")
Back to Journals » International Journal of Nanomedicine » Volume 13
Regulation of in vivo behavior of TAT-modified liposome by associated protein corona and avidity to tumor cells
Authors Amin M, Bagheri M, Mansourian M, Jaafari MR , ten Hagen TLM
Received 5 April 2018
Accepted for publication 3 July 2018
Published 15 November 2018 Volume 2018:13 Pages 7441—7455
DOI https://doi.org/10.2147/IJN.S170274
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Delayed nuclear delivery of DXR at a density of 400 peptides
Views: 115
Mohamadreza Amin,1–3 Mahsa Bagheri,3 Mercedeh Mansourian,3 Mahmoud Reza Jaafari,3 Timo LM ten Hagen1
1Laboratory of Experimental Surgical Oncology, Section of Surgical Oncology, Department of Surgery, Erasmus Medical Center, Rotterdam, the Netherlands; 2Cellular and Molecular Research Center, Faculty of Medicine, Sabzevar University of Medical Sciences, Sabzevar, Iran; 3Biotechnology Research Center, Pharmaceutical Technology Institute, Mashhad University of Medical Sciences, Mashhad, Iran
Introduction: PEGylated liposomes are widely used and studied as carriers for chemotherapeutics. While pharmacokinetics of the encapsulated drug is drastically altered resulting in favorable circulation time, improved tumor accumulation, and better manageable or reduced side effects, therapeutic efficacy has been disappointing. Major drawbacks are a failure to reach the tumor cell, limited penetration depth, and impaired uptake by tumor cells.
Materials and methods: Here, we study the implication of HIV-1 transactivator of transcription (TAT)-derived peptides inserted on PEGylated liposomal doxorubicin (PLD) and followed in vitro and in vivo fate. PLDs were installed with 25–400 TAT peptides per liposome without an effect on PLD stability.
Results: While TAT peptides facilitate active endocytosis of the carriers, we observed that these peptides did not promote endosomal escape or enhanced intracellular availability of doxorubicin. Interestingly, incorporation of TAT peptides did not change pharmacokinetics or biodistribution, which we found to result from a dysopsonization of the TAT-modified liposomes by serum proteins. A protein corona (PC) on TAT peptide-modified PLDs shields the active moieties and effectively reduces clearance of the TAT peptide containing nanoparticles. However, intratumoral activity was influenced by the number of TAT peptides present. The best antitumor efficacy was observed with a TAT peptide density of 100, while lower amounts showed results comparable to unmodified PLDs. At 200 TAT peptides, the preparation appeared to be least effective, which likely results from augmented interaction with tumor cells directly upon extravasation.
Conclusion: We conclude that by optimizing TAT-modified PLDs, the occurring PC balances pharmacokinetics and tumor penetration through interference with avidity.
Keywords: PEGylated liposomal doxorubicin, TAT peptide, ligand-modified liposomes, protein corona, dysopsonization
Introduction
New agents, molecules, and therapies for cancer are being developed with mixed success. However, quite a few classic compounds, with profound in vitro proven antitumor activity, are available but cannot be used optimally in patients. Poor water solubility, poor pharmacokinetics, and severe dose-limiting side effects hinder their usability in patients and adversely affect therapeutic outcome. Nanomedicines are employed to improve solubility, tailor release rates, and improve pharmacokinetics of the cytotoxic agents, while minimizing side effects.1,2 Doxil®/Caelyx® (PEGylated liposomal doxorubicin [PLD, Janssen Pharmaceuticals NV, Beerse, Belgium]) was therefore the first US FDA-approved nano-device.3–5
PEGylated nano-sized particles like Caelyx present a stealthy behavior, and therefore are not recognized by the reticuloendothelial system (RES), and circulate long in blood. It is believed that liposomes extravasate into tumor tissue through the leaky tumor vasculatures and retain there because of the impaired lymphatic drainage in tumors, a phenomenon known as the enhanced permeation and retention (EPR) effect.6 However, the PEGylated nature and rigid lipid composition of Caelyx dramatically decrease direct interaction with tumor cells, and doxorubicin (DXR) delivery to tumor cells mainly relies on DXR release from destabilized liposomes which in turn is affected by phospholipases or acidic tumor environment.3,4 Therefore, ligand-modified liposomes have attracted attention to increase the intracellular delivery of liposomal drugs into tumor cells7 and reduce the retrograde movement of the extravasated liposomes back to the blood stream.8 However, this is an ideal scenario. Although active targeting dramatically enhances in vitro cellular delivery of liposomes, tremendous efforts on designing ligand-targeted liposomes against tumors resulted in no preclinical accepted preparation that progressed beyond Phase I or II of clinical trials.9 This shifted the scientific society’s concerns to analyzing the causes of these failures.7,10–12
It is well-recognized that once a nanoparticle (NP) enters the bloodstream, a layer of serum proteins named “protein corona” (PC) forms rapidly around the NP, which obscures the NP properties and confers a new identity to the NP surface. Afterward, it is the PC, rather than the pristine NP formulation, that governs the circulation fate and the biological response of the NP. Formation of opsonin-enriched PC fully explains the increased clearance rate of ligand-modified liposomes compared to the non-targeted counterparts due to the RES function, which is now well known as great hurdle in passive delivery of targeted liposomes to tumor interstitium where these liposomes should actively deliver their payload into cells.7,9 Besides, the question arises on whether the surface-anchored ligand would be functional when a liposome is covered by a serum-derived PC. Formation of PC may, in part, be linked to failure of ligand-targeted liposomes to emerge on the market,10 as a result of augmented clearance rate or loss of functionality.
TAT peptide derived from the transactivator of transcription (TAT) of HIV-1 is a cell-penetrating peptide (CPP) that has drawn considerable attention to enhance cellular uptake of nanomedicines.14–17 The simplicity of sequence, ease, and low cost of preparation and conjugation, and most importantly, activity against a wide variety of cancer cells makes the TAT peptide a great candidate for ligand modification of liposomes.18,19 The exact mechanisms of cellular uptake, intracellular trafficking, and role of cell surface molecules in the internalization process of CPPs are still under debate.14,20,21 The electrostatic interaction of the positively charged TAT with negatively charged cell-surface glycoproteins was found to be a prerequisite for passage across the plasma membrane.22 However, proposed internalization pathways of TAT-modified cargoes into cells are controversial and both energy-independent translocation23 or energy-dependent endocytosis24 were said to be involved in TAT regulated internalization. Although the capability of TAT-modified liposomes in association and internalization into tumor cells has been extensively studied in vitro,25–28 in vivo therapeutic efficacy and biodistribution properties have not been clearly addressed in literature.
The positively charged TAT peptide may make liposomes prone to RES-mediated clearance. To avoid or mitigate this, optimizing ligand density on liposomes is the primary approach with respect to optimizing the efficacy and pharmaceutical manufacturing.
The current study was conducted to delineate the biological fate and activity of TAT-modified liposomes with respect to formulation optimization, stability, in vitro and in vivo drug delivery capacity and efficacy.
Materials and methods
Materials
Hydrogenated soya phosphatidylcholine (HSPC) and methoxypolyethyleneglycol (MW 2000)-distearylphosphatidylethanolamine (mPEG2000-DSPE) were purchased from Lipoid (Ludwigshafen, Germany), maleimide-PEG2000 distearoylphosphatidylethanolamine (Mal-PEG2000-DSPE) was purchased from Avanti Polar Lipids (Alabaster, AL, USA). 3,3′-Dioctadecyloxacarbocyanine perchlorate (DiO), 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine, and 4-chlorobenzenesulfonate salt (DiD) were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Ammonium sulfate, cholesterol, α-tocopherol, XTT, PMS and DXR hydrochloride were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). TAT peptide analog equipped with 3G as spacer and a C for conjugation (CGGG-RKKRRQRRRGYG) was synthesized by Peptron Inc. (Daejeon, South Korea) with purity of 98%. Commercially available Caelyx was purchased from pharmacy. All other solvents and reagents were used as chemical grade.
Preparation of TAT-modified liposomes
Fluorescent-labeled PEGylated liposomes (FPLs) composed of HSPC, mPEG2000-DSPE, cholesterol, α-tocopherol, and the lipophilic dye (56.1:5.5:38.2:0.1:0.1 mol%) was prepared by solvent evaporation and hydration in HEPES 10 mM and NaCl 135 mM (pH 7.4), then downsized by 10 minutes bath sonication and extrusion through polycarbonate membranes of 200 and 80 nm sequentially, using LIPEX™ (Northern lipid Inc., Vancouver, BC, Canada).
For post-insertion of the ligand, the conjugated DSPE-PEG2000-TAT (for conjugation details see supplementary material and Figure S1) was first dispersed in distilled water at a concentration of 840 nmol/mL followed by 10 minutes agitation to form micellar lipopeptide. Considering that liposomes of 100 nm size are composed of 80,000 phospholipid molecules, the amounts of liposomes and micellar DSPE-PEG2000-TAT were adjusted to prepare liposomes with different surface densities of TAT (Table S1). A calculated amount of micellar lipopeptide was then added to preformed FPL (FPL-TATs) or Caelyx (PLD-TATs) in a glass tube under argon atmosphere and incubated at 50°C for 30 minutes with gentle agitation.
Characterization and stability assessments of liposomes
Liposomes were characterized with respect to size, polydispersity index, and ζ-potential by a dynamic light scattering instrument (Nano-ZS; Malvern Instruments, Malvern, UK) at 25°C with a scattering angle of 173°. The phospholipid content of preparations was measured by a method based on Bartlett phosphate assay.29 Leakage stability of PLDs was assessed either through post-insertion treatment or during 48 hours of incubation at 37°C in the presence of 30% fetal calf serum (FCS) (Supplementary material).
Evaluation of in vitro behavior of TAT-modified liposomes
Flow cytometry analysis
The capability of TAT-modified liposomes in interaction with C26 (Cell Line Services GmbH, Eppelheim, Germany) and B16F0 (Sigma-Aldrich Co.) cells was evaluated by flow cytometry using FACS Caliber (BD Biosciences, San Jose, CA, USA). Briefly, 105 cells were seeded in 24-well plates and incubated overnight at culturing condition (Supplementary material). Then, the medium was replaced by liposomal preparations diluted in complete culture medium (100 nmol liposomal phospholipid/0.5 mL) and incubated for 3 hours at either 37°C or 4°C. Cells were then detached by trypsinization and washed three times with PBS (pH 7.4) containing 1% FCS. Five hundred microliters of cell suspensions was incubated with 5 μL of propidium iodide (PI) (100 μg/mL) for 15 minutes at room temperature. Ten thousand cells gated on live cells by forward and side scatters ( FSC/SSC) and PI exclusion and assayed for the intensity of green DiO fluorescent dye.
Confocal imaging
Internalization of FPL-TAT200 and DiO-labeled PLD-TAT200 (Supplementary material) into tumor cells was also visualized using living cell confocal microscopy. Briefly, cells were seeded in cell culture chambers equipped with a fibronectin coated cover glass insert. After overnight incubation, cells were exposed to 100 nmol liposomal phospholipid (0.5 mL) for 3 hours at 37°C, washed with pre-warmed complete medium, and were analyzed on a Zeiss LSM 510 META confocal laser scanning microscope.
The internalization pathway of FPL-TAT200 into C26 cells was also tracked in cells pre-incubated with Lysotracker® Red (Thermo Fisher Scientific (Waltham, MA, USA), a dye that specifically stains the acidified cell organelles.
Cytotoxicity of PLDs against tumor cells
The antiproliferative effects of PLDs as well as free DXR were assessed using XTT assay. C26 and B16F0 cells were seeded at the density of 2,500 cells/well in 96-well microplates, incubated overnight at 37°C, and exposed to serially diluted liposomal DXR for 3 hours. Cells were then washed three times with pre-warmed complete culture medium and allowed to proliferate at 37°C for 72 hours in their complete culture medium. Finally, the culture medium was replaced with 100 μL of XTT (1 mg/mL)/PMS (7.66 μg/mL) solution freshly prepared in phenol red and serum free culture medium. After 1 hour of incubation at 37°C in a CO2 incubator, the absorbance at 490 nm was recorded and the relative cell death (R) was calculated as follows: R=1-[(Atest − Ablank)/(Acontrol − Ablank)], where Atest and Acontrol were the absorbances of the cells treated with the test solutions and the culture medium (negative control), respectively. Ablank was the absorbance of XTT/PMS solution added in cell free wells. IC50 values were then calculated based on dose–response curve using CalcuSyn version 2 software (Biosoft, Cambridge, UK).
Evaluation of in vivo behavior of TAT-modified liposomes
Mice model of subcutaneous C26 colon carcinoma tumor model
This animal experiments were approved by the Institutional Ethical Committee and Research Advisory Committee of Mashhad University of Medical Sciences (research project code 913286). Female BALB/c mice aged 4–6 weeks were inoculated by a subcutaneous injection of 3×105 C26 colon carcinoma cells in the right flank. Animals with proper tumor, as specified later, were used for therapeutic efficacy and biodistribution studies.
Dorsal skin-fold window chamber model of B16 melanoma
Animal experiments were approved by the Instantie voor Dierenwelzijn Erasmus Medical Center and conducted with permissions granted by the Nederlandse Dierexperimentencommissie. The experiments were performed according to the European Directive 2010/63/EU on the protection of animals used for scientific purposes. Ten million B16 cells were injected subcutaneously into the flanks of C57Bl6 mice; the animals were housed at 20°C–22°C with a humidity of 50%–60%. Bulk tumors of 10 mm in diameter were used for transplantation into C57Bl6 mice, expressing an eNos-tag-GFP fusion protein constitutively in their vascular endothelium. Tumor pieces were implanted in a dorsal skin flap window chamber for intravital imaging.30–32 Window chamber-bearing mice were used for experiments after 8–12 days of tumor implantation when the tumor size reached 4–6 mm in diameter. These mice were housed in an incubator room with a humidity of 70% and temperature of 30°C–32°C.
Biodistribution of PLDs in mice model of C26 tumor
Biodistribution of PLD-TATs was evaluated in two doses of 10 and 15 mg/kg liposomal DXR, in two separate experiments. On day 14 post-tumoring, when the tumor size was ~5 mm wide, mice (eight per group) received liposomal DXR via the tail vein. Blood was collected via the orbital sinus 6 (four of eight mice) and 12 (other four mice) hours after dosing. At 24- or 48-hour time points, mice (four mice) were deeply anesthetized by ketamine-xylazine cocktail and euthanized. Blood was collected by heart puncture, and the tumor, kidneys, spleen, lungs, and liver were dissected, weighed, and less than 300 mg of tissues were transferred into 2 mL polypropylene microvials (Biospec Products, Bartlesville, OK, USA) containing 1 mL of acidified alcohol (90% isopropanol/0.075 M HCl) and zirconia beads and homogenized by Mini-Beadbeater-1 (Biospec Products). Blood was allowed to coagulate at 4°C and then centrifuged to separate the serum. Sera were separated by centrifuging at 14,000× g for 10 minutes33 for mice receiving 10 mg/kg and at 5,000× g for 5 minutes for mice receiving 15 mg/kg of liposomal DXR. Then an adequate amount of serum was diluted in 1 mL of acidified isopropanol. To extract DXR, homogenized tissue samples and sera were stored at 4°C overnight. Finally, all samples were centrifuged to collect the supernatant for DXR assay using spectrofluorimetric method (excitation: 470 nm, emission: 590 nm). The calibration curves were obtained by preparing serial dilutions of DXR in tissue and sera extracts of control mice.
Therapeutic efficacy of PLDs against subcutaneous C26 tumor model
On day 7 post-inoculation, mice with palpable tumor received a single tail vein injection of either sucrose 10% solution as negative control or DXR at 10 or 15 mg/kg encapsulated in PLDs. The tumor volume was estimated by measuring the three orthogonal diameters of tumors using the (a×b×c)/2 formula. Mice were monitored for up to 60 days post-inoculation or until one of the following conditions for euthanasia was met: 1) their body weight dropped below 20% of their initial mass; 2) their tumor was greater than 2.0 cm across in any dimension or the tumor volume was greater than 1 cm3; 3) they became lethargic or sick and unable to feed; or 4) they were found dead.33,34
In vivo visualization of intratumoral behavior of TAT-modified liposomes
In vivo behavior of fluorescently labeled TAT-modified liposomes (FPL-TAT-200) in tumor was observed by intravital confocal microscopy on dorsal skin-fold window chamber-bearing mice after an iv injection through tail vein at a dose of 5 μmol of lipid. The mice were then anesthetized with isoflurane (Nicholas Piramal, London, UK) and placed on a heated stage (37°C) under the confocal microscope (Zeiss LSM 510 META).
Impact of mice serum on colloidal properties of PLD-TATs
Mimicking in vivo serum/PLD ratio after an iv injection of 15 mg/kg liposomal DXR, 160 μL of different combinations of PLD-TATs were diluted in 1,200 μL of normal mice serum (Abcam, Cambridge, UK) and incubated at 37°C for 6 hours.
To assess the impact of serum on colloidal properties of PLD-TATs, 100 μL of serum-treated or untreated PLD-TATs were diluted in 1,900 μL of Tris-HCl 10 mM and NaCl 135 mM (pH 7.4), and the size, ζ-potential, and polydispersity index (PDI) were measured by the dynamic light scattering instrument.
SDS-PAGE analyses of serum-treated liposomes
Liposomes incubated with mouse serum were passed through Sepharose CL-4B (Pharmacia, Uppsala, Sweden) size-exclusion column (24×2.5 cm) equilibrated with Tris-buffered saline at pH 7.435 to separate liposomes from bulk serum protein. Column-recovered liposomes were collected, concentrate by freeze drying, and resuspended in distilled water. DXR concentration was assayed and equal amount of liposomal DXR in adjusted volume of 25 μL was diluted with 25 μL of 2× Laemmli sample buffer (Bio-Rad) supplemented with 5% mercaptoethanol (Sigma-Aldrich Co.) and heated in a thermal shaker for 10 minutes at 99°C. Fifty microliters of samples was then applied on precast 4%–20% gradient Mini-Protean TGX gel (Bio-Rad). The electrophoresis buffer was composed of 25 mM Tris, 192 mM glycine, and 0.1% SDS (pH 8.3). After electrophoresis, the gel was silver-stained with SilverQuest staining kit (Thermo Fisher Scientific).
Statistical analysis
Statistical analyses were performed using GraphPad Prism version 6 (GraphPad Software, Inc., La Jolla, CA, USA). Survival data were analyzed by the log-rank test. For other comparisons, one-way ANOVA and Newman–Keuls multiple comparisons test were employed.
Results and discussion
Characterization of liposomes
The particle size of various liposomes was around 100 nm (PDI <0.1). While TAT had no impact on the observed liposome size, its presence on liposome surface reduced the negative ζ-potential of liposomes (Tables S1 and S2). Importantly, insertion of TAT at elevated temperature caused no significant DXR release (Figure 1 inset). Liposomes were also stable in the presence of serum proteins and no pronounced leakage or instability was found when liposomes were incubated with FCS (Figure 1).
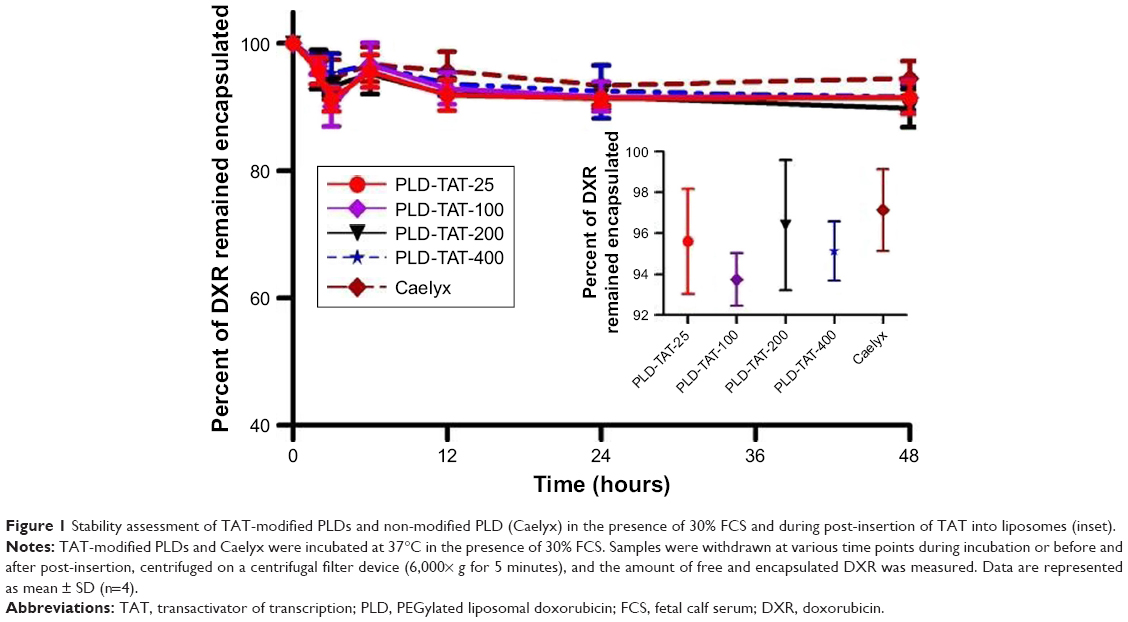 | Figure 1 Stability assessment of TAT-modified PLDs and non-modified PLD (Caelyx) in the presence of 30% FCS and during post-insertion of TAT into liposomes (inset). |
In vitro assessment of intracellular fate of TAT liposomes
In vitro association of TAT-modified liposomes with cells including C26 and B16 revealed an increased liposome–cell association by increasing the TAT density with identical pattern at both 4°C and 37°C (Figures 2A and S2). While plain liposomes were barely observable associated with cells (Figure S3), TAT was effective in improving liposome–cell association even at a density of 25 moieties per liposome, which in turn resulted in elevated cytotoxicity of PLD-TATs (Figure 2B). Insertion of 25, 50, 100, 200, 300 or 400 TAT peptides on Caelyx increased the cytotoxicity of Caelyx by 2, 6.1, 14.1, 26.4, 35.8, and 49.6 fold against C26 cells, respectively, and by 1.7, 3.8, 7.8, 16.6, 21.3, and 33 fold against B16 cells, respectively. However, the enhanced association or cytotoxicity did not follow a linear correlation indicating saturation. Flow cytometry and cytotoxicity analysis revealed the following trends. The densities of 0–100 TAT peptides/liposome produced a sharp increase in association (Figure 2A) and cytotoxicity (Figure 2B), whereas for densities of 100–400 TAT peptides/liposome, the increased association or cytotoxicity was diminished. Based on these data, densities above 200 TAT peptides/liposome were excluded from further in vivo experiments.
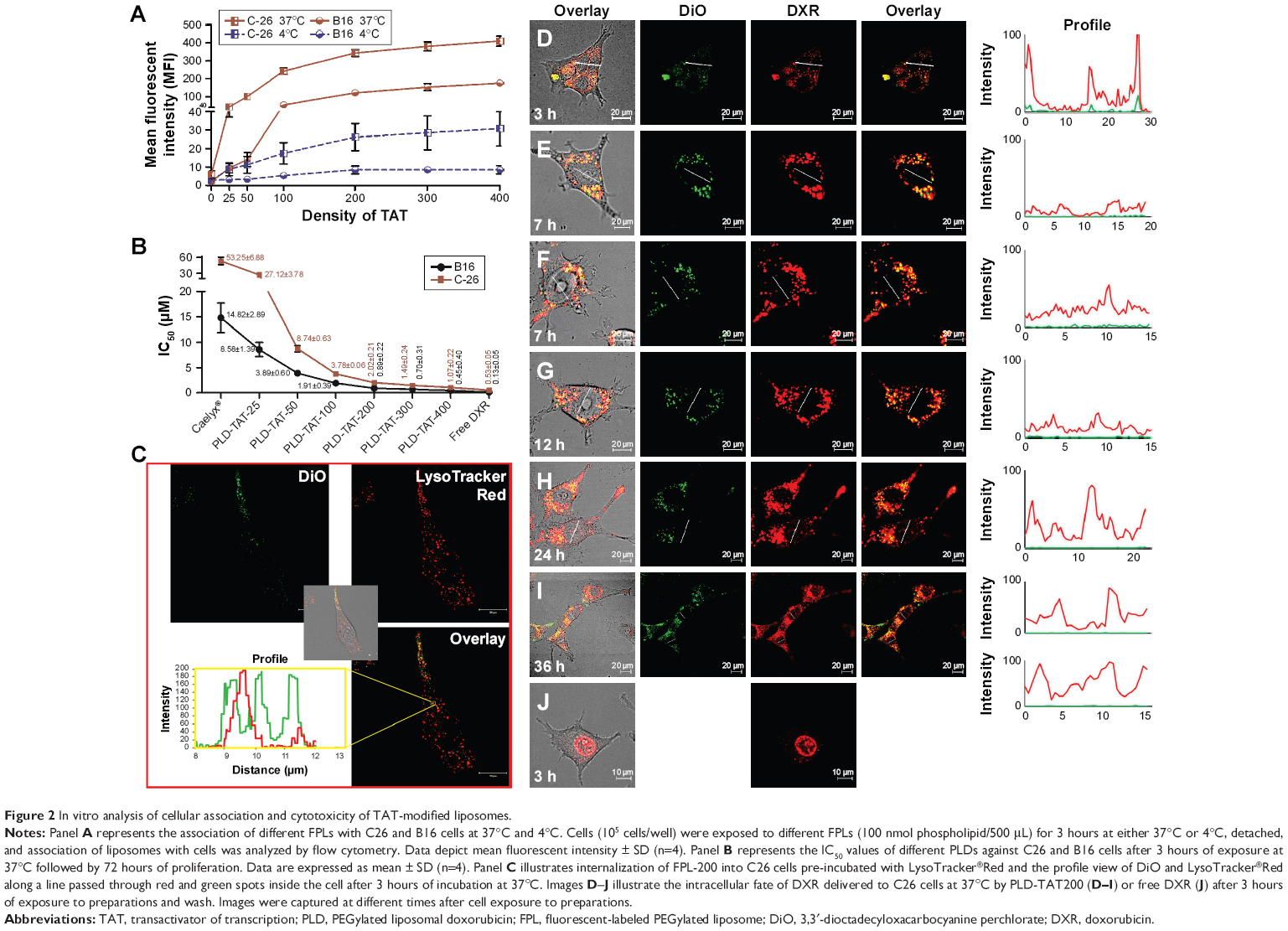 | Figure 2 In vitro analysis of cellular association and cytotoxicity of TAT-modified liposomes. |
Analyzed by ZEN 2012 SP2 blue edition (Carl Zeiss Microscopy GmbH), co-localization of green DiO liposomal dye with the red fluorescence of acidified endosomal compartments labeled with LysoTracker®Red (Figure 2C) indicated that TAT enabled liposomes to be internalized into C26 via receptor-mediated endocytosis. Internalization of TAT liposomes into B16 cells was also found to be mediated by endocytosis (Figure S2C). Alternatively, no free TAT-modified liposome was detected in cell cytoplasm, indicating the absence of direct translocation of TAT liposomes into cells.
To analyze the intracellular fate of PLD-TATs, DiO-labeled PLD-TAT200 was prepared and its intracellular fate was monitored over 36 hours (Figure 2D–I) in C26 cultured cells. TAT remarkably enhanced the internalization of PLD into cells (Figure S3I and J). However, the most significant observation in this experiment was the delayed nuclear delivery of DXR by PLD-TAT200 compared to free DXR. While free DXR promptly reached the cell nucleus (Figure 2J), no significant amount of DXR could be detected along the lines crossing the nucleus of the cells incubated with PLD-TAT and imaged between 3 and 7 hours post-incubation (Figure 2D–G) as liposomal DXR was mainly entrapped in the endosomal system. At 12 hours post-exposure, minor nuclear delivery of DXR could be detected, while significant levels of DXR staining was only detected in the nucleus 24–36 hours later (Figure 2H and I), which probably resulted from slow DXR release from the endosomal system (Figure S2H and I). Comparable delayed nuclear delivery was also observed with a density of 400 peptides (Figure S4 and supplementary video). These results on the one hand indicate that TAT does not regulate liposome–cell fusion or endosomal escape, and on the other hand reveal that, although TAT dramatically promotes PLD internalization into cells, it does not affect intracellular availability. We observed that internalized liposomes were mainly retained in lysosomes and only provided a source of slow-release DXR, which was found to be effective in killing the cells (Figure 2B).
In fact, this delayed release is attributed to the high concentration of intraliposomal DXR sulfate (>200 mM). DXR molecules tend to form aggregates and a visible precipitation of DXR sulfate occurs at a concentration of <2 mM.36 Therefore, at least 100-fold dilution is required for DXR monomers to occur and escape the lysosome and reach the nucleus. Besides, the acidic pH of lysosomes further reduces the dissolution rate of DXR.
Biodistribution
Biodistribution of PLD-TATs as well as Caelyx was extensively evaluated at doses of 10 and 15 mg/kg in mice bearing C26 tumor, in two separate experiments with minor modification in the protocol involving the separation of sera.
In the first experiment, in which mice received 10 mg/kg liposomal DXR and sera were separated by 14,000× g centrifugation, it was observed that the plasma clearance rate of liposomes increased gradually with increased TAT density (Figure 3A). However, all preparations created statistically identical levels in liver, spleen, lungs, and kidneys (Figure 3B and C) and accumulated to the same extent inside the tumor (Figure 3D) 48 hours post-injection (P>0.05). Based on our previous observations,13,32 it was first speculated that reduction of plasma DXR levels should be attributed to fast clearance of the RES by cells, directed against PLD-TATs because of the cationic charge of TAT molecules. This should have been reflected in higher levels of DXR in spleen and liver of mice that received short circulating liposomes.13.32 Yet, analysis of DXR levels in liver and spleen revealed no significant differences in uptake of TAT-modified and non-modified PLDs by these two organs, which argue against a dominant function of the RES against PLD-TATs. On the other hand, PLD-TATs showed no leakage instability in biological fluid in vitro (Figure 1), and DXR levels in kidneys of different groups were statistically identical which reject the hypothesis that liposomes become more unstable and release DXR during circulation, as well. Besides, identical DXR levels in lungs of mice receiving these preparations indicate no aggregation upon administration. Interestingly, despite the rapid drop in serum DXR levels of mice receiving PLD-TATs, tumor accumulation of different PLDs was identical and the apparent differences in plasma clearance rates of different PLDs did not impact on their extravasation and tumor accumulation.
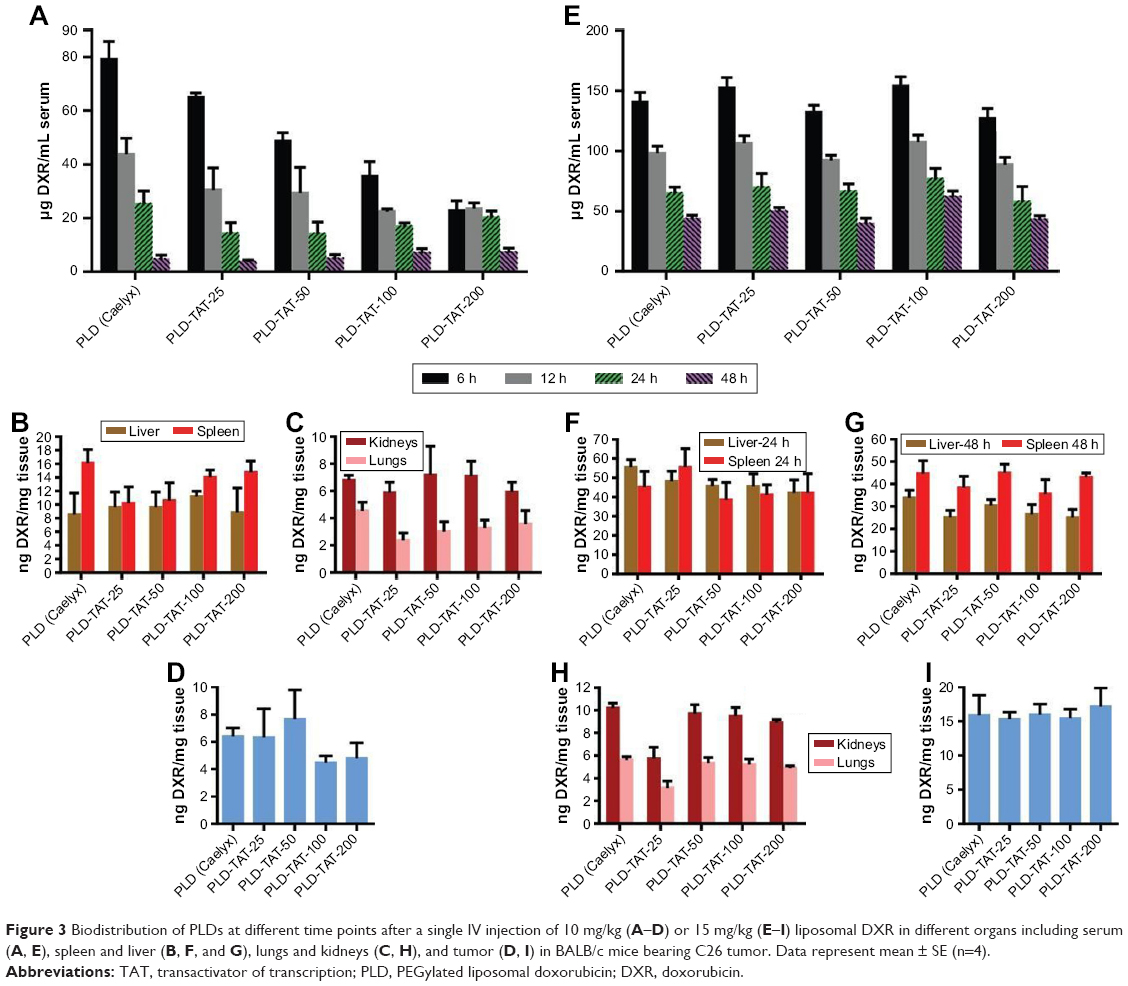 | Figure 3 Biodistribution of PLDs at different time points after a single IV injection of 10 mg/kg (A–D) or 15 mg/kg (E–I) liposomal DXR in different organs including serum (A, E), spleen and liver (B, F, and G), lungs and kidneys (C, H), and tumor (D, I) in BALB/c mice bearing C26 tumor. Data represent mean ± SE (n=4). |
We were doubtful as to whether 14,000× g centrifugation is sufficient for serum separation; therefore, in the second experiment, in which mice received 15 mg/kg liposomal DXR, sera were separated with a lower centrifugation force of 5,000× g. In contrast to the first experiment, no significant differences were found in plasma levels of PLD-TATs and Caelyx (Figure 3E). Consistent with the first experiment, DXR concentration in liver, spleen, lungs, kidneys (Figure 3F–H), and tumor (Figure 3I) showed virtually similar values for all preparations (P>0.05).
It was also possible that equal levels in liver and spleen at 48 or 24 hours after administration could be attributed to the metabolizing function of these two organs. However, at 6 hours post-injection, DXR levels in spleens of mice receiving Plain-PLD or PLD-TATs were identical, and a slight increase in hepatic levels of DXR in mice receiving PLD-TATs (Figure S5) could not justify the observed rapid clearance rate of first experiment (Figure 3A).
It is worth mentioning that once the mice received 15 mg/kg DXR as PLD-TATs and sera were separated by the high centrifugation force (Figure S6), the correlation between serum DXR level and TAT density was identical, as shown in Figure 3A. Based on the above-mentioned results, we concluded that the different pattern of serum DXR observed in the current experiments is not attributed to dosing.
Taken together, we hypothesize that the reduced plasma levels observed in the first experiment must be attributed to the centrifugation forces. To confirm this finding, PLD-TATs were subjected to centrifugation at both 14,000× g for 10 minutes and 4,000× g for 5 minutes. Since none of the PLD-TATs sedimented by these forces (data not shown), we hypothesized that protein binding on PLD-TATs during circulation might have increased PLD-TATs density which, in turn, caused sedimentation by high centrifugal forces. This prompted us to investigate the protein binding of PLD-TATs in the presence of mouse serum, which is presented in the following section.
Impact of mice serum on colloidal properties of TAT-modified liposomes
PLD-TAT-25, 100, 200, 400, and Caelyx were incubated with freshly prepared mouse serum for 6 hours at 37°C and were then compared against untreated counterpart with respect to their colloidal properties and protein binding.
Analysis of colloidal properties of PLDs before and after serum treatment (Figures 4A, B) (Figure S7) revealed a reduction in mean diameter of Caelyx after serum treatment. This was consistent with others studies investigating PC formation on liposomes37–39 that have related this to an osmotically-driven shrinkage due to high elastic deformation of liposomes. While in bare liposomes, incorporation of TAT did not cause significant size changes (P>0.05), serum-treated PLD-TATs revealed a direct correlation between their size and their TAT density, indicating the thickness of the PC increased with increasing TAT moieties on liposome surface (P<0.05). Nonetheless, all serum-treated PLDs showed unimodal size distribution without serious aggregation (Figure S7). Interestingly, while the positive charge of TAT reduced the negative charge of PLDs, formation of PC normalized their ζ-potentials as all serum-treated PLDs had similar ζ-potential values (P>0.05). Evidently, serum tries to neutralize the TAT moieties by increasing the number of macromolecules adsorbed on liposomes; the more TAT is inserted, the thicker PC is formed.
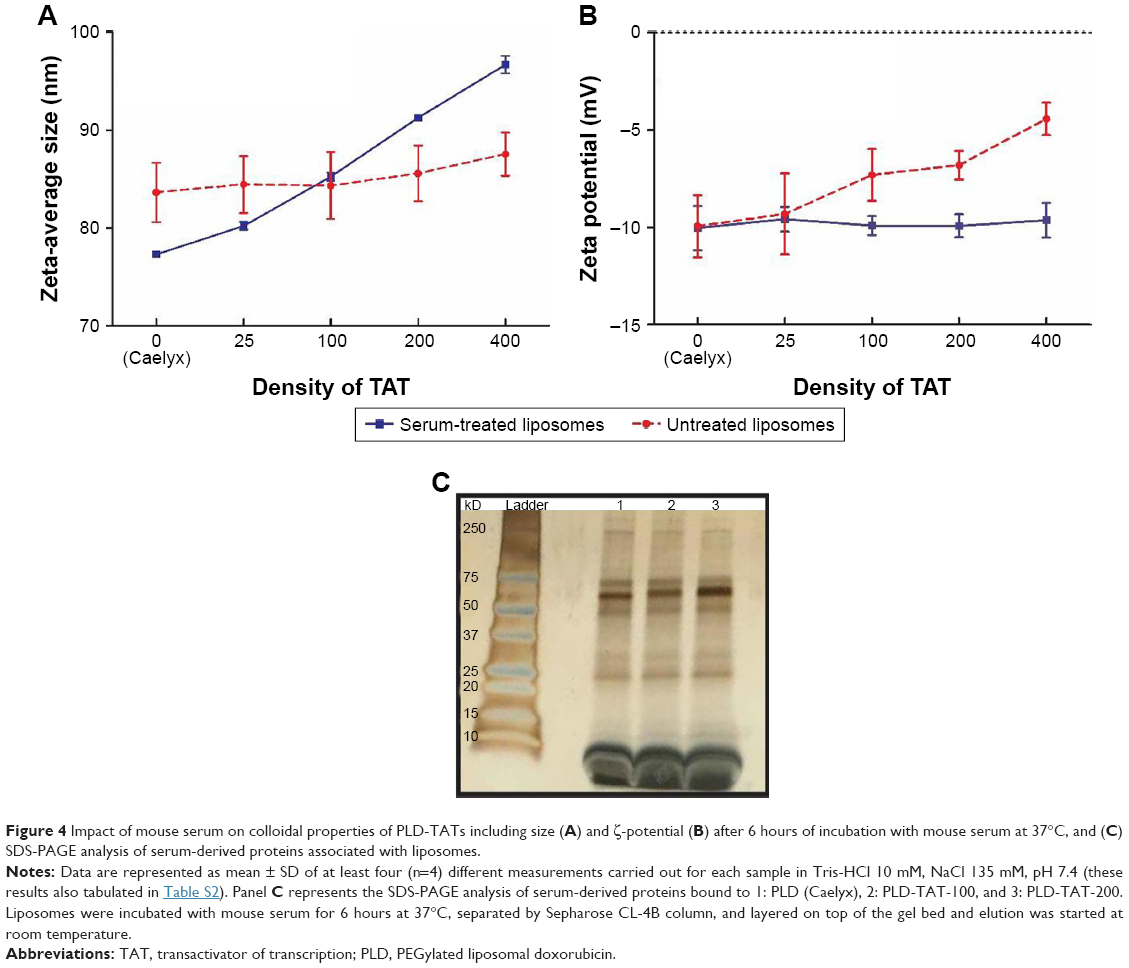 | Figure 4 Impact of mouse serum on colloidal properties of PLD-TATs including size (A) and ζ-potential (B) after 6 hours of incubation with mouse serum at 37°C, and (C) SDS-PAGE analysis of serum-derived proteins associated with liposomes. |
The increased thickness of PC as a function of TAT density also reflected in enhanced sedimentation at high centrifuge force of 14,000× g proportional to TAT density, ie, Caelyx < PLD-TAT-25 < PLD-TAT-100 < PLD-TAT-200. However, 5,000× g centrifugation caused no notable sedimentation. Although the magnitude of differences in in tubo condition was not as large as what was observed in vivo, these data were consistent with the biodistribution results, demonstrating the impact of serum-derived components on sedimentation of PLD-TATs during serum separation.
Liposomes recovered from serum were also run on SDS-PAGE. As could be seen in Figure 4C, while all serum-treated PLDs indicated identical band patterns, the intensity of the bands, especially between 50 and 75 kD, was directly proportional to TAT density on PLDs. SDS-PAGE results were consistent with the results obtained by size distribution analysis where thickness of PC increased by increasing surface-anchored TAT, while no massive protein binding and aggregation had taken place upon exposure of liposomes with serum (Figure S7 and Table S2).
Judging from the main different SDS-PAGE band (between 50 and 75 kD) and the direct correlation of the band intensity with TAT density (Figure 4C), we concluded that the presence of TAT moieties on PLD surface is associated with TAT-dependent adsorption of proteins that represent band of molecular weight around 65 kD. With regard to the biodistribution properties of PLD-TATs, eg, the stealth-like behavior during circulation, it is most likely that surface association of these proteins on PLD-TATs shields TAT residues from RES function by inhibiting opsonins adsorption on particle surface.10,40
While our findings indicate that albumin (MW 67 kD)41 is involved in dysopsonization of PLD-TATs,10,41,42 thorough identification and comparison of PC associated with PLD-TAT vs PLD-Plain is needed. To this end, liquid chromatography mass spectrometry-based proteomics analysis is in process.
Visualization of tumor cell association of TAT-modified liposomes in vivo
In vivo behavior of fluorescently labeled TAT-modified liposomes (FPL-TAT-200) in tumor was observed by intravital confocal microscopy on dorsal skin-fold window chamber-bearing mice after iv injection at a dose of 5 μmol of lipid. As can be seen in Figure 5A–F, TAT-modified liposomes strongly associated with tumor cells after extravasation into tumor as early as 12 hours post-injection and at later time points either in regions with established tumor vessels (Figure 5A, C, and E) or the growing front (Figure 5B, D, and F). Meanwhile, plain liposomes (Figure 5G and H) showed minor association with cells and diffusely distributed inside tumor. Intravital imaging also shows the limited movement of FPL-TAT from the extravasation site. This is more clear in the tumor growing fronts (5A, D, and F) in which extravasated liposomes vastly interact with cells surrounding neovasculatures while no detectable liposome–cell association was observable in non-vascularized regions. Figure 5C and F also revealed the prolonged circulation time of FPL-TAT as it was observable inside tumor vasculature 24 and 48 hours post-injection, respectively. This brings the question of how the protein coated PLD-TATs were capable of interacting with tumor cells inside the tumor interstitium. Although serum treatment of FPLs reduced the extent of liposome–cell association in static condition (Figure S8), interaction of serum treated and non-treated FPL-TATs was blocked equally in the presence of heparin (Figure S9) and cells starved of serum did not uptake PC-associated liposomes more than what normal cells did in spite of their higher demand on serum nutrients (Figure S10). These results reveal that association of TAT liposomes is only mediated by the exposing TAT residues and PC only reduces the exposure rate of TAT peptide rather than blocking it.
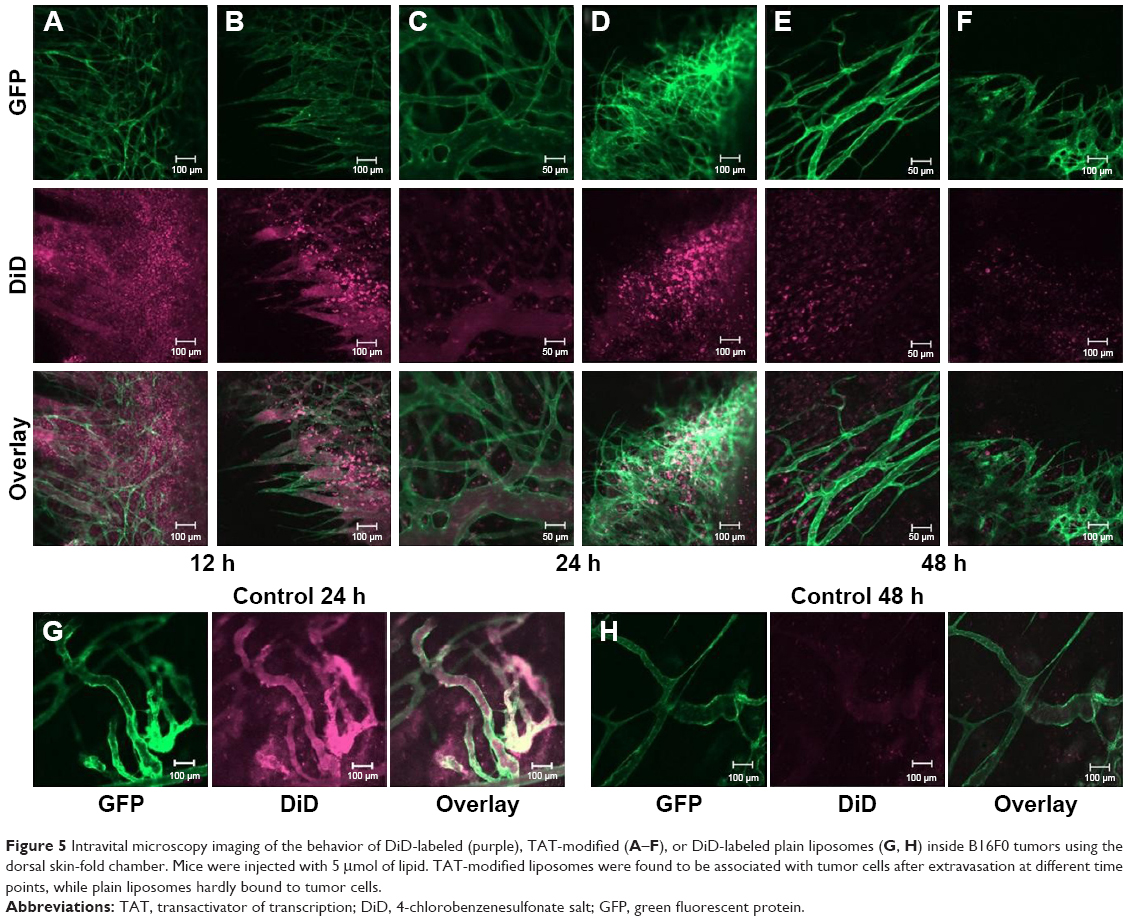 | Figure 5 Intravital microscopy imaging of the behavior of DiD-labeled (purple), TAT-modified (A–F), or DiD-labeled plain liposomes (G, H) inside B16F0 tumors using the dorsal skin-fold chamber. Mice were injected with 5 μmol of lipid. TAT-modified liposomes were found to be associated with tumor cells after extravasation at different time points, while plain liposomes hardly bound to tumor cells. |
Hadjidemetriou et al38,39 showed an incomplete formation of PC around PEGylated liposomes modified with IgG ligand which obviously justifies the interaction of PLD-TAT with tumor cells. However, given the fact that PC formation is a thermodynamic response of blood pool for diminution of the Gibbs free energy and compensation of the high free energy protruded by NP surface, the heterogeneous formation of PC seems not convincing. Whereas, it is more likely that regions observed intact were artifacts created during extensive washing steps or sample preparation for electron microscopy imaging. Considering the ability of PC-associated TAT liposomes in interacting with cells in static conditions of in vitro experiments (Figures S8 and S10) or in vivo tumor interstitium and that they were not recognized by RES in dynamic conditions of circulation, we concluded that differences in evolution of PC in static vs dynamic conditions determine the targeting ability of TAT-modified liposomes.
In fact, composition, thickness, and decoration of PC is dependent on a variety of factors including physicochemical properties of NP surface (surface chemistry, size, charge, shape, topography, and curvature) and environmental factors such as protein source and incubation protocol.43,44 Pozzi et al45 demonstrated that even in in vitro conditions, the composition of PC formed under static conditions is different from what is formed under dynamic conditions. In addition, the PC is initially composed of proteins with high concentration and high association rate constant that, depending on the surface characteristics, may then dissociate to be replaced by other proteins with lower concentration, lower exchange rate, and higher affinity.46,47 These exchange processes are important when particles redistribute from one compartment or organ to another. For instance, as speculated earlier, albumin could be one of the potential dysopsonizing candidates. At physiological pH, the difference between the negative charge of albumin (–19) and the positive charge of TAT (+8) enforces albumin–TAT binding. However, lowering the pH from 7.4 to 6 reduces the negative charge of albumin to −6,48 whereas basic TAT (pI TAT: 12.13) does not undergo significant charge changes in this pH range. Theoretically, it is likely that reduced pH of the tumor interstitium49 loosens the electrostatic interaction between albumin and TAT and allows albumin to be dissociated by proteases or through competitive interactions with tumor-associated stromal matrix such as secreted protein acidic rich in cysteine (SPARC), a prototypic matricellular protein expressed around many cancer cells50,51 which particularly has high affinity to albumin molecule,52–54 making TAT residues accessible and functional in the proximity of tumor cells.
Therapeutic efficacy
Antitumor efficacy of different PLDs was evaluated in an sc murine C26 colon carcinoma tumor model after a single dose of either 10 or 15 mg/kg liposomal DXR. Tumor growth rates and survival results are depicted in Figure 6. Median survival time (MST), time to reach end point (TTE), and % tumor growth delay (TGD) for each treatment group are also summarized in Table 1. When mice received 10 mg/kg liposomal DXR, no significant differences were observed amongst different treatments (Table 1). However, administration of PLD-TAT-100 indicated a superior antitumor trend compared to other treatments and cured one mouse by complete elimination of the tumor (Figure 6A) and resulted in a slower tumor growth rate (Figure 6B).
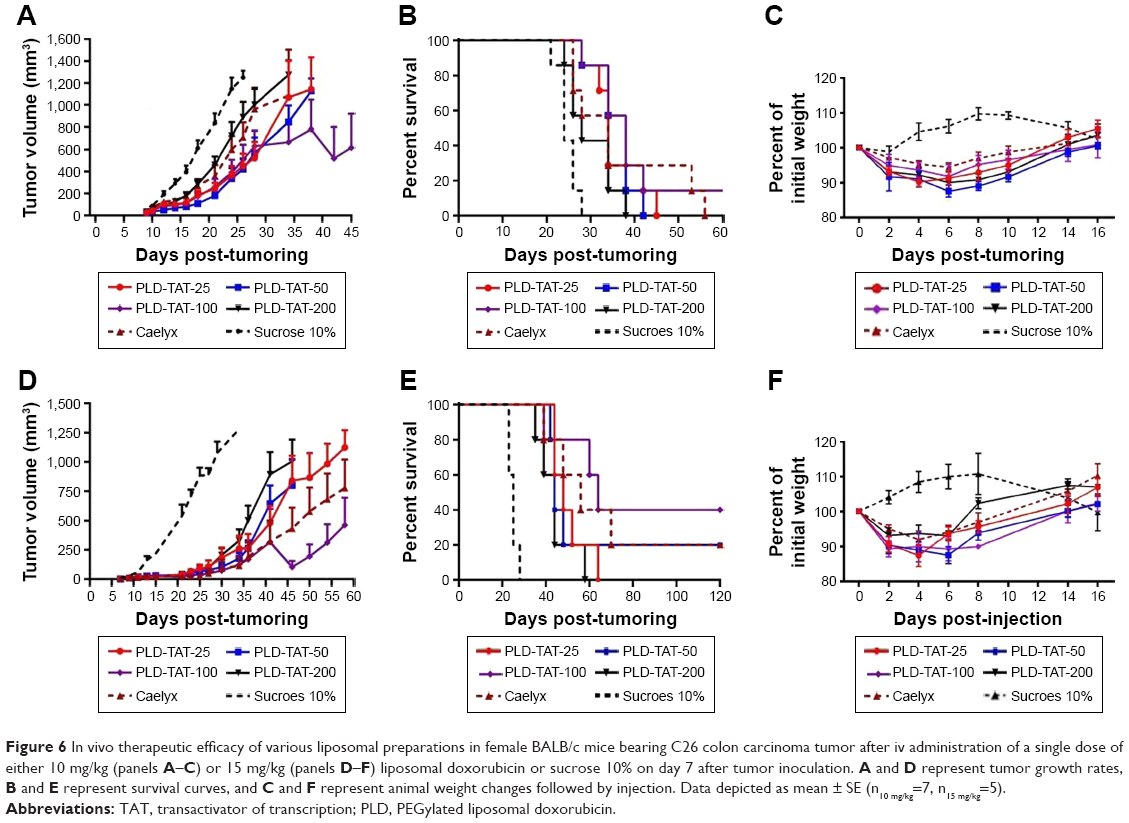 | Figure 6 In vivo therapeutic efficacy of various liposomal preparations in female BALB/c mice bearing C26 colon carcinoma tumor after iv administration of a single dose of either 10 mg/kg (panels A–C) or 15 mg/kg (panels D–F) liposomal doxorubicin or sucrose 10% on day 7 after tumor inoculation. A and D represent tumor growth rates, B and E represent survival curves, and C and F represent animal weight changes followed by injection. Data depicted as mean ± SE (n10 mg/kg=7, n15 mg/kg=5). |
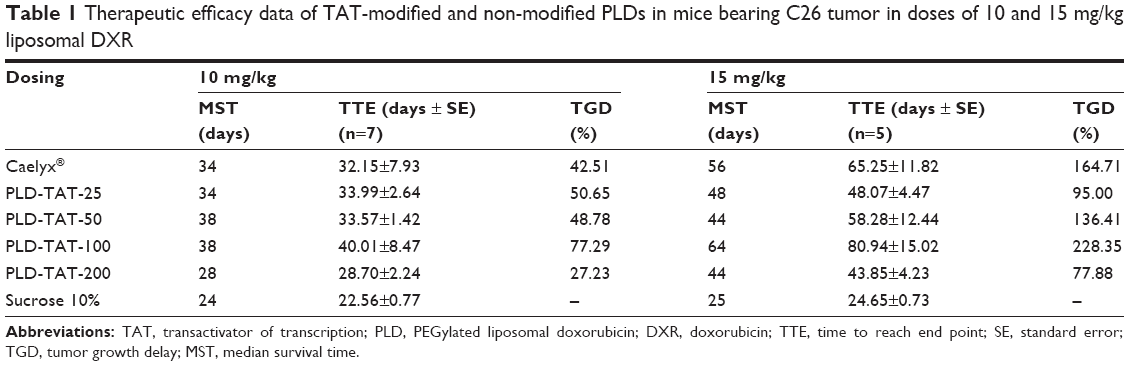 | Table 1 Therapeutic efficacy data of TAT-modified and non-modified PLDs in mice bearing C26 tumor in doses of 10 and 15 mg/kg liposomal DXR |
Treatment with 15 mg/kg liposomal DXR magnified the differences between the treated groups. While modification of Caelyx with 25 or 50 TAT lipopeptides did not enhance the therapeutic efficacy of Caelyx, PLD-TAT-100 showed a superior antitumor effect and caused the longest MST, TTE, and the highest %TGD. Forty percent of mice treated with this preparation were cured (Figure 6D), and this group exhibited a slower tumor growth rate (Figure 6E). In contrast, PLD-TAT-200 exhibited significantly (P<0.05) lower therapeutic values (Table 1) and significantly faster tumor growth rate compared to PLD-TAT-100 (Figure 6E).
In contrast to the in vitro toxicity of PLD-TATs, where increasing the TAT density directly resulted in increased toxicity against tumor cells, this trend was not observed in vivo. While all PLD-TATs indicated comparable tumor accumulation, PLD-TAT-100 was the most effective preparation and PLD-TAT-200 was repeatedly found to be the least effective preparation at both dosages.
As visualized in Figure 5, while FPL-TAT shows homogenous interaction with cells around established tumor vessels (Figure 5A, C and E) – representing tumor periphery where tumor vessels are distributed in high density – in growing fronts (Figure 5B, D, and F), interaction of extravasated FPL-TAT is greatly restricted to tumor cells located close to vessels. Limited associations could be seen in dark regions of central tumor mass where no vasculature is present.
In fact, extravasation of NPs from functional blood vessels in central region of tumor, if there is any, is negligible due to high interstitial fluid pressure,55 and tumor accumulation of NPs mainly relies on peripheral extravasation.56–58 Therefore, penetration of NPs from tumor periphery into deeper tumor regions is crucial to effectively eliminating cancer cells. The lack of convective movement inside tumors restricts NP transport mainly to diffusion. However, intratumoral diffusion of NPs is strongly hindered by the dense complex structure of the extracellular matrix (ECM) in solid tumors.56,57 Moreover, specific affinity of macromolecules or NPs for tumor cells may further reduce the effective diffusion rate of targeted liposomes inside tumor interstitium.55,59 This is consistent with intravital imaging analysis where FPL-TAT movement from extravasate site was limited to surrounding cells. From this, it could be concluded that the antitumor activity of PLD-TAT200 is affected by its poor spatial distribution throughout the tumor as a function of its high avidity to tumor cells. In addition to this, PLD-TATs showed strong cell internalization but delayed DXR delivery, as they are mainly retained in lysosomal system for a long period of time (Figure 2). Therefore, due to a higher avidity, PLD-TAT-200 delivered a large amount of DXR to cells immediately surrounding blood vessels in peripheral tumor regions, leaving deep regions of the tumor untouched by the therapy. Another evidence that supports the indirect relationship of avidity with the spatial distribution of PLD-TATs was the occurrence of skin necrosis when mice bearing dermal C26 tumor received different PLD-TATs (Supplementary material), where increasing the avidity of PLD-TATs by increasing TAT density was accompanied by an increase in the occurrence of skin necrosis (Figure S11 and Table S3). In dermal tumor, the dermis is tightly in contact with tumor periphery due to pressure derived from tumor growth. Therefore, the poor spatial distribution of peripherally extravasated liposomes, due to liposome avidity, might have resulted in localization of sufficient levels of DXR to cause local cell kill and damage in the periphery of the tumor and adjacent skin which was reflected as necrosis in skin. These reveal that limited penetration depth, either as a result of dense ECM or NP avidity, plays more crucial role compared to EPR.
Another striking observation of the therapeutic efficacy of PLD-TATs was their safety, as we observed no remarkable systemic side effects. As represented in Figure 6C and F, post-injection monitoring of animal weights, which is an indicator of incidence of systemic side effects, revealed no difference between treatment with PLD-TATs and Caelyx. The safety of PLD-TATs may further underscore the crucial role of PC composition that, in this study, covered TAT moieties from off-target interactions during circulation.
Taken together, our results indicate that insertion of 100 TAT peptides on PLD provides a superior balance between intracellular drug delivery and tumor penetration compared to other preparations, thereby inflicting a better antitumor effect.
Conclusion
Herein we show a comprehensive investigation of the biological performance of TAT-modified liposomes. TAT regulates cellular internalization of liposomes via endocytosis rather than direct translocation or fusion. PLDs modified with different densities of TAT peptide were found to be safe and long circulating. However, to gain a proper antitumor therapy, their avidity should be optimized to balance between cellular uptake and penetration depth into tumor. We observed an enhanced therapeutic efficacy of a commercial PLD, Caelyx, against C26 colon carcinoma when 100 TAT moieties were present. Since, PLD-TATs are easily prepared in house and impose no serious manufacturing complexity, further development is warranted.
The current study, which to the best of our knowledge is the first report in literature, provides a vivid example showing the effect of PC formation on in vivo and in vitro behavior of TAT-modified liposomes. TAT moieties on liposomes seem to adsorb dysopsonins rather than opsonins, which results in protection of liposomes from clearance and off-target interaction during dynamic condition of circulation. Importantly, targeting ability of TAT-modified liposomes in static condition of the tumor interstitium was maintained in spite of the presence of PC. We conclude that formation of a PC on TAT liposomes did not obscure the targeting moiety for interaction with tumor cells. We speculate that the difference in kinetic on and off rates (or equilibrium binding constants) of PC components in static condition vs dynamic condition may regulate this behavior of TAT liposomes.
In contrast to the general consensus that ligand modification does not necessarily improve tumor localization of liposomes as a result of enhanced clearance, some reports have shown an improved tumor accumulation due to active targeting. This important contradiction might be resolved by considering that PC formation balances between improved circulation time and better uptake by tumor cells.
Finally, our observations highlight the need to take PC formation into account during preclinical evaluation of NPs, particularly ligand-modified NPs. Optimization of ligand density and performance of modified NPs should be thoroughly tested in vitro and in vivo to establish the optimally balanced product.
Acknowledgments
The financial support of the Biotechnology Research Center and Nanotechnology Research Center, Mashhad University of Medical Sciences (MUMS), is gratefully acknowledged.
Disclosure
The authors report no conflicts of interest in this work.
References
Gregoriadis G. Liposomology: delivering the message. J Liposome Res. 2018;28(1):1–4. | ||
Venditto VJ, Szoka FC Jr. Cancer nanomedicines: so many papers and so few drugs! Adv Drug Deliv Rev. 2013;65(1):80–88. | ||
Biswas S, Deshpande PP, Perche F, Dodwadkar NS, Sane SD, Torchilin VP. Octa-arginine-modified pegylated liposomal doxorubicin: an effective treatment strategy for non-small cell lung cancer. Cancer Lett. 2013;335(1):191–200. | ||
Barenholz Y. Doxil® – the first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160(2):117–134. | ||
Bulbake U, Doppalapudi S, Kommineni N, Khan W. Liposomal formulations in clinical use: an updated review. Pharmaceutics. 2017;9(2):12. | ||
Maeda H. Macromolecular therapeutics in cancer treatment: the EPR effect and beyond. J Control Release. 2012;164(2):138–144. | ||
Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Deliv Rev. 2013;65(1):36–48. | ||
Gabizon A, Shmeeda H, Horowitz AT, Zalipsky S. Tumor cell targeting of liposome-entrapped drugs with phospholipid-anchored folic acid-PEG conjugates. Adv Drug Deliv Rev. 2004;56(8):1177–1192. | ||
van der Meel R, Vehmeijer LJC, Kok RJ, Storm G, van Gaal EVB. Ligand-targeted particulate nanomedicines undergoing clinical evaluation: current status. Adv Drug Deliv Rev. 2013;65(10):1284–1298. | ||
Caracciolo G. Liposome-protein corona in a physiological environment: challenges and opportunities for targeted delivery of nanomedicines. Nanomedicine. 2015;11(3):543–557. | ||
Anchordoquy TJ, Barenholz Y, Boraschi D, et al. Mechanisms and barriers in cancer nanomedicine: addressing challenges, looking for solutions. ACS Nano. 2017;11(1):12–18. | ||
Lammers T, Kiessling F, Ashford M, Hennink W, Crommelin D, Storm G. Cancer nanomedicine: Is targeting our target? Nat Rev Mater. 2016;1(9):16069. | ||
Amin M, Badiee A, Jaafari MR. Improvement of pharmacokinetic and antitumor activity of PEGylated liposomal doxorubicin by targeting with N-methylated cyclic RGD peptide in mice bearing C-26 colon carcinomas. Int J Pharm. 2013;458(2):324–333. | ||
Wang F, Wang Y, Zhang X, Zhang W, Guo S, Jin F. Recent progress of cell-penetrating peptides as new carriers for intracellular cargo delivery. J Control Release. 2014;174:126–136. | ||
Sarko D, Beijer B, Garcia Boy R, et al. The pharmacokinetics of cell-penetrating peptides. Mol Pharm. 2010;7(6):2224–2231. | ||
Bechara C, Sagan S. Cell-penetrating peptides: 20 years later, where do we stand? FEBS Lett. 2013;587(12):1693–1702. | ||
Brooks H, Lebleu B, Vivès E. Tat peptide-mediated cellular delivery: back to basics. Adv Drug Deliv Rev. 2005;57(4):559–577. | ||
Tseng YL, Liu JJ, Hong RL. Translocation of liposomes into cancer cells by cell-penetrating peptides penetratin and tat: a kinetic and efficacy study. Mol Pharmacol. 2002;62(4):864–872. | ||
Rizzuti M, Nizzardo M, Zanetta C, Ramirez A, Corti S. Therapeutic applications of the cell-penetrating HIV-1 Tat peptide. Drug Discov Today. 2015;20(1):76–85. | ||
Pae J, Säälik P, Liivamägi L, et al. Translocation of cell-penetrating peptides across the plasma membrane is controlled by cholesterol and microenvironment created by membranous proteins. J Control Release. 2014;192:103–113. | ||
Amand HL, Boström CL, Lincoln P, Nordén B, Esbjörner EK. Binding of cell-penetrating penetratin peptides to plasma membrane vesicles correlates directly with cellular uptake. Biochim Biophys Acta. 2011;1808(7):1860–1867. | ||
Koren E, Apte A, Jani A, Torchilin VP. Multifunctional PEGylated 2C5-immunoliposomes containing pH-sensitive bonds and TAT peptide for enhanced tumor cell internalization and cytotoxicity. J Control Release. 2012;160(2):264–273. | ||
Torchilin VP, Rammohan R, Weissig V, Levchenko TS. TAT peptide on the surface of liposomes affords their efficient intracellular delivery even at low temperature and in the presence of metabolic inhibitors. Proc Natl Acad Sci U S A. 2001;98(15):8786–8791. | ||
Chung SK, Maiti KK, Lee WS. Recent advances in cell-penetrating, non-peptide molecular carriers. Int J Pharm. 2008;354(1–2):16–22. | ||
Fretz MM, Koning GA, Mastrobattista E, Jiskoot W, Storm G. OVCAR-3 cells internalize TAT-peptide modified liposomes by endocytosis. Biochim Biophys Acta. 2004;1665(1–2):48–56. | ||
Gump JM, Dowdy SF. TAT transduction: the molecular mechanism and therapeutic prospects. Trends Mol Med. 2007;13(10):443–448. | ||
Gupta B, Levchenko TS, Torchilin VP. Intracellular delivery of large molecules and small particles by cell-penetrating proteins and peptides. Adv Drug Deliv Rev. 2005;57(4):637–651. | ||
Torchilin VP. Tat peptide-mediated intracellular delivery of pharmaceutical nanocarriers. Adv Drug Deliv Rev. 2008;60(4–5):548–558. | ||
Bartlett GR. Phosphorus assay in column chromatography. J Biol Chem. 1959;234(3):466–468. | ||
Seynhaeve AL, Hoving S, Schipper D, et al. Tumor necrosis factor alpha mediates homogeneous distribution of liposomes in murine melanoma that contributes to a better tumor response. Cancer Res. 2007;67(19):9455–9462. | ||
Li L, Ten Hagen TL, Hossann M, et al. Mild hyperthermia triggered doxorubicin release from optimized stealth thermosensitive liposomes improves intratumoral drug delivery and efficacy. J Control Release. 2013;168(2):142–150. | ||
Amin M, Mansourian M, Koning GA, Badiee A, Jaafari MR, Ten Hagen TLM. Development of a novel cyclic RGD peptide for multiple targeting approaches of liposomes to tumor region. J Control Release. 2015;220(Pt A):308–315. | ||
Huang Z, Szoka FC. Sterol-modified phospholipids: cholesterol and phospholipid chimeras with improved biomembrane properties. J Am Chem Soc. 2008;130(46):15702–15712. | ||
Huang Z, Jaafari MR, Szoka FC. Disterolphospholipids: nonexchangeable lipids and their application to liposomal drug delivery. Angew Chem Int Ed Engl. 2009;48(23):4146–4149. | ||
Johnstone SA, Masin D, Mayer L, Bally MB. Surface-associated serum proteins inhibit the uptake of phosphatidylserine and poly(ethylene glycol) liposomes by mouse macrophages. Biochim Biophys Acta. 2001;1513(1):25–37. | ||
Lasic DD, Frederik PM, Stuart MC, Barenholz Y, Mcintosh TJ. Gelation of liposome interior. A novel method for drug encapsulation. FEBS Lett. 1992;312(2–3):255–258. | ||
Wolfram J, Suri K, Yang Y, et al. Shrinkage of pegylated and non-pegylated liposomes in serum. Colloids Surf B Biointerfaces. 2014;114:294–300. | ||
Hadjidemetriou M, Al-Ahmady Z, Kostarelos K. Time-evolution of in vivo protein corona onto blood-circulating PEGylated liposomal doxorubicin (DOXIL) nanoparticles. Nanoscale. 2016;8(13):6948–6957. | ||
Hadjidemetriou M, Al-Ahmady Z, Mazza M, Collins RF, Dawson K, Kostarelos K. In vivo biomolecule corona around blood-circulating, clinically used and antibody-targeted lipid bilayer nanoscale vesicles. ACS Nano. 2015;9(8):8142–8156. | ||
Mahon E, Salvati A, Baldelli Bombelli F, Lynch I, Dawson KA. Designing the nanoparticle-biomolecule interface for “targeting and therapeutic delivery”. J Control Release. 2012;161(2):164–174. | ||
Maclaren IA, Petras ML. Isolation and properties of serum albumin from the house mouse Mus musculus. Biochim Biophys Acta. 1976;427(1):238–250. | ||
Piao L, Li H, Teng L, et al. Human serum albumin-coated lipid nanoparticles for delivery of siRNA to breast cancer. Nanomedicine. 2013;9(1):122–129. | ||
Bigdeli A, Palchetti S, Pozzi D, et al. Exploring cellular interactions of liposomes using protein corona fingerprints and physicochemical properties. ACS Nano. 2016;10(3):3723–3737. | ||
Saha K, Rahimi M, Yazdani M, et al. Regulation of macrophage recognition through the interplay of nanoparticle surface functionality and protein corona. ACS Nano. 2016;10(4):4421–4430. | ||
Pozzi D, Caracciolo G, Digiacomo L, et al. The biomolecular corona of nanoparticles in circulating biological media. Nanoscale. 2015;7(33):13958–13966. | ||
Cedervall T, Lynch I, Foy M, et al. Detailed identification of plasma proteins adsorbed on copolymer nanoparticles. Angew Chem Int Ed Engl. 2007;46(30):5754–5756. | ||
Lynch I, Salvati A, Dawson KA. Protein-nanoparticle interactions: What does the cell see? Nat Nanotechnol. 2009;4(9):546–547. | ||
Fogh-Andersen N, Bjerrum PJ, Siggaard-Andersen O. Ionic binding, net charge, and Donnan effect of human serum albumin as a function of pH. Clin Chem. 1993;39(1):48–52. | ||
Danhier F, Feron O, Préat V. To exploit the tumor microenvironment: passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J Control Release. 2010;148(2):135–146. | ||
Podhajcer OL, Benedetti LG, Girotti MR, Prada F, Salvatierra E, Llera AS. The role of the matricellular protein SPARC in the dynamic interaction between the tumor and the host. Cancer Metastasis Rev. 2008;27(4):691–705. | ||
Tai IT, Tang MJ. SPARC in cancer biology: its role in cancer progression and potential for therapy. Drug Resist Updat. 2008;11(6):231–246. | ||
Yardley DA. nab-Paclitaxel mechanisms of action and delivery. J Control Release. 2013;170(3):365–372. | ||
Desai N, Trieu V, Damascelli B, Soon-Shiong P. SPARC expression correlates with tumor response to albumin-bound paclitaxel in head and neck cancer patients. Transl Oncol. 2009;2(2):59–64. | ||
Clark CJ, Sage EH. A prototypic matricellular protein in the tumor microenvironment – where there’s SPARC, there’s fire. J Cell Biochem. 2008;104(3):721–732. | ||
Jain RK. Physiological barriers to delivery of monoclonal antibodies and other macromolecules in tumors. Cancer Res. 1990;50(3 Suppl):814s–819s. | ||
Tang L, Gabrielson NP, Uckun FM, Fan TM, Cheng J. Size-dependent tumor penetration and in vivo efficacy of monodisperse drug-silica nanoconjugates. Mol Pharm. 2013;10(3):883–892. | ||
Waite CL, Roth CM. Nanoscale drug delivery systems for enhanced drug penetration into solid tumors: current progress and opportunities. Crit Rev Biomed Eng. 2012;40(1):21–41. | ||
Hu CM, Zhang L. Therapeutic nanoparticles to combat cancer drug resistance. Curr Drug Metab. 2009;10(8):836–841. | ||
Adams GP, Schier R, Mccall AM, et al. High affinity restricts the localization and tumor penetration of single-chain fv antibody molecules. Cancer Res. 2001;61(12):4750–4755. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.