")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 17
Reexamining Ophthalmic Drugs, Safety and Tolerability in Phase 1 Clinical Trials
Authors Muñoz-Villegas P , Navarro-Sánchez AA, Sánchez-Ríos A, Olvera-Montaño O , Baiza-Durán LM
Received 29 July 2021
Accepted for publication 7 October 2021
Published 21 October 2021 Volume 2021:17 Pages 1123—1134
DOI https://doi.org/10.2147/TCRM.S331294
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Garry Walsh
Patricia Muñoz-Villegas, Andrea A Navarro-Sánchez, Alejandra Sánchez-Ríos, Oscar Olvera-Montaño, Leopoldo M Baiza-Durán
Medical Affairs Department, Laboratorios Sophia, S.A. de C.V., Zapopan, Jalisco, México
Correspondence: Patricia Muñoz-Villegas
Laboratorios Sophia, S.A. de C.V., Paseo del Norte 5255, Guadalajara Technology Park, Zapopan, 45010, Jalisco, México
Tel +52 33 3001 4200, Ext: 1018
Email [email protected]
Purpose: The purpose of this study was to evaluate the safety and tolerability profile of drugs used for treating common eye disorders when applied to normal healthy volunteers (NHVs) as explored in phase 1 trials.
Subjects and Methods: A total of 166 NHVs were identified in six phase 1 trials, examined in a retrospective analysis. The primary endpoints were visual comfort (by ocular comfort index, OCI) and safety (laboratory evaluations, vital signs (VS), visual acuity (VA), intraocular pressure (IOP), lissamine green and fluorescein staining, conjunctival hyperemia, chemosis, and adverse events’ incidence (AE)).
Results: Compared to baseline, 75.9%, 40.4% and 73.7% of NHV (for lubricant, hypotensive and antibiotic treatments, respectively) improved their OCI score by their final visit. Laboratory evaluations and VS were within normal ranges in 88% of NHV. Similar results were found for VA, corneal and conjunctival staining, and chemosis. IOP decreased significantly in the hypotensive agents’ group, trace to mild hyperemia was reported in 32.1%, 27.1%, and 6.8%, respectively. Additionally, lubricant and hypotensive investigational drugs (ID) had a lower risk of incidence of AE than approved drugs (OR 0.856, 95% CI [0.365, 1.999] and 0.636, 95% CI [0.096, 4.197], respectively). Meanwhile, on antibiotic drugs, the risk for ID-related AE was higher (OR 1.313, 95% CI [0.309, 5.583]).
Conclusion: Phase 1 trials are important in order to ensure the safety and tolerability of ophthalmic medications. This study demonstrates that NHVs do not face a significant risk of harm in these studies, since 98% of the reported AE were mild, and all AE were resolved by the end of the study in which they appeared.
Trial Registration: This is a retrospective study of six previously conducted clinical trials, registered on clinicaltrials.gov with the following registration IDs: NCT04081610, NCT03524157, NCT03520348, NCT03966365, NCT03965052 and, NCT03519516.
Keywords: healthy volunteer, phase 1 trial, safety, tolerability, ophthalmic drug
Introduction
Clinical research can be divided into 4 phases, and the basic standards and technical requirements of each phase are strictly regulated. Phase 1 trials are performed on new drugs which have already cleared a previous pre-clinical safety and toxicology assessment.1 During their participation in phase 1 trials, normal healthy volunteers (NHVs) play an important role in identifying side effects of investigational drugs (ID).2 Any physical change, harmful or unpleasant reactions that a participant experiences while in such a trial, regardless of severity (mild, moderate, or severe), is considered an adverse event (AE) rather than an “effect” because any detected symptoms may or may not be directly related to the experimental drug.2,3 In general, the overall risk of serious/severe AE has been estimated to be very low for NHV participating in phase 1 trials. Some common side effects such as headache, diarrhea, nausea, skin rashes, among others are frequently reported mild-AEs in trials evaluating systemic drugs.2,4 The phase 1 trials of ophthalmic medications included in this study varied in terms of the types of ID being tested, study procedures, and protocol designs. However, inclusion criteria generally matched or overlapped, including characteristics such as therapeutic area, and treatment-specific safety and tolerability parameters. They also included the evaluation of vital signs, laboratory tests, and matching protocol-defined parameters.
Despite the great importance of this research stage, since it links preclinical studies and the first human exposure to a certain formulation, the helpful information provided by phase 1 studies is not usually published or made available in any way to both the rest of the scientific community and general public. Even when recently there has been a slight increase in the number of published phase 1 trials,5–8 these are mostly cancer trials, leaving a significant gap of available data regarding the development of other specialty’s drugs during this essential stage of initial exposure of the target species of such medications.9
In order to present a wider view of NHV who have undergone ophthalmology trials, our study describes relevant elements of phase 1 clinical trials, including drugs used for common eye disorders: dry eye therapy, glaucoma, and antibiotic agents.10
Furthermore, the results obtained in ophthalmic phase 1 clinical trials may provide valuable information in regards of the potential adherence to be expected for a topically applied solution. As has been stated earlier in this document, there are many ophthalmological diagnoses that require a specific dosage of a topically applied medication, including cases of multiple daily instillations for a long period of time. In order to continue protecting users from possible serious and irreversible consequences of an undertreated pathology, safety and tolerability profiles of common ophthalmic drugs must be studied.
The purpose of this study was to evaluate the tolerability, visual comfort, and safety profile after ophthalmic drug instillation (ID and approved drugs [AD]) in NHV enrolled in phase 1 clinical trials.
Subjects and Methods
Study Design
This is a retrospective study of six previously conducted clinical trials, registered on clinicaltrials.gov with the following registration IDs: NCT04081610, NCT03524157, NCT03520348, NCT03966365, NCT03965052 and, NCT03519516. The design of this study was based on previous similar published works.11–14 A retrospective analysis of the former phase 1 clinical trials executed by Laboratorios Sophia, S.A. de C.V. was performed. Six studies met the criteria to be considered in the analysis, all of which were completed as planned, with a median duration of 8.5 (7 to 10) days. The studies included were randomized, parallel, prospective, controlled, and single-center trials. They were conducted in six centers in Mexico after an ethics committee for each trial study reviewed and approved said study’s protocol and its corresponding informed consent form (see Ethics approval and Consent to Participate section). All studies were conducted in compliance with the Declaration of Helsinki and in accordance with Good Clinical Practices Standards. All volunteers that participated provided written and signed informed consent. NHVs were recruited between July 2017 and October 2019, see Table 1. The data obtained in these prospective studies from their respective subjects were further analyzed retrospectively in order to evaluate the safety and tolerability profile of ophthalmic drugs when applied to healthy volunteers. The drugs used in these studies belonged to one of the following therapeutic families: lubricant, hypotensive agents and antibiotics. See Table 1 for details.
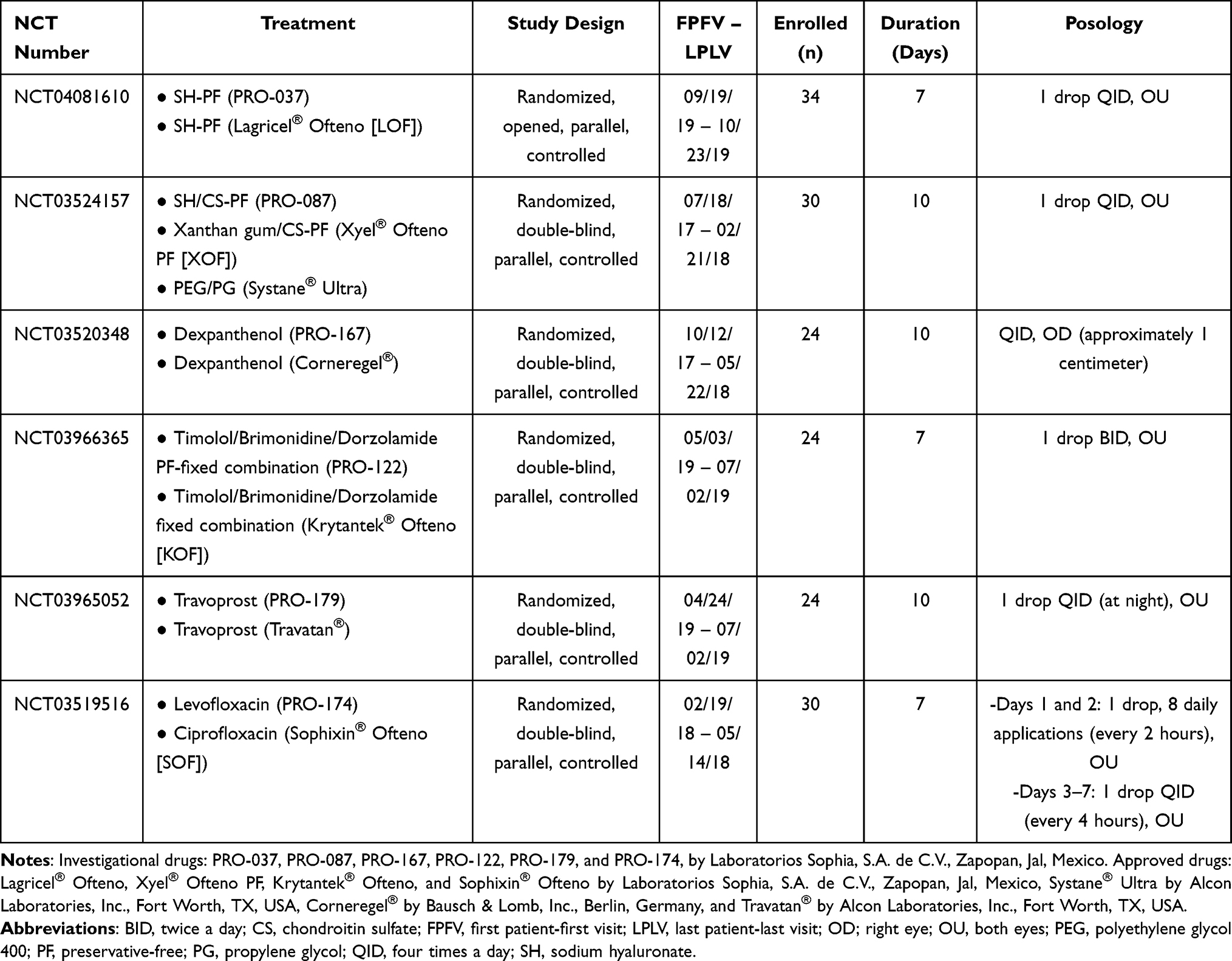 |
Table 1 Characteristics of Studies |
Subjects
Inclusion criteria included normal healthy male or female volunteers (aged 18–45 years), visual acuity of 20/30 or better, IOP ≥10 and ≤21 mmHg, normal vital signs, normal laboratory evaluation with a 20% margin. In the case of women of childbearing age, a birth control method was necessary, because pregnancy/breastfeeding or high childbearing potential without a birth control method before inclusion were considered exclusion criteria. Other exclusion criteria were the prevalent use of topical ocular drugs, trial participation <90 days before signing informed consent, current pharmacological medication or herbology treatments (that may affect the study’s outcomes) by any other route of administration and being a contact lens user. For more information about inclusion and exclusion criteria see Supporting Information (Tables S1 and S2).
Treatment and Evaluations
One hundred and sixty-six NHVs were randomized in a 1:1 ratio to receive either a lubricant (ID: n = 38 or AD: n = 50), a hypotensive drug (ID: n = 24 or AD: n = 24), or an antibiotic drug (ID: n = 15 or AD, n = 15) through computer software randomization numbers (SAS Institute, Inc, Cary, NC, USA). The posology and experimental time of each trial are represented in Table 1.
For the analysis of the variables described during the ophthalmological exploration (VA, IOP, ocular surface staining, ocular symptomatology, and conjunctival impression cytology [CIC]), for each subject, the average value of right and left eyes was considered an individual case (single data point per individual).15,16
Primary Endpoints
Visual Comfort
Ocular surface irritation was examined using the Ocular Comfort Index (OCI). It is a questionnaire designed for an objective measurement of ocular surface irritation. It produces estimates in a linear interval scale (score 0 to 100). This tool evaluates symptoms encompassing the discomfort associated with alterations of the ocular surface. Every item in this questionnaire evaluates both frequency and severity of each symptom.17 The OCI questionnaire is suitable for assessing the impact of ocular surface disease and changes in severity to design therapeutic strategies. Only the extreme responses (0, never; 6, always) were labeled, in order to minimize the effects of differences in subjective interpretation.18
Safety Assessments
Safety was evaluated through changes on specified laboratory evaluations of interest including liver-associated enzymes (LAE), vital signs (VS), visual acuity (VA), intraocular pressure (IOP), fluorescein and lissamine green corneal and conjunctival staining, conjunctival hyperemia, chemosis and incidence of adverse events. The laboratory evaluations included glucose, creatinine, hematic cytometry, and LAE (AST, ALT, direct bilirubin (conjugated), and total bilirubin). To pool the data from different studies, LAE levels were converted to multiples of the upper limit of normal value (ULN).19 The measured VS were the heart and respiratory rate (HR and RR, respectively), and systolic and diastolic blood pressures (SBP and DBP, respectively). Potential safety signals for most laboratory evaluations were identified based on values that fell outside of normal reference ranges in Mexican population. The VA was determined with a Snellen chart. The IOP was measured using a calibrated Goldmann applanation tonometer. The ocular surface was evaluated with a slit lamp, aided by fluorescein and green lissamine staining (GLS). Surface dye staining was classified in a scale from 0 to V in accordance with the percentage of the affected area (Oxford scale). Changes in conjunctival hyperemia were assayed with the Efron scale and incidence of chemosis was recorded. For AE evaluation, any method used in the clinical trials to elicit participant/reported AE, such as a diary, checklist, memory aid, etc., whether applied face-to-face or otherwise were considered. AEs occurring during each clinical protocol were recorded and included in this analysis.
Secondary Endpoints
Satisfaction
NHV under lubricants and antibiotic trials were questioned about their satisfaction with their respective treatments (discomfort), and it was measured through the ocular symptomatology post-installation determined by burning, itching, and foreign body sensation (FBS) using a survey questionnaire. Additionally, only in trials involving lubricants goblet cells density and Nelson’s grades were determined by CIC. Kendall’s Tau-b was conducted to determine whether a linear association existed between the goblet cell density and CIC grades score.
Data Analysis
A sample size was not calculated based on statistical power calculations for this retrospective study since the subjects included were those respectively recruited for each individual study included in this analysis. We investigated the association of the type of treatment (lubricant, hypotensive or antibiotic drug) and their safety profile. In a second analysis, we stratified data by type of drugs: ID versus AD (pre-specified in each protocol). The categorization was based on the conclusion of the study (eg, the ID was than safer as AD). All the participants who were enrolled in each study were included in the analyses (intent-to-treat population, ITT; n = 166 NHV). The continuous variables were assessed using a general linear model (GLM) multivariate analysis and repeated measures for data collected at least three times. Bonferroni’s comparisons were used when required for the post hoc analyses. The ordinal variables were analyzed using p × q contingency tables and the differences were calculated with Pearson Chi-square test or Fisher’s exact test. For the AE analysis, a logistic regression was used to calculate the odd ratio (OR) and 95% confidence interval (CI) for the association between determinants and studies. ORs were used to determine whether the ID exposure may be a risk factor for the incidence of AE, and to compare the magnitude of type-of-drug risk factor for that outcome as follows: OR = 1, exposure does not affect odds of outcome; OR > 1, exposure associated with higher odds of outcome, and OR < 1, exposure associated with lower odds of outcome.20 All data analyses were in SPSS 19.0 software for Windows (SPSS Inc., Chicago, IL, USA). A p-value ≤ 0.05 was considered significant.
The datasets generated and/or analyzed during the current study are available in the Open Science Framework (https://osf.io) repository, as DOI 10.17605/OSF.IO/GZU6J.
Results
Characteristics of the participants: A total of 166 NHV, were enrolled, two subjects discontinued their participation because of either AE (ocular hypotonia and rhinitis), one for protocol deviations, and another due to poor adherence to the indicated treatment (<80%). Therefore, 162 NHVs completed their entire protocol without deviations up to the safety call, instilling ID or AD during a study from different therapeutic areas. There were no demographic or clinically relevant differences at baseline between treatment groups. Mean age ± standard deviation (SD) was 27.41 ± 6.6 years, 53.6% of the NHVs were female (Pearson Chi-square test, p = 0.413), see Table 2.
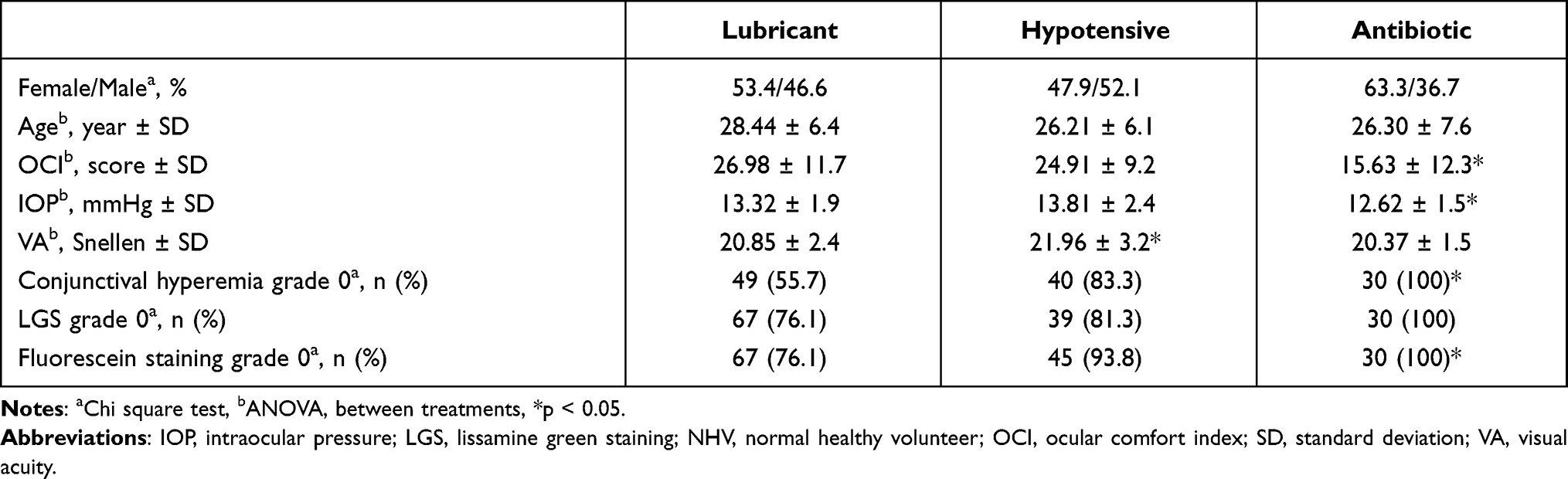 |
Table 2 Initial Characteristics of Each Group (n = 166 NHV) |
Primary Endpoints
Visual Comfort
Baseline OCI score (mean ± SD) was similar between the lubricant and hypotensive treatment groups (see Table 2); meanwhile, the antibiotic group had a lower basal OCI score (F(2,165)=11.760; p = 0.0001). On the final visit, 75.9%, 40.4%, and 73.7% of the NHVs in each group improved their initial score (Pearson Chi-square test, p = 0.0001). The mean of change ± SD from baseline to final visit was −7.29 ± 12.9 for lubricant, 2.17 ± 10.9 for hypotensive and, −5.05 ± 11.8 for antibiotic groups. The hypotensive group showed a significant increase in its score compared to the lubricant group (Bonferroni test, p = 0.0001). Additionally, no significant differences between the type of drug (ID or AD) were observed (p = 0.752), also the between-factor interaction (treatment × type of drug) was not significant (p = 0.374), see Table 3.
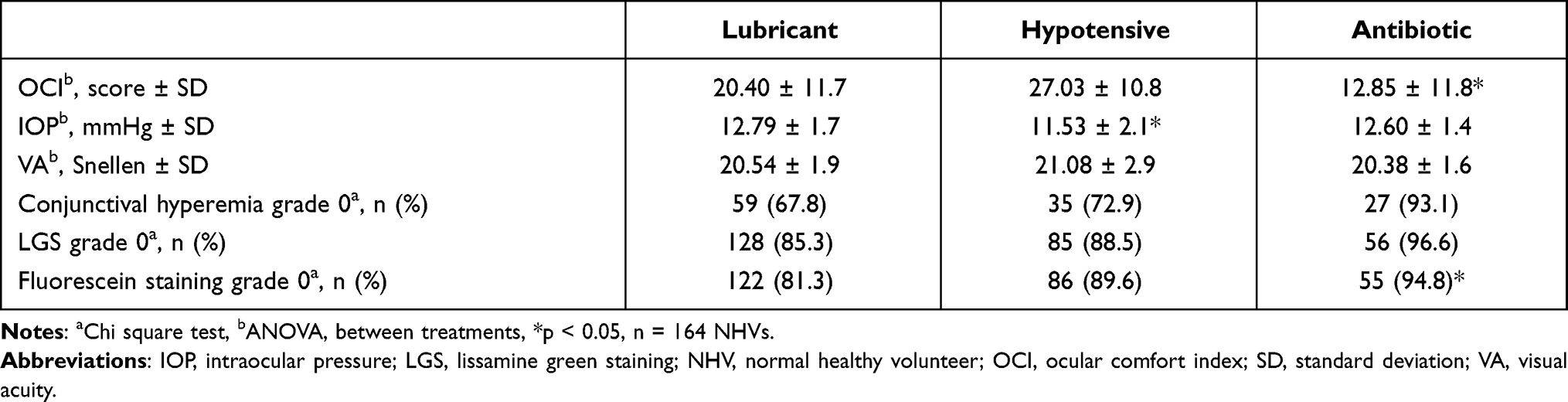 |
Table 3 Ophthalmological Exploration at Final Visit |
Laboratory Evaluations
As expected for NHV, laboratory results for hematological and biochemical parameters were within normal ranges at baseline. We did, however, find statistically significant differences on hematocrit (HTC), hemoglobin (HGB), and mean corpuscular volume (MCV) levels (all of them, under ULN values). The hypotensive group’s levels were higher than that of lubricant and antibiotic groups (Bonferroni test, p < 0.05). On the final visit, the MCV level was significantly different between treatments (p = 0.0001). Also, differences were evidenced between type of drug on AST and total bilirubin (p <0.05); however, between-factor interaction (visit × treatment × type of drug) was not significant (p > 0.05), see Table 4. ALT and AST elevations were observed in three subjects under lubricants and another in the hypotensive study group (2.4%), from baseline to final visit (reported as an AE). Two subjects (AD) had values greater than 1.2 times ULN-AST. For ALT levels, two NHVs had values 1.5 times higher, and another 2.6 times higher, meanwhile only one NHV in ID had one value greater than 1.02 times ULN during the treatment. LAE were resolved by the end of the follow-up study period and no safety additional issues were raised.
 |
Table 4 Hematological and Biochemical Parameters |
Vital Signs (VS)
As expected for NHV vital signs parameters were within normal ranges at baseline. Relatively few VS values were statistically lower between treatments at baseline. The RR in hypotensive group (F(2,164)= 4.209; p = 0.017), the SBP in antibiotic (F(2,164)= 3.446; p = 0.034) and, the DBP in lubricant group (F(2,164)= 4.180; p = 0.017) were consistently and significantly lower at the final visit (p-values: 0.001, 0.049 and, 0.006 respectively). However, the interactions between-factors (visit × treatment and visit × type of drug) were not significant (p > 0.05). On the final visit, only 1 (0.6%) NHV had high HR, 12 (7.3%) NHV had high SBP, and 3 (1.8%) NHV had high DBP. However, these findings were not reported as AEs.
Visual Acuity (VA) and Intraocular Pressure (IOP)
Baseline VA was similar between groups however, the mean value for the hypotensive group was higher compared with the antibiotic treatment (Bonferroni, p = 0.025, 95% CI [0.15, 3.04]). The type of drugs was not different (p = 0.378), and the interaction (treatment × type of drug) was also not significant (p = 0.109). The VA on the final visit was different versus the baseline values (F(1, 161)= 6.021, p = 0.015). The visit × treatment interaction was not significant (p = 0.107) and the visit × type of drug interaction was not significant too (0.446), see Table 3.
Baseline IOP was similar between treatments; however, the IOP from the hypotensive group was higher compared with the antibiotic group (Bonferroni, p = 0.034, 95% CI [0.073, 2.314]). The type of drug and the interaction between treatments versus the type of drug was not different (p-values: 0.644 and 0.402, respectively). On the final visit, as expected, the IOP decreased significantly by 2.3 mmHg in the hypotensive agent’s group (p = 0.0001), without differences between the type of drug (p = 0.210). The visit × treatment interaction was significant (p = 0.0001) but visit × type of drug and between-factors interactions were not significant (0.210 and 0.692, respectively), see Table 3.
Fluorescein and Lissamine Green Staining
On the final visit, fluorescein staining was graded as absent (grade 0, Oxford scale) for 77% and as minimal-to-mild (grade I or II) for 23% of NHV exposed to lubricants. Meanwhile, 87.5% and 89.7% were absent and 12.5% and 10.3% were grade minimal for hypotensive and antibiotic treatments, respectively (Pearson Chi-square test, p = 0.400). No differences were observed between the factor type of drug on baseline and at the final visit (p-values; 0.898 and 0.588). Similar percentages for LGS were observed on the final visit, 79.3% were absent and 20.7% minimal-to-mild for lubricant, 85.4% absent and, 14.6% was minimal-to-mild for hypotensive, meanwhile, 93.1% was absent and 6.9% minimal for the antibiotic group (p = 0.342). No differences were observed between the factor type of drug on baseline and at the final visit (p-values; 0.375 and 0.645), see Table 3.
Conjunctival Hyperemia and Chemosis
Similar findings were observed in the analysis of conjunctival hyperemia; after the intervention time and compared with baseline, there was no significant improvement in all groups, it was graded as “trace to mild” hyperemia for lubricant (32.1%), hypotensive (27.1%) and antibiotic groups (6.8%) (Pearson Chi-square test, p = 0.078). The type of drug was significant on baseline (p = 0.022), but this finding was not retained in the final visit (p = 0.968). Finally, no participants presented chemosis before or after their respective treatments, see Table 3.
Adverse Events
A total of 205 AE occurred in 59.6% of NHV (99/166). The hypotensive group had a higher incidence of AE than that the other treatments (Pearson Chi-square test, p = 0.0001). A total of 65 AEs/39 NHVs were reported for lubricants (46.2% for ID vs 53.8% in AD, p = 0.781), 117 AE/43 NHV for hypotensive agents (47% for ID vs 53% in AD, p = 0.705), and 23 AE/17 NHV (56.5% for ID vs 43.5% in AD, p = 0.559) for antibiotic drugs. The most common class of reported AE was burning (30.7% [26.2%, 27.4%, and 60.9% in lubricant, hypotensive and antibiotic treatments, respectively]), followed by conjunctival hyperemia (11.7% [19.7% and 4.3% in hypotensive and antibiotic treatments]), and itching (10.7% [15.4%, 6.8%, and 17.4% in lubricant, hypotensive and antibiotic treatments, respectively]), see Figure 1. There were a total of 201 mild AE (98% of the total AE), 3 moderates (1.4%), and one serious AE (0.5%, ocular hypotonia), without differences between treatments (p = 0.667). Only 14.6% of the AE were deemed unrelated to the study medication. No deaths were reported. The logistic regression (used to calculate ORs), found that in lubricant trials, the ID had a lower risk of incidence of AE than AD, OR 0.856, 95% CI, [0.365, 1.999]. Similar results occurred in hypotensive drugs, OR 0.636, 95% CI, [0.096, 4.197], nevertheless for antibiotic drugs, the risk of incidence for ID was higher than AD, OR 1.313, 95% CI, [0.309, 5.583]. For all groups, no differences between the risk for occurrence of AE in ID versus AD were observed, OR 1.008, 95% CI, [0.541, 1.887].
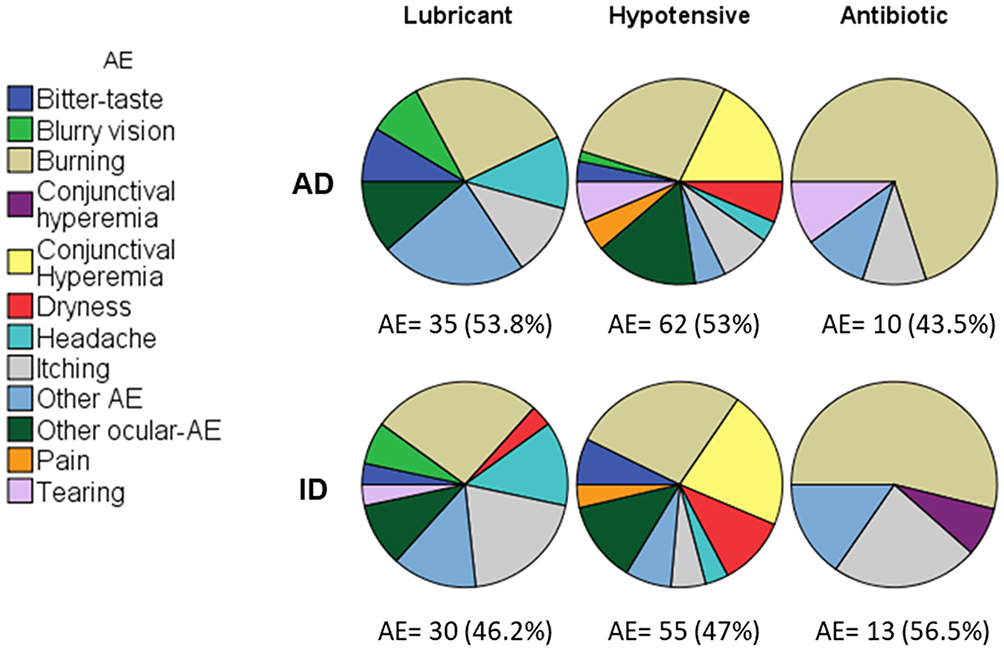 |
Figure 1 Incidence of adverse events (AE) in 166 NHV for approved drugs (AD, upper polar graphs) and investigational drugs (ID, lower polar graphs). A total of 205 adverse events were presented in 59.6% NHV (99/166). For another AE the incidence was <2%. Presence of AE in hypotensive drugs > lubricant and antibiotic drugs, Chi square test, p = 0.0001. |
Secondary Endpoints
Satisfaction
Burning, itching, and FBS, were considered the parameters to evaluate discomfort. In all studies, lubricants and antibiotics were well tolerated. At the final visit, “burning sensation” was reported in 22.6% NHV for lubricants and 27.6% for the antibiotic group (Pearson Chi-square test, p = 0.321). No significant differences were observed between types of drugs in each treatment (p > 0.05). Findings were similar for itching between treatments: 28.3% for lubricant and 17.2% for the antibiotic group reported itching at the final visit (p = 0.537). For each treatment, there were no differences between ID and AD (p-values; 0.240 and 0.681). Additionally, only 15.1% of NHV exposed to lubricants reported FBS at the final visit (Fisher exact test, p = 0.046); however, these findings were not statistically different in the analysis between types of drugs (p = 1.000). Finally, compared to baseline, there was a significant increase in conjunctival goblet cell density (304.59 ± 143.7 cell/mm2 vs 349.81± 125.2 cell/mm2, p = 0.015) by the final visit in the lubricants group. At baseline, 37.7% NHVs were classified as having normal CIC grade-0 (classification of Nelson), by the final visit, 39.6% had grade 0, without significant differences (p = 0.234). No significant differences were observed between type of drugs on baseline and final visit (p > 0.05). There was a negative association between goblet cell density and CIC grading score, t = −0.778, p < 0.001.
Discussion
Phase 1 trial participants are typically healthy volunteers who pass health screenings and have no identifiable medical conditions related to the investigative drugs.2 Additionally, because these volunteers are in “good health”, they gain no direct medical benefit from research participation; also, they are typically recurring participants enrolling serially in phase 1 trials.21 The main purpose for executing phase 1 trials is to evaluate a drug’s safety, with a low risk of originating numerous and/or severe AEs in unaffected parties. Participating in such trials requires every NHV to report any symptoms experienced during the study in order to identify all the adverse events potentially related to the ID.2 In this case, the six trials included were comparative studies with the approved counterpart of each ID, therefore allowing the confirmation of previously described AE or the addition of newly discovered unfavorable related symptoms. More so, evaluating the tolerability of such ophthalmic products in NHV may translate to a clinically and statistically significant conjecture on how the symptoms associated to their application may affect the user’s adherence and consequently their efficacy once they are commercialized.
The OCI was selected to evaluate tolerability since it provides a valid measurement on the basis of Rasch analysis, more so than other questionnaires like the Ocular Surface Disease Index (OSDI) or McMonnies. However, all these tools have shown weak correlations with objective dry eye disease clinical tests like VA, tear break-up time or corneal fluorescein staining.22 Because of this, it is advisable to use both subjective and objective markers as endpoints in clinical trials for ophthalmic lubricants. On the other hand, topical hypotensive agents, of any drug families available today, characterize themselves for giving rise to ocular surface symptoms in a more prevalent and severe fashion than other ophthalmic products.23 This study confirms this presumption as shown by the decrease in OCI scores by the final visit of NHVs in the hypotensive agents’ group, being this decrease significantly lesser for them in comparison to those in the antibiotics and lubricants groups (40.4%, 73.7%, and 75.9%, respectively).
For other variables regarding ocular surface evaluation, such as hyperemia, NHVs exposed to hypotensive agents and lubricants did not present a statistically significant difference (27.1% and 32.1%, respectively), whereas the antibiotic group presented a lower incidence (6.8%). Both fluorescein and lissamine green staining did not show a difference when comparing basal and final visits for any type of medications.
A total of 205 AE in 99 NHVs (59.6%; 99/166) were reported. For the hypotensive agents’ group 117 AE were present in 43 NHVs (98.3% considered mild), a significantly greater number than that of lubricants and antibiotics (p = 0.0001). As shown in Figure 1, for every group of medications, there was no difference between ID and AD groups (lubricants, p = 0.781; hypotensive agents, p = 0.705; antibiotics, p = 0.559). However, it is relevant to point out that for the ocular hypotensive agents’ group, all the studied variables were also considered AE; whereas for the lubricants and antibiotics groups the AE and the findings of the ophthalmological explorations were analyzed separately in the original trials. It is also worth mentioning that none of the total moderate (3) AE were considered related to the use of the studied ophthalmic products. Concerning the only reported severe AE, as expected with the hypotensive agents, the IOP decreased significantly in one NHV who experienced a decrease of ~8 mmHg. However, by the final follow-up visit, this parameter had normalized, and no safety additional issues were raised. Similarly, decrease in IOP has been observed in other phase 1 trials where NHV receiving hypotensive agents like beta-blockers or prostaglandin analogues also reported a significant IOP drop.24,25
The VS are fundamental to assess any drug’s general safety profile because they are objective measurements of essential physiological functions. Even though the systemic effects of ophthalmic solutions are rare, some compounds have been reported to cause systemic alterations after absorption into the bloodstream, particularly those used to lower de IOP since their mechanism of action may influence receptors in the cardiovascular and pulmonary systems. Blood pressure, heart, and respiratory frequencies were measured. Basal values were normal, and by the final visit, there were no clinical or statistically significant differences, observing only a slight raise in HR, SBP, and DBP (0.6%, 7.3%, and 1.8%, respectively). Despite the reported potential variations of VS after instillation, NHV exposed to hypotensive agents presented an increase of DBP equivalent to a significant p = 0.006, while the rest of the parameters being either equal or lower than those belonging to the other two study groups.
Regarding the laboratory evaluations, some participants were considered to suffer transaminitis and were considered to present an AE for such results. However, it is important to mention that none of those NHVs had any other related signs or symptoms, and that all cases remitted within the 7-day follow-up without clinical relevance. Furthermore, a slight increment of ALT (10% above de ULN value) and AST or bilirubin (20% above the ULN value) has been reported in the literature as acceptable as long as no other signs or symptoms of apparent disease are present.26 It has also been reported that ALT elevation above the ULN can occur in participants of Phase I trials with no history of significant disease being treated only with placebo. Out of the volunteers receiving placebo for 14 days, 20% reported at least one value between one and two times the ULN value, and some even obtained values higher than twice the UNL. The probability of having an increased value raises with repeated measurements.19 Such increments in healthy participants can be explained by genetic polymorphisms, intraindividual short-term (1–7 days) and long-term variations, weight, body mass index, age, sex, physical activity and maintained calorie intake.19,26–28 Hence, the results of transaminases in phase 1 trials should be analyzed. and interpreted carefully to avoid misdiagnosed hepatotoxicity.19
Finally, goblet cell density was evaluated for the lubricant group, and an increase, though not statistically significant, was observed. This increase coincides with reports portrayed in other studies and may translate in a clinically meaningful enhancement.
Summarizing, all the data analyzed in this study, from the subjective OCI questionnaire to the objective evaluation of ophthalmological variables, systemic examinations and laboratory results, ascertains that ophthalmic medications from three different pharmaceutical families are safe and tolerable when applied on the ocular surface of healthy volunteers. No new adverse events were detected beyond those previously described in the literature for each group; however, for both lubricants and hypotensive agents, the investigative drugs proved to have a lesser risk to produce adverse events, in comparison to their approved counterparts.14
Even when the clinical relevance of phase 1 studies may be questioned and therefore their publication is unfortunately often relegated, the public availability of the information compiled in such studies entails ethical and methodical grounds to justify their issuance. Regardless of the final results of phase 1 trials, both negative and positive outcomes should be accessible for ethics committees, final users and the scientific community in general since indication and treatment options may be affected by the adverse events described in this stage of development of any formulation. Within the context of any given diagnosis, social and economic considerations, comorbidities, etc., the data collected in phase 1 studies could translate into significant outcomes for a particular patient’s adherence to treatment and quality of life, for example.
Several reviewers have previously assessed the safety of research studies and publication bias, but none have focused on phase 1 trials for ophthalmology drugs.13,14,29 After a reviewed cohort of available studies in the literature, the analyzed database included ophthalmology phase 1 clinical trial protocols (clinicaltrials.gov, searched to February 2021), and published phase 1 controlled trials as scientific papers (PubMed, searched to February 2021). A total of 95 eligible controlled clinical trials for ophthalmic drugs, that enrolled NHV were found. Among these complete trials, 18 had available results (18.9%). On the secondary cohort, a total of 266 clinical trials were published as scientific papers. Out of the 33 studies with NHV (12.4%), 29 are available, see Figure 2.
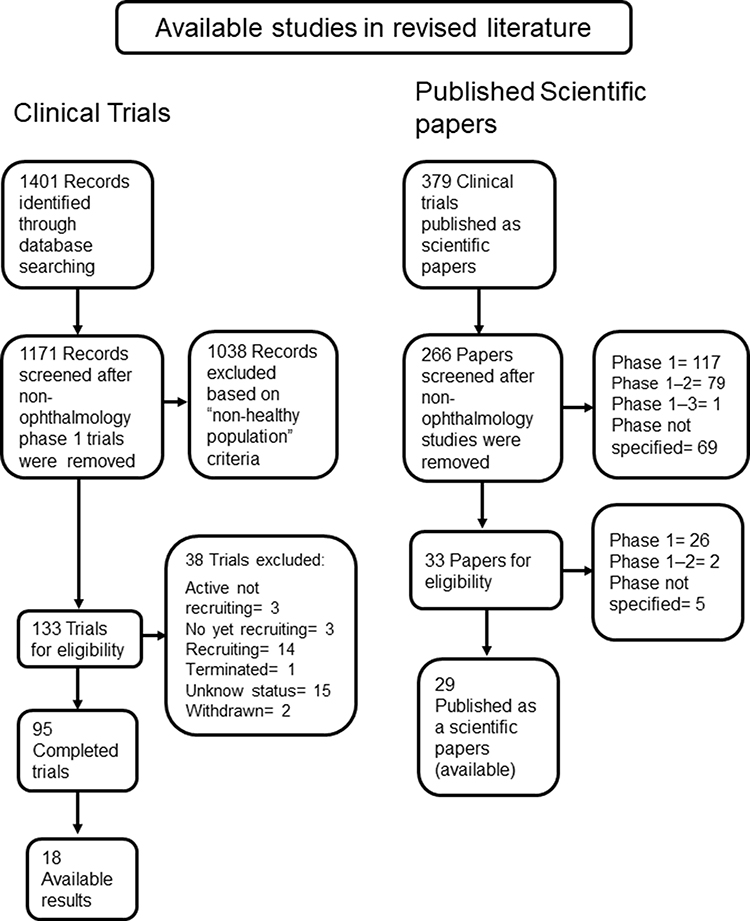 |
Figure 2 Flow chart of studies. |
This study had several limitations. First, the data presented belonged to NHV under pharmacological treatment, but no participants assigned to placebo were included. This means that interpretations of safety and tolerability may be biased.2,4 Second, due to ethical reasons, the trials included in this study were executed following a single dose/concentration schedule, in order to reduce the unnecessary risk of possible harm, especially since the active ingredients are known compounds and their posology has already been established. However, despite these limitations, the overall strength of our statistical analyses and the data management allowed mitigation of these risk perceptions.
Conclusion
In conclusion, phase 1 clinical trials of ophthalmologic medications provide meaningful information with a small risk of yielding any severe or long-lasting adverse events. Furthermore, it was also demonstrated that the instillation of ID is as safe and tolerable as that of AD in NHV. Safety and tolerability profiling can not only protect future patients of deleterious local or systemic signs or symptoms induced by ophthalmic drugs but also assess the tolerability associated with them and therefore their potential effectiveness both through their actions on the ocular surface and the prospective adherence for medium and long-term treatment periods.
Abbreviations
AD, approved drug; AE, adverse event; CIC, conjunctival impression cytology; FBS, foreign body sensation; GLS, green lissamine staining; ID, investigational drug; IOP, intraocular pressure; LAE, liver associated enzymes; NHV, normal healthy volunteer; OCI, ocular comfort index; VA, visual acuity.
Ethics Approval and Consent to Participate
The studies’ protocols were approved by their respective Institutional Review Boards, as follows: Comité de Ética en Investigación Hospital Real San José; Comité de Investigación Instituto Jalisciense de Investigación Clínica S.A. de C.V.; Comité de Ética en Investigación Centro Hospitalario Vicor, S.A. de C.V., CHG Hospitales; and Comité de Ética en Investigación de la Unidad de Bioequivalencia, S. de R.L. de C.V. All volunteers that participated provided written and signed informed consent.
Acknowledgments
The authors thank Dr. Alfredo Gazca (Biomedical Research G&L, S. de R.L. de C.V.), Dr. Mariana Díaz (Private office), Dr. Anel De Luca (Centro Oftalmológico San Ángel), Dr. Manuel Zepeda (Unidad Clínica de Bioequivalencia, S. de R.L. de C.V.), Dr. José Navarro (Private Office), and Dr. Miguel Padilla (Private Office).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
This study was sponsored by Laboratorios Sophia, S.A. de C.V. (Zapopan, Jalisco, Mexico). The funder provided support in the form of salaries for authors, but this commercial affiliation did not have any additional role in the data collection.
References
1. Wei Y, Li H, Wang H, Zhang S, Sun Y. Psychological status of volunteers in a Phase I clinical trial assessed by Symptom Checklist 90 (SCL-90) and Eysenck Personality Questionnaire (EPQ). Med Sci Monit. 2018;24:4968–4973. doi:10.12659/MSM.909524
2. McManus L, Fisher JA. To report or not to report: exploring healthy volunteers’ rationales for disclosing adverse events in Phase I drug trials. AJOB Empir Bioeth. 2018;9(2):82–90. doi:10.1080/23294515.2018.1469552
3. Edwards IR, Aronson JK. Adverse drug reactions: definitions, diagnosis, and management. Lancet. 2000;356(9237):1255–1259. doi:10.1016/S0140-6736(00)02799-9
4. Johnson RA, Rid A, Emanuel E, Wendler D. Risks of phase I research with healthy participants: a systematic review. Clin Trials. 2016;13(2):149–160. doi:10.1177/1740774515602868
5. Tanihara H, Inoue T; K-115 Clinical Study Group. Phase 1 clinical trials of a selective Rho kinase inhibitor, K-115. JAMA Ophthalmol. 2013;131(10):1288–1295. doi:10.1001/jamaophthalmol.2013.323
6. Teuchner B, Schmid E, Ulmer H, Gottardi W, Nagl M. Tolerability of N-chlorotaurine plus ammonium chloride in the rabbit and human eye–a phase 1 clinical study. Graefes Arch Clin Exp Ophthalmol. 2008;246(12):1723–1730. doi:10.1007/s00417-008-0900-x
7. Ackert J, Mohamed K, Slakter JS, et al. Randomized placebo-controlled trial evaluating the ophthalmic safety of single-dose tafenoquine in healthy volunteers. Drug Saf. 2019;42(9):1103–1114. doi:10.1007/s40264-019-00839-w
8. Quiroz-Mercado H, Ivri E, Gonzalez-Salinas R, et al. Clinical evaluation of a novel electromechanical topical ocular drug delivery system: two Phase 1 proof of concept studies. Clin Ophthalmol. 2020;14:139–147. doi:10.2147/OPTH.S221749
9. Buoen C, Bjerrum OJ, Thomsen MS. How first-time-in-human studies are being performed: a survey of phase I dose-escalation trials in healthy volunteers published between 1995 and 2004. J Clin Pharmacol. 2005;45(10):1123–1136. doi:10.1177/0091270005279943
10. Drugs for Some Common Eye Disorders. JAMA. 2020;323(5):470–471. doi:10.1001/jama.2019.20663
11. Cai Z, Christianson AM, Ståhle L, Keisu M. Reexamining transaminase elevation in Phase I clinical trials: the importance of baseline and change from baseline. Eur J Clin Pharmacol. 2009;65(10):1025–1035. doi:10.1007/s00228-009-0684-x
12. van den Bogert CA, Souverein PC, Brekelmans CT, et al. Non-publication is common among Phase 1, single-center, not prospectively registered, or early terminated clinical drug trials. PLoS One. 2016;11(12):e0167709. doi:10.1371/journal.pone.0167709
13. Allen EN, Chandler CI, Mandimika N, Leisegang C, Barnes K. Eliciting adverse effects data from participants in clinical trials. Cochrane Database Syst Rev. 2018;1(1):MR000039.
14. Decullier E, Chan AW, Chapuis F. Inadequate dissemination of phase I trials: a retrospective cohort study. PLoS Med. 2009;6(2):e1000034. doi:10.1371/journal.pmed.1000034
15. Gilger BC. Concerns with analysis of correlated eye data. Vet Ophthalmol. 2011;14(3):214. doi:10.1111/j.1463-5224.2011.00893.x
16. Armstrong RA. Statistical guidelines for the analysis of data obtained from one or both eyes. Ophthalmic Physiol Opt. 2013;33(1):7–14. doi:10.1111/opo.12009
17. Michel M, Sickenberger W, Pult H. The effectiveness of questionnaires in the determination of contact lens induced dry eye. Ophthalmic Physiol Opt. 2009;29(5):479–486. doi:10.1111/j.1475-1313.2009.00658.x
18. Johnson ME, Murphy PJ. Measurement of ocular surface irritation on a linear interval scale with the ocular comfort index. Invest Ophthalmol Vis Sci. 2007;48(10):4451–4458. doi:10.1167/iovs.06-1253
19. Rosenzweig P, Miget N, Brohier S. Transaminase elevation on placebo during phase I trials: prevalence and significance. Br J Clin Pharmacol. 1999;48(1):19–23. doi:10.1046/j.1365-2125.1999.00952.x
20. Szumilas M. Explaining odds ratios. J Can Acad Child Adolesc Psychiatry. 2010;19(3):227–229.
21. Walker RL, Cottingham MD, Fisher JA. Serial participation and the ethics of Phase 1 healthy volunteer research. J Med Philos. 2018;43(1):83–114.
22. McAlinden C, Gao R, Wang Q, et al. Rasch analysis of three dry eye questionnaires and correlates with objective clinical tests. Ocul Surf. 2017;15(2):202–210. doi:10.1016/j.jtos.2017.01.005
23. Zhang X, Vadoothker S, Munir WM, Saeedi O. Ocular surface disease and glaucoma medications: a clinical approach. Eye Contact Lens. 2019;45(1):11–18. doi:10.1097/ICL.0000000000000544
24. Mottow-Lippa LS, Lippa EA, Naidoff MA, Clementi R, Bjornsson T, Jones K. 008% timolol ophthalmic solution. A minimal-effect dose in a normal volunteer model. Arch Ophthalmol. 1990;108(1):61–64. doi:10.1001/archopht.1990.01070030067030
25. Sponsel WE, Mensah J, Kiel JW, et al. Effects of latanoprost and timolol-XE on hydrodynamics in the normal eye. Am J Ophthalmol. 2000;130(2):151–159. doi:10.1016/S0002-9394(00)00401-3
26. Breithaupt-Groegler K, Coch C, Coenen M, et al. Who is a ‘healthy subject’?-consensus results on pivotal eligibility criteria for clinical trials. Eur J Clin Pharmacol. 2017;73(4):409–416. doi:10.1007/s00228-016-2189-8
27. Piton A, Poynard T, Imbert-Bismut F, et al. Factors associated with serum alanine transaminase activity in healthy subjects: consequences for the definition of normal values, for selection of blood donors, and for patients with chronic hepatitis C. MULTIVIRC Group. Hepatology. 1998;27(5):1213–1219. doi:10.1002/hep.510270505
28. Pacifico L, Ferraro F, Bonci E, Anania C, Romaggioli S, Chiesa C. Upper limit of normal for alanine aminotransferase: quo vadis? Clin Chim Acta. 2013;422:29–39. doi:10.1016/j.cca.2013.03.030
29. Chan AW, Hróbjartsson A, Haahr MT, Gøtzsche PC, Altman DG. Empirical evidence for selective reporting of outcomes in randomized trials: comparison of protocols to published articles. JAMA. 2004;291(20):2457–2465. doi:10.1001/jama.291.20.2457
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.