")
Back to Journals » Stem Cells and Cloning: Advances and Applications » Volume 8
Regenerative medicine in the treatment of idiopathic pulmonary fibrosis: current position
Authors Alvarez D, Levine M, Rojas M
Received 28 June 2014
Accepted for publication 11 December 2014
Published 15 April 2015 Volume 2015:8 Pages 61—65
DOI https://doi.org/10.2147/SCCAA.S49801
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Bernard Binetruy
Diana Álvarez,1,2 Melanie Levine,1 Mauricio Rojas1–3
1Dorothy P and Richard P Simmons Center for Interstitial Lung Disease, 2Pulmonary, Allergy, and Critical Care Medicine, 3McGowan Institute for Regenerative Medicine, University of Pittsburgh Medical Center, Pittsburgh, PA, USA
Abstract: Idiopathic pulmonary fibrosis (IPF) is a progressive, irreversible disease of the lung that has no lasting option for therapy other than transplantation. It is characterized by replacement of the normal lung tissue by fibrotic scarring, honeycombing, and increased levels of myofibroblasts. The underlying causes of IPF are still largely unknown. The focus of the current review is the possible use of stem cell therapy, specifically mesenchymal stem cells (MSCs), a multipotent stromal cell population, which have demonstrated promising data in multiple animal models of pulmonary fibrosis (PF). The most studied source of MSCs is the bone marrow, although they can be found also in the adipose tissue and umbilical cord, as well as in the placenta. MSCs have immunomodulatory and tissue-protective properties that allow them to manipulate the local environment of the injured tissue, ameliorating the inflammation and promoting repair. Because IPF primarily affects older patients, the issue of aging is intrinsically linked to many aspects of the disease, including the age of the stem cells. Animal models have shown the success of MSC therapy in mitigating the fibrotic effects of bleomycin-induced PF. However, bleomycin, the most commonly used model for PF, is imperfect in mimicking IPF as it presents in humans, as the duration of the illness is not parallel or reversible, and honeycombing is not produced. Furthermore, the time of MSC dosage has proven to be critical in determining whether the cells will ultimately have a positive or negative effect on disease progression, since it has been demonstrated that the maximal beneficial effect of MSCs occurs during the early inflammatory phase of the disease and that there is no or negative effect during the late fibrotic phase. Therefore, all the current clinical trials of MSCs and IPF, though promising, should proceed with caution as we move toward true stem cell therapy for this disease.
Keywords: aging, cell therapy, idiopathic pulmonary fibrosis, lung disease, lung fibrosis, mesenchymal stem cells
Introduction
Idiopathic pulmonary fibrosis (IPF) is a lung-restricted disease characterized by a chronic, progressive, and irreversible course, and is the most common of the interstitial lung diseases. The mean survival time of the disease is about 3 years after diagnosis, and to date the only definitive treatment is lung transplantation.1 However, multiple new therapies are under investigation, including the use of stem cells.2
Mesenchymal stem cells (MSCs) have been defined as multipotent cells with the potential of transdifferentiation in vitro, clonality, and self-renewal, with actions that include immunomodulation, epithelial repair, bactericidal activity, and mitochondrial transfer, as well as secretion of growth factors and microvesicles.1,3
Animal models have demonstrated the positive effects of MSCs in ameliorating inflammation and moderating the remodeling of the fibrotic lung, but only when they are administered during the early phase of bleomycin-induced lung injury.4 However, because bleomycin is not a perfect model in mimicking IPF, as well as the lack of a complete knowledge of the pathogenesis of IPF, the use of MSCs remains controversial and under study.5 In this review, we discuss different considerations regarding MSCs in the treatment of IPF.
Characterization of IPF
IPF is the most common form of interstitial lung disease, representing 45% of the total.1,6,7 The prevalence of IPF in the US is estimated to be between 20 and 60 cases per 100,000 inhabitants.8 It is calculated that around 14,000–34,000 are new patients each year.9 This disease preferentially affects adults older than 60 years, and its presentation has been shown to be greater in men compared to women (ratio 1.5:1).8,10,11 A diagnosis of IPF begins with a restrictive lung pattern, and includes radiological studies and histopathology showing a typical pattern of usual interstitial pneumonia with honeycombing and areas of fibrosis intercalated with normal parenchyma with basal predominance.8
Genetic susceptibility has been studied, where the MUC5B allele, which encodes for a macromolecule fundamental in mucociliary clearance, has been associated with IPF and has raised interest in the possible role of MUC5B as a biomarker for risk for the disease. The mechanisms by which MUC5B plays a role in IPF are not completely understood, but seem to involve the alteration of mucosal host defense and local cytokine production, leading to a decrease in the ability of lung cells to respond to injury.6,12 However, the MUC5B variant is absent in about 40% of IPF cases, meaning that it is only one of many factors that contribute to disease risk.10,12,13 Furthermore, polymorphisms in genes regulating the telomere length have also been implicated in the disease.14
Two theories have driven the possible pathogenesis of IPF, epithelial injury, and inflammation. The epithelial theory suggests that the disease is a result of multiple injuries that result in the combination of loss of epithelial integrity with various epigenetic changes, promoting an aberrant reaction of the alveolar epithelial cells to the epithelium.15–17 The abnormal reepithelialization promotes the deposition of myofibroblasts and fibroblasts, which are the major effector cells in remodeling,9 increasing the synthesis of such extracellular matrix proteins as collagen,15,17 further contributing to the loss of function that is characterized by low compliance and scarring of the lung parenchyma.18,19 Additionally, matrix metalloproteinases (MMPs), such as MMP1, MMP2, MMP3, MMP7, MMP8, and MMP9, have been implicated in IPF.13,18,20
However, although the epithelium is believed to be key in the pathogenesis of IPF, inflammation also plays a critical role in the disease. Several cytokines, such as IL-4, IL-5, IL-13, IL-18, and MIP-1α, have been found higher in cellular cultures and bronchoalveolar lavage of IPF patients compared to normal individuals. The recruitment to the lung of neutrophils and macrophages among other inflammatory cells contributes to the activation of profibrotic proteins, such as TGFβ and CTGF, together contributing in the release of collagen from tissue fibroblasts.14 All these factors suggest that the epithelial and inflammatory theories work together in explaining the disease and perpetuating the fibrotic process. However, there is still a lack of complete understanding of the pathogenesis of IPF, although repetitive alveolar epithelial cell injury seems to be essential.19
In the specific case of IPF, in a series of preclinical studies, we and others used bleomycin-induced lung fibrosis as a model to evaluate the ability of stem cells (SCs) to prevent the development of pulmonary fibrosis (PF) by decreasing the collagen content, thus reducing the severity of the injury.21 Additionally, we demonstrated that mice in which bleomycin was administered presented an increase in proinflammatory cytokines, such as IFNγ, IL-2, IL-1β and IL-4, and low levels of GM-CSF. Once MSCs were administered, the levels of cytokines returned to normal values and circulating levels of GM-CSF were significantly elevated. Increased levels of GM-CSF have been related with the mobilization and recruitment of endogenous SCs.21 However, the possible effects on the total reparative process are still not totally understood.1,22
Aging and stem cell exhaustion in IPF
Normal aging has been related to cellular apoptosis, shortening of telomeres, and oxidative stress (Figure 1).16,23 Some of those cellular changes have also been found in IPF patients, prompting a connection being drawn between IPF and diseases of premature aging. This theory has been supported by several investigations of IPF patients that have found mutations affecting telomerase activity.6,12 Moreover, the presence of an abnormal shortening of telomeres also leads to impaired autophagy, apoptosis, and accumulation of senescent cells, and may contribute to fibrosis.6,9,23
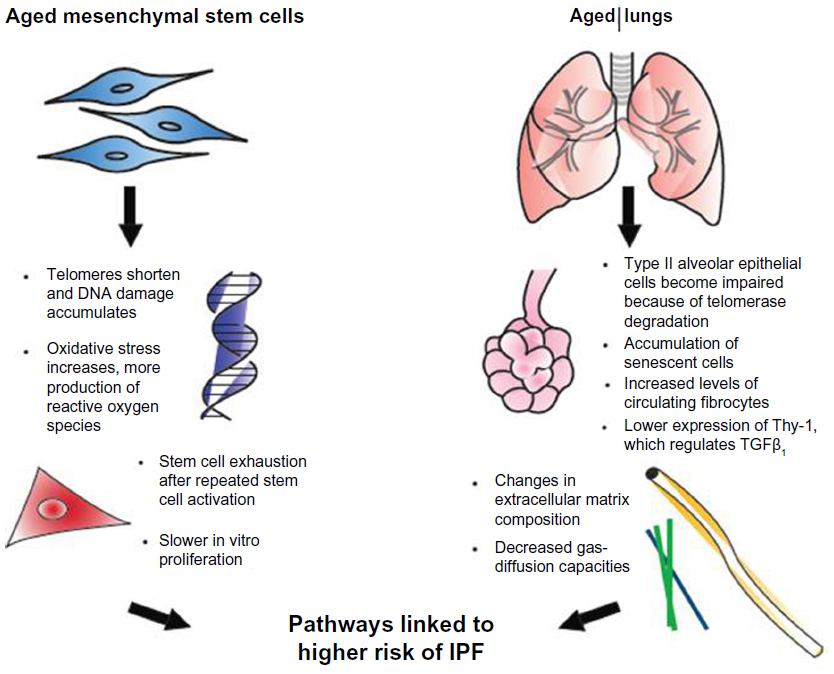 | Figure 1 Effects of aging on mesenchymal stem cells and the lungs. |
We compared the response to bleomycin-induced lung injury in young versus old mice, and found that after bleomycin administration in old mice, there was more severe lung fibrosis that was associated with an increase in local TGFβ.7 We compared the response to bleomycin between old and young mice at day 7 and day 14 after lung injury, where an increase in vulnerability to injury and a decrease in protector mechanisms and repair ability in elderly mice were observed.16,24 These studies demonstrate the better ability of young mice in responding to the fibrotic process.
Not much is known about the consequences of age on MSC activity. However, its behavior has several age-related variations. MSCs from old mice proliferate more slowly in vitro, and fail to protect in lipopolysaccharide-induced acute respiratory distress-syndrome models.1,24–26
In addition, persistent SC activation and proliferation has a negative effect of decrease in telomere length, leading to cell exhaustion and contributing to an accelerated decline in cellular functions and premature senescence.27 What exactly causes the continuous SC activation is still unknown. However, some chronic insults have been proposed as triggers, in which silent chronic viral infections, toxic substances, gastroesophageal reflux (leading to microaspiration), and telomere dysfunction or genetic abnormalities are included.8,16,23,28
In one study, we compared the effect of a single dose of intratracheal bleomycin in a murine model of accelerated aging on 6-month-old senescence-accelerated-prone mice (SAMP) and SA-resistant mice (SAMR) with 12 month-old mice. Fourteen days after the insult, we observed a decrease in lung-repair ability in SAMP after bleomycin-induced lung injury, resulting in an increase in lung fibrosis when compared to SAMR. In SAMP, these changes were associated with higher levels of TGFβ in the lung, a higher number of fibroblast progenitor cells (or fibrocytes) in circulation, and a decrease in the ability of MSCs to respond to the soluble signals of injury.29 Published data by our group suggest that the decrease in the ability of MSCs to respond to injury is associated with a reduction in the expression of TNFα and IFNγ receptors required for MSC activation.
Approach to treatment options for IPF and the role of MSCs
Because of the devastating course of the disease, and the lack of a definitive therapy for IPF other than transplantation, there is a continuous need to find new options for IPF treatment. Although the management of comorbid conditions, such as obesity, gastroesophageal reflux, obstructive apnea, pulmonary hypertension, and emphysema, may help to improve symptoms and quality of life of these patients, there is no evidence of a related minimizing fibrotic process on IPF.15
Some pharmacological therapies (corticosteroids, azathioprine, N-acetylcysteine, and etanercept, among others)15 have also been explored, without a clear benefit. Therefore, cellular therapies have emerged as a promising option, and the use of MSCs is consistently being studied.1,30
The properties that make MSCs a promising therapy in this pathology are several, in which the secretion of such growth factors as KGF, HGF, VEGF, and Ang-1 play an important role in the repair of the alveolar epithelium and endothelium.1,30,31 Additionally, through nitric oxide and indoleamine 2,3-dioxygenase, MSCs suppress T-cell proliferation to avoid cytotoxic T cells and natural killer cells.30 Furthermore, their capability of restoration of the injured tissue has been demonstrated through mitochondrial transfer, which has been crucial, due to the existent knowledge that mitochondrial dysfunction is linked to cellular senescence and inability to protect the lung from injury.32
MSCs also induce an anti-inflammatory response by increasing IL-10 and soluble IL-1β receptor, which results in a decrease in the levels of TNFα, IFNγ and IL-2.1 MSCs are immunoprivileged, escaping the immune response through the lack of expression of HLA class II and weak expression of HLA class I, which do not induce proliferation and activation of T lymphocytes, making them also ideal in the setting of therapy, because these cells can be administered to an immunocompetent patient without the need for further immunosuppression.3
As we mentioned before, it has been demonstrated through animal models that MSCs have a beneficial effect on PF if they are administered early in the disease course and not when the fibrotic changes already exist. This may be explained by the ability of MSCs to secrete TGFβ to modulate immune responses, which helps to ameliorate inflammation. However, at the same time, MSCs can act as a target for TGFβ, which results in an increase in collagen expression and deposition. Furthermore, other research has demonstrated that clonally derived GFP-MSCs administered to mice during the fibrotic phase of radiation-induced lung injury coexpressed vimentin, which is a marker of fibroblasts, implying a transdifferentiation into a fibroblast phenotype and promoting the fibrosis.25
Because of these concerns, there is a need to continue focusing on the evaluation of the safety profile and beneficial outcomes of MSCs in IPF patients to clarify any doubts and provide a better understanding of possible treatment options. As of June 2014, there were three ongoing clinical trials on clinicaltrials.gov trying to evaluate the safety of MSC therapy in IPF.
In Australia, Prince Charles Hospital conducted a study using placental-derived MSCs.33 The preliminary data support a good safety profile and a marginal improvement in walking abilities and forced vital capacity after 6 months of follow-up. Another clinical trial is in the recruitment phase at the University of Navarra, where researchers plan to test the endobronchial infusion of bone marrow-derived autologous SCs. Finally, in the Interdisciplinary Stem Cell Institute of University of Miami, the “AETHER” study is currently recruiting, and will evaluate the safety and effects on lung function with intravenous delivery of allogeneic human MSCs.28
Conclusion
IPF is a devastating disease with a poor prognosis, and although many strategies have been developed to clarify its pathologic progression and mechanisms, there is still a lack of complete knowledge on the details of the disease.
MSCs are a promising option in regenerative medicine for their ability to be isolated and expanded in culture, and are thus crucial to study for a better understanding of their properties and probable uses. A large risk lies in determining the appropriate time to deliver the dosage of MSCs, as the cells can in fact exacerbate the fibrotic process in IPF.
A promising observation in clinical trials is the good safety profile using MSCs, which is an essential aspect that supports the use of MSCs in IPF. However, the weak preclinical data that support the use of MSCs in IPF creates the need for caution in future investigations that concern appropriate timing to use MSCs, the ideal dosage, and route of administration.
Finally, it is important to continue enhancing the basic knowledge of the disease, due to the increased need to understand the exact pathogenesis in humans suffering from IPF. In our opinion, an important weakness in the study of IPF is the lack of knowledge of lung aging, which is a main risk factor in IPF. Until now, most of the mouse models have included young animals (less than 3 months), and it is clear that in humans IPF is a pathology of the elderly, and as the association between aging, IPF, and SCs is well developed, this variable must be taken into account in evaluating preclinical results and in translation to human applications.
Disclosure
The authors report no conflicts of interest in this work.
References
Huleihel L, Levine M, Rojas M. The potential of cell-based therapy in lung diseases. Expert Opin Biol Ther. 2013;13(10):1429–1440. | |
Sueblinvong V, Weiss DJ. Stem cells and cell therapy approaches in lung biology and diseases. Transl Res. 2010;156(3):188–205. | |
Sinclair K, Yerkovich ST, Chambers DC. Mesenchymal stem cells and the lung. Respirology. 2013;18(3):397–411. | |
Weiss DJ, Ortiz LA. Cell therapy trials for lung diseases: progress and cautions. Am J Respir Crit Care Med. 2013;188(2):123–125. | |
McNulty K, Janes SM. Stem cells and pulmonary fibrosis: cause or cure? Proc Am Thorac Soc. 2012;9(3):164–171. | |
Selman M, Pardo A. Revealing the pathogenic and aging-related mechanisms of the enigmatic idiopathic pulmonary fibrosis. An integral model. Am J Respir Crit Care Med. 2014;189(10):1161–1172. | |
Mora AL, Rojas M. Adult stem cells for chronic lung diseases. Respirology. 2013;18(7):1041–1046. | |
Ryu JH, Moua T, Daniels CE, et al. Idiopathic pulmonary fibrosis: evolving concepts. Mayo Clin Proc. 2014;89(8):1130–1142. | |
Royce SG, Moodley Y, Samuel CS. Novel therapeutic strategies for lung disorders associated with airway remodelling and fibrosis. Pharmacol Ther. 2014;141(3):250–260. | |
King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011;378(9807):1949–1961. | |
Nalysnyk L, Cid-Ruzafa J, Rotella P, Esser D. Incidence and prevalence of idiopathic pulmonary fibrosis: review of the literature. Eur Respir Rev. 2012;21(126):355–361. | |
Barkauskas CE, Noble PW. Cellular mechanisms of tissue fibrosis. 7. New insights into the cellular mechanisms of pulmonary fibrosis. Am J Physiol Cell Physiol. 2014;306(11):C987–C996. | |
Sbardella D, Fasciglione GF, Gioia M, et al. Human matrix metalloproteinases: an ubiquitarian class of enzymes involved in several pathological processes. Mol Aspects Med. 2012;33(2):119–208. | |
Bringardner BD, Baran CP, Eubank TD, Marsh CB. The role of inflammation in the pathogenesis of idiopathic pulmonary fibrosis. Antioxid Redox Signal. 2008;10(2):287–301. | |
Loomis-King H, Flaherty KR, Moore BB. Pathogenesis, current treatments and future directions for idiopathic pulmonary fibrosis. Curr Opin Pharmacol. 2013;13(3):377–385. | |
Kapetanaki MG, Mora AL, Rojas M. Influence of age on wound healing and fibrosis. J Pathol. 2013;229(2):310–322. | |
Parker MW, Rossi D, Peterson M, et al. Fibrotic extracellular matrix activates a profibrotic positive feedback loop. J Clin Invest. 2014;124(4):1622–1635. | |
Dancer RC, Wood AM, Thickett DR. Metalloproteinases in idiopathic pulmonary fibrosis. Eur Respir J. 2011;38(6):1461–1467. | |
Pardo A, Selman M. Idiopathic pulmonary fibrosis: new insights in its pathogenesis. Int J Biochem Cell Biol. 2002;34(12):1534–1538. | |
Pardo A, Selman M. Role of matrix metaloproteases in idiopathic pulmonary fibrosis. Fibrogenesis Tissue Repair. 2012;5 Suppl 1:S9. | |
Rojas M, Xu J, Woods CR, et al. Bone marrow-derived mesenchymal stem cells in repair of the injured lung. Am J Respir Cell Mol Biol. 2005;33(2):145–152. | |
Toonkel RL, Hare JM, Matthay MA, Glassberg MK. Mesenchymal stem cells and idiopathic pulmonary fibrosis. Potential for clinical testing. Am J Respir Crit Care Med. 2013;188(2):133–140. | |
Chilosi M, Carloni A, Rossi A, Poletti V. Premature lung aging and cellular senescence in the pathogenesis of idiopathic pulmonary fibrosis and COPD/emphysema. Transl Res. 2013;162(3):156–173. | |
Bustos ML, Huleihel L, Kapetanaki MG, et al. Aging mesenchymal stem cells fail to protect because of impaired migration and antiinflammatory response. Am J Respir Crit Care Med. 2014;189(7):787–798. | |
Mora AL, Rojas M. Aging and lung injury repair: a role for bone marrow derived mesenchymal stem cells. J Cell Biochem. 2008;105(3):641–647. | |
Sueblinvong V, Neveu WA, Neujahr DC, et al. Aging promotes pro-fibrotic matrix production and increases fibrocyte recruitment during acute lung injury. Adv Biosci Biotechnol. 2014;5(1):19–30. | |
López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194–1217. | |
Antunes MA, Laffey JG, Pelosi P, Rocco PR. Mesenchymal stem cell trials for pulmonary diseases. J Cell Biochem. 2014;115(6):1023–1032. | |
Xu J, Gonzalez ET, Iyer SS, et al. Use of senescence-accelerated mouse model in bleomycin-induced lung injury suggests that bone marrow-derived cells can alter the outcome of lung injury in aged mice. J Gerontol A Biol Sci Med Sci. 2009;64(7):731–739. | |
Lavoie JR, Rosu-Myles M. Uncovering the secretes of mesenchymal stem cells. Biochimie. 2013;95(12):2212–2221. | |
Shi Y, Su J, Roberts AI, Shou P, Rabson AB, Ren G. How mesenchymal stem cells interact with tissue immune responses. Trends Immunol. 2012;33(3):136–143. | |
Islam MN, Das SR, Emin MT, et al. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat Med. 2012;18(5):759–765. | |
Ilić N, Atkinson K. Manufacturing and use of human placenta-derived mesenchymal stromal cells for phase I clinical trials: establishment and evaluation of a protocol. Vojnosanit Pregl. 2014;71(7):651–659. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.