 during the process of pancreatic fibrosis.17–20 However, few reports on exosomal miR-130a-3p in pancreatic fibrosis are available. A recent study also found that exosomal miR-103-3p from lipopolysaccharide (LPS)-activated THP-1 macrophages contributes to activation of hepatic stellate cells.21 Therefore, exosomal miR-130a-3p was selected as an miRNA target for pancreatic fibrosis research.
PSC Uptake of Exosomes Secreted by Acinar Cells and Exosomal miRNA
To investigate the distribution of miR-130a-3p, we used RT-PCR to detect miR-130a-3p in the complete culture medium of control group and activated group cells. As expected, acinar cells released more miR-130a-3p after activation (Figure 1C). To further understand the distribution of miR-130a-3p, we centrifuged complete culture medium from the two groups to obtain exosomes and supernatant and then assessed the miR-130a-3p levels in both. Surprisingly, we found that miR-130a-3p was mainly distributed in exosomes from both the control and activation groups, but the miR-130a-3p level in the activated group was substantially higher than that in the control group, which was consistent with our microarray analysis results (Figure 1D). To observe the trajectory of miRNAs in exosomes and exosomal miRNA, we labeled exosomes and the miRNAs in exosomes with fluorescent markers and observed them with a confocal fluorescence microscope. We observed no fluorescence in PSCs after adding Dil-C18-labeled supernatant to the PSC medium. In contrast, red fluorescence appeared in the PSCs (Figure 1E) after incubation with Dil-C18-labeled exosomes. This result confirmed that the exosomes were successfully extracted and were ingested by PSCs after incubation. AO was used to label miRNA in the supernatant group and the exosome group, followed by incubation with PSCs. In the AO-labeled supernatant group, no red fluorescence appeared in the PSCs, while in the AO-labeled exosome group, red fluorescence appeared in the PSCs (Figure 1E). This result confirmed that miRNAs were abundantly enriched in the exosome group and entered the PSCs through exosomes. In summary, we confirmed that miR-130a-3p was highly expressed in pancreatic acinar cells after injury and is mainly distributed in exosomes that transport miRNAs into PSCs.
Exosomal miR-130a-3p Derived from Pancreatic Acinar Cells Can Activate PSCs and Promote Pancreatic Fibrosis
To study the biological functions of acinar cell-derived exosomal miR-130a-3p after it enters PSCs, we extracted exosomes from acinar cells with overexpression or knockdown of miR-130a-3p and incubated PSCs with the exosomes. RT-PCR was used to test the efficiency of cell transfection with miR-130a-3p. The results showed that the exosome mimic group had higher miR-130a-3p expression, while the exosome inhibitor group had lower miR-130a-3p expression than the exosome group, indicating successful in vitro control of miR-130a-3p expression (Figure 2A). We examined the expression of the PSC activation marker α-SMA protein and the fibrosis markers collagen I and collagen III at the RNA and protein levels and found that the α-SMA, collagen Ⅰ, and collagen ш mRNA (Figure 2B–D) and protein (Figure 2E) levels in PSCs were significantly increased in the exosome group and mimic exosome group compared with the control group. Interestingly, the expression levels of α-SMA, collagen Ⅰ, and collagen ш in PSCs showed no differences at the mRNA level or protein level between the exosome group and the exosome mimic group. However, with miR-130a-3p knockdown in the exosome inhibitor group, the α-SMA, collagen Ⅰ, and collagen ш mRNA (Figure 2B–D) and protein (Figure 2E) expression levels were significantly reduced and were close to normal levels in the PSCs, indicating that acinar cell-derived exosomal miR-130a-3p can activate PSCs and promote collagen overexpression by cells, leading to CP development. However, this activation was initiated with only a certain amount of miR-130a-3p and did not rely on the expression level. Furthermore, knockdown of exosomal miR-130a-3p significantly inhibited PSC activation and fibrosis (Figure 2B–D). More surprisingly, in the mimic control group, we directly overexpressed miR-130a-3p in PSCs without exosome involvement. Although miR-130a-3p expression was highest in this group (Figure 2A), the PSCs were not activated (Figure 2B) and did not show higher expression of fibrosis markers (Figure 2C and D), indicating that PSC activation depends on exosomal miR-130a-3p derived from acinar cells.
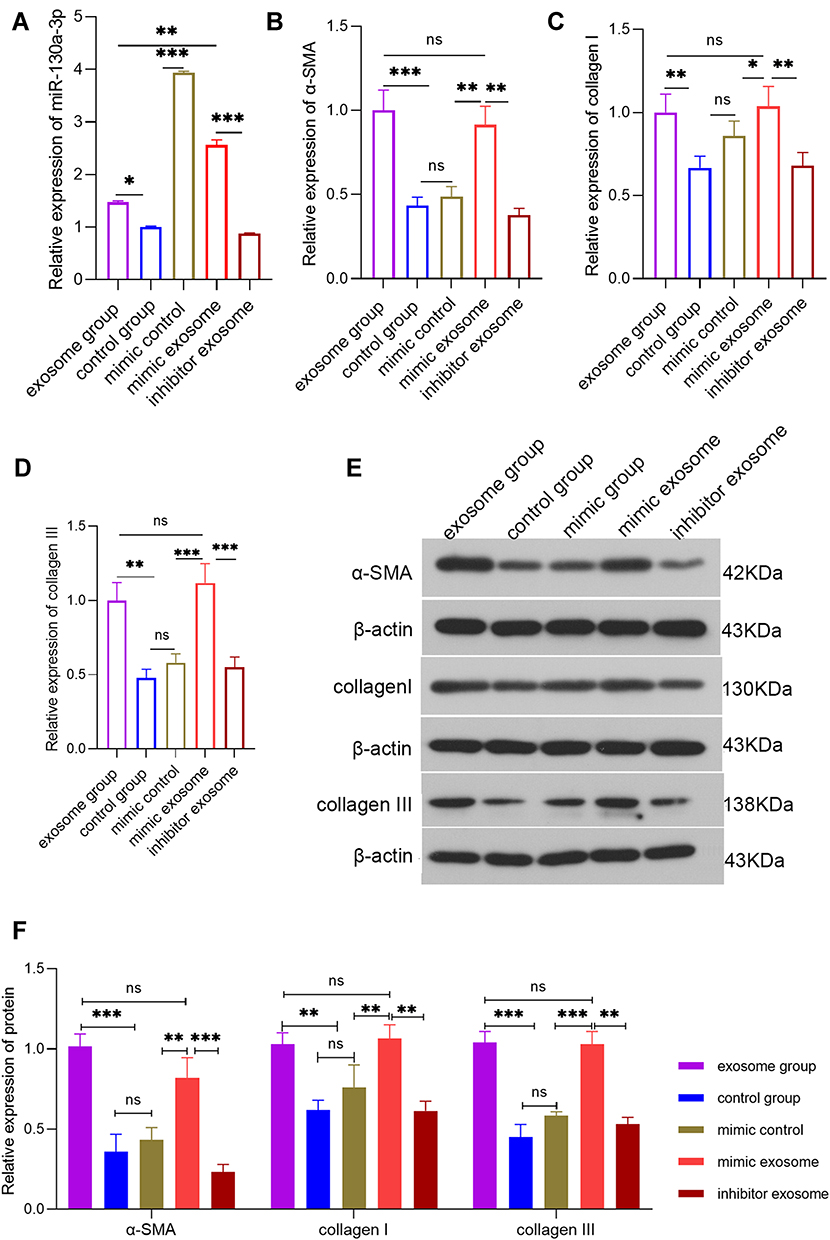 |
Figure 2 Detection of the various indicators in each group of PSCs at the cellular level via PCR or Western blot (WB). (A) PCR detection of the relative expression level of miR-130a-3p in each group of PSCs. (B) PCR detection of α-SMA expression levels in each group of PSCs. (C) PCR detection of the collagen I expression level in each group of PSCs. (D) PCR detection of the collagen ш expression level in each group of PSCs. (E and F) WB detection of the α-SMA, collagen I, and collagen III protein levels in PSCs (***p <0.001, **p <0.01, *p <0.05, NS denotes no significant difference). |
To further explore the biological effects of miR-130a-3p in vivo, we divided rats into four groups: a sham operation group, a CP group, an inhibitor control group, and an inhibitor group. Among these groups, CP rats in the inhibitor group were transfected with miR-130a-3p inhibitor lentivirus in the pancreas. To test whether the model was successful, we used RT-PCR to detect miR-130a-3p expression in pancreatic tissues in each group. The results showed that miR-130a-3p expression in the CP group and the inhibitor control group was significantly higher than that in the inhibitor group or the sham operation group. The inhibitor group had the lowest miR-130a-3p expression. This result indicates that we achieved in vivo control of miR-130a-3p expression (Figure 3A). We examined the expression of the PSC activation marker α-SMA and the fibrosis markers collagen Ⅰ and collagen ш in pancreatic tissue and found that the expression of α-SMA, collagen Ⅰ, collagen ш mRNA (Figure 3B–D) and protein (Figure 3E) was significantly increased in the CP group compared with the sham operation group. However, α-SMA, collagen Ⅰ, and collagen ш expression was significantly reduced in the inhibitor group compared with the CP group (Figure 3B–E). H&E (Figure 4A) and Masson (Figure 4B) staining confirmed that the degree and area of fibrosis in the inhibitor group were significantly smaller than those in the CP group or the inhibitor control group (Figure 4D), indicating that pancreatic tissue fibrosis is associated with CP. Knockdown of miR-130a-3p in vivo significantly inhibited pancreatic tissue fibrosis. Immunohistochemistry also confirmed that knockdown of miR-130a-3p in vivo can inhibit activation of PSCs and reduce the expression of the specific protein α-SMA (Figure 4C and E). In summary, PSCs are significantly activated upon the occurrence of CP, and a significant increase in miR-130a-3p is associated with obvious pancreatic fibrosis. Moreover, knockdown of miR-130a-3p can inhibit PSC activation and prevent pancreatic fibrosis.
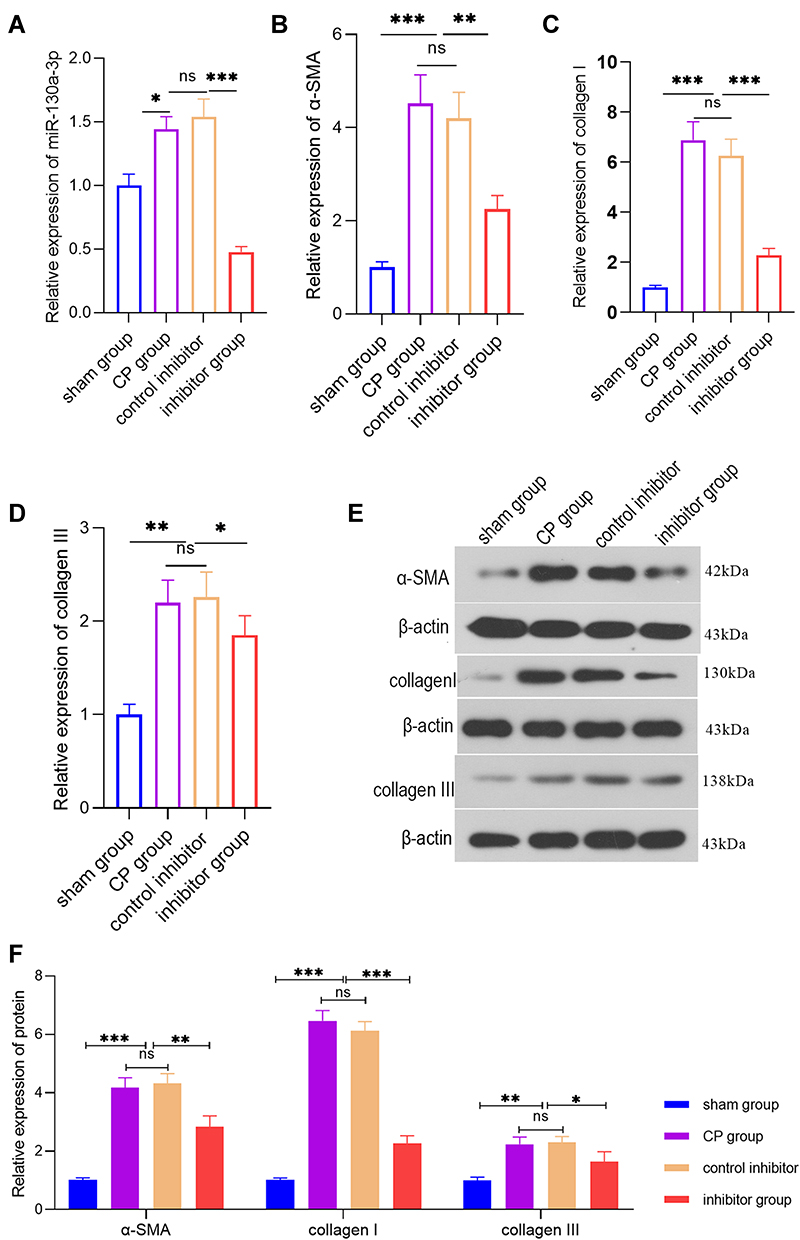 |
Figure 3 PCR or WB detection of the various indicators in pancreatic tissue from each group at the animal level. (A) PCR detection of the relative miR-130a-3p expression level in pancreatic tissue from each group. (B) PCR detection of the α-SMA expression level in pancreatic tissues from each group. (C) PCR detection of the collagen I expression level in pancreatic tissues from each group. (D) PCR detection of the collagen ш expression level in pancreatic tissues from each group. (E and F) WB detection of the α-SMA, collagen Ⅰ, and collagen ш protein levels in pancreatic tissue (***p <0.001, **p <0.01, *p <0.05, NS denotes no significant difference). |
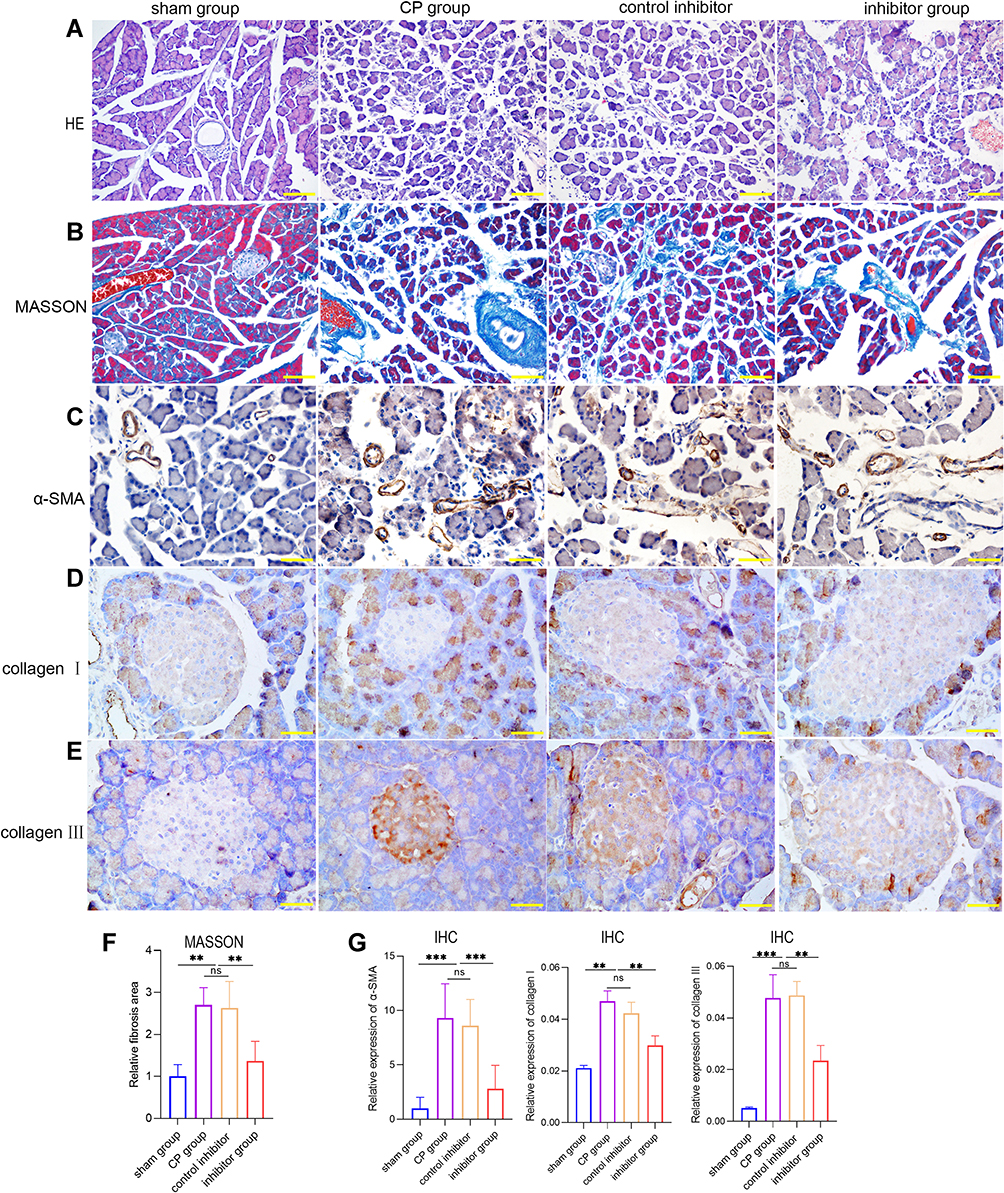 |
Figure 4 (A and B) H&E and Masson staining of pancreatic tissues from each group. (C–E) Immunohistochemistry of α-SMA, collagen Ⅰ, and collagen ш in pancreatic tissues from each group. (F) Masson staining of fibrosis areas in pancreatic tissues from each group. (G) The relative expression levels of α-SMA, collagen Ⅰ, and collagen ш in pancreatic tissues from each group (***p <0.001, **p <0.01, NS denotes no significant difference). |
Knockdown of miR-130a-3p Not Only Improves Pancreatic Endocrine and Exocrine Functions but Also Protects Endothelial Cells
To investigate the role of miR-130a-3p in the recovery of the pancreas after pancreatic injury, we tested multiple indicators in the peripheral blood of four groups of animals. Among these indicators, the C-peptide level reflects the endocrine function of the pancreas, with a lower level indicating worse function. The β-amylase level reflects the exocrine function of the pancreas, with a higher level indicating worse function. As expected, compared with the sham operation group, the CP group had a significantly lower C-peptide level and a significantly higher β-amylase level, indicating that endocrine and exocrine functions in the CP group were severely damaged. Surprisingly, however, knockdown of miR-130a-3p led to a significantly higher C-peptide level and a significantly lower β-amylase level in the inhibitor group than in the CP group (Figure 5A and B). Therefore, knockdown of miR-130a-3p significantly improved pancreatic endocrine and exocrine functions. Similarly, we tested the serum HA content in the four groups. The HA content reflects the function of endothelial cells, with a higher content indicating better function. We found that the HA content in the inhibitor group was significantly higher than that in the CP group (Figure 5C). This result confirmed that knockdown of miR-130a-3p can effectively protect endothelial cell function. In summary, knockdown of miR-130a-3p can not only effectively improve pancreatic endocrine and exocrine functions but also protect endothelial cells and may have played a certain role in promoting recovery in the pancreatic group.
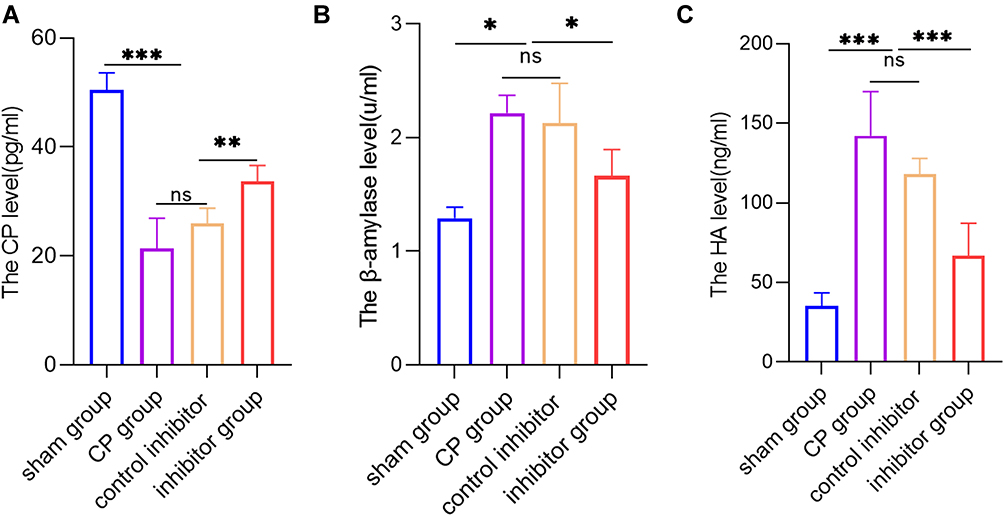 |
Figure 5 Knockdown of miR-130a-3p not only improves pancreatic endocrine and exocrine functions but also protects endothelial cells. (A) ELISA detection of C-peptide in serum from mice in each group. (B) ELISA detection of hyaluronic acid (HA) in serum from mice in each group. (C) DNS method for calculation of the β-amylase activity in serum from mice in each group (***p <0.001, **p <0.01, *p <0.05, NS denotes no significant difference). |
miR-130a-3p Promotes Pancreatic Fibrosis by Inhibiting PPAR-γ Expression
In the early stage, we confirmed that miR-130a-3p is involved in the activation of PSCs and promotes pancreatic fibrosis. To investigate the underlying mechanisms and identify the downstream target genes of miR-130a-3p, we analyzed differentially expressed mRNAs before and after PSC activation (Figure 6A) and constructed a PPI network (Figure 6B). We analyzed the centrality of the PPI network through CytoNCA and identified 10 essential proteins (Hub genes) in the biological network (Figure 6B). Then, the TargetScan and miRanda databases were used to predict the target genes of miR-130a-3p and to identify intersections of the target genes and the mRNAs encoding the 10 essential proteins. Finally, we identified that PPAR-γ (also known as PPARG) was significantly underexpressed, which was consistent with the predicted trend of miRNA inhibition (Figure 6C). A literature review revealed that the PPAR-γ pathway has been confirmed to be involved in PSC activation and that PPAR-γ is a “star molecule”. Therefore, we performed an in-depth study of the miR-130a-3p-PPAR-γ pair. To verify that miR-130a-3p can bind to the target gene PPAR-γ and the regulatory relationship between them, we conducted a dual-luciferase reporter gene experiment. The results showed that the fluorescence activity difference between the WT-NC and MUT-NC groups was not significant, but with overexpression of miR-130a-3p, fluorescence activity in the WT+miR-130a-3p mimic group was significantly lower than that in the WT-NC group, indicating that PPAR-γ expression was significantly suppressed. When the 3ʹUTR sequence of PPAR-γ was mutated, fluorescence activity in the WUT+miR-130a-3p mimic group was restored to a higher level than that in the WT+miR-130a-3p mimic group, indicating that PPAR-γ expression was restored. This result confirms that miR-130a-3p entering PSCs can significantly inhibit PPAR-γ gene expression by directly binding to the PPAR-γ 3ʹUTR (Figure 6D).
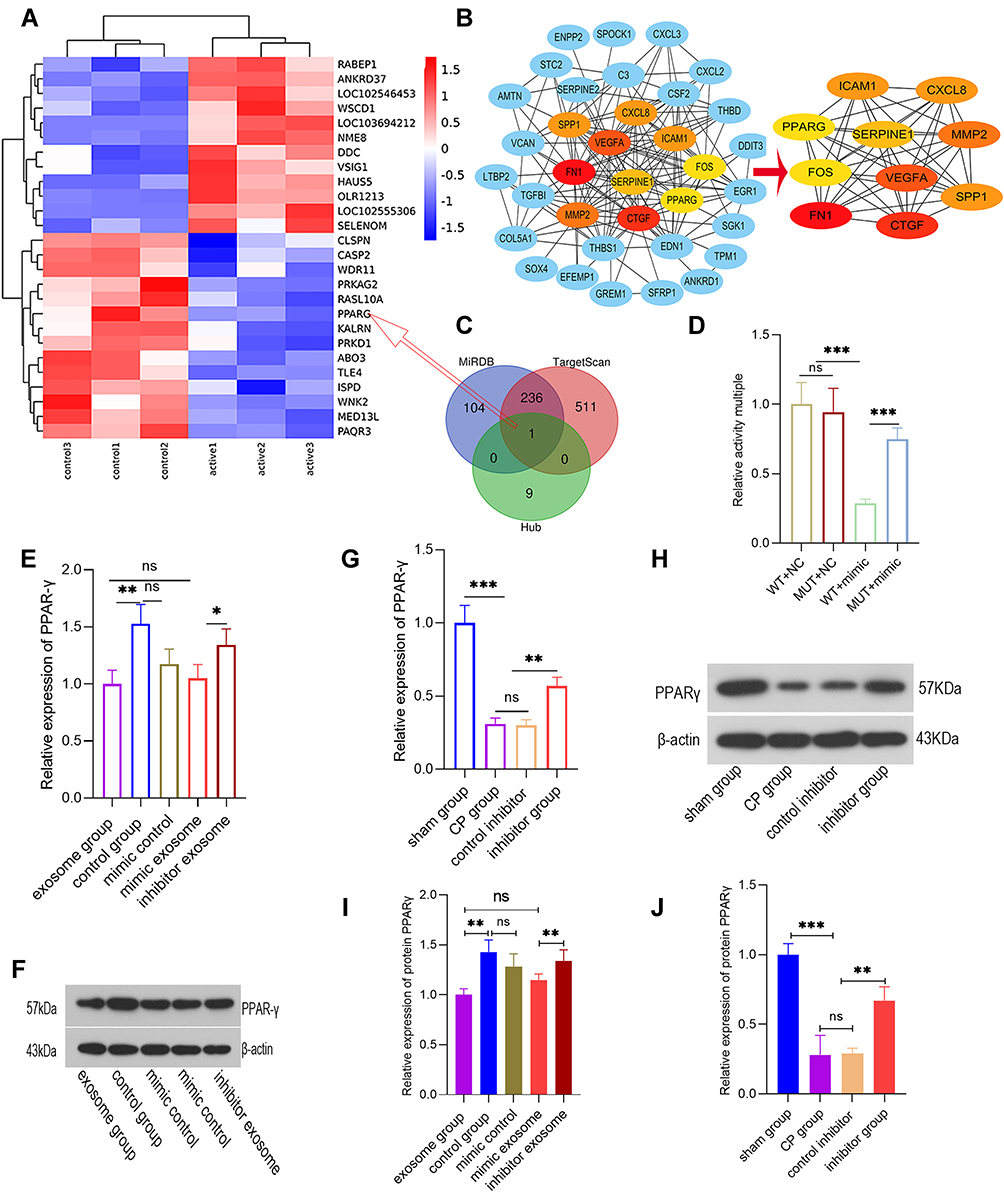 |
Figure 6 miR-130a-3p promotes pancreatic fibrosis by inhibiting PPAR-γ expression. (A) mRNA expression before and after PSC activation. (B) Based on the STRING database, a protein-protein interaction (PPI) network map was plotted for significantly differentially expressed genes, and a centrality analysis of the PPI network through CytoNCA was conducted to obtain 10 essential proteins (hub genes). (C) Venn diagram showing miR-130a-3p target genes and 10 essential protein mRNAs (hub genes). (D) Dual-luciferase reporter gene assay for determination of relative PPAR-γ activity. (E and F) The relative expression levels of PPAR-γ mRNA and protein detected in cells. (G and H) The relative expression levels of PPAR-γ mRNA and protein detected in tissues. (I) The quantification of the density of the blotting bars of PPARγ in cell. (J) The quantification of the density of the blotting bars of PPARγ in pancreatic tissue (***p <0.001, **p <0.01, *p <0.05, NS denotes no significant difference). |
To further verify PPAR-γ regulation by miR-130a-3p, exosomes were extracted from acinar cells with overexpression or knockdown of miR-130a-3p and then incubated with PSCs. Then, we examined the expression level of PPAR-γ. RT-PCR and Western blotting showed that PPAR-γ expression was significantly lower in the exosome group than in the control group, while PPAR-γ expression was increased in the exosome group with miR-130a-3p knockdown (Figure 6E and F). Once again, our results confirmed an inhibitory effect of exosomal miR-130a-3p on PPAR-γ expression. Subsequently, we further verified PPAR-γ regulation by miR-130a-3p at the animal level. The results showed that PPAR-γ expression in the CP group was significantly lower than that in the sham operation group. However, when miR-130a-3p was knocked down, PPAR-γ expression was significantly increased. PPAR-γ is known to maintain the lipogenic properties of PSCs and to maintain these cells in a quiescent state. Decreased PPAR-γ expression can cause PSC activation, leading to pancreatic fibrosis; however, knockdown of miR-130a-3p can significantly upregulate PPAR-γ expression (Figure 6G and H).
Discussion
In recent years, miRNA has received increasing attention for its role in fibrotic diseases due to its ability to regulate gene expression through RNA interference. Before and after PSC activation, the expression of nearly 100 miRNAs changed significantly, including the let-7 family, miRNA-21, miRNA-98, the miRNA-29 family, the miRNA-200 family, miRNA-140, miRNA-34a, etc.22–24 The multiple miRNAs cited above have been confirmed to regulate various signaling pathways in the process of PSC activation and to change the expression of endothelial-mesenchymal transition (EMT)-related genes, indicating that they play an important regulatory role in PSC activation. RNA sequencing (RNA-seq) has shown that miRNAs are abundant in exosomes. Notably, the miRNA repertoire of exosomes may differ from that of the producer cell.25 Changing the expression of exosomal miRNA to enable PSCs to maintain a resting phenotype, prevent conversion to a fibroblast phenotype, or even reverse this process will facilitate the treatment of pancreatic fibrosis.
Pancreatic fibrosis is a chronic pathological process involving excessive accumulation of ECM in pancreatic tissue, and pancreatic mesenchymal PSCs play a key role in this process. PSCs are activated after stimulation by a variety of factors, and activated PSCs are transformed into myofibroblasts and migrate to injured sites to secrete excessive ECM, which eventually leads to pancreatic fibrosis. In this pathological process, exosomes and the miRNAs that they carry play an important role. In the process of liver fibrosis, miR-181-5p carried by exosomes can regulate the STAT3/Bcl-2/Beclin 1 signaling pathway and reduce the expression of collagen fibers, thereby alleviating liver fibrosis.26 In pancreatic cancer, cancer cells secrete a large number of exosomes, and the miRNA carried by these exosomes produces a fibrotic microenvironment that is conducive to cancer cell metastasis, thereby promoting liver metastasis of pancreatic cancer.27,28 Masamune et al found that exosomes secreted by pancreatic cancer cell lines (SUIT-2, Panc-1) contain a large amount of miR-1246 and miR-1290, which can stimulate PSC migration and activation and promote the expression of α-SMA and fibrosis-related genes.29 These studies have verified the important role of exosomes and the miRNAs that they carry in the occurrence and development of diseases. In vitro fluorescence labeling and dynamic tracing showed that exosomes carry miRNAs into PSCs. Further studies confirmed that exosomal miR-130a-3p mediates information transmission between acinar cells and PSCs and activates PSCs, thus promoting pancreatic fibrosis. Previous studies have demonstrated that the miRNA-130 family is a key regulator of fibrosis pathways in 137 diseases and that inhibiting its expression can prevent ECM accumulation.30 Gooch et al31 confirmed that miR-130 is involved in cyclosporine A-mediated renal fibrosis. Chu et al confirmed that miR-130 can promote nuclear factor (NF) kappa B-mediated inflammation and aggravate transforming growth factor (TGF)-β1-mediated fibrosis by inhibiting the protective effect of the target gene PPAR-γ in a rat acute myocardial infarction model.32 These findings suggest that miR-130 is involved in the pathological process of kidney33 and heart tissue fibrosis. How does this mRNA affect the process of fibrosis in pancreatic tissue? Our study shows that the occurrence of CP is associated with higher miR-130a-3p expression in acinar cells, which is mainly distributed in exosomes (Figure 1D). After incubation with PSCs, exosomes can carry miR-130a-3p into PSCs (Figure 1E) and stimulate PSC activation, leading to the secretion of more collagen fibers. However, initiation of this activation requires only a certain amount of miR-130a-3p and does not rely on the level of miR-130a-3p expression. However, knockdown of exosomal miR-130a-3p can significantly inhibit PSC activation and inhibit fibrosis (Figure 2B–E, Figures 3 and 4). Notably, in the mimic control group, direct overexpression of miR-130a-3p in PSCs without exosome involvement did not activate PSCs or significantly increase ECM production, indicating that PSC activation depends on acinar cell-derived exosomal miR-130a-3p and does not rely on exogenous miRNA (Figure 2B–D). In addition, knockdown of miRNA-130a-3p in animal models reduced the levels of serum HA and β-amylase but increased the C-peptide content (Figure 5A–C), indicating that knockdown of miR-130a-3p can not only inhibit pancreatic fibrosis but also protect pancreatic endocrine, exocrine, and endothelial cell functions.
Which molecules are targets of miR-130a-3p? To identify this pathway, we conducted a series of bioinformatics analyses and determined from the entire network that PPAR-γ is a target of miR-130a-3p (Figure 6B and C). Kei Asukai et al34 found that transfection of miR-130a-3p mimic suppressed the expression of PPAR-γ and increased gemcitabine resistance in cholangiocarcinoma. Studies have shown that interleukin (IL)-4 downregulates miR-130a-3p by histone deacetylation in profibrogenic macrophages, leading to upregulation of PPAR-γ expression and causing macrophages to secrete a large amount of TGF-β.22 TGF-β is the key driving force of fibrosis and can activate liver stellate cells. These data suggest that PPAR-γ may also be the target gene of miR-130a-3p in PSCs. PPAR-γ plays an important role in maintaining PSC quiescence. PPAR-γ can maintain the adipogenic properties of PSCs and inhibit PSC proliferation, collagen synthesis, and α-SMA expression.35 High PPAR-γ expression can inhibit PSC activation and the promotion of fibrosis induced by PDGF and thus has potential application value in the treatment of CP.36,37 The PPAR-γ receptor agonist troglitazone inhibits PSC proliferation, collagen synthesis, and α-SMA expression by blocking the G1 phase of the PSC cell cycle and reduces CP fibrosis.38,39 These previous studies have shown that PPAR-γ is a key molecule in pancreatic fibrosis.40,41 However, PPAR-γ is significantly underexpressed in CP (Figure 6A). How is PPAR-γ regulated? Through a dual-luciferase gene reporter experiment, we confirmed that exosomal miRNA-130a-3p can directly bind to PPAR-γ and inhibit its expression (Figure 6D). To verify this conclusion, we performed experiments to overexpress miRNA-130a-3p in vitro and in vivo, and the results showed that PPAR-γ expression decreased at both the transcriptional and protein levels and that pancreatic fibrosis was aggravated compared with the control group. With miRNA-130a-3p knockdown, PPAR-γ expression significantly increased at both the transcriptional and protein levels, and the degree of fibrosis significantly improved (Figure 6E–H). Shao‑ce Zhi et al42 found that rosiglitazone can alleviate hepatic fibrosis through a specific mechanism; upregulation of miR-124-3p expression resulted in a reduction in HOTAIR mRNA, significantly increasing PPAR-γ levels. However, whether rosiglitazone can improve pancreatic fibrosis through the miR-130a-3p/PPAR-γ axis requires further verification in future studies.
ECM is regulated by a balance between matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs). MMPs are a family of calcium-dependent proteinases with an important role in ECM degradation. The activity of MMPs is regulated at the level of transcription, proenzyme activation, or inhibition of activated enzyme TIMPs.43 Pancreatic mesenchymal PSCs play a key role in this process. Activated PSCs also express activated TGF-1, which upregulates collagen-1 expression while reducing the expression of MMP-3 and MMP-9. Research has demonstrated that PSCs in both tissue culture models and in vivo during fibrosis are sources of TIMP-1 and TIMP-2. Both TIMP-1 and TIMP-2 have potent inhibitory effects on all activated MMPs, including those with interstitial collagenase activity.44 The clear localization of TIMP-1 and TIMP-2 to the areas of fibrosis in each example of chronic pancreatitis suggests that these proteins are expressed by activated PSCs. The dual PPAR-α/γ agonist saroglitazar can inhibit TIMP-1 and improve liver fibrosis.45 However, TIMP-4 induced by PPAR-γ in vascular smooth muscle is an endogenous protective mechanism that inhibits the migration of vascular smooth muscle cells.46 Another study showed that tryptase can upregulate MMP-1 and MMP-2 and inhibit TIMP-1 and TIMP-2 through the PPAR-γ pathway to promote atrial fibrosis.47 PPAR-γ and TIMP clearly exhibit tissue specificity; therefore, miR-130a-3p derived from acinar cells in pancreatic tissue may change the balance of TIMP and MMP by inhibiting PPAR-γ and further lead to pancreatic fibrosis, which is simply our conjecture that we must verify through experiments.
Conclusions
In summary, this study demonstrated that damaged pancreatic acinar cells release exosomes that carry miR-130a-3p and transport miR-130a-3p to PSCs. By inhibiting PPAR-γ expression, miR-130a-p promotes PSC activation and collagen formation, leading to fibrosis of pancreatic tissue, whereas knockdown of miR-130a-3p can significantly inhibit pancreatic fibrosis (Figure 7). CP fibrosis is an important pathological feature of pancreatic cancer.48,49 Investigating the mechanism of pancreatic fibrosis has far-reaching clinical significance. This study provides new ideas for the prevention and treatment of CP fibrosis from the perspective of exosome-mediated intercellular communication.
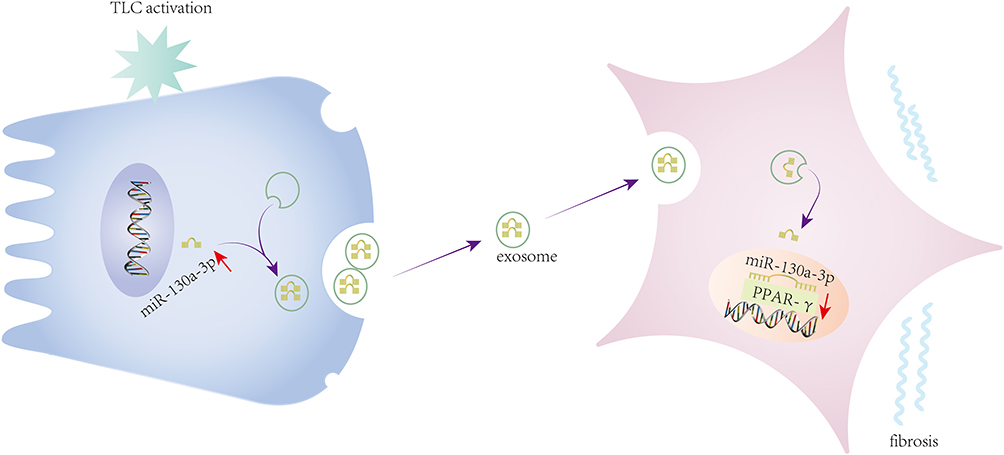 |
Figure 7 After acinar cell activation, the cells’ secretion of exosomal miR-130a-3p was upregulated, and exosomal miR-130a-3p promoted PSC activation and collagen formation by inhibiting stellate cell PPAR-γ. |
Ethical Statement
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee of The First Affiliated Hospital of Harbin Medical University.Animal experiments are conducted in accordance with the National Animal Welfare Law.The National Animal Welfare Law is specific to laboratory animals, which includes the purpose of animal experiments, experiment qualification, operation procedure, and legal responsibility.
Acknowledgments
The authors would like to acknowledge the helpful comments regarding this paper received from the reviewers. This study was supported by a grant from the National Natural Science Foundation of China (81570579).
These authors are regarded as co-first authors: Qiang Wang and Hao Wang.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bachem MG, Schneider E, Gross H, et al. Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology. 1998;115(2):421–432. doi:10.1016/s0016-5085(98)70209-4
2. Jaskiewicz K, Nalecz A, Rzepko R, et al. Immunocytes and activated stellate cells in pancreatic fibrogenesis. Pancreas. 2003;26(3):239–242. doi:10.1097/00006676-200304000-00006
3. Yang Y, Yu X, Huang L, et al. GLP-1R agonist may activate pancreatic stellate cells to induce rat pancreatic tissue lesion. Pancreatology. 2013;13(5):498–501. doi:10.1016/j.pan.2013.07.281
4. Siech M, Zhou Z, Zhou S, et al. Stimulation of stellate cells by injured acinar cells: a model of acute pancreatitis induced by alcohol and fat (VLDL). Am J Physiol Gastrointest Liver Physiol. 2009;297(6):G1163–1171. doi:10.1152/ajpgi.90468.2008
5. Patel M, Fine DR. Fibrogenesis in the pancreas after acinar cell injury. Scand J Surg. 2005;94(2):108–111. doi:10.1177/145749690509400205
6. Lee HB, Park HK, Choi HJ, et al. Evaluation of circulating microRNA biomarkers in the acute pancreatic injury dog model. Int J Mol Sci. 2018;19(10):10. doi:10.3390/ijms19103048
7. Lux A, Kahlert C, Grutzmann R, et al. c-Met and PD-L1 on circulating exosomes as diagnostic and prognostic markers for pancreatic cancer. Int J Mol Sci. 2019;20(13):13. doi:10.3390/ijms20133305
8. Sendler M, Beyer G, Mahajan UM, et al. Complement component 5 mediates development of fibrosis, via activation of stellate cells, in 2 mouse models of chronic pancreatitis. Gastroenterology. 2015;149(3):765–776 e710. doi:10.1053/j.gastro.2015.05.012
9. Doiron B, Hu W, Norton L, et al. Lentivirus shRNA Grb10 targeting the pancreas induces apoptosis and improved glucose tolerance due to decreased plasma glucagon levels. Diabetologia. 2012;55(3):719–728. doi:10.1007/s00125-011-2414-z
10. Tang Y, Li M, Wang J, et al. CytoNCA: a cytoscape plugin for centrality analysis and evaluation of protein interaction networks. Biosystems. 2015;127:67–72. doi:10.1016/j.biosystems.2014.11.005
11. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. doi:10.1101/gr.1239303
12. Wong N, Wang X. miRDB: an online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2015;43(Databaseissue):D146–152. doi:10.1093/nar/gku1104
13. Alexiou P, Maragkakis M, Papadopoulos GL, et al. The DIANA-mirExTra web server: from gene expression data to microRNA function. PLoS One. 2010;5(2):e9171. doi:10.1371/journal.pone.0009171
14. Snyder-Talkington BN, Dong C, Sargent LM, et al. mRNAs and miRNAs in whole blood associated with lung hyperplasia, fibrosis, and bronchiolo-alveolar adenoma and adenocarcinoma after multi-walled carbon nanotube inhalation exposure in mice. J Appl Toxicol. 2016;36(1):161–174. doi:10.1002/jat.3157
15. Ai K, Zhu X, Kang Y, et al. miR-130a-3p inhibition protects against renal fibrosis in vitro via the TGF-beta1/Smad pathway by targeting SnoN. Exp Mol Pathol. 2020;112:104358. doi:10.1016/j.yexmp.2019.104358
16. Wang Y, Du J, Niu X, et al. MiR-130a-3p attenuates activation and induces apoptosis of hepatic stellate cells in nonalcoholic fibrosing steatohepatitis by directly targeting TGFBR1 and TGFBR2. Cell Death Dis. 2017;8(5):e2792. doi:10.1038/cddis.2017.10
17. Asama H, Suzuki R, Hikichi T, et al. MicroRNA let-7d targets thrombospondin-1 and inhibits the activation of human pancreatic stellate cells. Pancreatology. 2019;19(1):196–203. doi:10.1016/j.pan.2018.10.012
18. Ji T, Feng W, Zhang X, et al. HDAC inhibitors promote pancreatic stellate cell apoptosis and relieve pancreatic fibrosis by upregulating miR-15/16 in chronic pancreatitis. Hum Cell. 2020;33(4):1006–1016. doi:10.1007/s13577-020-00387-x
19. Wang W, Dong R, Guo Y, et al. CircMTO1 inhibits liver fibrosis via regulation of miR-17-5p and Smad7. J Cell Mol Med. 2019;23(8):5486–5496. doi:10.1111/jcmm.14432
20. Wang H, Wang B, Zhang A, et al. Exosome-mediated miR-29 transfer reduces muscle atrophy and kidney fibrosis in mice. Mol Ther. 2019;27(3):571–583. doi:10.1016/j.ymthe.2019.01.008
21. Chen L, Yao X, Yao H, et al. Exosomal miR-103-3p from LPS-activated THP-1 macrophage contributes to the activation of hepatic stellate cells. FASEB J. 2020;34(4):5178–5192. doi:10.1096/fj.201902307RRR
22. Su S, Zhao Q, He C, et al. miR-142-5p and miR-130a-3p are regulated by IL-4 and IL-13 and control profibrogenic macrophage program. Nat Commun. 2015;6(1):8523. doi:10.1038/ncomms9523
23. Mei Y, Bian C, Li J, et al. miR-21 modulates the ERK-MAPK signaling pathway by regulating SPRY2 expression during human mesenchymal stem cell differentiation. J Cell Biochem. 2013;114(6):1374–1384. doi:10.1002/jcb.24479
24. Roderburg C, Urban GW, Bettermann K, et al. Micro-RNA profiling reveals a role for miR-29 in human and murine liver fibrosis. Hepatology. 2011;53(1):209–218. doi:10.1002/hep.23922
25. Squadrito ML, Baer C, Burdet F, et al. Endogenous RNAs modulate microRNA sorting to exosomes and transfer to acceptor cells. Cell Rep. 2014;8(5):1432–1446. doi:10.1016/j.celrep.2014.07.035
26. Qu Y, Zhang Q, Cai X, et al. Exosomes derived from miR-181-5p-modified adipose-derived mesenchymal stem cells prevent liver fibrosis via autophagy activation. J Cell Mol Med. 2017;21(10):2491–2502. doi:10.1111/jcmm.13170
27. Yu Z, Zhao S, Ren L, et al. Pancreatic cancer-derived exosomes promote tumor metastasis and liver pre-metastatic niche formation. Oncotarget. 2017;8(38):63461–63483. doi:10.18632/oncotarget.18831
28. Zhang Y, Wang XF. A niche role for cancer exosomes in metastasis. Nat Cell Biol. 2015;17(6):709–711. doi:10.1038/ncb3181
29. Masamune A, Yoshida N, Hamada S, et al. Exosomes derived from pancreatic cancer cells induce activation and profibrogenic activities in pancreatic stellate cells. Biochem Biophys Res Commun. 2018;495(1):71–77. doi:10.1016/j.bbrc.2017.10.141
30. Bertero T, Cottrill KA, Annis S, et al. A YAP/TAZ-miR-130/301 molecular circuit exerts systems-level control of fibrosis in a network of human diseases and physiologic conditions. Sci Rep. 2015;5(1):18277. doi:10.1038/srep18277
31. Gooch JL, King C, Francis CE, et al. Cyclosporine A alters expression of renal microRNAs: new insights into calcineurin inhibitor nephrotoxicity. PLoS One. 2017;12(4):e0175242. doi:10.1371/journal.pone.0175242
32. Chu X, Wang Y, Pang L, et al. miR-130 aggravates acute myocardial infarction-induced myocardial injury by targeting PPAR-gamma. J Cell Biochem. 2018;119(9):7235–7244. doi:10.1002/jcb.26903
33. Apte M, McCarroll J, Pirola R, et al. Pancreatic MAP kinase pathways and acetaldehyde. Novartis Found Symp. 2007;285:200–211. doi:10.1002/9780470511848.ch15
34. Asukai K, Kawamoto K, Eguchi H, et al. Micro-RNA-130a-3p regulates gemcitabine resistance via PPARG in cholangiocarcinoma. Ann Surg Oncol. 2017;24(8):2344–2352. doi:10.1245/s10434-017-5871-x
35. Luo G, Long J, Cui X, et al. Highly lymphatic metastatic pancreatic cancer cells possess stem cell-like properties. Int J Oncol. 2013;42(3):979–984. doi:10.3892/ijo.2013.1780
36. Masamune A, Shimosegawa T. Signal transduction in pancreatic stellate cells. J Gastroenterol. 2009;44(4):249–260. doi:10.1007/s00535-009-0013-2
37. Jaster R, Lichte P, Fitzner B, et al. Peroxisome proliferator-activated receptor gamma overexpression inhibits pro-fibrogenic activities of immortalised rat pancreatic stellate cells. J Cell Mol Med. 2005;9(3):670–682. doi:10.1111/j.1582-4934.2005.tb00497.x
38. Figliolini F, Cantaluppi V, De Lena M, et al. Isolation, characterization and potential role in beta cell-endothelium cross-talk of extracellular vesicles released from human pancreatic islets. PLoS One. 2014;9(7):e102521. doi:10.1371/journal.pone.0102521
39. Pelli H, Lappalainen-Lehto R, Piironen A, et al. Risk factors for recurrent acute alcohol-associated pancreatitis: a prospective analysis. Scand J Gastroenterol. 2008;43(5):614–621. doi:10.1080/00365520701843027
40. Agostinho P, Cunha RA, Oliveira C. Neuroinflammation, oxidative stress and the pathogenesis of alzheimer’s disease. Curr Pharm Des. 2010;16(25):2766–2778. doi:10.2174/138161210793176572
41. Shimizu K, Shiratori K, Hayashi N, et al. Thiazolidinedione derivatives as novel therapeutic agents to prevent the development of chronic pancreatitis. Pancreas. 2002;24(2):184–190. doi:10.1097/00006676-200203000-00010
42. Zhi SC, Chen SZ, Li YY, et al. Rosiglitazone inhibits activation of hepatic stellate cells via up-regulating micro-RNA-124-3p to alleviate hepatic fibrosis. Dig Dis Sci. 2019;64(6):1560–1570. doi:10.1007/s10620-019-5462-8
43. Duarte S, Baber J, Fujii T, et al. Matrix metalloproteinases in liver injury, repair and fibrosis. Matrix Biol. 2015;44–46:147–156. doi:10.1016/j.matbio.2015.01.004
44. Shek FW, Benyon RC, Walker FM, et al. Expression of transforming growth factor-beta 1 by pancreatic stellate cells and its implications for matrix secretion and turnover in chronic pancreatitis. Am J Pathol. 2002;160(5):1787–1798. doi:10.1016/s0002-9440(10)61125-x
45. Makled MN, Sharawy MH, El-Awady MS. The dual PPAR-alpha/gamma agonist saroglitazar ameliorates thioacetamide-induced liver fibrosis in rats through regulating leptin. Naunyn Schmiedebergs Arch Pharmacol. 2019;392(12):1569–1576. doi:10.1007/s00210-019-01703-5
46. Forrester SJ, Eguchi S. Vascular matrix metalloproteinase inhibition, a new mechanism for how peroxisome proliferator-activated receptor-gamma protects target organ damage. Hypertension. 2016;67(1):36–37. doi:10.1161/HYPERTENSIONAHA.115.06532
47. Tan H, Chen Z, Chen F, et al. Tryptase promotes the profibrotic phenotype transfer of atrial fibroblasts by PAR2 and PPARgamma pathway. Arch Med Res. 2018;49(8):568–575. doi:10.1016/j.arcmed.2018.12.002
48. Thomas D, Radhakrishnan P. Tumor-stromal crosstalk in pancreatic cancer and tissue fibrosis. Mol Cancer. 2019;18(1):14. doi:10.1186/s12943-018-0927-5
49. Tandon M, Coudriet GM, Criscimanna A, et al. Prolactin promotes fibrosis and pancreatic cancer progression. Cancer Res. 2019;79(20):5316–5327. doi:10.1158/0008-5472.CAN-18-3064
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.