")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Reductive metabolism of oxymatrine is catalyzed by microsomal CYP3A4
Authors Liu W, Shi J, Zhu L, Dong L, Luo F, Zhao M, Wang Y, Hu M, Lu L, Liu Z
Received 12 July 2015
Accepted for publication 23 September 2015
Published 30 October 2015 Volume 2015:9 Pages 5771—5783
DOI https://doi.org/10.2147/DDDT.S92276
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Wenqin Liu,1,2,* Jian Shi,1,2,* Lijun Zhu,2 Lingna Dong,1 Feifei Luo,2 Min Zhao,2 Ying Wang,2 Ming Hu,2,3 Linlin Lu,2 Zhongqiu Liu1,2
1Department of Pharmaceutics, School of Pharmaceutical Sciences, Southern Medical University, Guangzhou, Guangdong, People’s Republic of China; 2International Institute for Translational Chinese Medicine, Guangzhou University of Chinese Medicine, Guangzhou, Guangdong, People’s Republic of China; 3Department of Pharmacological and Pharmaceutical Sciences, College of Pharmacy, University of Houston, Houston, TX, USA
*These authors contributed equally to this work
Abstract: Oxymatrine (OMT) is a pharmacologically active primary quinolizidine alkaloid with various beneficial and toxic effects. It is confirmed that, after oral administration, OMT could be transformed to the more toxic metabolite matrine (MT), and this process may be through the reduction reaction, but the study on the characteristics of this transformation is limited. The aim of this study was to investigate the characteristics of this transformation of OMT in the human liver microsomes (HLMs) and human intestinal microsomes (HIMs) and the cytochrome P450 (CYP) isoforms involved in this transformation. The current studies demonstrated that OMT could be metabolized to MT rapidly in HLMs and HIMs and CYP3A4 greatly contributed to this transformation. All HLMs, HIMs, and CYP3A4 isoform mediated reduction reaction followed typical biphasic kinetic model, and Km, Vmax, and CL were significant higher in HLMs than those in HIMs. Importantly, different oxygen contents could significantly affect the metabolism of OMT, and with the oxygen content decreased, the formation of metabolite was increased, suggesting this transformation was very likely a reduction reaction. Results of this in vitro study elucidated the metabolic pathways and characteristics of metabolism of OMT to MT and would provide a theoretical basis and guidance for the safe application of OMT.
Keywords: oxymatrine, matrine, reduction reaction, CYP3A4, metabolism, microsomes
Introduction
Oxymatrine (OMT) is a primary effective and toxic quinolizidine alkaloid derived from several traditional Chinese medicines (TCMs) and is a well-known active component of Sophorae flavescentis Radix (Kushen) and Sophora subprostate Root (Shandougen), which are widely distributed in Asia and the Pacific islands.1–3 Recently, these medicines, which are generally used for the treatment of hepatitis and cardiac diseases, have held more and more attraction in the scientific field due to their many pharmacological activities, such as antiviral, anti-hepatitis B virus, neuroprotective, cardioprotective, antioxidant, and antitumor activities.4 Quinolizidine alkaloids, which have powerful and wide pharmacological activities, have been proven to be the major bioactive constituents in S. flavescens. Among them, OMT is of great pharmacological interest because it possesses various pharmacological activities including protection against apoptosis, tumor and fibrotic tissue development, and inflammation.5–7 Furthermore, OMT has been shown to decrease cardiac ischemia8 (decreased blood perfusion), myocardial injury,9 arrhythmias (irregular heartbeats),10 and improve heart failure by increasing cardiac function.11 Therefore, OMT has been widely used as human drugs or animal feed additives for treatment and health care.12–14
Recently, except the studies on its molecular mechanisms of the pharmacological activities, much attention has been focused on the absorption and metabolism of OMT. Our group has demonstrated that the oral absolute bioavailability of OMT was only 6.79%±2.52% and that most of OMT was converted to its metabolite matrine (MT) in vivo.15 This result was consistent with other reports, which confirmed that OMT could be rapidly metabolized to MT in the gastrointestinal tract and liver after oral administration.16,17 The AUC value of MT was much higher than that of OMT, thus suggesting that MT may also play a significant role in the pharmacological action of orally administered OMT. OMT and MT have been demonstrated to function as the foremost toxic alkaloids, with the nervous system as the main target of MT.2 The therapeutic and toxic potential of OMT and MT has been extensively discussed in literatures for years. Because MT has higher toxicity and different pharmacological effects as compared with those of OMT,18,19 the metabolism of OMT could affect its safety and efficacy in clinical treatment. Therefore, understanding the disposition of OMT is the first and key step to solving the main challenge associated with OMT development. The fast metabolism of OMT in vivo lets us make a hypothesis that first-pass metabolism was the major reason the oral bioavailability of OMT was very low, with substantial amount of MT found in the plasma. Although the metabolic characteristics of OMT in rats have been reported,20 the valid information about the characterization of the metabolic mechanism in humans is poorly understood.
Cytochrome P450 (CYP) enzymes are involved in the metabolism of many currently available drugs21 and these significant Phase I drug-metabolizing enzymes participate in xenobiotic metabolism and detoxification.22 It has been estimated that most human drug metabolism can be attributed to eleven main enzymes (CYP1A1, CYP1A2, CYP1B1, CYP2A6, CYP2B6, CYP2C8, CYP2C19, CYP2D6, CYP2E1, CYP3A4, and CYP3A5).23 The increased polarity of drugs by CYP metabolism increases the water solubility of these drugs and leads to their more rapid elimination from the body. In addition to catalyzing the oxidation of endogenous/xenobiotics compounds, CYP has also been shown to play a role in the reduction of many drugs, such as triarylmethanes, azodyes, nitro compounds, and N-acetyl-p-benzoquinone imine. Particularly, under anaerobic conditions, CYP has been shown to function in a reductive rather than in an oxidative manner. Because OMT has a reducible N-oxide group and could be metabolized to MT in vivo, this metabolism of OMT to MT could be a reduction reaction,24,25 which is likely to be mediated by CYP enzymes. Recently, multiple drug therapy has become a common therapeutic practice, particularly in patients with various diseases or conditions.26 Therefore, CYP enzymes could be inhibited or induced by concomitant drug treatment. These changes in enzymes activities could markedly change the exposure of drug or its metabolites at possibly toxic levels in the body. Both OMT and MT are potentially highly toxic, and MT has higher toxicity. Consequently, changes in the enzyme activities, which mediate the reduction action, through simultaneous administration with other drugs that possess potent inhibitory effects may significantly change the concentrations of OMT or MT in plasma. Therefore, the characteristics of metabolism of OMT would be significantly useful for clinical use. However, the role of CYP in the metabolism of OMT and the characteristics of OMT are unknown.
The liver and intestine are considered the main sites of metabolism of drugs after oral administration. Therefore, the aim of this in vitro study was to investigate the metabolic characteristics of OMT in human liver microsomes (HLMs), human intestinal microsomes (HIMs), and in recombinant CYP450 enzymes. The selective chemical inhibitors of specific CYP enzymes (CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4) were used to confirm and identify the CYP isoforms that were mainly responsible for the OMT metabolism. Moreover, the metabolism of OMT under different oxygen content conditions was compared to confirm the characteristics of the metabolic reaction involved. The prospective results would provide helpful and in-depth information on OMT metabolism in humans that may be useful in the clinical application of OMT and OMT-containing herbs to help avoid the toxic effects and to control the pharmacological effects. The information on OMT transformation by CYP enzymes would be valuable for the development of applications for toxicological, pharmacological, and clinical use. The protocol used in the present study was approved by the Southern Medical University’s Ethics Committee. The study was undertaken after the obtainment of informed consent.
Materials and methods
Chemicals and reagents
OMT (C15H24N2O2, ≥98%) and MT (C15H24N2O, ≥98%) were purchased from Chengdu Mansite Pharmaceutical Co. Ltd. (Chengdu, People’s Republic of China). The recombinant human CYP isoforms (CYP1A1, CYP1A2, CYP1B1, CYP2A6, CYP2B6, CYP2C8, CYP2C19, CYP2D6, CYP2E1, CYP3A4, and CYP3A5, 4 mg/mL), pooled HIMs, pooled HLMs (20 mg/mL, including reductase), solution A (β-NADP, glucose-6-phosphate and magnesium chloride), solution B (glucose-6-phosphate dehydrogenase in sodium citrate), potassium phosphate dibasic (K2HPO4), potassium dihydrogen phosphate (KH2PO4), and the selective chemical inhibitors of CYP isoforms (fluvoxamine maleate, gemfibrozil, amiodarone hydrochloride, omeprazole, quinidine, ketoconazole, and diethyldithiocarbamic acid) were purchased from BD Gentest Corp. (Woburn, MA, USA). Testosterone (TES, >98%) was used as an internal standard (IS) and purchased from Nacalai Tesque (Kyoto, Japan). Acetonitrile, dichloromethane, and formic acid were HPLC grade. Ultrapure water was used for all analyses. All of the other chemicals and reagents commercially available were of the highest analytical grade.
UPLC analysis of OMT and metabolite and identification by LC/MS
The following UPLC conditions were used to analyze OMT and MT: systems, Agilent 1290 Infinity LC system (Agilent Technologies, Santa-Clara, CA, USA), which consisted of a solvent degasser, a binary pump, an autosampler, and a column oven; column, Agilent ZORBAX RRHD SB-C18 column (100 mm ×3 mm, 1.8 μm); mobile Phase B, 100% acetonitrile; mobile Phase A, 0.1% formic acid in water; flow rate, 0.3 mL/min; gradient, 95%–85% A for 0–1.8 min, 85%–64% A for 1.8–4.0 min, 64%–55% A for 4.0–4.5 min, 55%–5% A for 4.5–5.0 min, 5%–5% A for 5.0–6.5 min, 5%–95% A for 6.5–7.5 min, and 95%–95% A for 7.5–8.0 min; wavelengths, 210 nm for OMT and MT, and 243 nm for TES; injection volume, 10 μL. The MS/MS detector used was an Agilent 6540 quadrupole-time of flight (Q-TOF) mass spectrometer in combination with an Agilent 1,290 Infinity ultrahigh-performance liquid chromatography system. Samples were analyzed using Dual AJS ESI (Agilent Technologies) in the positive model. The main working parameters were set as follows: capillary voltage, 3,500 V; temperature of the dry heater, 300°C; nebulizer voltage, 35 (psig); dry gas, 8.0 L/min; sheath gas temperature, 350°C; and sheath gas flow, 11 L/min. Data were collected and analyzed by the Qualitative Analysis software (version B.06.00, Agilent Technologies).
Method validation
To ensure that the method was feasible, the testing of accuracy, precision, and stability of MT in potassium phosphate buffer (KPI, pH 7.4) solution were conducted. The intra- and interday accuracy and precision of the analytes were determined by analyzing six sets of spiked plasma samples at three quality control concentrations on the same day or 3 consecutive days, respectively. Accuracy (%) was obtained by calculating the percentage deviation from the theoretical concentration [(observed value of concentration)/true value of concentration ×100%]. Precision (%) was determined by calculating the relative standard deviation (% RSD) for inter- and intraday replicates. The stability in KPI solution was tested after storage at 0.5, 1, 1.5, and 2 hours incubation at 37°C using the QC samples at three concentrations (six samples for each concentration).
Incubation of OMT with HLMs
A typical Phase I incubation mixture contained 0.1 mg/mL protein, 3.3 mM glucose-6-phosphate, 3.3 mM MgCl2, 0.4 U/mL glucose-6-phosphate dehydrogenase, and OMT (50, 100, or 200 μM) in the 50 mM potassium phosphate buffer (KPI, pH 7.4).27 The incubation mixture was preincubated at 37°C for 5 min, and the reaction was initiated with the addition of an NADP to form a NADPH-regenerating system. Incubation without the addition of NADP was regarded as the control. The final mixture (500 μL) was incubated in a shaking water bath (150 rpm) at 37°C for 1.5 hours to obtain more metabolites. Subsequently, the reaction was terminated by adding 4 mL ice-cold CH2Cl2, then 200 μL of TES (100 μM, dissolved in acetonitrile, used as an IS). The mixture was vortexed for 8 min, then centrifuged at 1,000 rpm for 10 min to remove precipitated protein. The organic phase was drawn into another tube and dried with a gentle stream of nitrogen gas. The residue was reconstituted with 150 μL of 50% methanol in water. Each sample was vortexed for 2 min, and then centrifuged at 13,000 rpm for 30 min for UPLC analysis. All reactions were performed in triplicate.
Metabolism of OMT in recombinant human CYP enzymes
Incubation of OMT (50 μM) with eleven recombinant human CYP enzymes (CYP1A1, CYP1A2, CYP1B1, CYP2A6, CYP2B6, CYP2C8, CYP2C19, CYP2D6, CYP2E1, CYP3A4, and CYP3A5) was conducted as described above for HLMs, except that the concentration of enzymes used was 0.08 mg/mL. All reactions were performed in triplicate.
CYP450 inhibition experiments in HLMs
The effects of specific chemical inhibitors for CYP enzymes on OMT metabolism were investigated in HLMs. Well-characterized inhibitors of specific CYP enzymes, ie, fluvoxamine maleate for CYP1A2,28,29 gemfibrozil for CYP2C8,28,29 amiodarone hydrochloride for CYP2C9,28,30 omeprazole for CYP2C19,28,30 quinidine for CYP2D6,29 diethyldithiocarbamic acid for CYP2E1,31 and ketoconazole for CYP3A29 were preincubated for 5 min with microsomes at 37°C and an NADPH-regenerating system before the addition of OMT (50 μM) to initiate the reaction. Two concentrations of each inhibitor were used in the experiment. The Ki value of each inhibitor was the low concentration, and the twice Ki value was the high concentration. The concentrations of each inhibitor were 0.2 and 0.4 μM for CYP1A2, 70 and 140 μM for CYP2C8, 2 and 4 μM for CYP2C9, 5 and 10 μM for CYP2C19, 0.4 and 0.8 μM for CYP2D6, 10 and 20 μM for CYP2E1, and 0.2 and 0.4 μM for CYP3A4. Incubation without the addition of inhibitors was regarded as the control. The chemical inhibition test of TES (50 μM) was used as a positive control of the CYP3A4 inhibitor. All reaction were performed in triplicate. The incubation procedures and extraction methods were similar to that for HLMs.
Kinetics of OMT metabolism in HLMs, HIMs, and CYP3A4 isoform
The incubation procedures for measuring kinetic profiling of OMT in HLMs, HIMs, and CYP3A4 were the same as described earlier. The substrate concentrations used for HLMs were 5, 10, 50, 75, 200, 500, and 800 μM; the substrate concentrations used for HIMs were 5, 10, 50, 75, 100, 200, 500, and 800 μM; and the substrate concentrations used for HIMs were 5, 10, 50, 75, 100, 200, 500, and 800 μM. The time of incubation for all concentrations was 2 h. Rates of OMT metabolism by HLMs, HIMs, and CYP3A4 isoform were expressed as amounts of metabolites formed per min per mg protein (nmol/min/mg). Kinetic parameters were then obtained according to the profile of Eadie–Hofstee plots.32,33 All reactions were performed in triplicate. A comparison was made between these kinetic parameters from the three different catalysts. If the Eadie–Hofstee plot was linear, formation rates (V) of MT at respective substrate concentrations (C) were fit to the standard Michaelis–Menten equation:
|
|
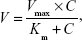
where Km is the Michaelis–Menten constant, and Vmax is the maximum rate of forming MT.
If Eadie–Hofstee plots showed characteristic profiles of atypical kinetics, including autoactivation, biphasic kinetics, and substrate inhibition kinetics, the data from these atypical profiles were fit to the equations, using the ADAPT II program.34,35 To determine the best-fit model, the model candidates were discriminated using the Akaike’s information criterion (AIC),36 and the rule of parsimony was applied. Therefore, using this minimum AIC estimation, a negative AIC value would be considered a better representation of the data versus a set of data having a positive AIC value. According to these rules, Eadie–Hofstee plots of OMT in HLMs, HIMs, and CYP3A4 showed characteristic profiles of biphasic kinetics. Therefore, the kinetic parameters were fit to Equation 2, which described enzyme reactions with biphasic kinetics:
|
|
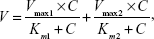
where Vmax1 is the maximum enzyme velocity of the high-affinity phase, Vmax2 is the maximum velocity of the low-affinity phase, Km1 is concentration of substrate to achieve half of Vmax1 for high-affinity phase, and Km2 is concentration of substrate to achieve half of Vmax2 for high-affinity phase.
Effects of different oxygen contents on the metabolism of OMT in HLMs
The effect of different oxygen contents on metabolism OMT of was assessed by comparing the amount of metabolite formed in four experimental groups: open group (the samples in the opened glass tube were incubated in the recycled air), closed group (the samples in the closed glass tube were incubated in the stale air), ventilation for 2 s group (oxygen in the glass tube was driven out by helium or nitrogen for 2 s, then the samples were incubated in the closed glass tube), and ventilation for 30 s group (oxygen in the glass tube was driven out by helium or nitrogen for 30 s, then the samples were incubated in the closed glass tube). Three concentrations (50, 100, and 200 μM) of OMT were used to investigate the effect of different oxygen contents on its metabolism. Furthermore, OMT (50 μM) was incubated with the inactivated enzyme to determine whether there is a chemical reaction involved. The enzyme was inactivated by incubation in boiled water for 2 h, and hence cannot metabolize any substrate. The procedures were the same as ventilation for 30 s group before incubation. All reactions were performed in triplicate. The incubation procedures and extraction methods were the same as described above for HLMs.
Effects of different oxygen contents on the oxidative metabolism of TES in HLMs
According to previous reports, TES could be oxidized to 6-β-OH-testosterone by CYP3A4.37 Therefore, TES was used as a positive control in this study to investigate the effect of different oxygen contents on oxidative metabolism.38,39 The amount of oxidative metabolite formed in four experimental groups was compared as described earlier for OMT. Three concentrations (50, 100, and 200 μM) of TES were tested to confirm the effect of different oxygen contents on its metabolism. All reactions were performed in triplicate. The incubation procedures and extract methods were the same as described earlier.
Statistical analysis
Data were expressed as mean ± SD. One-way ANOVA with or without Tukey’s multiple comparison (post hoc) tests was used to evaluate the statistical differences. Differences were considered significant when P<0.05.
Results
Identification of OMT metabolite in HLMs
The HPLC method developed for OMT and MT had a run time of 4.4 min (Figure 1). The tested linear response range was 0.39–400 μM (for a total of 11 concentrations) with the lower limit of quantification of 0.2 μM.
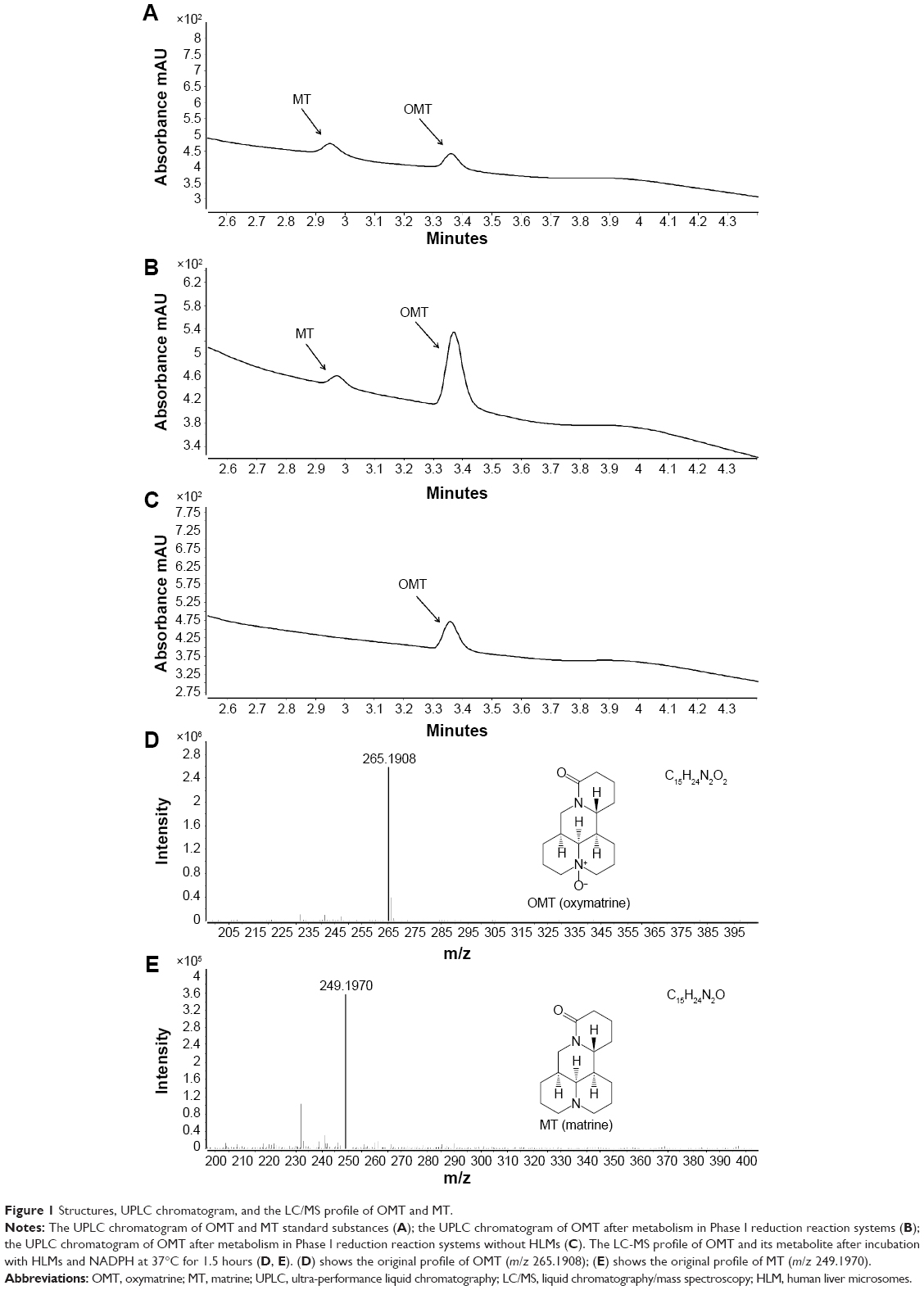 | Figure 1 Structures, UPLC chromatogram, and the LC/MS profile of OMT and MT. |
Compared with control (Figure 1C), only one metabolite was found in HLMs (Figure 1B). The metabolite was identified by its chromatographic behavior and characteristic mass spectrometric fragmentation feature. The prototype peak eluted at 3.36 min corresponded to unmodified OMT, whereas the metabolite peak eluted at 2.95 min was identified as MT by comparing it with standard substances (Figure 1A). A UPLC-ESI-Q-TOF-MS running at a positive ion mode was used to confirm the MS spectrum of the OMT and MT. The peak eluting at 3.36 min OMT showed a pseudo-molecule ion [M+H]+ at m/z 265.1908 (Figure 1D) in the full scan mass spectrum, which confirmed the smart molecular formula (C15H24N2O2). Metabolite MT had HPLC retention time of 2.95 min. A pseudo-molecule ion [M+H]+ at m/z 249.1970 (Figure 1E) confirmed the smart molecular formula (C15H24N2O) and a loss of 16 Da (O) from OMT, hence suggesting that it was the reduzate of OMT.
Method validation
The intra- and interday precisions and accuracies of QC samples are presented in Table S1. The intraday precision (RSD) ranged between 1.0% and 4.3%, and accuracy (RE) ranged between 98.3% and 102.7%. Interday precision (RSD) ranged between 1.5% and 3.6%, and accuracy (RE) ranged between 97.7% and 103.7%, respectively. The results of stability experiments showed that no significant degradation occurred, and these results are summarized in Table S2. All inter- and intraday precision and accuracy and stability were acceptable for working in KPI solution.
Metabolism of OMT in recombinant human CYP enzymes
To determine which CYP isoforms primarily contributed to the metabolism of OMT, 11 recombinant human CYP enzymes were incubated with OMT. Figure 2 showed that CYP3A4 contributed greatly to the formation of MT, whereas only a small amount of MT could be detected after the incubation of other cDNA-expressed CYP enzymes.
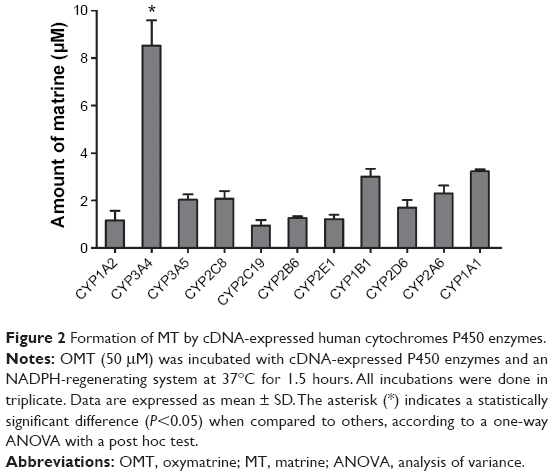 | Figure 2 Formation of MT by cDNA-expressed human cytochromes P450 enzymes. |
Effect of CYP-specific chemical inhibitors on MT formation in HLMs
To further determine which isoform of CYP could be involved in OMT metabolism, the effect of CYP-specific chemical inhibitors on OMT metabolism was investigated. The results in Figure 3A showed that the formation rate of MT was significantly inhibited by the inhibitors of CYP1A2, CYP3A, CYP2C9, CYP2C19, and CYP2E1 at both concentrations, and the inhibitor of CYP3A was the most effective inhibitor. Compared with the control, the other chemical inhibitors (CYP2C8 and CYP2D6) had weak inhibitory effects on metabolite formation, but the effects were not significant. In addition to this, the chemical inhibition test of TES was used as a positive control of the CYP3A4 inhibitor. Results indicated that TES metabolism was inhibited by over 95%, which confirmed the strong inhibitory effect on CYP3A of the selected inhibitor (Figure 3B).
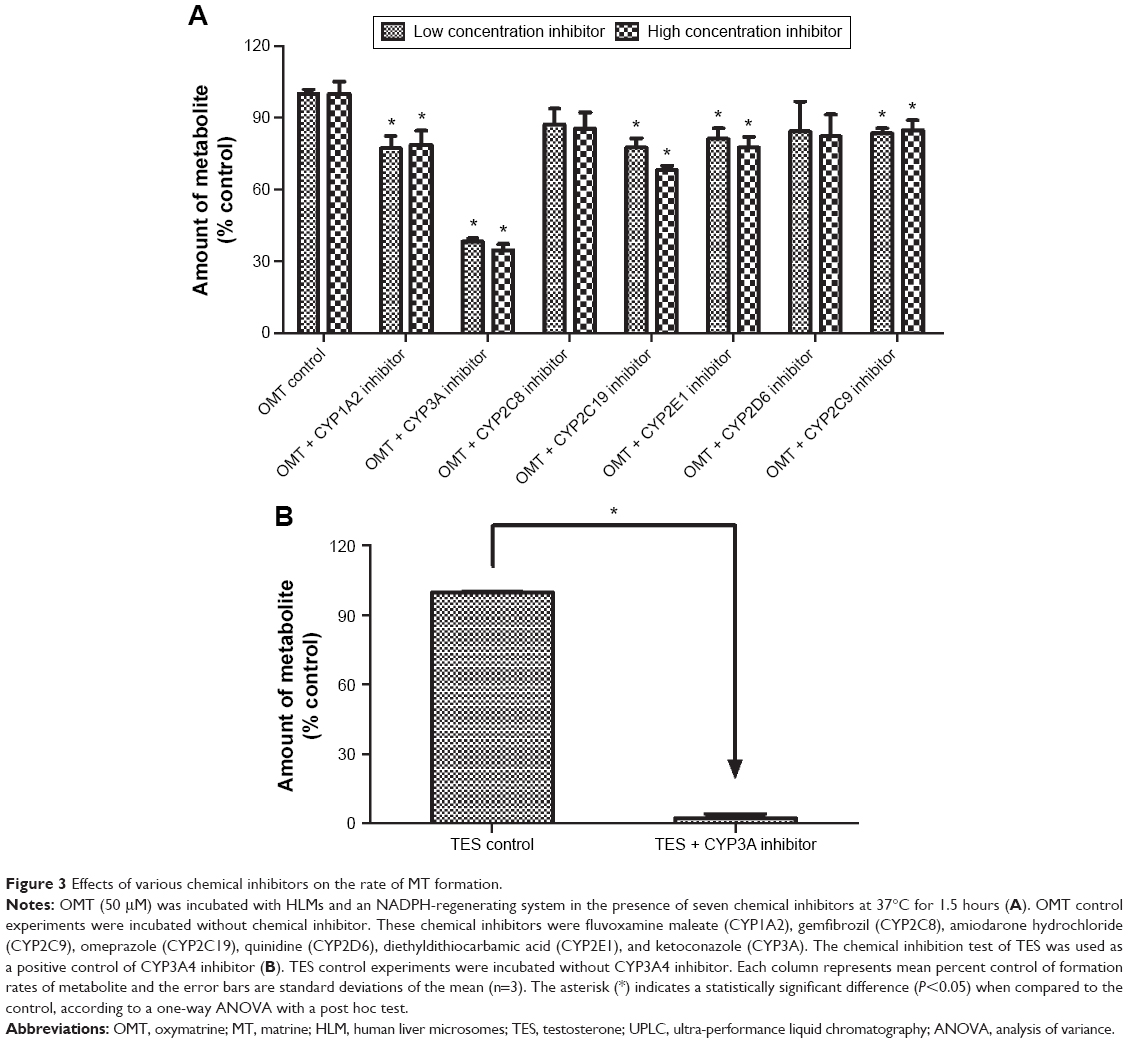 | Figure 3 Effects of various chemical inhibitors on the rate of MT formation. |
Kinetics of OMT metabolism in HLMs, HIMs, and CYP3A4 isoform
The effects of the concentration of OMT on its metabolism activity by HLMs, HIMs, and CYP3A4 are depicted by the direct plots and the Eadie–Hofstee plots (Figure 4). The Vmax and Km as well as the CLint, which were calculated from the formations of MT, are summarized in Table 1. The OMT metabolism catalyzed by HLMs, HIMs, and CYP3A4 all followed the biphasic kinetics equation. In general, the Vmax and CLint values were much higher in HLMs than in HIMs (1.49±0.24 versus 0.46±0.05 nmol/min/mg and 0.0067 versus 0.0031 mL/min/mg).
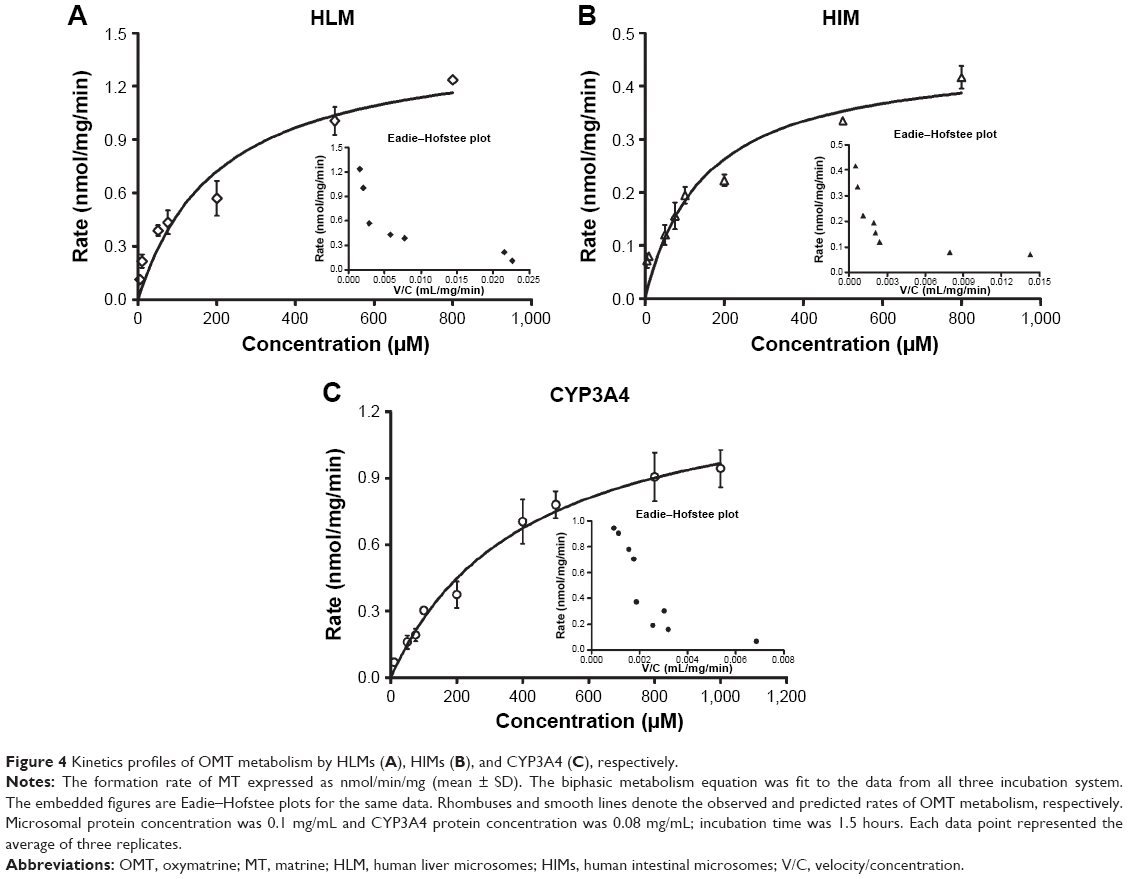 | Figure 4 Kinetics profiles of OMT metabolism by HLMs (A), HIMs (B), and CYP3A4 (C), respectively. |
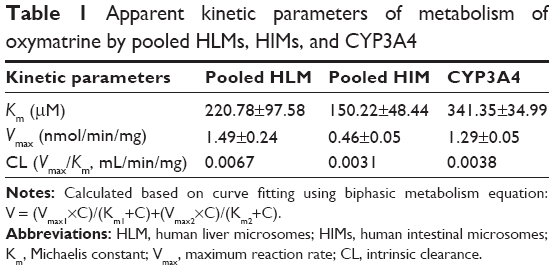 | Table 1 Apparent kinetic parameters of metabolism of oxymatrine by pooled HLMs, HIMs, and CYP3A4 |
Effects of different oxygen contents on OMT metabolism in HLMs
A comparison of the amount of metabolite formed in the four experimental groups indicated that with the increase of ventilation time, the amount of metabolite of OMT formed was increased (Figure 5). This illustrated that reduced oxygen content would increase the formation of metabolite. The ventilation for the 30 s group generated the most metabolite among all three concentration groups. Compared to the open group, the amount of MT in the ventilation for 30 s group was increased by 50%–290% (Figure 5A and B). However, no metabolites were observed in the inactivated enzyme test (Figure 5C).
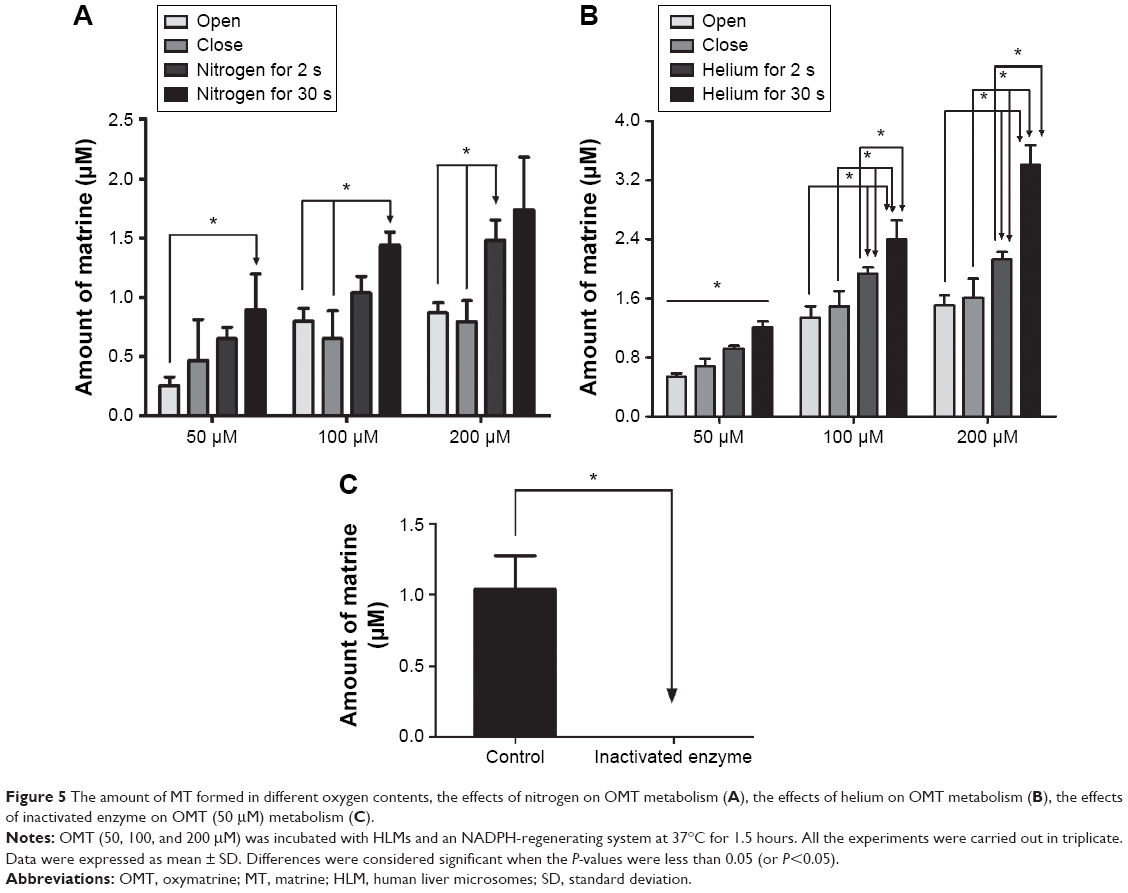 | Figure 5 The amount of MT formed in different oxygen contents, the effects of nitrogen on OMT metabolism (A), the effects of helium on OMT metabolism (B), the effects of inactivated enzyme on OMT (50 μM) metabolism (C). |
Effects of different oxygen contents on TES metabolism in HLMs
TES could be oxidized by CYP3A to generate 6-β-OH-TES through the oxidative pathway.39 Therefore, TES was used as a positive control to investigate the effect of different oxygen contents on oxidative metabolism. A comparison of the amount of oxidative metabolite formed in four experimental groups indicated that oxygen in the incubations was reduced as the ventilation time was increased; consequently, the amount of oxidative metabolite formed was decreased (Figure S1). The open group generated the most metabolite among the three concentration groups, whereas the ventilation for 30 s group generated the least metabolite. Compared with the open group, the amount of 6-β-OH-TES in the ventilation for 30 s group was reduced by 20%–60%.
Discussion
OMT is the main active ingredient in the traditional Chinese herbal medicine Sophora subprostrata; this compound has been used as an essential agent for several years because of its extremely excellent effects against hepatitis and cardiovascular disease.40,41 However, aside from its useful pharmacological activities, OMT causes side effects and liver toxicity.42,43 To understand the mechanism of its pharmacological action, several reports studied the pharmacokinetic behavior of OMT and found that, after oral administration, OMT could be metabolized to MT, which is more toxic than OMT, in humans and other animal species.24,44 However, the knowledge on the metabolic pathways and metabolism of OMT was limited. In this study, the characteristics and mechanism of metabolism of OMT were determined using HLMs and HIMs and the CYP isoforms responsible for the metabolism with chemical inhibitors. In addition, we confirmed the metabolic pathway of OMT was a reduction reaction first through changing the oxygen contents in the incubation system.
Results of this study showed that OMT could be metabolized to MT in HLMs (Figure 1). To further confirm which CYP isoforms were responsible for the metabolism of OMT, eleven recombinant human CYP enzymes (CYP1A1, CYP1A2, CYP1B1, CYP2A6, CYP2B6, CYP2C8, CYP2C19, CYP2D6, CYP2E1, CYP3A4, and CYP3A5) were assayed. The CYPs responsible for this were identified because most of human drug metabolism may be attributed to these eleven enzymes.45 The results showed that CYP3A4 greatly contributed to the formation of MT, whereas other enzymes only played minor roles (Figure 2). To further confirm our results, a CYP-specific chemical inhibitor study was conducted to investigate which CYP isoforms were primarily involved in the metabolism of OMT. The results showed that the CYP3A4 inhibitor had a strong inhibitory effect, whereas the CYP1A2, CYP2C9, CYP2C19, and CYP2E1 inhibitors only had a modest inhibitory effect. However, because the specific chemical inhibitor could only block or reduce the rate of reaction and because their concentrations were not very high, the reaction was not stopped completely and some MT was generated in the experiments. By contrast, although the inhibitors of CYP2C8 and CYP2D6 could weakly inhibit the formation of MT, the inhibitory effects were not significant (Figure 3). Taken together, the results of recombinant human CYP enzymes and chemical inhibitors imply that CYP3A4 is the major isoform responsible for the metabolism of OMT.
After oral administration, OMT could be metabolized to MT in the gastrointestinal tract and liver.24,25 Therefore, to determine the metabolic behavior of OMT in the liver and the intestine, we used HLMs, HIMs, and CYP3A4 isoform to analyze the enzyme kinetics of the reaction. Results showed that Km, Vmax, and CL were significantly higher in HLMs than that in HIMs (Figure 4, Table 1), and the formation of MT mediated by these three systems best fit the biphasic kinetics model. This atypical kinetic profile has been described as a two-site model, with one high-affinity binding site and another low-affinity binding site at high substrate concentrations, thereby causing the reaction profile to become linear with eventually increasing substrate concentration.35 On the other hand, Km’s for HLMs, HIMs, and CYP3A4 were different, and Km for CYP3A4 was the highest. HLMs and HIMs were microsomal preparations and contained various isoforms, and the proportions of these isoforms were different. Although CYP3A4 was the major isoform responsible for the metabolism of OMT, other isoforms played minor roles on its metabolism and the affinity between them and OMT was different. Therefore, the metabolism characteristics of OMT in HLMs and HIMs were more complex than in the recombinant CYP3A4 isoform. This could be the reason for the Km for recombinant CYP3A4 being higher than that for the HLMs and HIMs.
A comparison of the chemical structures of OMT with MT revealed that they had similar structures and that MT only lacks one oxygen atom to make them identical. In addition, previous studies demonstrated that the metabolism of OMT to MT could be a reduction reaction.24,25 Because the rates of reduction reaction were related to the oxygen content, different oxygen contents conditions were designed by driving out oxygen with nitrogen or helium to investigate the effect of oxygen contents on the metabolism of OMT and confirm the characteristics of the CYP3A4 metabolic reaction. Surprisingly, the increased metabolite formed during incubation increased with the reduced oxygen contents. The ventilation of the 30 s group produced the most metabolites because the decreased oxygen levels promoted the generation of MT. Meanwhile, the reduction of oxygen in the inactivated enzyme group did not affect the production of OMT metabolites. Therefore, the metabolism of OMT to MT is probably a reduction reaction mainly mediated by CYP3A4, rather than any other chemical reaction (Figure 5). In addition, TES was used as a positive control to confirm the metabolic mechanisms involved, because its metabolism characteristics were studied extensively. TES is a typical substrate for CYP3A4 oxidative metabolism. If the change trend of metabolism of OMT and TES under different oxygen contents were opposite, it would further indicate that the metabolism of OMT was a reduction reaction. The results showed that when the amount of oxygen was reduced during incubation, the amount of oxidative metabolites formed was decreased. This not only showed that the metabolic characteristic of OMT was reduction reaction, but also demonstrated the viability of this experimental model. Given the aforementioned results, we can conclude that the metabolism of OMT to MT was a Phase I reduction reaction mediated by CYP enzymes, specifically CYP3A4.
These results suggested that metabolic change could significantly occur when drugs containing OMT are administrated orally in combination with other drugs which are inducers of CYP3A4. Indeed, CYP3A4 is the primary CYP isoenzyme subfamily and is involved in the metabolism of over 50% of marketed drugs that undergo metabolic elimination.46,47 The high level of CYP3A4 expression in the liver, as well as its broad substrate specificity, may explain its susceptibility to drug–drug interactions.48,49 The metabolic drug–drug interactions could modulate CYP3A4 activity, and some drug interactions can be life threatening for the human body.50,51 In some cases, the orally administrated drugs were shown to inhibit CYP3A4 activity and, as a result, increase the concentration of its active metabolite form in the blood circulation, which may cause unfavorable and long-lasting interactions.52,53 Therefore, after oral administration simultaneously with a drug which potently induces effect on CYP3A4, the concentration of MT, the metabolite of OMT, in the plasma and liver would be increased significantly at therapeutic doses. OMT is considered to have stronger activity than MT,54 and both of them could cause hepatic toxicity, of which MT toxicity is greater than OMT.55 This suggested that the increased metabolism of OMT would lead to more severe liver damage and could further affect the expression and activity of CYP3A4 in the liver. It means that many factors should be considered when OMT is orally administered simultaneously with a drug which has a potently inducing effect on CYP3A4. Therefore, the clarification of the metabolism could be helpful in investigating the TCMs toxicity, and our findings indicated that more attention should be paid to when OMT was orally administered with other drugs, especially those inducers of CYP3A4.21
Conclusion
This study indicated that OMT could be primarily transformed to MT in vitro in HLMs and HIMs by Phase I reduction reaction. The MT formation was mainly catalyzed by CYP3A4 in humans. The findings of this study provides basic and detailed information to support further studies on the metabolism of OMT and implication of potential drug interactions of OMT with other CYP3A4 substrates for the safe application of OMT and the OMT-containing herbs. Meanwhile, further studies focusing on drug–drug interactions are necessary.
Acknowledgment
This work was supported by the grants of National Natural Science Foundation of China (81473410) and Science and Technology Project of Guangzhou City (201509010004) and Guangdong Natural Science Foundation (2015A030312012).
Disclosure
The authors report no conflicts of interest in this work. Publication of this article will not benefit or adversely affect the financial situations of the authors.
References
Chui CH, Lau FY, Tang JC, et al. Activities of fresh juice of Scutellaria barbata and warmed water extract of Radix Sophorae Tonkinensis on anti-proliferation and apoptosis of human cancer cell lines. Int J Mol Med. 2005;16:337–341. | ||
Rabea EI, Nasr HM, Badawy ME. Toxic effect and biochemical study of chlorfluazuron, oxymatrine, and spinosad on honey bees (Apis mellifera). Arch Environ Contam Toxicol. 2010;58:722–732. | ||
Wong S, Tsui S, Kwan S. Analysis of proprietary Chinese medicines for the presence of toxic ingredients by LC/MS/MS. J Pharm Biomed Anal. 2002;30:161–170. | ||
He X, Fang J, Huang L, et al. Sophora flavescens Ait.: Traditional usage, phytochemistry and pharmacology of an important traditional Chinese medicine. J Ethnopharmacol. 2015;172:10–29. | ||
Ma L, Wen S, Zhan Y, et al. Anticancer effects of the Chinese medicine matrine on murine hepatocellular carcinoma cells. Planta Med. 2008; 74:245–251. | ||
Jiang H, Hou C, Zhang S, et al. Matrine upregulates the cell cycle protein E2F-1 and triggers apoptosis via the mitochondrial pathway in K562 cells. Eur J Pharmacol. 2007;559:98–108. | ||
Yamazaki M. The pharmacological studies on matrine and oxymatrine. Yakugaku Zasshi. 2000;120:1025–1033. | ||
Hong LS, Lei L, Lei S, et al. Cardioprotective effects and underlying mechanisms of oxymatrine against Ischemic myocardial injuries of rats. Phytother Res. 2008; 22:985–989. | ||
Zhang M, Wang X, Wang X, et al. Oxymatrine protects against myocardial injury via inhibition of JAK2/STAT3 signaling in rat septic shock. Mol Med Rep. 2013;7:1293–1299. | ||
Cao YG, Jing S, Li L, et al. Antiarrhythmic effects and ionic mechanisms of oxymatrine from Sophora flavescens. Phytother Res. 2010;24: 1844–1849. | ||
Hu ST, Tang Y, Shen YF, et al. Protective effect of oxymatrine on chronic rat heart failure. J Physiol Sci. 2011;61:363–372. | ||
Chai NL, Fu Q, Shi H, et al. Oxymatrine liposome attenuates hepatic fibrosis via targeting hepatic stellate cells. World J Gastroenterol. 2012; 18:4199–4206. | ||
Runtao G, Guo D, Jiangbo Y, et al. Oxymatrine, the main alkaloid component of Sophora roots, protects heart against arrhythmias in rats. Planta Med. 2011;77:226–230. | ||
Unahabhokha T, Sucontphunt A, Nimmannit U, et al. Molecular signalings in keloid disease and current therapeutic approaches from natural based compounds. Pharm Biol. 2014;53:457–463. | ||
Tang L, Dong LN, Peng XJ, et al. Pharmacokinetic characterization of oxymatrine and matrine in rats after oral administration of radix Sophorae tonkinensis extract and oxymatrine by sensitive and robust UPLC-MS/MS method. J Pharm Biomed Anal. 2013;83:179–185. | ||
Wang S, Wang G, Li X, et al. Simultaneous determination of oxymatrine and its active metabolite matrine in dog plasma by liquid chromatography-mass spectrometry and its application to pharmacokinetic studies. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;817:319–325. | ||
Fan RX, Liu R, Ma R, et al. Determination of oxymatrine and its active metabolite matrine in human plasma after administration of oxymatrine oral solution by high-performance liquid chromatography coupled with mass spectrometry. Fitoterapia. 2013;89:271–277. | ||
Wan XY, Luo M, Li XD, et al. Hepatoprotective and anti-hepatocarcinogenic effects of glycyrrhizin and matrine. Chem Biol Interact. 2009;181:15–19. | ||
Wang XY, Liang L, Chang JL, et al. Toxicity of matrine in Kunming mice. Nan fang yi ke da xue xue bao. 2010;30:2154–2155. | ||
Yuan F, Chen J, Wu WJ, et al. Effects of matrine and oxymatrine on catalytic activity of cytochrome p450s in rats. Basic Clin Pharmacol Toxicol. 2010;107:906–913. | ||
Bachmann KA, Ghosh R. The use of in vitro methods to predict in vivo pharmacokinetics and drug interactions. Curr Drug Metab. 2001;2:299–314. | ||
Omura T. Forty years of cytochrome P450. Biochem Biophys Res Commun. 1999;266:690–698. | ||
Ye L, Tang L, Gong Y, et al. Characterization of metabolites and human P450 isoforms involved in the microsomal metabolism of mesaconitine. Xenobiotica. 2011;41:46–58. | ||
Xie MZ, Zhou WZ, Zhang Y. Oxymatrine metabolic fate. Chin Med J (Engl). 1983;96:145–150. | ||
Wang ML, Zhou QL, Wang BX. Studies on metabolism of oxymatrine by human intestinal bacteria. Zhongguo Zhong Yao Za Zhi. 2001;26: 272–274. | ||
Al-Lazikani B, Banerji U, Workman P. Combinatorial drug therapy for cancer in the post-genomic era. Nat Biotechnol. 2012;30:679–692. | ||
Ye L, Wang T, Yang C, et al. Microsomal cytochrome P450-mediated metabolism of hypaconitine, an active and highly toxic constituent derived from Aconitum species. Toxicol Lett. 2011;204:81–91. | ||
Eagling VA, Tjia JF, Back DJ. Differential selectivity of cytochrome P450 inhibitors against probe substrates in human and rat liver microsomes. Br J Clin Pharmacol. 1998;45:107–114. | ||
Kobayashi K, Urashima K, Shimada N, et al. Selectivities of human cytochrome P450 inhibitors toward rat P450 isoforms: study with cDNA-expressed systems of the rat. Drug Metab Dispos. 2003;31:833–836. | ||
Ando Y, Fuse E, Figg WD. Thalidomide metabolism by the CYP2C subfamily. Clin Cancer Res. 2002;8:1964–1973. | ||
Schmider J, Greenblatt DJ, Fogelman SM, et al. Metabolism of dextromethorphan in vitro: involvement of cytochromes P450 2D6 and 3A3/4, with a possible role of 2E1. Biopharm Drug Dispos. 1997; 18:227–240. | ||
Liu X, Tam VH, Hu M. Disposition of flavonoids via enteric recycling: determination of the UDP-glucuronosyltransferase isoforms responsible for the metabolism of flavonoids in intact Caco-2 TC7 cells using siRNA. Mol Pharm. 2007;4:873–882. | ||
Wang SW, Chen J, Jia X, et al. Disposition of flavonoids via enteric recycling: structural effects and lack of correlations between in vitro and in situ metabolic properties. Drug Metab Dispos. 2006;34: 1837–1848. | ||
Houston JB, Kenworthy KE. In vitro-in vivo scaling of CYP kinetic data not consistent with the classical Michaelis-Menten model. Drug Metab Dispos. 2000;28:246–254. | ||
Hutzler JM, Tracy TS. Atypical kinetic profiles in drug metabolism reactions. Drug Metab Dispos. 2002;30:355–362. | ||
Yamaoka K, Nakagawa T, Uno T. Application of Akaike’s information criterion (AIC) in the evaluation of linear pharmacokinetic equations. J Pharmacokinet Biopharm. 1978;6:165–175. | ||
Dai D, Tang J, Rose R, et al. Identification of variants of CYP3A4 and characterization of their abilities to metabolize testosterone and chlorpyrifos. J Pharmacol Exp Ther. 2001;299:825–831. | ||
Lee JY, Lee SY, Oh SJ, et al. Assessment of drug-drug interactions caused by metabolism-dependent cytochrome P450 inhibition. Chem Biol Interact. 2012;198:49–56. | ||
Baati T, Horcajada P, Gref R, et al. In vitro determination of the CYP 3A4 activity in rat hepatic microsomes by liquid-phase extraction and HPLC-photodiode array detection. J Pharmacol Toxicol Methods. 2012;66(1):29–34. | ||
Yao N, Wang X. In vitro Immunomodulatory Activity of Oxymatrine on Toll-Like Receptor 9 Signal Pathway in Chronic Hepatitis B. Am J Chin Med. 2014;42:1399–1410. | ||
Zhang W, Zhang J, Kang Y, et al. Cardioprotective effects of oxymatrine on isoproterenol-induced heart failure via regulation of DDAH/ADMA metabolism pathway in rats. Eur J Pharmacol. 2014;745:29–35. | ||
Mao L, Henderson G. Antifeedant activity and acute and residual toxicity of alkaloids from Sophora flavescens (leguminosae) against formosan subterranean termites (Isoptera: Rhinotermitidae). J Econ Entomol. 2007;100:866–870. | ||
Li X, Luan Y, Li X, et al. Study on anti-inflammatory efficacy accompanied by side effects of different components of Sophorae Tonkinensis Radix et Rhizoma. Zhongguo Zhong yao za zhi. 2012;37: 2232–2237. | ||
Lu ZG, Li MH, Wang JS, et al. Developmental toxicity and neurotoxicity of two matrine-type alkaloids, matrine and sophocarpine, in zebrafish (Danio rerio) embryos/larvae. Reprod Toxicol. 2014;47:33–41. | ||
Guengerich FP. Role of cytochrome P450 enzymes in drug-drug interactions. Adv Pharmacol. 1997;43:7–35. | ||
Topletz AR, Dennison JB, Barbuch RJ, et al. The relative contributions of CYP3A4 and CYP3A5 to the metabolism of vinorelbine. Drug Metab Dispos. 2013;41:1651–1661. | ||
Stresser DM, Kupfer D. Prosubstrates of CYP3A4, the major human hepatic cytochrome P450: transformation into substrates by other P450 isoforms. Biochem Pharmacol. 1998;55:1861–1871. | ||
Lee KS, Kim SK. Direct and metabolism-dependent cytochrome P450 inhibition assays for evaluating drug-drug interactions. J Appl Toxicol. 2013;33:100–108. | ||
Zhou ZW, Zhou SF. Application of mechanism-based CYP inhibition for predicting drug-drug interactions. Expert Opin Drug Metab Toxicol. 2009;5:579–605. | ||
Markowitz JS, Donovan JL, deVane CL, et al. Effect of St John’s Wort on drug metabolism by induction of cytochrome P450 3A4 enzyme. JAMA. 2003;290:1500–1504. | ||
Chow HH, Garland LL, Hsu CH, et al. Resveratrol modulates drug- and carcinogen-metabolizing enzymes in a healthy volunteer study. Cancer Prev Res. 2010;3:1168–1175. | ||
Chen XW, Sneed KB, Pan SY, et al. Herb-drug interactions and mechanistic and clinical considerations. Curr Drug Metab. 2012; 13:640–651. | ||
Boinpally R, Gad N, Gupta S, et al. Influence of CYP3A4 induction/inhibition on the pharmacokinetics of vilazodone in healthy subjects. Clin Ther. 2014;36:1638–1649. | ||
Zhao B, Rong D, Wang X, et al. Relationship of activity with electron structure for matrine and oxymatrine. J Mol Sci. 2000;16:88–93. | ||
Zhang Q, Li R, Jin R, et al. Study on the hepatotoxicity induced by matrine and oxymatrine. Chin Arch Tradit Chin Med. 2011;29:1222–1225. |
Supplementary materials
 | Table S1 Intra- and interday precision and accuracy for MT in KPI solution (n=6) |
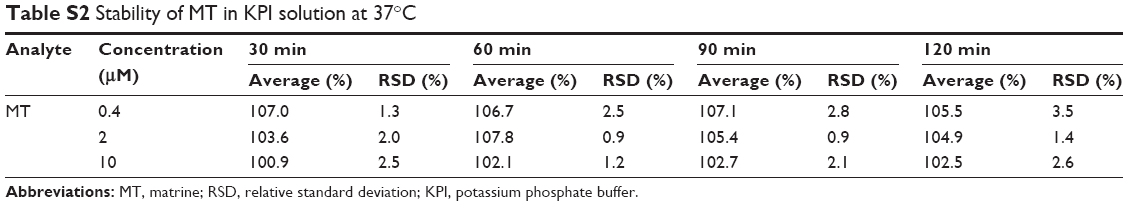 | Table S2 Stability of MT in KPI solution at 37°C |
 | Figure S1 The amount of 6-β-OH-testosterone formed in different oxygen contents, the effects of nitrogen on TES metabolism (A), and the effects of helium on TES metabolism (B). |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.