")
Back to Journals » Cancer Management and Research » Volume 12
Regulation of Autophagy by Non-Steroidal Anti-Inflammatory Drugs in Cancer
Received 10 March 2020
Accepted for publication 12 May 2020
Published 16 June 2020 Volume 2020:12 Pages 4595—4604
DOI https://doi.org/10.2147/CMAR.S253345
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Kenan Onel
Xiangjie Fu,1 Tan Tan,2 Peijun Liu3
1Cholestatic Liver Diseases Center and Department of Gastroenterology, Southwest Hospital, Third Military Medical University, Chongqing, People’s Republic of China; 2Translational Medicine Institute, The First Affiliated Hospital of Chenzhou, University of South China, Hunan, People’s Republic of China; 3Center for Translational Medicine, The First Affiliated Hospital of Xi’an Jiaotong University, Shanxi, People’s Republic of China
Correspondence: Tan Tan Tel +86-18975523383
Fax +86-735-2343902
Email [email protected]
Peijun Liu Tel +86-18991232306
Fax +86-29-85324628
Email [email protected]
Abstract: Cancer is the leading cause of death, placing a substantial global health burden. The development of the most effective treatment regimen is the unmet clinical need for cancer. Inflammation plays a role in tumorigenesis and progression, and anti-inflammation may be a promising option for cancer management and prevention. Emerging studies have shown that non-steroidal anti-inflammatory drugs (NSAIDs) display anticarcinogenic and chemopreventive properties through the regulation of autophagy in certain types of cancer. In this review, we summarize the pharmacological functions and side effects of NSAIDs as chemotherapeutic agents, and focus on its mode of action on autophagy regulation, which increases our knowledge of NSAIDs and cancer-related inflammation, and contributes to a putative addition of NSAIDs in the chemoprevention and treatment of cancer.
Keywords: NSAIDs, non-steroidal anti-inflammatory drugs, pharmacological function, side effects, anticancer activity, autophagy
Introduction
Approximately 1.81 million new cancer diagnoses and 606,520 cancer-related deaths are projected to occur in the United States in 2020.1 Cancer research has focused on the identification of less toxic and more efficient therapeutic approaches to improve the overall prognosis of patients with cancer. Studies have shown that multiple modes of cell death and cell inhibition, other than apoptosis, contributing to chemotherapeutic efficacy.2 Autophagy is an evolutionarily conserved proteolytic process that involves lysosomal degradation and recycling of damaged cellular components and energy to maintain homeostasis.3,4 Autophagy is a survival mechanism of self-rescuing that favors the degradation of harmful and unwanted substances under stress conditions.5,6 Of note, autophagy is crucial for cell metabolism mechanism eliminating damaged organelles, proteins, lips and like that to maintain homeostasis and energy savings.7 By contrast, autophagic dysfunction contributes to various disorders including cancers, immune diseases, metabolic diseases, and neurological diseases.8–10 This review focuses on the recent progress of cancer in the regulation of autophagy.
Non-steroidal anti-inflammatory drugs (NSAIDs) exert antipyretic, analgesic, and anti-inflammatory effects.11 Although the chemical structures of these drugs differ, the therapeutic effects of NSAIDs are fundamentally similar due to decreased production of prostaglandins (PGs) through the inhibition of cyclooxygenases (COXs).12 Many experimental, epidemiological, and clinical studies have suggested that NSAIDs, particularly selective COX-2 inhibitors, may reduce cancer risk.13–15 It has been well established a mode of action that NSAIDs exert anticancer effects through induction of apoptosis, DNA damage repair, immune surveillance, and inhibition of tumor proliferation and invasion via COX-dependent or/and -independent pathways.16–20 NSAIDs regulate autophagy via Beclin1-dependent and Beclin1-independent pathways, which may result in anti-cancer effects.12,21 However, NSAIDs for cancer chemoprevention is on debate because the safety, efficacy, and doses of NSAIDs limit the clinical use in terms of cancer patients. Furthermore, the exact effects of NSAIDs on the regulation of autophagy are divergent according to the works of literature in different types and stages of cancer. Herein, we reviewed the pharmacological functions, side effects, and effects and mechanisms of NSAIDs including celecoxib, aspirin, sulindac sulfide, and meloxicam on autophagy regulation in cancer. This review may provide clues for the potential use and clinical trials of NSAIDs in the setting of clinical cancer treatment.
Pharmacological Activity of NSAIDs
Two forms of COX are present in tissues: constitutive COX-1 and inducible COX-2. The biological effects of COXs vary with tissue specificity, synthases, and the expression and regulation patterns. Both forms of COX are rate-limiting enzymes for the conversion of arachidonic acids and unsaturated fatty acids to PGs. COX enzymes convert arachidonic acid to PGH2, and generation of PGD2, PGE2, PGF2α, PGI2 or thromboxanes (TXA2).22,23 PGs produced by COX-1 regulate physiological processes such as hemostasis, stomach and kidney blood flow, and gastric acid secretion. COX-1 acts as a housekeeping gene and constitutively expressed in normal cells and tissues of platelet, blood vessel, mesothelial cells, stomach, and kidney. However, the COX-2 gene is not expressed in normal tissues (although expressed in the kidney). COX-2 expression is induced in inflammatory tissues by cytokines, lipopolysaccharides, and TNF-α. In tumors, COX-2 expression increased by up to 80%.24–26 PGs produced by COX-2 have important roles in inflammation, cell proliferation, angiogenesis, invasiveness, extracellular matrix adhesion, immune escape, and apoptosis inhibition in cancer.27,28 NSAIDs can be divided into two categories: non-selective COX inhibitors which inhibit COX-1/2, and selective COX-2 inhibitors. Non-selective COX inhibitors include aspirin, sulindac sulfide, acetaminophen, indomethacin, diclofenac acid, ibuprofen, naproxen, flurbiprofen, loxoprofen, hydroxybutanone, and piroxicam. Selective COX-2 inhibitors include celecoxib, meloxicam, rofecoxib, nimesulide, etc.29,30
Side Effects of NSAIDs
NSAIDs are also associated with a range of side effects that vary according to their COX inhibition selectivity.31–33 The side effects can be attributed to the systemic synthesis of PGs and local production of carboxylic acid. The major side effects in different systems are reviewed as follows. (1) In the gastrointestinal system, it was reported that serious gastrointestinal toxicity including upper gastrointestinal ulcers, gross bleeding, or perforation appeared to occur in patients for both long-term and short-term, with or without warning symptoms in the duration of NSAIDs administration. Minor upper gastrointestinal adverse effects such as dyspepsia, are common and occur at any time during NSAID therapy. Mechanically, the use of NSAID results in a reduction in the level of PGs that protects the membrane of the stomach from its acidic environment, making it susceptible to lesions. (2) Glomerular filtration rate and renal hemodynamics depend on COX-1, whereas salt and water excretion is mainly under the control of COX-2. NSAIDs treatment-related side effects of kidney damage through promotion of sodium and water retention, hypoxia, and ischemia via inhibition of PG production. Renal PGs have predominantly vasodilator effects on the kidneys. NSAIDs, especially indomethacin, have potential uses in various types of glomerulonephritis and nephrotic syndrome. The increased risk of analgesic nephropathy of chronic nephritis and renal papillary necrosis may be observed in the long term use of NSAIDs. (3) Hypertension, thrombosis, and increased risk of stroke, myocardial infarction, and heart failure are the main side effect of the cardiovascular system in the long term use of NSAIDs. COX-2 selective inhibitors have been shown to disrupt homeostasis between TXA2, which is produced by COX-1 and promotes platelet release and aggregation, and PGI2, which is produced by COX-2 and inhibits platelet release and aggregation from the endothelium. A large number of clinical trials and studies have aimed to determine safe doses of NSAIDs and evaluate their clinical potential.34 To minimize the potential risk for an adverse event, the lowest effective dose should be found in clinical practice and high-risk patients should be considered to receive alternate therapies that do not involve NSAIDs.
Decreased Cancer Risk and NSAIDs
Numerous experimental, epidemiological, and clinical studies have shown that NSAIDs decrease the risk of certain types of cancer.
Aspirin
Primary guidelines of NSAIDs for prevention of cardiovascular diseases in patients with colorectal cancer were established in the United States and worldwide, based on the high-level evidence of aspirin acting a beneficial role in cancer prevention.35 The results of six randomized trials and 26 case-controlled studies showed that aspirin was associated with reduced risk of colorectal cancer over a 20-year period.36 Recent epidemiological studies have shown that low-dose aspirin is more effective than high-dose aspirin at reducing morbidity and mortality associated with colorectal cancer.37 In 2011, a clinical trial by Rothwell et al (25,570 patients) showed significant correlations between long-term aspirin use and a lower risk of death resulting from gastrointestinal and solid tumors.38 Furthermore, other studies have demonstrated the beneficial effects of 5 or more years of regular aspirin use against colorectal, esophageal, pancreatic, stomach, cholangiocarcinoma, prostate, breast, pancreatic, and ovarian cancers.39–41
Non-Aspirin NSAIDs
Several non-aspirin NSAIDs, including celecoxib, sulindac sulfide, piroxicam, loxoprofen, and ibuprofen, have been shown to have anticancer effects. A randomized trial of patients with familial adenomatous polyposis (FAP) showed that sulindac sulfide and celecoxib inhibited the growth of adenomatous polyps and eliminated polyps in some cases.42 Celecoxib was approved by the US Food and Drug Administration in 1995 for prevention of colorectal adenomas and adenocarcinomas in patients with FAP.43 Besides, celecoxib can be used to prevent and treat breast, lung, prostate, stomach, head, and neck cancers.44 Similarly, loxoprofen was shown to inhibit the growth of implanted Lewis lung carcinoma in vivo.11 Furthermore, a study showed that ibuprofen treatment resulted in 40–82% inhibition of tumor growth and reduced liver metastases of colorectal cancer in vitro and in vivo. 45 Thereafter, the relationship between chronic inflammation and cancer has long been discovered in the retrospective studies of the use of NSAIDs in cancer treatment and prevention. The mechanisms of action by which NSAIDs exhibits its unique anticancer activity include inducing apoptosis and inhibiting proliferation and invasion of tumors by COX-dependent and COX-independent inhibition pathway are shown in Figure 1.
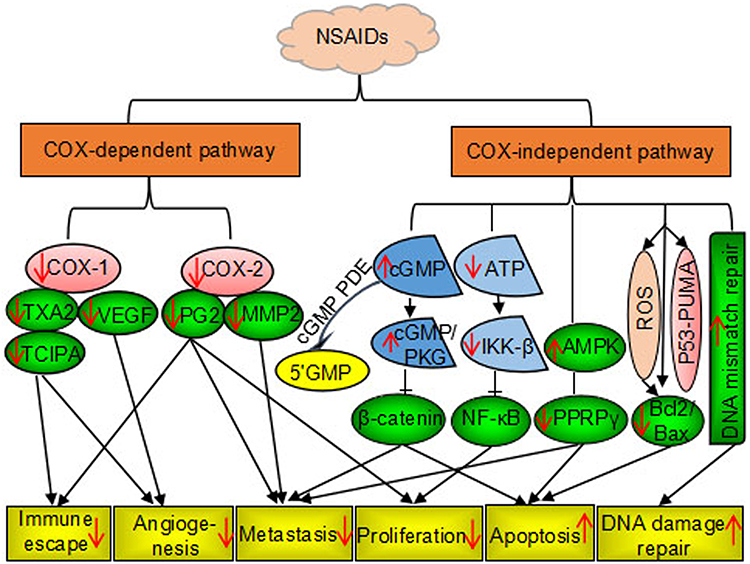 |
Figure 1 Nonsteroidal anti-inflammatory drug-induced anticancer effects via COX-dependent and COX-independent pathways. Notes: Up arrow: promotion; down arrow: inhibition. |
NSAID-Mediated Regulation of Autophagy
Many studies have shown that regulation of autophagy is an important mechanism associated with antitumor treatments.46,47 Therefore, we reviewed NSAID-mediated regulation of autophagy in cancer. Autophagy comprises the following four stages.48
- Autophagy initiation. Extracellular or intracellular stress factors (hunger, insulin, growth factors, hypoxia, and endoplasmic reticulum stress) activate the PKA and PI3K/AKT signaling pathways; this results in inactivation of the AMPK signaling pathway and inhibition of mTOR, which can cause the release of ULK complexes (ULK1, ULK2, FIP200, ATG101, and ATG13) and activation of autophagy.
- Vesicular nucleation. Phosphorylation of ULK complexes results in the activation of Vps34/Beclin-1 complexes. Class III PI3K forms a complex with Beclin-1 and other proteins (AMBRA1, ATG14, UVRAG) to form the phagophore. Binding of AMBRA1 to Beclin-1 stabilizes the class III PI3K complex, and binding of ATG14 and UVRAG to Beclin-1 enhances binding of Beclin-1 to VPS34, resulting in the formation of a double phagophore.
- Vesicle elongation and completion. The phagophore complex drives nucleation and elongation to form annular vesicles; during this process, LC3-I is continuously converted to LC3-II under two types of ubiquitin-like conjugation system (Atg8/LC3-PE and Atg12-Atg5).
- Autolysosome formation and degradation. In the presence of STX17, autophagosomes promote transport of degradable substances to lysosomes, resulting in the formation of autolysosomes. Autolysosomes play an important part in the degradation and circulation of damaging substances and energy. In vitro and in vivo studies have suggested that NSAIDs modulate autophagy in cancer via the classical Beclin-1-dependent pathway and a non-classical independent pathway. Specific mechanisms are shown in Figure 2.
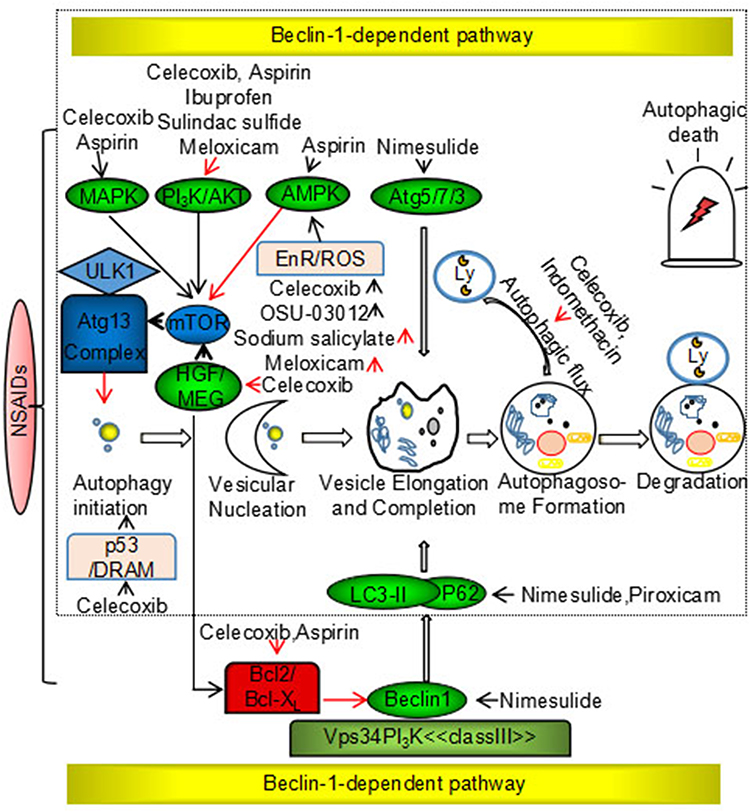 |
Figure 2 Nonsteroidal anti-inflammatory drugs induce cytoprotection and suppress autophagy via Beclin-1-dependent and Beclin-1-independent pathways. Notes: Black arrow: promotion; red arrow: suppression. |
Classical Beclin-1-Dependent Signaling Pathway
BECN1, a homolog of yeast ATG6, is a candidate tumor suppressor gene and encodes the Beclin-1 protein, which is a well-established regulator of the autophagic pathway. Deletion mutations of the Beclin-1 gene occur in ovarian (75%), breast (50%), and prostate (40%) cancers.49 Beclin-1 is a component of the class III PI3K complex that promotes autophagy through regulation of autophagosome nucleation. Beclin-1 is regulated by the Bcl-2 anti-apoptotic family of proteins, and Beclin-1 binds to the BH3 domain of Bcl-2/Bcl-xL.50 Signaling of NSAIDs through the classical Beclin-1 dependent pathway interferes with the expression of Beclin-1 (in the case of nimesulide) and the Bcl-2 family of proteins (celecoxib and aspirin), dissociation of Beclin-1 from the Vps34/class III PI3K complex, conversion of LC3-I to LC3-II, and induction of autophagy.51,52
Non-Classical Beclin-1-Independent Signaling Pathway
The non-classical Beclin-1 independent pathway is composed primarily of the mTOR signaling pathway, which has a pivotal role in the induction of autophagy.53 NSAID targets upstream of the mTOR signaling pathway include PI3K/AKT, AMPK, and MAP kinases. The PI3K/AKT pathway negatively regulates mTOR signaling in the presence of various growth factors. NSAIDs (celecoxib, aspirin, ibuprofen, sulindac sulfide, and meloxicam) induce autophagy through the PI3K/AKT/mTOR signaling pathway, resulting in anticancer effects.54–56 AMPK is a key cellular metabolic sensor of AMP/ATP homeostasis and negatively regulates autophagy via the mTOR signaling pathway.57 Nutrient deficiency, low levels of endoplasmic reticulum stress, and reactive oxygen species (ROS) induce protective autophagy to promote cell survival and stability. Many studies have shown that NSAIDs (sodium salicylate and meloxicam) inhibit ER stress and ROS production, and disrupt the balance between cell death and cell survival via autophagy regulation in cancer. By contrast, some NSAIDs (celecoxib and OSU-03012) have been shown to induce ER stress and ROS production, resulting in autophagic tumor cell death.58 MAP kinases inhibit autophagy via the mTOR signaling pathway. Studies have shown that celecoxib activates JNK-mediated autophagic death, and aspirin promotes conversion from protective autophagy to autophagic death through p38.59,60 In addition, NSAIDs directly target the LC3-II, p62, p53, Atg5, Atg12, and HGF genes. p53, the most commonly mutated tumor suppressor gene in human cancers, positively regulates autophagy in DNA-damaged cells.61 Celecoxib directly upregulates p53-dependent DRAM to induce autophagy.62 Furthermore, NSAIDs have been shown to inhibit Bcl-2 and mTOR kinase through inhibition of the HGF/MET autocrine loop, which results in the induction of autophagy and decreased drug resistance.63 Nimesulide has been shown to directly upregulate LC3-II, Beclin-1, p62, Atg5-12, and p53, and induces autophagy to promote anticancer effects.64 Autophagy is a dynamic process in which autophagosomes are continuously formed and degraded, and studies have shown that NSAIDs (celecoxib and indomethacin) regulate autophagy by modulating autophagic flux in cancer.65,66
Regulation of Autophagy by NSAIDs in Cancer Therapy
Many studies have shown that induction of autophagy inhibits the development of tumors from premalignant lesions, and the initiation and inhibition of autophagy may be promising strategies to treat advanced cancers. In cancer cells, autophagy is a dynamic process that is regulated by the intracellular microenvironment in different tumor stage and tumor tape.67 Interestingly, NSAIDs induce anticancer effects and increase chemotherapy/radiotherapy sensitivity by inducing tumor-suppressive autophagy, and also regulate cytoprotective autophagy in combination with chemotherapeutic drugs to enable more efficacious treatment of apoptosis-resistant or drug-resistant tumors.
Celecoxib and Its Derivatives
Celecoxib selectively inhibits COX-2. In vivo and in vitro studies have shown that celecoxib has significant potential as an anticancer drug. Many studies have focused on the role of autophagy in celecoxib-induced apoptosis. These studies have evaluated whether celecoxib induces protective or deleterious effects based on cancer type, which has provided insights into its use as an anticancer agent.4,44,68
Celecoxib further enhances apoptosis induced by Bcl-2/Bcl-XL antagonists (ABT-737 and sabutoclax) via inducing autophagy in colon cancer and oral squamous cells.69,70 Celecoxib induces apoptosis and autophagy through the inhibition of the PI3K/Akt/mTOR signaling pathway, resulting in the inhibition of proliferation of tumor cells, including multidrug-resistant hepatoma cells.71,72 In addition, combination treatment with celecoxib and CPT-11 inhibited tumor growth in neuroblastoma through induction of autophagy.73 Moon reported that celecoxib induced autophagic death of p53 wild-type glioma cells through direct upregulation of p53, inhibition of DNA synthesis, and arrest of the cell cycle at the G1 phase.74 The ER stress response system has two distinct roles in NSAID-mediated autophagy. Under moderate stress, NSAID-induced autophagy is self-protective, such as in radio-resistant or drug-resistant tumor cells. Under strong cellular ER stress, NSAID-induced autophagy results in induction of apoptosis.75–78 Dimethyl celecoxib, a derivative of celecoxib, effectively inhibits colon cancer, triple-negative breast cancer, and glioma cell growth through induction of ER stress and in combination with chloroquine, bortezomib, and radiotherapy to play synergic effects on the induction of tumor cell autophagy.79,80 Celecoxib targets the pro-oxidative state of highly metastatic cancer cells and cancer stem cells and drives excess ROS production in these cells, resulting in cell death.81 OSU-03012 inhibits the proliferation of liver cancer through ROS-induced tumor-suppressive autophagy.59 Recently, studies have shown that celecoxib, 2,5-dimethyl-celecoxib, and OSU-03012 increase the sensitivity of multidrug-resistant cancer cells, including CD-44-overexpressing cancer cells, to Hsp90 inhibitors through activation of autophagy.82
By contrast, celecoxib in combination with chemotherapeutic drugs may be a more effective approach for cancer treatment through inhibition of autophagy. For example, inhibition of autophagy (3-MA or bafilomycin A1) increased celecoxib-induced apoptosis in human urothelial carcinoma cells. In addition, induction of autophagy (by rapamycin or GFP-LC3 transfection) alleviated celecoxib-induced cytotoxicity.83 Zhou et al reported that inhibition of autophagy enhanced celecoxib-induced apoptosis in osteosarcoma.85 Moreover, celecoxib inhibited autophagy through modulation of lysosomal function, which resulted in anticancer effects in HL-60 cells and increased the sensitivity of imatinib-resistant chronic granulocytes.66
Aspirin
Aspirin has been widely used as a clinical treatment. Regular use of aspirin can reduce the risk of cancer incidence, recurrence, metastasis, and mortality.17,85,86 Recent prospective studies showed that aspirin reduced the incidence of cancer through suppressive or cytoprotective autophagy and acted synergistically with chemotherapeutic agents. A number of studies have shown that aspirin downregulates Bcl-2, resulting in the autophagic death of human hepatoma, colon, and breast cancer cells. Huang et al reported that aspirin induced Beclin-1-dependent autophagy in human hepatocellular carcinoma cells.52 In addition, aspirin suppressed growth through induction of apoptosis and autophagy in PIK3CA-mutant colorectal cancer cells, a mouse liver cancer and sarcoma model, colorectal cancer, pancreatic cancer cells, and PI3K-mutant breast cancer.55,71,87 A recent study reported that 5-FU and anti-EGFR antibodies in combination with aspirin increased autophagy in a three-dimensional sphere culture system comprising HCT116 and HT29 colorectal cancer cells through increased autophagy.88 Autophagy has dual roles in cancer. Many studies have shown that autophagy can induce the development of tumors from premalignant lesions. By contrast, promotion or inhibition of autophagy may be an appropriate cancer treatment strategy during late-stage disease.89–91 For example, the long-term use of aspirin in combination with ABT-737 can induce lethal autophagy in lung cancer through the p38/MAPK signaling pathway. Furthermore, aspirin has been shown to induce protective autophagy in the early stages of non-small-cell lung cancer and autophagic death in the late stages of this disease.60 However, another study showed that aspirin inhibited autophagy and enhanced metformin-induced apoptosis of TPC-1 thyroid cancer cells.92
Sulindac Sulfide
Sulindac sulfide is clinically used for the treatment of rheumatoid arthritis, osteoarthritis, and ankylosing spondylitis. It is also used to treat gout and certain types of bursitis and tendonitis. A previous study showed that sulindac sulfide suppressed the growth of colorectal carcinoma HCA-7 and HCT-116 cells and xenografts.93 Regulation of autophagy by sulindac sulfide may be a promising novel cancer therapeutic strategy. Sulindac sulfide has been shown to inhibit the growth of lung adenocarcinoma cells through the induction of autophagy via the Akt/mTOR signaling pathway.94 Furthermore, sulindac sulfide increased the sensitivity of multidrug-resistant cancer cells to Hsp90 inhibitors through the induction of autophagy.75 Sodium salicylate inhibited the growth of A549 cells through the transformation from tumor necrosis to tumor-suppressive autophagy.95 Moreover, Bauvy et al reported that inhibition of autophagy increased sulindac sulfide-induced apoptosis in HT-29 cells.96,97
Meloxicam
Meloxicam has been shown to have anticancer effects against various malignancies.98 A previous study in hepatoma cells showed that meloxicam prevented apoptosis through induction of cytoprotective autophagy, and the autophagy inhibitor 3-MA promoted meloxicam-induced toxicity.99 Similarly, Zhong et al reported that meloxicam induced apoptosis and cytoprotective autophagy under conditions of low ER stress, and inhibition of autophagy increased the toxicity of meloxicam against hepatoma cells.100
Other NSAIDs
Other NSAIDs have been shown to regulate autophagy in cancer cells. For example, ibuprofen increased the sensitivity of multidrug-resistant cancer cells to Hsp90 inhibitors through the induction of autophagy.95 Nimesulide induced apoptosis and autophagy, resulting in aggravation of MPTP-induced neuroblastoma cell death.65 Piroxicam increased the cytotoxicity of carboplatin in human bladder cancer cells through the induction of autophagy.101 Consistent with these findings, indomethacin blocked autophagic flux through inhibition of lysosomal function, resulting in increased sensitivity of gastric cancer cells to indomethacin.67 These results demonstrate that the regulation of autophagy by NSAIDs plays an important part in anticancer therapy. Thus, the use of NSAIDs in combination with chemotherapeutic drugs may be a promising strategy for cancer treatment.
Conclusion and Future Perspectives
Anti-inflammation is believed to play a role in cancer management and chemoprevention. In the past few decades, research has shown that NSAIDs decrease the risk of certain types of cancer. The key mechanism of the protective action of NSAIDs is the inhibition of COX, which catalyzes the synthesis of PGs in inflammatory processes. In addition, NSAIDs exert anticancer properties and inhibition of tumor proliferation and invasion by inducing apoptosis, DNA damage repair, and immune surveillance in a COX-independent manner.20 However, epidemiological evidence of the association between NSAIDs uptake and the risk of cancer remains inconsistent and controversial. Based on the data that patients with non-aspirin NSAIDs uptake (HR = 0.81, 95% CI: 0.70–0.94), but not aspirin (HR = 0.77, 95% CI: 0.58–1.02), showed a statistically reduced the risk in hepatocellular carcinoma.102 Of note, the mechanism of NSAIDs in cancer prevention and treatment is still not clear, NSAIDs induce autophagy is a newly identified mechanism to explain the complicated effect of NSAIDs in cancer cells respond to stress induced by chemoradiotherapy. From the update results, a clear portrait emerges that divergent effect of autophagy induced by NSAIDs depends on tumor type, stage of tumorigenesis, tumor microenvironment, as well as genetic and epigenetic factors. Our laboratory data showed 2.5-dimethyl-celecoxib increases radiosensitivity in nasopharyngeal carcinoma by inhibiting autophagic flux (data not shown). Nowadays, persuasive evidence was reported to determine the fate of tumor cells treated with NSAIDs by molecular biology assays such as the interfering of autophagy inducer or inhibitor, autophagy-related gene over-expression and down-expression by plasmid, autophagy flux detection, and transgenic animal model in vivo. The relation between autophagy regulation and NSAIDs in cancer needs to more comprehensive laboratory investigation, and importantly a cluster of prospective, randomized controlled trials to determine the efficacy and safety of NSAIDs in common types of malignancy would be established to provide high-level evidence for clinical decision to support the combination treatment of NSAIDs and chemoradiotherapy or NSAIDs alone, which may be presenting a promising approach in the treatment of chemo- and radio-resistance tumors.
Acknowledgments
This work was financially supported by grants from Key Laboratory Open Project of Shanxi Provincial [KLTPM-SX2018-B5], Chenzhou City Science, T Technology Plan Key Project [JSYF2017021]. We thank Dr. Jiaquan Qu for the valuable reviews and comments.
Disclosure
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30. doi:10.3322/caac.21590
2. Marino G, Niso-Santano M, Baehrecke EH, et al. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15:e81. doi:10.1038/nrm3735
3. Yazdani HO, Huang H, Tsung A. Autophagy: dual response in the development of hepatocellular carcinoma. Cells. 2019;8:e91. doi:10.3390/cells8020091
4. Wilde L, Tanson K, Curry J, et al. Autophagy in cancer: a complex relationship. Biochem J. 2018;475:1939–1954. doi:10.1042/BCJ20170847
5. Kondo Y, Kanzawa T, Sawaya R, et al. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer. 2005;5(9):e726. doi:10.1038/nrc1692
6. Yu L, Strandberg L, Lenardo MJ. The selectivity of autophagy and its role in cell death and survival. Autophagy. 2008;4(5):567–573. doi:10.4161/auto.5902
7. Rabinowitz JD, White E. Autophagy and metabolism. Science. 2010;330(6009):1344–1348. doi:10.1126/science.1193497
8. Doherty J, Baehrecke EH. Life, death and autophagy. Nat Cell Biol. 2018;20:1110–1117. doi:10.1038/s41556-018-0201-5
9. Singh SS, Vats S, Chia AYQ, et al. Dual role of autophagy in hallmarks of cancer. Oncogene. 2018;37(9):e1142. doi:10.1038/s41388-017-0046-6
10. Lorente J, Velandia C, Leal JA, et al. The interplay between autophagy and tumorigenesis: exploiting autophagy as a means of anticancer therapy. Biol Rev. 2018;93(1):152–165. doi:10.1111/brv.12337
11. Wong RSY. Role of Nonsteroidal Anti-Inflammatory Drugs (NSAIDs) in cancer prevention and cancer promotion. Adv Pharmacol Sci. 2019;e2019.
12. Ulrich CM, Bigler J, Potter JD. Non-steroidal anti-inflammatory drugs for cancer prevention: promise, perils and pharmacogenetics. Nat Rev Cancer. 2006;6(2):e130. doi:10.1038/nrc1801
13. Wakabayashi K. NSAIDs as cancer preventive agents. Asian Pac J Cancer Prev. 2000;1(2):97–113.
14. Cuzick J, Otto F, Baron JA, et al. Aspirin and non-steroidal anti-inflammatory drugs for cancer prevention: an international consensus statement. Lancet Oncol. 2009;10(5):501–507. doi:10.1016/S1470-2045(09)70035-X
15. Zhang Z, Chen F, Shang L. Advances in antitumor effects of NSAIDs. Cancer Manag Res. 2018;10:e4631. doi:10.2147/CMAR.S175212
16. Lucotti S, Cerutti C, Soyer M, et al. Aspirin blocks formation of metastatic intravascular niches by inhibiting platelet-derived COX-1/thromboxaneA2. J Clin Invest. 2019;129:5. doi:10.1172/JCI121985
17. Jana NR. NSAIDs and apoptosis. Cell Mol Life Sci. 2008;65(9):1295–1301. doi:10.1007/s00018-008-7511-x
18. Goel A, Chang DK, Ricciardiello L, et al. A novel mechanism for aspirin-mediated growth inhibition of human colon cancer cells. Clin Cancer Res. 2003;9(1):383–390.
19. Zelenay S, Van Der Veen AG, Böttcher JP, et al. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell. 2015;162(6):1257–1270. doi:10.1016/j.cell.2015.08.015
20. Gurpinar E, Grizzle WE, Piazza GA. NSAIDs inhibit tumorigenesis, but how? Clin Cancer Res. 2014;20(5):1104–1113. doi:10.1158/1078-0432.CCR-13-1573
21. Yu C, Li W, Liu J, et al. Autophagy: novel applications of nonsteroidal anti-inflammatory drugs for primary cancer. Cancer Med. 2018;7(2):471–484. doi:10.1002/cam4.1287
22. Gunaydin C, Bilge SS. Effects of nonsteroidal anti-inflammatory drugs at the molecular level. Eurasian j Med. 2018;50(2):e116. doi:10.5152/eurasianjmed.2018.0010
23. Rouzer CA, Marnett LJ. Cyclooxygenases: structural and functional insights. J Lipid Res. 2009;50(Supplement):S29–S34. doi:10.1194/jlr.R800042-JLR200
24. Brune K, Patrignani P. New insights into the use of currently available non-steroidal anti-inflammatory drugs. J Pain Res. 2015;8:e105. doi:10.2147/JPR.S75160
25. Frölich JC. A classification of NSAIDs according to the relative inhibition of cyclooxygenase isoenzymes. Trends Pharmacol Sci. 1997;18(1):30–34. doi:10.1016/S0165-6147(96)01017-6
26. Morita I. Distinct functions of COX-1 and COX-2. Prostaglandins Other Lipid Mediat. 2002;68:165–175. doi:10.1016/S0090-6980(02)00029-1
27. Mohammed NA, El-Aleem SA, El-Hafiz HA, et al. Distribution of constitutive (COX-1) and inducible (COX-2) cyclooxygenase in postviral human liver cirrhosis: a possible role for COX-2 in the pathogenesis of liver cirrhosis. J Clin Pathol. 2004;57(4):350–354. doi:10.1136/jcp.2003.012120
28. Crofford LJ. COX-1 and COX-2 tissue expression: implications and predictions. J Rheumatol Suppl. 1997;49:15–19.
29. Díaz-González F, Sánchez-Madrid F. NSAIDs: learning new tricks from old drugs. Eur J Immunol. 2015;45(3):679–686. doi:10.1002/eji.201445222
30. Badri W, Miladi K, Nazari QA, et al. Encapsulation of NSAIDs for inflammation management: overview, progress, challenges and prospects. Int J Pharm. 2016;515(1–2):757–773. doi:10.1016/j.ijpharm.2016.11.002
31. Ngo SNT, Addison CJ. Are COX-2 selective NSAIDs associated with less GI, renal, and cardiovascular side effects: evidence from animals treated with NSAIDs. Annu Res Rev Biol. 2018;1–8.
32. Domiati S, El-Mallah A, Ghoneim A, et al. Evaluation of anti-inflammatory, analgesic activities, and side effects of some pyrazole derivatives. Inflammopharmacology. 2016;24(4):163–172. doi:10.1007/s10787-016-0270-7
33. Flauaus C, Schmidtko A. Gastrointestinal and cardiovascular side effects of NSAIDs. PHARMAKON. 2017;5(1):61–68.
34. Shah V. NSAIDs: are they all the same? Pharmacology. 2018;e4.
35. Bibbins-Domingo K. Aspirin use for the primary prevention of cardiovascular disease and colorectal cancer: US preventive services task force recommendation statement. Ann Intern Med. 2016;164(12):836–845. doi:10.7326/M16-0577
36. Rothwell PM, Wilson M, Elwin CE, et al. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet. 2010;376(9754):1741–1750. doi:10.1016/S0140-6736(10)61543-7
37. Avivi D, Moshkowitz M, Detering E, et al. The role of low-dose aspirin in the prevention of colorectal cancer. Expert Opin Ther Targets. 2012;16(sup1):S51–S62. doi:10.1517/14728222.2011.647810
38. Lochhead PJ, Chan AT. Aspirin and the Prevention of Colorectal Cancer[M]//Nsaids and Aspirin. Cham: Springer; 2016:219–240.
39. Hua H, Zhang H, Kong Q, et al. Complex roles of the old drug aspirin in cancer chemoprevention and therapy. Med Res Rev. 2019;39(1):114–145. doi:10.1002/med.21514
40. Xu XR, Yousef GM, Ni H. Cancer and platelet crosstalk: opportunities and challenges for aspirin and other antiplatelet agents. Blood. 2018;131(16):1777–1789. doi:10.1182/blood-2017-05-743187
41. Tsoi KKF, Ho JMW, Chan FCH, et al. Long-term use of low-dose aspirin for cancer prevention: a 10-year population cohort study in Hong Kong. Int j Cancer. 2019;145(1):267–273.
42. Samadder NJ, Kuwada SK, Boucher KM, et al. Association of sulindac and erlotinib vs placebo with colorectal neoplasia in familial adenomatous polyposis: secondary analysis of a randomized clinical trial. JAMA oncol. 2018;4(5):671–677. doi:10.1001/jamaoncol.2017.5431
43. Barnett RM, Borras E, Samadder NJ, et al. Chemoprevention in hereditary colorectal cancer syndromes. Hereditary Colorectal Cancer. 1999;86(11): 2551–2563.
44. Tołoczko-Iwaniuk N, Dziemiańczyk-Pakieła D, Nowaszewska BK, et al. Celecoxib in cancer therapy and prevention–review. Curr Drug Targets. 2019;20(3):302–315. doi:10.2174/1389450119666180803121737
45. Kanda A, Ebihara S, Takahashi H, et al. Loxoprofen sodium suppresses mouse tumor growth by inhibiting vascular endothelial growth factor. Acta Oncol (Madr). 2003;42(1):62–70. doi:10.1080/0891060310002258
46. Yao M, Zhou W, Sangha S, et al. Effects of nonselective cyclooxygenase inhibition with low-dose ibuprofen on tumor growth, angiogenesis, metastasis, and survival in a mouse model of colorectal cancer. Clin Cancer Res. 2005;11(4):1618–1628. doi:10.1158/1078-0432.CCR-04-1696
47. Ko J-H, Lee S-G, Yang W, et al. The application of embelin for cancer prevention and therapy. Molecules. 2018;23(3):e621. doi:10.3390/molecules23030621
48. Hasima N, Ozpolat B. Regulation of autophagy by polyphenolic compounds as a potential therapeutic strategy for cancer. Cell Death Dis. 2014;5(11):e1509–e1509. doi:10.1038/cddis.2014.467
49. Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nature Reviews Molecular Cell Biology. 2018:1.
50. Marquez RT, Xu L. Bcl-2: beclin 1 complex: multiple, mechanisms regulating autophagy/apoptosis toggle switch. Am J Cancer Res. 2012;2(2):e214.
51. Hill SM, Wrobel L, Rubinsztein DC. Post-translational modifications of Beclin 1 provide multiple strategies for autophagy regulation. Cell Death Differ. 2018;1.
52. Huang Z, Fang W, Liu W, et al. Aspirin induces Beclin-1-dependent autophagy of human hepatocellular carcinoma cell. Eur J Pharmacol. 2018;823:58–64. doi:10.1016/j.ejphar.2018.01.031
53. Qian HR, Shi ZQ, Zhu HP, et al. Interplay between apoptosis and autophagy in colorectal cancer. Oncotarget. 2017;8(37):e 62759. doi:10.18632/oncotarget.18663
54. Yu L, Chen Y, Tooze SA. Autophagy pathway: cellular and molecular mechanisms. Autophagy. 2018;14(2):207–215. doi:10.1080/15548627.2017.1378838
55. Din FVN, Valanciute A, Houde VP, et al. Aspirin inhibits mTOR signaling, activates AMP-activated protein kinase, and induces autophagy in colorectal cancer cells. Gastroenterology. 2012;142(7):1504–1515. doi:10.1053/j.gastro.2012.02.050
56. Wang XW, Zhang YJ. Targeting mTOR network in colorectal cancer therapy. World J Gastroenterol. 2014;20(15):e4178. doi:10.3748/wjg.v20.i15.4178
57. Zhao Q, Wang Z, Wang Z, et al. Aspirin may inhibit angiogenesis and induce autophagy by inhibiting mTOR signaling pathway in murine hepatocarcinoma and sarcoma models. Oncol Lett. 2016;12(4):2804–2810. doi:10.3892/ol.2016.5017
58. Yue W, Yang CS, DiPaola RS, et al. Repurposing of metformin and aspirin by targeting AMPK-mTOR and inflammation for pancreatic cancer prevention and treatment. Cancer Prev Res. 2014;7(4):388–397. doi:10.1158/1940-6207.CAPR-13-0337
59. Ralph SJ, Nozuhur S, Moreno-Sánchez R, et al. NSAID celecoxib: a potent mitochondrial pro-oxidant cytotoxic agent sensitizing metastatic cancers and cancer stem cells to chemotherapy. J Cancer Metastasis Treat. 2018;4(9):e49. doi:10.20517/2394-4722.2018.42
60. Zhu X, Zhou M, Liu G, et al. Autophagy activated by the c-Jun N-terminal kinase-mediated pathway protects human prostate cancer PC3 cells from celecoxib-induced apoptosis. Exp Ther Med. 2017;13(5):2348–2354.
61. Zhang C, Shi J, Mao S, et al. Role of p38 MAPK in enhanced human cancer cells killing by the combination of aspirin and ABT-737. J Cell Mol Med. 2015;19(2):408–417. doi:10.1111/jcmm.12461
62. Crighton D, Wilkinson S, O’Prey J, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126(1):121–134. doi:10.1016/j.cell.2006.05.034
63. Kang KB, Zhu C, Yong SK, et al. Enhanced sensitivity of celecoxib in human glioblastoma cells: induction of DNA damage leading to p53-dependent G 1 cell cycle arrest and autophagy. Mol Cancer. 2009;8(1):e66. doi:10.1186/1476-4598-8-66
64. Mazzanti R, Platini F, Bottini C, et al. Down-regulation of the HGF/MET autocrine loop induced by celecoxib and mediated by P-gp in MDR-positive human hepatocellular carcinoma cell line. Biochem Pharmacol. 2009;78(1):21–32. doi:10.1016/j.bcp.2009.03.013
65. Niranjan R, Mishra KP, Thakur AK. Inhibition of Cyclooxygenase-2 (COX-2) initiates autophagy and potentiates MPTP-induced autophagic cell death of human neuroblastoma cells, SH-SY5Y: an inside in the pathology of parkinson’s disease. Mol Neurobiol. 2018;55(10):8038–8050. doi:10.1007/s12035-018-0950-y
66. Zhou P, Li Y, Li B, et al. Autophagy inhibition enhances celecoxib-induced apoptosis in osteosarcoma. Cell Cycle. 2018;17(8):997–1006. doi:10.1080/15384101.2018.1467677
67. Vallecillo-Hernández J, Barrachina MD, Ortiz-Masiá D, et al. Indomethacin disrupts autophagic flux by inducing lysosomal dysfunction in gastric cancer cells and increases their sensitivity to cytotoxic drugs. Sci Rep. 2018;8(1):e3593. doi:10.1038/s41598-018-21455-1
68. Johnsen JI, Lindskog M, Ponthan F, et al. Cyclooxygenase-2 is expressed in neuroblastoma, and nonsteroidal anti-inflammatory drugs induce apoptosis and inhibit tumor growth in vivo. Cancer Res. 2004;64(20):7210–7215. doi:10.1158/0008-5472.CAN-04-1795
69. Lynch PM, Burke CA, Phillips R, et al. An international randomised trial of celecoxib versus celecoxib plus difluoromethylornithine in patients with familial adenomatous polyposis. Gut. 2016;65(2):286–295. doi:10.1136/gutjnl-2014-307235
70. Huang S, Sinicrope F. Celecoxib-induced apoptosis is enhanced by ABT-737 and by inhibition of autophagy in human colorectal cancer cells. Autophagy. 2010;6(2):256–269. doi:10.4161/auto.6.2.11124
71. Quinn BA. Novel Therapeutic Strategies for Pancreatic Cancer. VCU Scholars Compass. 2014:e3.
72. Park GB, Jin DH, Kim D. Sequential treatment with celecoxib and bortezomib enhances the ER stress-mediated autophagy-associated cell death of colon cancer cells. Oncol Lett. 2018;16(4):4526–4536. doi:10.3892/ol.2018.9233
73. Zhu RT, Gutkind JS, Johnson DE, et al. PIK3CA mutations in colorectal and breast cancer: impact on oncogenesis and response to non-steroidal anti-inflammatory drugs. Targeting Cell Survival Pathways to Enhance Response to Chemotherapy. 2019:123–144.
74. Nitulescu GM, Van De Venter M, Nitulescu G, et al. The Akt pathway in oncology therapy and beyond. Int J Oncol. 2018;53(6):2319–2331. doi:10.3892/ijo.2018.4597
75. Moon H-J, Kim H-B, Lee S-H, et al. Sensitization of multidrug-resistant cancer cells to Hsp90 inhibitors by NSAIDs-induced apoptotic and autophagic cell death. Oncotarget. 2018;9(13):e11303. doi:10.18632/oncotarget.24130
76. Raza MH, Siraj S, Arshad A, et al. ROS-modulated therapeutic approaches in cancer treatment. J Cancer Res Clin Oncol. 2017;143:1789–1809. doi:10.1007/s00432-017-2464-9
77. Simon HU, Haj-Yehia A, Levi-Schaffer F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis. 2000;5:415–418. doi:10.1023/A:1009616228304
78. de Sa Junior PL, Camara DAD, Porcacchia AS, et al. The roles of ROS in cancer heterogeneity and therapy. Oxid Med Cell Longev. 2017;e2467940.
79. Poillet-Perez L, Despouy G, Delage-Mourroux R, Boyer-Guittaut M. Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biol. 2015;4:184–219. doi:10.1016/j.redox.2014.12.003
80. Zhou H, Yuan M, Yu Q, et al. Autophagy regulation and its role in gastric cancer and colorectal cancer. Cancer Biomark. 2016;17(1):1–10. doi:10.3233/CBM-160613
81. Thomas S, Sharma N, Golden EB, et al. Preferential killing of triple-negative breast cancer cells in vitro and in vivo when pharmacological aggravators of endoplasmic reticulum stress are combined with autophagy inhibitors. Cancer Lett. 2012;325(1):63–71. doi:10.1016/j.canlet.2012.05.030
82. Gao M, Yeh PY, Lu YS, et al. OSU-03012, a novel celecoxib derivative, induces reactive oxygen species–related autophagy in hepatocellular carcinoma. Cancer Res. 2008;68(22):9348–9357. doi:10.1158/0008-5472.CAN-08-1642
83. Moon H-J, Park S-Y, Lee S-H, et al. Nonsteroidal anti-inflammatory drugs sensitize CD44-overexpressing cancer cells to Hsp90 inhibitor through autophagy activation. Oncology Research Featuring Preclinical and Clinical Cancer Therapeutics. 2019;27(7):835–847. doi:10.3727/096504019X15517850319579
84. Huang KH, Kuo KL, Ho IL, et al. Celecoxib-induced cytotoxic effect is potentiated by inhibition of autophagy in human urothelial carcinoma cells. PLoS One. 2013;8(12):e82034. doi:10.1371/journal.pone.0082034
85. Lu Y, Liu XF, Liu TR, et al. Celecoxib exerts antitumor effects in HL-60 acute leukemia cells and inhibits autophagy by affecting lysosome function. Biomed Pharmacother. 2016;84:1551–1557. doi:10.1016/j.biopha.2016.11.026
86. Zhang X, Feng H, Du J, et al. Aspirin promotes apoptosis and inhibits proliferation by blocking G0/G1 into S phase in rheumatoid arthritis fibroblast-like synoviocytes via downregulation of JAK/STAT3 and NF-κB signaling pathway. Int J Mol Med. 2018;42(6):3135–3148. doi:10.3892/ijmm.2018.3883
87. Liu S, Tang Y, Yan M, et al. PIK3CA mutation sensitizes breast cancer cells to synergistic therapy of PI3K inhibition and AMPK activation. Invest New Drugs. 2018;36(5):763–772. doi:10.1007/s10637-018-0563-3
88. Olejniczak-Kęder A, Szaryńska M, Wrońska A, et al. Effects of 5-FU and anti-EGFR antibody in combination with ASA on the spherical culture system of HCT116 and HT29 colorectal cancer cell lines. Int J Oncol. 2019;55(1):223–242. doi:10.3892/ijo.2019.4809
89. Levy JMM, Thorburn A. Targeting autophagy during cancer therapy to improve clinical outcomes. Pharmacol Ther. 2011;131(1):130–141. doi:10.1016/j.pharmthera.2011.03.009
90. Baehrecke EH. Autophagy: dual roles in life and death? Nat Rev Mol Cell Biol. 2005;6(6):e505. doi:10.1038/nrm1666
91. Bhat P, Kriel J, Priya BS, et al. Modulating autophagy in cancer therapy: advancements and challenges for cancer cell death sensitization. Biochem Pharmacol. 2018;147:170–182. doi:10.1016/j.bcp.2017.11.021
92. Qi L, Ye JW, Xue WH, et al. The mechanism of aspirin combined with metformin induced apoptosis of thyroid cancer TPC-1 cells. Chin j Oncol. 2019;41(4):276–281. doi:10.3760/cma.j.issn.0253-3766.2019.04.006
93. Williams CS, Goldman AP, Sheng H, Morrow JD, DuBois RN. Sulindac sulfide, but not sulindac sulfone, inhibits colorectal cancer growth. Neoplasia. 1999;6(2):170–176. doi:10.1038/sj.neo.7900024
94. Gurpinar E, Grizzle WE, Shacka JJ, et al. A novel sulindac derivative inhibits lung adenocarcinoma cell growth through suppression of Akt/mTOR signaling and induction of autophagy. Mol Cancer Ther. 2013;12(5):663–674. doi:10.1158/1535-7163.MCT-12-0785
95. Lim SC, Kim SM, Choi JE, et al. Sodium salicylate switches glucose depletion-induced necrosis to autophagy and inhibits high mobility group box protein 1 release in A549 lung adenocarcinoma cells. Oncol Rep. 2008;19(5):1165–1171.
96. Bauvy C, Gane P, Arico S, et al. Autophagy delays sulindac sulfide-induced apoptosis in the human intestinal colon cancer cell line HT-29. Exp Cell Res. 2001;268(2):139–149. doi:10.1006/excr.2001.5285
97. Chiou SK, Hoa N, Ge L, et al. Nutrient availability alters the effect of autophagy on sulindac sulfide-induced colon cancer cell apoptosis. Gastroenterol Res Pract. 2012;2012.
98. Kola V, Mondal S, Mondal P. Investigation of cytotoxic activity of prepared PLGA nanoparticle formulations of meloxicam in HT29 colon cancer cell lines. Lat Am J Pharm. 2017;36(12):2379–2385.
99. Dong X, Li R, Xiu P, et al. Meloxicam executes its antitumor effects against hepatocellular carcinoma in COX-2-dependent and-independent pathways. PLoS One. 2014;9(3):e92864. doi:10.1371/journal.pone.0092864
100. Zhong J, Dong X, Xiu P, et al. Blocking autophagy enhances meloxicam lethality to hepatocellular carcinoma by promotion of endoplasmic reticulum stress. Cell Prolif. 2015;48(6):691–704. doi:10.1111/cpr.12221
101. Silva J, Arantes-Rodrigues R, Pinto-Leite R, et al. Synergistic effect of carboplatin and piroxicam on two bladder cancer cell lines. Anticancer Res. 2017;37(4):1737–1745.
102. Pang Q, Jin H, Qu K, et al. The effects of nonsteroidal anti-inflammatory drugs in the incident and recurrent risk of hepatocellular carcinoma: a meta-analysis. Onco Targets Ther. 2017;10(2017):4645–4656. doi:10.2147/OTT.S143154
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.