")
Back to Journals » Drug Design, Development and Therapy » Volume 16
Reduced the Food Effect and Enhanced the Oral Bioavailability of Ivacaftor by Self-Nanoemulsifying Drug Delivery System (SNEDDS) Using a New Oil Phase
Authors Miao Y, Zhao S, Zuo J, Sun J, Wang J
Received 17 January 2022
Accepted for publication 17 May 2022
Published 23 May 2022 Volume 2022:16 Pages 1531—1546
DOI https://doi.org/10.2147/DDDT.S356967
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Editor who approved publication: Professor Manfred Ogris
Yanfei Miao,1 Shihua Zhao,1 Jian Zuo,1 Jiqin Sun,1 Jingnan Wang2
1College of Chemistry and Chemical Engineering, Taishan University, Tai’an, People’s Republic of China; 2School of Biotechnology and Pharmaceutical Engineering, Nanjing Tech University, Nanjing, People’s Republic of China
Correspondence: Yanfei Miao, College of Chemistry and Chemical Engineering, Taishan University, Tai’an 271000, People’s Republic of China, Tel/Fax +86-0538-6713958, Email [email protected]
Purpose: The purpose of this work was to develop an ivacaftor self-nanoemulsion drug delivery system (IVA-SNEDDS) using the newly developed double headed miscellaneous lipid (DHML) as oil phase to reduce the food effect and inter-individual absorption variability of IVA.
Methods: The lipids with the greatest solubility to IVA were selected as the oil phase of IVA-SNEDDS by saturation solubility method. Then, among different surfactants and co-surfactants, those with good emulsifying ability for the selected oil phase were selected, and the proportion of surfactant and co-surfactant was further selected by pseudo-ternary phase diagram. The prepared IVA-SNEDDS were screened and evaluated in vitro and in beagle dogs.
Results: The optimized IVA-SNEDDS formulation consisting of DHML, Tween 80, and Transcutol HP with the weight ratio of 2:2:1 was physically stable and it was easy to disperse in water, pH 1.2 hydrochloric acid and pH 6.8 phosphate buffer solution, and generated a fine homogeneous nanoemulsion, with mean globule size less than 75 nm regardless of dilution ratio. In vitro drug release studies showed that the drug in IVA-SNEDDS could be completely released in a short time, while the drug release in IVA-suspension was less than 1% at 60 min. In vivo, using IVA-suspension (Fed) as a reference, the relative oral bioavailability of IVA-suspension (Fasted), IVA-SNEDDS (Fasted), and IVA-SNEDDS (Fed) were 23.35%, 153.63%, and 149.89%, respectively. This showed that IVA-SNEDDS could eliminate the positive food effect, improve the oral bioavailability, and reduce the IVA absorption difference between individuals.
Conclusion: As the oil phase of SNEDDS, DHML can significantly improve the drug solubility and drug loading of IVA-SNEDDS. Moreover, DHML was easily emulsified and can effectively form a nanoemulsion in vivo and in vitro. The prepared IVA-SNEDDS can reduce the inter-individual absorption variability of IVA, eliminate its food effect and improve its oral bioavailability.
Keywords: ivacaftor, food effect, self-nanoemulsion drug delivery system (SNEDDS), oral bioavailability, oil phase
Introduction
In the last three decades, oral dosage forms have been made available for about 75% of discovered active pharmaceutical ingredients (APIs). This is because most patients show better acceptance and treatment compliance with oral administration. In addition, there are some other benefits, for instance, ease of self-medication, no pain, flexibility of administration scheme, and low price.1,2 Effective oral administration is faced with the need for good dissolution of drugs in aqueous gastrointestinal fluid and distribution ability through lipophilic gastrointestinal membranes.3 For the past few years, more than 75% of new drug candidate compounds have poor solubility and are classified as class II or IV in the biopharmaceutical classification system (BCS). Therefore, they show poor dissolution and absorption in the gastrointestinal tract.4,5 Additionally, we found that about 25% of marketed oral preparations demonstrated higher oral bioavailability when they were taken with food, especially high-fat food.6 This is called the “positive food-dosage effect”, however, due to the influence of factors such as age, eating habits and beliefs (such as vegetarians), this is an inherent variable, which will be affected by the composition of a meal.7–10 Most importantly, patients and their family members have insufficient understanding of proper medication and often do not follow instructions strictly, which increases the number of cases in which the patient’s absorption is reduced, the efficacy of the drug is reduced, or the effective therapeutic concentration is not reached when the drug is administered outside the facility.
Cystic fibrosis (CF) is a serious autosomal recessive disease with shortened lifespan of patients. The main clinical manifestations include pancreatic insufficiency and chronic progressive suppurative lung disease.11 Ivacaftor (IVA) was first approved in the USA in January 2012 and is available as a tablet and granules for the treatment of CF. The molecular formula of IVA is C24H28N2O3 and the molecular weight is 392.49. The chemical structure of IVA is shown in Figure 1. However, one problem with oral IVA is that IVA must be taken with food to ensure its good absorption due to its poor water solubility (<0.05 microgram/mL).12 Besides that, IVA shows significant inter-individual variability because it is metabolized by the mixed function oxidase CYP3A4. It has been reported that many patients may maintain unsatisfactory blood drug levels during the administration interval, resulting in a decline in efficacy.13 When taken with fatty foods, the oral bioavailability of IVA increased by about 2.5–4 times.14 The reasons for the positive phenomenon of food dose effect include the formation of micelles in the presence of oil and fat in food (increasing the solubility of drugs), reducing gastric emptying in the presence of food (increasing drug absorption time), and reducing hepatic first pass.15 However, due to the different lifestyles of individual in the modern world, it sometimes seems difficult to follow specific instructions, such as taking medicine with food.
 |
Figure 1 Chemical structure of ivacaftor. |
Consequently, the development of drug delivery systems for these compounds is necessary to eliminate food effects and provide the optimal oral administration of hydrophobic drug compounds, so as to further improve the therapeutic effect and patient compliance. Many strategies have been proposed to increase the solubility and reduce the positive food effect of these APIs including but not limited to solid dispersion,16,17 lipid-based formulations,18,19 nanoparticles,20,21 liposomes,22 nanocrystals,23 and complexes.24
Among the lipid-based formulations, a self-nanoemulsifying drug delivery system (SNEDDS) has unique advantages in improving the solubility and absorption of poorly water-soluble drugs.25,26 SNEDDS is a pre-concentrated mixture of surfactant(s), co-surfactant(s), and oil(s), which produces fine droplets of emulsion (5–100 nm), when diluted with water upon mild agitation or under peristalsis of the gastrointestinal tract (GIT).27,28
Lipophilic API can be dissolved in the inner phase of oil/water emulsion while maintaining a dissolution state. Nano o/w emulsion provides a large surface area for drug absorption.29 In addition, drugs present in SNEDDS can reduce enzymatic hydrolysis in GIT, reduce its pre-system clearance, and bypass liver first pass metabolism through intestinal lymphatic system absorption.30 Moreover, SNEDDS has shown a significantly reduced food effect on the absorption of BCS II and IV drugs such as ziprasidone, itraconazole, and torcetrapib.31–33 One of the constraints to the further development of SNEDDS is the lack of an oil phase capable of dissolving a large amount of drugs. Although many natural and semi-synthetic oil phases are available, due to their relatively simple structure, these lipids are increasingly insufficient as the oil phase in SNEDDS to solve the delivery problem of new molecules with complex structure and diverse properties.34,35
As far as we know, no literature to reduce the food effect and inter-individual variability of IVA using SNEDDS has been reported. Hence, this work was aimed to fabricate an IVA self-nanoemulsifying drug delivery system (IVA-SNEDDS) to enhance its oral bioavailability under fasted state using the newly developed double headed miscellaneous lipid (DHML) as oil phase.36 The prepared IVA-SNEDDS was analyzed and evaluated by in vitro experiments to screen the optimal prescription. Then, the best IVA-SNEDDS prescription was used for the pharmacokinetics experiment in beagle dog, and IVA-suspension as a control to confirm the preparation of IVA-SNEDDS can achieve the expected effect.
Materials and Methods
Materials
Ivacaftor was purchased from Ideal biotechnology (Ideal, Nanjing, Jiangsu, China). Capmul GMO, Labrasol, soybean oil, isopropyl myristate (IPM), and erucic acid were obtained as a gift from Yihui Co., Ltd (Yihui, Liaocheng, Shandong, China). Ethyl oleate, Tween 80, and glycerol were bought from Nanjing Chemical Reagent Co., Ltd (Nanhua, Nanjing, Jiangsu, China). Polyethylene glycol 400 (PEG 400), Capmul MCM, Span 80, and Transcutol HP were purchased from the Anhui Shanhe Co., Ltd (Shanhe, Xuancheng, Anhui, China). Macrogolglycerol ricinoleate (Kolliphor EL) was purchased from Merck (Merck, Beijing, China). Double headed miscellaneous lipid (DHML) was obtained as gift from Taian Rutocel Co., Ltd (Tai’an, Shandong, China).
HPLC Analysis
The content of IVA in the samples was determined by HPLC (SHP LC1620A, Shanghai, China). The mobile phase composition was ammonium acetate buffer (pH 5.0) and acetonitrile in the volume ratio of 60:40. The flow rate was 1.0 mL/min and the separation of IVA was obtained on Waters C18 column (5 μm, 250×4.6 mm) and the detection was recorded using a UV detector at a wavelength of 225 nm.37
Selection of Excipients
Selection of excipients such as oil, surfactant, and co-surfactant were conducted by saturation solubility and % transmittance study.38,39 Saturation solubility studies were developed separately by adding 0.5 g IVA in 1.0 g various excipients, such as IPM, DHML, erucic acid, soybean oil, ethyl oleate, Capmul MCM, Capmul GMO (oil), Tween 80, Span 80, Labrasol and Kolliphor EL (surfactant), and PEG 400, glycerol, Transcutol HP (co-surfactants) in a glass vial and placed on a rotary shaker for mixing at 25 rpm for 24 h, at 37 ± 0.5℃. The samples were then taken out and centrifuged at 5000 rpm (RFC = 3435 g) for 10 min at room temperature. After that, the supernatant was removed with a straw and placed in a new tube. The solubility of IVA in different excipients was determined by HPLC. All the experiments were performed in triplicate.
Based on the saturation solubility, the selection of surfactant and co-surfactant were further studied by % transmittance. In brief, DHML and different surfactants (Tween 80, Span 80, Labrasol, and Kolliphor EL) in 2:1 ratio (w/w), diluted 100-fold with water and % transmittance was measured. For selecting of co-surfactant, surfactant and co-surfactant at 2:1 (w/w) were mixed with DHML, and diluted 100-fold with water and % transmittance was recorded using a UV spectrophotometer (UV-1650, Shimadzu, Japan) at 630 nm using distilled water as blank.40
Construction of Pseudo-Ternary Phase Diagrams
After the selection of excipients, oil with good solubilization capacity for IVA, surfactant and co-surfactant with good emulsification efficiency were harnessed for further study. Pseudo-ternary phase diagrams were carried out for selection of the best ratio of surfactant and co-surfactant using a water titration method.41 Firstly, the surfactant and co-surfactant obtained from the above experiments were evenly mixed according to the weight ratios of 3:1, 2:1, 1:1, and 1:2. Then, the oil phase and Smix (surfactant/co-surfactant mixture) were mixed evenly in the weight ratio of 1:9 to 9:1 respectively. Finally, the above mixtures were placed in a conical flask, stirred at 100 rpm by magnetic stirrers (IKA, Staufen, Germany) at room temperature and titrated with water. Visual observation was done during titration and the endpoint of titration was marked when an isotropic and optically clear solution was obtained. The pseudo-ternary phase diagrams were generated using OriginPro 8.6 software (OriginLab Corporation, Northampton, MA, USA).
Preparation of IVA-SNEDDS
According to the experimental results of pseudo-ternary phase diagram, Smix, which can form the largest area of nanoemulsion, was selected for further preparation of IVA-SNEDDS. IVA-SNEDDS were prepared from the screened oil phase and Smix according to different weight ratios (Table 1). The preparation steps were as follows: weighed the oil phase and Smix (surfactant and co-surfactant) accurately according to the data in Table 1 and placed it in a triangular flask, vortex and mixed for one minute to form a homogeneous liquid. Then, the excess IVA was added to the above liquid in an incremental manner, mixed by turbine for 30 min and centrifuged at 5000 rpm for 15 min. The supernatant was taken with a disposable pipette, placed in a test tube with a cover, and placed at room temperature until it was used for testing.
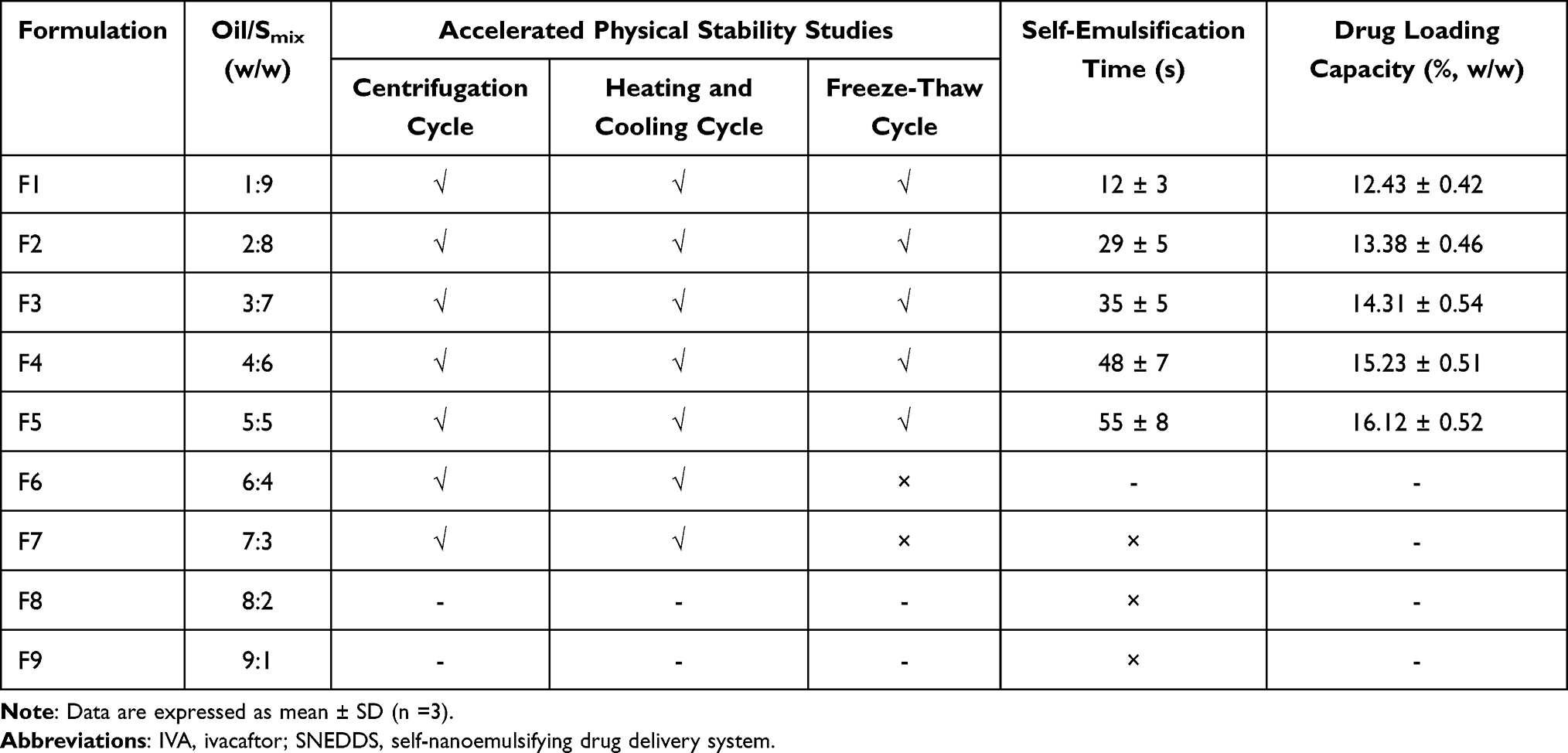 |
Table 1 Composition of the IVA-SNEDDS Formulations and Evaluation Parameters |
Accelerated Physical Stability Studies
The prepared IVA-SNEDDS formulations after dilution with 100-fold water (v/v), were subjected to centrifugation cycle, heating-cooling cycle, and freeze-thaw cycle as follows.
Centrifugation Cycle
10 mL of the above samples were placed in centrifuge tube respectively and centrifuged at 5000 rpm for 15 min to verify that they remained stable without phase separation under centrifugation. The stable formulations were used for the next studies.
Heating and Cooling Cycle
10 mL samples were put into test tubes with lids, placed in a refrigerator at 4℃ for 12 h, and then taken out and placed in an oven at 45℃ for another 12 h.42 The above steps were repeated three times. Through visual observation, formulations that passed through the heating and cooling cycles without flocculation, phase change, or drug precipitation were used for further study.
Freeze-Thaw Cycle
The samples were stored at −20℃ in the refrigerator for 48 h, and then stored at 25℃ in a water bath for another 48 h.43 Those IVA-SNEDDS which showed good performance in accelerated physical stability experiments, with no phase separation, drug precipitation or any droplet size as well as PDI (polymer dispersity index) changes were used for further studies on self-emulsification efficiency.44
Self-Emulsification Time
The time required for the spontaneous formation of nanoemulsion of IVA-SNEDDS was determined by a type II dissolution instrument of Chinese Pharmacopoeia (Tianda Tianfa, Tianjin, China). 1.0 mL of IVA-SNEDDS was dropped into the dissolution vessel containing 500 mL distilled water with a paddle speed of 50 rpm at 37 ± 0.5℃. The time required to visually observe the disappearance of IVA-SNEDDS and the formation of a clear dispersion was recorded as the self-emulsification time.45
Effect of pH and Robustness to Dilution
Robustness of IVA-SNEDDS to dilution was carried out by diluting IVA-SNEDDS with 50, 100, and 1000-fold (v/v) in various media such as water, pH 1.2 HCl and pH 6.8 PBS (phosphate buffer solution). All tested samples were placed in test tubes and stored at room temperature for 48 h to observe whether there were unstable phenomena such as drug precipitation and phase transformation.46
Droplet Size Analysis and Zeta Potential
The droplet sizes of nanoemulsion formed by IVA-SNEDDS in different solutions (water, pH 1.2 HCl and pH 6.8 PBS) and different dilution ratios (1:50, 1:100, and 1:1000 v/v) were studied. The particle size distribution, PDI and zeta potential of droplets were measured by dynamic light scattering (Malvern Zetasizer, Nano ZS-90, Worcestershire, UK). Average droplet size, PDI, and zeta potential values were recorded.
Drug Content Determination
Precisely weighed an appropriate amount of IVA-SNEDDS into a 100 mL volumetric flask, methanol added to dissolve, shaken well, and filtered through 0.45 μm membrane (Jinteng, Tianjin, China).47 The above samples were used to determine the content of IVA according to the external standard method under “HPLC analysis”. All the experiments were performed in triplicate.
Transmission Electron Microscopy (TEM)
The IVA-SNEDDS was diluted 100-fold with water and a drop of sample was placed on a copper grid. The sample was stained with 1% phosphotungstic acid solution for 40 s and finally put under an electron microscope (Philips Tecnai 12, Eindhoven, the Netherlands) to visualize the particle morphology.
In vitro Drug Release Study
The drug release profiles of IVA-SNEDDS and IVA suspension were conducted by dialysis bag technique.40 In short, IVA-SNEDDS (equivalent to IVA 150 mg) and IVA suspension (150 mg IVA was added to 1 mL water and mixed by vortex for 10 min) were filled in the dialysis bag (12,000–14,000 Da). These dialysis bags were put into dissolution vessels containing different dissolution media. The study was carried out in 900 mL water, pH 1.2 HCl and pH 6.8 PBS at 37 ± 0.5℃ with the paddle speed of 50 rpm separately. At defined time intervals (5, 10, 15, 30, 45, and 60 min), 5 mL aliquots were withdrawn and replaced with fresh medium. The concentrations of drug diffusing through the bag were determined by HPLC.
Storage Stability Studies
The optimal IVA-SNEDDS was filled in vials and placed in a stability chamber (Yiheng, Shanghai, China) at 40 ± 2℃ and 65 ± 5% relative humidity (RH) for storage stability experiments. At different times, such as 0, 1, 2, 3, and 6 months, some vials were taken out, and the appearance, self-emulsifying ability, particle size, etc. of IVA-SNEDDS were measured.
Pharmacokinetic Studies in Beagle Dogs
The pharmacokinetic studies described here were performed according to the animal care protocols approved by Taishan University Ethics Committee under approval number 20201001 for the use of experimental animals, in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals. An open-label, four-cycle, four-sequence, two-treatment, cross-feeding, and fasting study was carried out in male beagle dogs, weighing 10–12 kg.48,49 Twenty-four beagle dogs were randomly divided into 4 groups of 6 dogs each. Table 2 details the design method of the pharmacokinetic experiments with IVA-SNEDDS (equivalent to IVA 150 mg) and IVA suspension (containing 150 mg IVA). The washout time between two periods was 7 days.
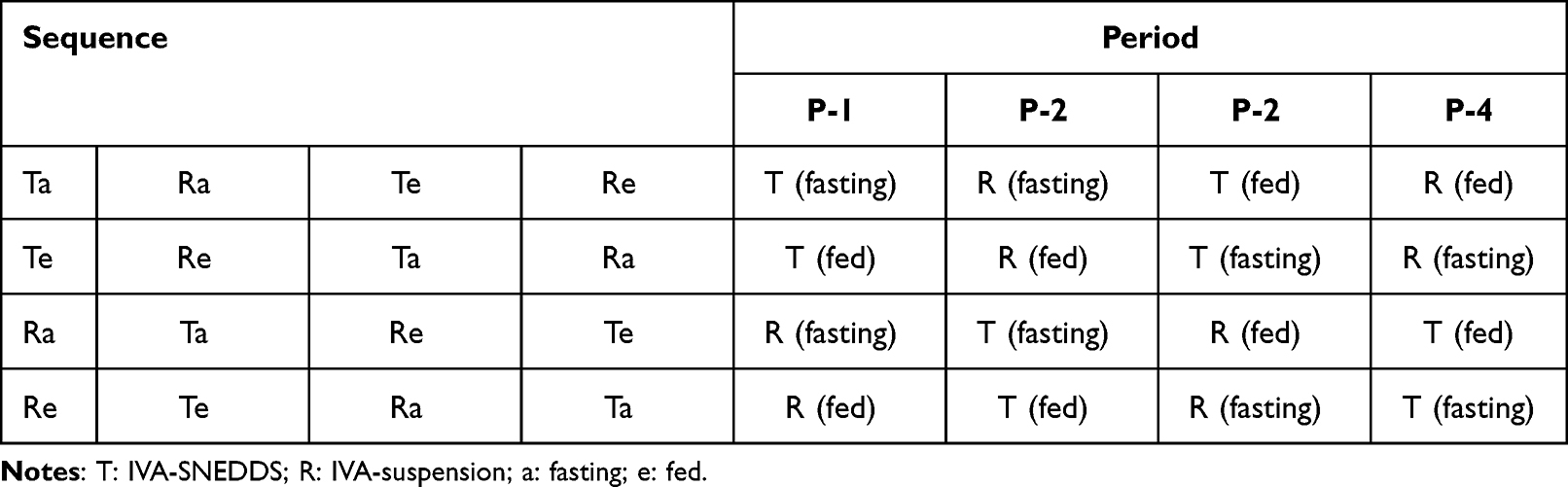 |
Table 2 Design of in vivo Pharmacokinetic Study in Beagle Dogs |
In the fasting study, dosing was administered to dogs in the morning that had been fasted for 12 h, followed immediately by oral gavage with 50 mL of water. In the feeding study, dogs that were fasted for 12 h were fed a diet consisting of 150 g of dry dog food (20% crude protein; 10% crude fat; 10% water; 9% crude fiber) in the morning. They were dosed (IVA-SNEDDS or IVA suspension, both of them containing 150 mg IVA) and then immediately given 50 mL of water by oral gavage.
4 mL of blood was collected from the forelimb vein using a vacuum blood collection tube containing heparin sodium at time points 0.5, 1.0, 1.5, 2, 2.5, 3, 4, 6, 8, 12, and 24 h after administration, respectively. The collected whole blood samples were centrifuged for 10 min at 4000 rpm immediately, and the supernatant was aspirated with a disposable pipette into a new plastic centrifuge tube with a lid and stored in the refrigerator at −20℃.
Determination of Plasma Concentration of IVA
First, the samples were taken out of the refrigerator and thawed at room temperature. Then precisely measured 1.0 mL of plasma sample, 40 μL of tezacaftor methanol solution (1.0 μg/mL, internal standard solution) and 5.0 mL of ethyl acetate placed into the same conical centrifuge tube were vortexed for 10 min. After that the samples were centrifuged at 4000 rpm for 10 min, the supernatants were taken into the centrifuge tube, and then the solvent removed by vacuum rotary evaporation. Lastly, precisely measured 100 μL of mobile phase solution was added to the above centrifuge tube to dissolve the residue. The drug content in the samples was determined by HPLC. The results of the assay were used to draw the serum concentration-time profile. Pharmacokinetic analyses were conducted using DAS 2.0 software package (China Food and Drug Administration, Beijing, China). The peak concentration (Cmax) and its time (Tmax) were obtained directly from serum concentration-time profile.
Statistical Analysis
Data were expressed as mean ± standard deviation (SD). Statistical analysis was done by Student’s t-test using GraphPad Prism 5 software (GraphPad Software Inc., San Diego, CA, USA). A p value of < 0.05 was considered statistically significant.
Results and Discussion
Selection of Excipients
For SNEDDS, the oil phase plays an important role in improving the solubility and bioavailability of drugs. Among various oils, IVA shows better solubility in IPM with 211.2 ± 9.8 mg/g, however, IVA has the highest solubility in DHML (255.4 ± 12.3 mg/g, Figure 2). Therefore, it was selected as the oil phase for the development of IVA-SNEDDS. DHML is a new lipidic biocompatible and safe material for SNEDDS. As an oil phase, the structure and properties of DHML are diverse. The molecular structure of DHML comprises a hydrocarbon chain (fat soluble tail) and poly (propyl ether imine) dendron head group linked through an ester bond and propylene spacer (to easily form hydrogen bonds with drugs).36 The structural formula of DHML is shown in Figure 3.
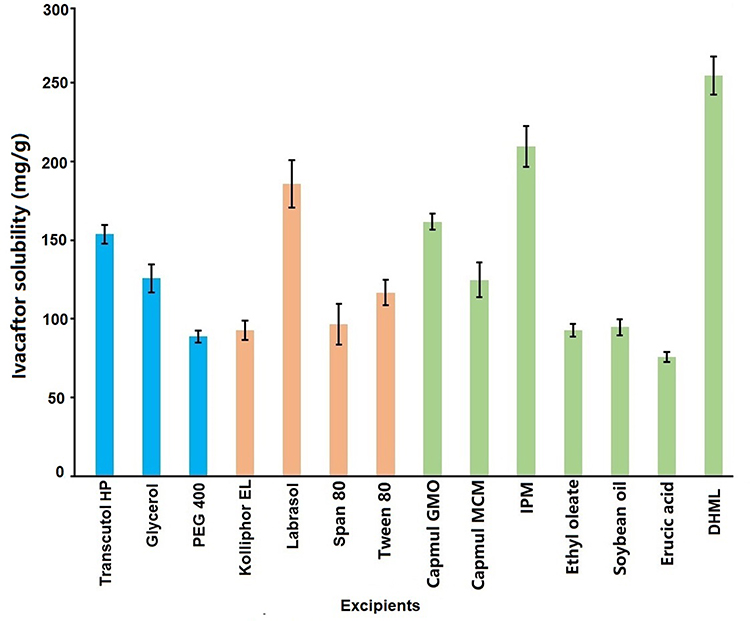 |
Figure 2 IVA solubility in different excipients (mean ± SD, n = 3). Abbreviations: IPM, isopropyl myristate; DHML, double headed miscellaneous lipid; PEG, polyethylene glycol. |
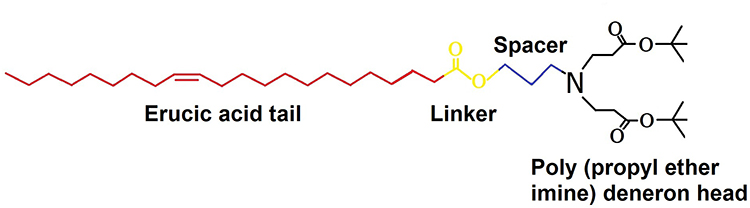 |
Figure 3 Chemical structures of DHML. Abbreviation: DHML, double headed miscellaneous lipid. |
The selection criteria of the surfactant and co-surfactant were on the strength of their emulsification efficiency for DHML instead of their ability to solubilize IVA. Highest solubility among the surfactants of IVA is Labrasol (186.7 ± 15.3 mg/g, Figure 2) but it had less capacity to emulsify DHML as we can see from the % transmittance value of 65.54 ± 2.81%. Tween 80 is a partial fatty acid ester of sorbitol and its anhydride contains 20 units of oxyethylene. The fatty acid composition is 70% oleic acid with several other fatty acids such as palmitic acid. This had maximum capacity to emulsify DHML (% transmittance value of 98.46 ± 1.52%) and the second highest solubility on IVA (117.6 ± 8.5 mg/g, Figure 2). Therefore Tween 80 was selected as the surfactant for further study.50,51 Transcutol HP was demonstrated with the highest solubility (154.3 ± 6.2 mg/g, Figure 2) of IVA and maximum ability to emulsify DHML (% transmittance value of 97.88 ± 1.66%). Besides that, Transcutol HP has the ability to alter the viscosity of aqueous phase and affect the mass transport kinetics of surfactant and oil.52 The combination of surfactant and co-surfactant with high and low hydrophilic lipophilic balance (HLB) values results in the rapid formation of a stable emulsion with fine emulsion globule size upon dispersion in water.53 Hence Tween 80 (HLB 15) and Transcutol-HP (HLB 4) were chosen as surfactant mixture in this study. Therefore, based on the above experimental results, in order to prepare a SNEDDS with high drug solubility and easy emulsification, DHML, Tween 80, and Transcutol HP were finally selected as the oil phase, surfactant, and co-surfactant for preparing IVA-SNEDDS.
Construction of Pseudo-Ternary Phase Diagrams
Pseudo-ternary phase diagrams of DHML (oil) and Smix (Tween 80 as surfactant and Transcutol HP as co-surfactant) were used to study the emulsifying ability of Smix to DHML and the range of forming nanoemulsion (Figure 4). The colored areas in Figure 4 represent the range in which DHML and different ratios of Smix can form nanoemulsions. The larger the area, the stronger the ability to spontaneously form microemulsions. It can be clearly seen from Figure 4 that when Smix is 2:1, that is, when the weight ratio of Tween 80 and Transcutol HP is 2:1, the microemulsion area formed is the largest. Thus, the optimal Tween 80 and Transcutol HP ratio of 2:1 was fixed for further investigation.
 |
Figure 4 Pseudo-ternary phase diagram of systems containing Smix ratios 1:1 (A), 2:1 (B), 3:1 (C) and 1:2 (D) for Tween 80/ Transcutol HP using DHML as oil and water as titrant (colored domain represents nanoemulsion existence region). Abbreviation: DHML, double headed miscellaneous lipid. |
Accelerated Physical Stability Studies
Tables 1 and 3 present the accelerated stability test results of formulations F1–F7. As can be seen from Table 1, formulation F6 and F7 failed to pass the freeze-thaw test due to drug precipitation occurring. All other formulations F1–F5 showed good stability with no indication of drug precipitation, phase separation, and no significant change in droplet size and PDI (Table 3). This will prove that IVA-SNEDDS can remain stable after nanoemulsions are formed in vivo.
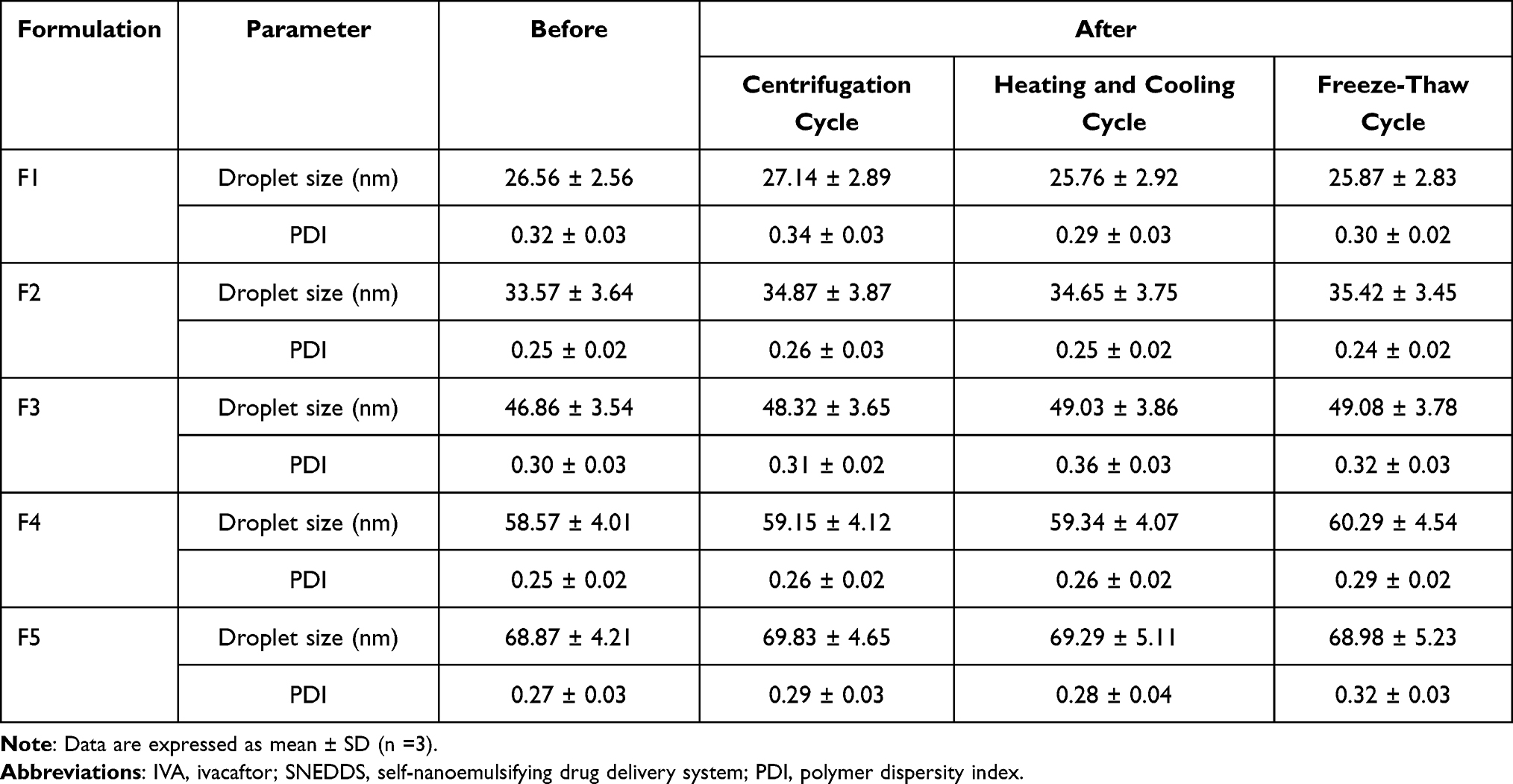 |
Table 3 Accelerated Stability for IVA-SNEDDS |
Self-Emulsification Time
With SNEDDS formulation, whether a microemulsion can be formed rapidly and spontaneously after oral administration is the premise to ensure the advantages of the drug delivery system. Therefore, the self-emulsification time is a key index to evaluate the prepared IVA-SNEDDS. The study showed that formulations F1–F5 (Table 1) could disperse totally and rapidly when diluted under mild agitation, which indicates that the IVA-SNEDDS will readily self-nanoemulsify and disperse in GIT fluids. The emulsification times of formulations F1–F5 were 12 ± 3 s, 29 ± 5 s, 35 ± 5 s, 48 ± 7 s, and 55 ± 8 s, respectively. The combination of non-ionic surfactant with high hydrophilic lipophilic balance value 15 (Tween 80) and low hydrophilic lipophilic balance value 4 (Transcutol HP) bring out the rapid formation of a stable emulsion. Those visual observations illustrated that with increased proportion of Smix in the system, the self-emulsification time was shorter.
Drug Loading Efficiency and Optimization of IVA-SNEDDS
The drug loading efficiencies in prescription F1–F5 are shown in Table 1. As can be seen from the table, drug content increases with the increase of oil phase. This was consistent with the solubility test results. Among the excipients screened, IVA has the highest solubility in DHML (Figure 2). However, because F9 and F8 could not spontaneously form emulsions, and F7 and F6 produced drug precipitation in freeze-thaw experiments, these four prescriptions were eliminated. With the increase of oil phase ratio (from 1:9 to 5:5), there was a gradual increase in droplet size from 26.56 ± 2.56 to 68.87 ± 4.21 nm, but PDI has maintained under 0.4 (Table 3). Finally, F5 was selected as the best prescription for further in vitro characterization. The addition amounts of each component in prescription F5 were: IVA 0.579 g, DHML 1.5 g, Tween 80 1.0 g and Transcutol HP 0.5 g, respectively.
Effect of pH and Robustness to Dilution
Since the environment of the GIT changes greatly, whether SNEDDS can form a uniform and stable microemulsion in different environments is also very important for drug absorption. The drugs encapsulated in SNEDDS may cause precipitation and other phenomena due to changes in the environment.54 To investigate the pH and volume variation of the droplet size in the transition from stomach to intestine following oral administration, IVA-SNEDDS formulation (F5) was diluted in various media such as water, pH 1.2 HCl, and pH 6.8 PBS with different dilution ratios. Experimental data showed that IVA-SNEDDS was able to form uniform transparent nanoemulsions at different pH solutions even upon dilution of 1000-fold, with the droplet size in the range of 67.44–71.23 nm (Table 4, Figure 5). Besides that, the small PDI value (<0.3) and its little change upon dilution can prove that the nanoemulsion formed by IVA-SNEDDS is relatively stable.55
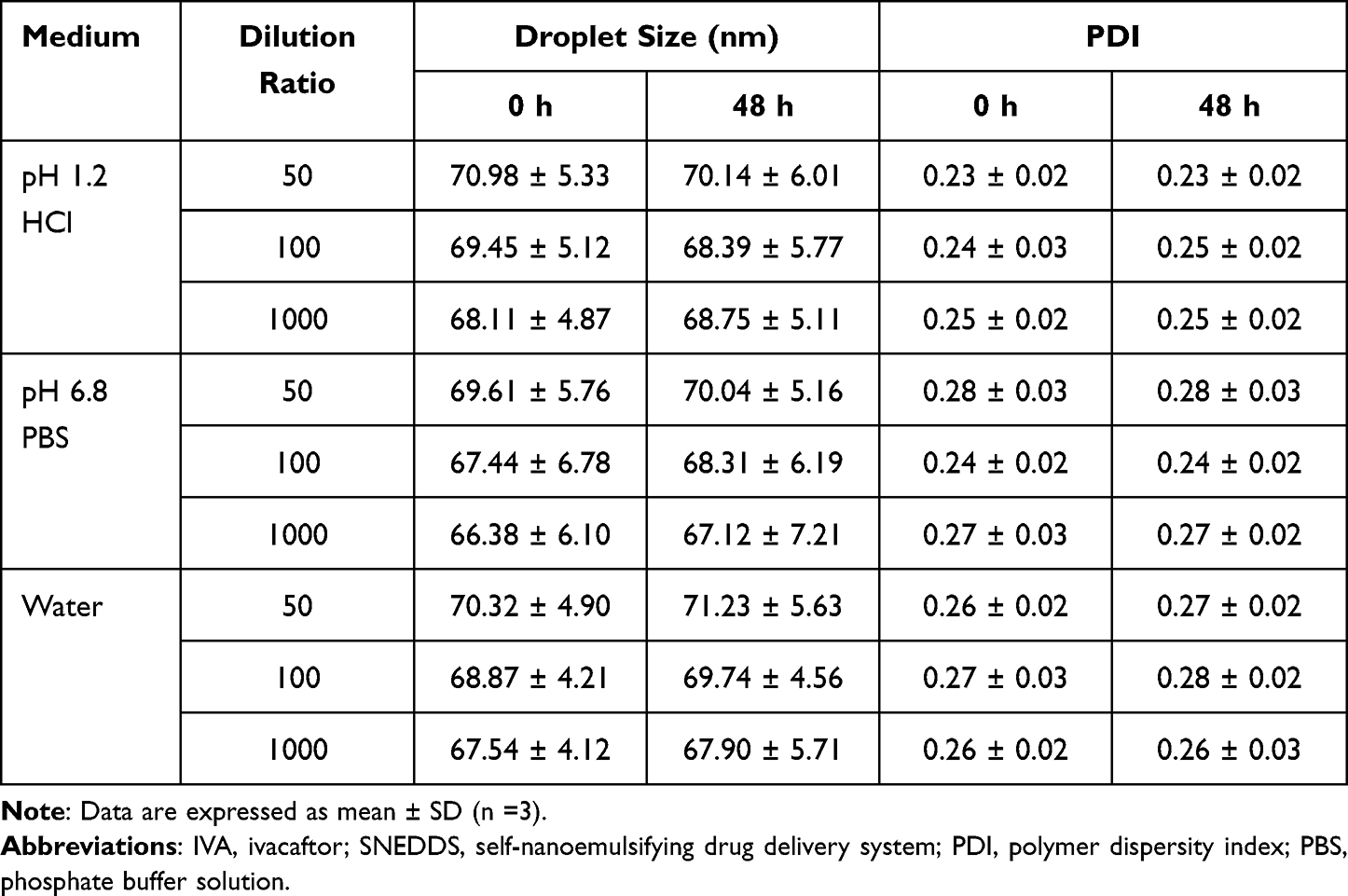 |
Table 4 Robustness to Dilution of IVA-SNEDDS with Different Medium |
 |
Figure 5 The droplet size distribution of IVA-SNEDDS. Abbreviations: IVA, ivacaftor; SNEDDS, self-nanoemulsifying drug delivery system. |
Since DHML has a tertiary amine structure, zeta potential values of IVA-SNEDDS upon dilution with water were found within the scope of 3.4–6.7 mV. Tween 80 and Transcutol HP are non-ionic surfactants that are not charged when forming microemulsions.19 According to Balakumar et al., the zeta potential value of ± 30 mV can offer stability to the system.54
Although the zeta potential of the optimal prescription is not within the above range, there were no significant differences in droplet size and PDI after storage in room temperature for 48 h. The small positive zeta potential values together with absorbed surfactant and co-surfactant layers will provide the required repulsive forces prevent globule coalescence.
Transmission Electron Microscopy (TEM)
Figure 6 portrays the transmission electron microscopic image, depicting the morphology of the reconstituted IVA-SNEDDS formulation. As illustrated in Figure 6, all formed nanoemulsions were spherical, with globule size of less than 100 nm which is in accordance with the results acquired from droplet size analysis. Besides that, Figure 6 clearly illustrates that there are no signs of coalescence, indicating thereby the enhanced physical stability of the formulation.
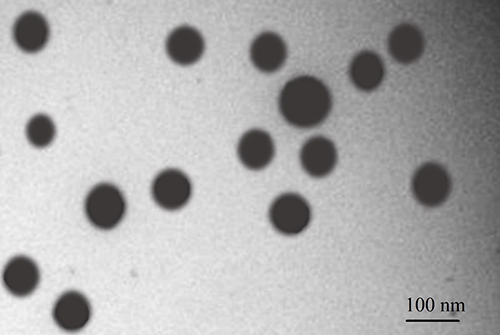 |
Figure 6 Transmission electron microscopy of IVA-SNEDDS. Abbreviations: IVA, ivacaftor; SNEDDS, self-nanoemulsifying drug delivery system. |
In vitro Drug Release Study
In order to increase the oral absorption of insoluble drugs and reduce the influence of food on their absorption, it is important to increase the dissolution of drugs in GIT. IVA is a poorly soluble drug with a solubility of less than 0.05 μg/mL in water. As shown in Figure 7, the drug release profiles of IVA from IVA-suspension were incomplete. Less than 1% of the drug released from the three different pH dissolution media. On the other hand, more than 90% of IVA was released from IVA-SNEDDS in three different pH media, showing faster and complete drug release profiles compared with IVA-suspension (Figure 7). This is because IVA is already dissolved in the formulation of SNEDDS. When it is put into the dissolution vessel, a nanoemulsion is spontaneously formed along with agitation. Moreover, the nanoemulsion can remain stable under different pH and dilution ratios. Thus, based on the results of in vitro drug release studies, we predicted that IVA-SNEDDS would reduce the food effect, and increase IVA oral bioavailability, especially in fasted state.
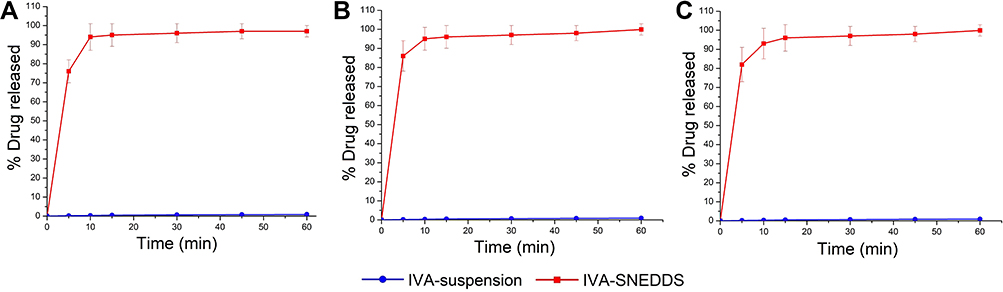 |
Figure 7 In vitro drug release profiles of IVA-SNEDDS and IVA-suspension from (A) water; (B) pH 1.2 HCl; (C) pH 6.8 PBS. Data are expressed as mean ± SD (n = 6). Abbreviations: IVA, ivacaftor; SNEDDS, self-nanoemulsifying drug delivery system; PBS, phosphate buffer solution. |
Storage Stability Studies
The storage stability test results for formulation F5 are shown in Table 5. It can be seen from Table 5 that the content of the drug in IVA-SNEDDS is maintained between 99.63–99.85%, which proves that the drug dissolved in SNEDDS can still remain stable under the experimental conditions for at least 6 months. Further, the appearance, particle size, PDI, and other key parameters of IVA-SNEDDS did not change significantly compared with the beginning, which indicated the formulation exhibited a good shelf life.
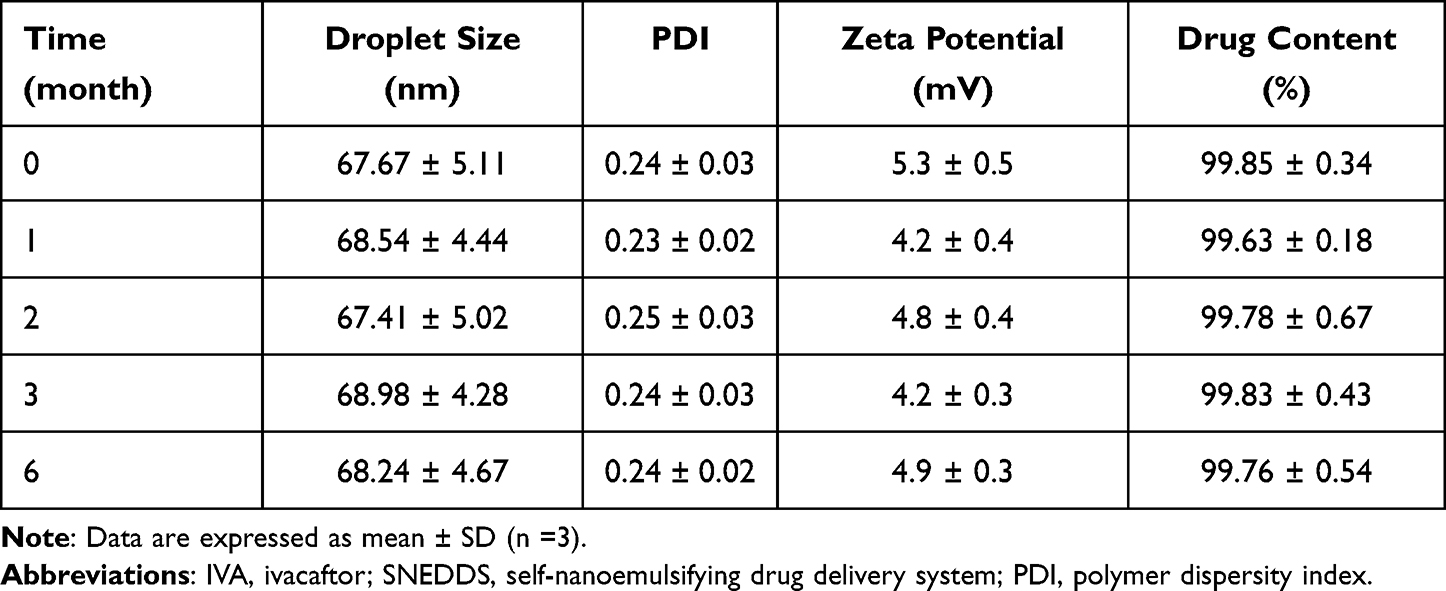 |
Table 5 Stability Data of IVA-SNEDDS |
Pharmacokinetic Studies in Beagle Dogs
Mean plasma IVA concentration-time profiles following oral administration of IVA-SNEDDS and IVA suspension in fasted and fed dogs are shown in Figure 8. The noncompartmental pharmacokinetic parameters were calculated to evaluate the absorption behavior of IVA from IVA-suspension and IVA-SNEDDS. Table 6 presents the relevant mean pharmacokinetic parameters for IVA suspension and IVA-SNEDDS.
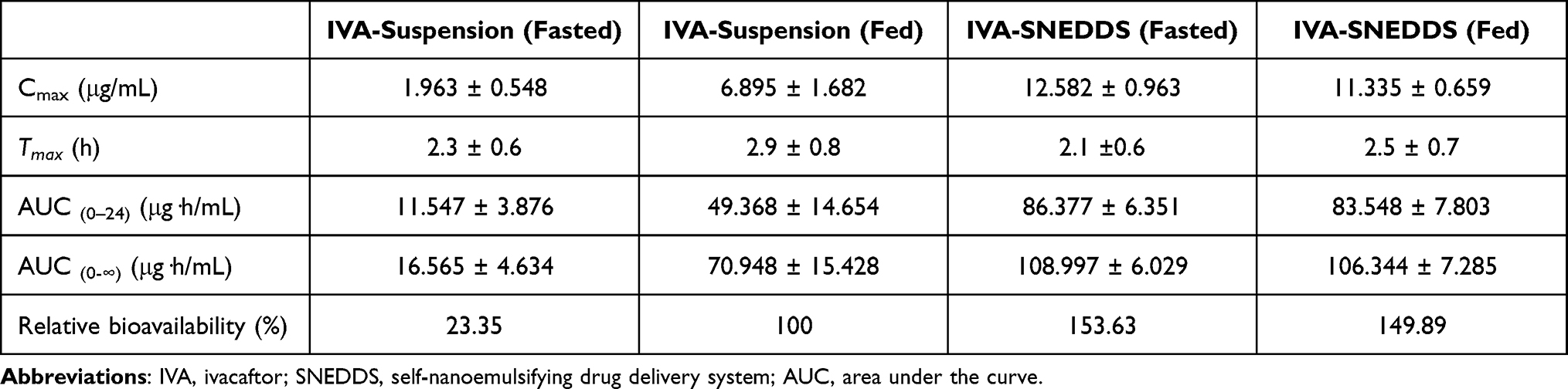 |
Table 6 Pharmacokinetic Parameters of IVA in Beagle Dogs (n = 24) After Oral Administration of IVA-Suspension and the IVA-SNEDDS |
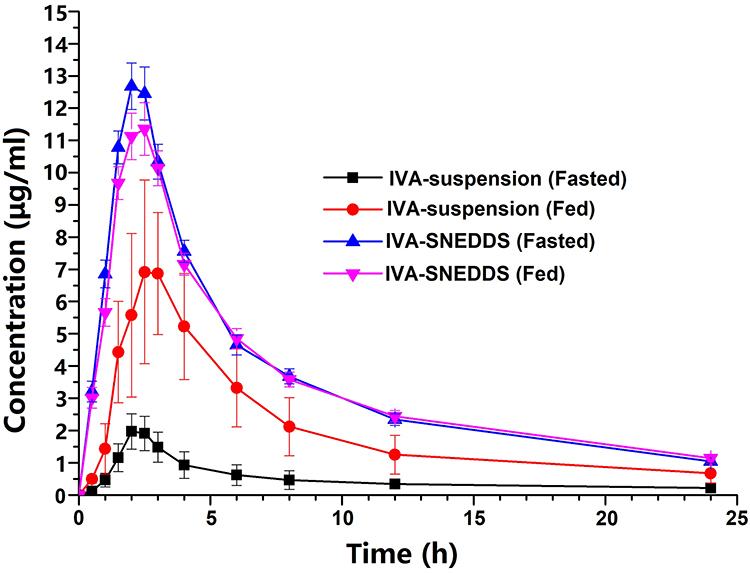 |
Figure 8 Plasma concentration vs time profile of IVA following a single oral administration of IVA-suspension and IVA-SNEDDS in the fasted and fed state. Data are expressed as mean ± SD (n = 24). Abbreviations: IVA, ivacaftor; SNEDDS, self-nanoemulsifying drug delivery system. |
Different from the results of the in vitro drug release study, the absorption of IVA in vivo is slow, and the Tmax is about 2.1–2.9 h. First, this is because the rate and extent of oral drug absorption is determined by a complex interaction between a drug’s physicochemical properties, gastrointestinal (GI) physiologic factors, and the nature of the formulation administered. Different regions of the GI tract have different drug absorptive properties. Thus, the transit time in each GI region and its variability between subjects may contribute to the variability in the rate and/or extent of drug absorption.56,57 Presently, no single in vitro drug release model exists which is able to fully and consistently predict the in vivo performance of SNEDDS.58 Second, self-nanoemulsion can promote the lymphatic absorption of drugs, and the speed of lymphatic circulation is much slower than that of blood circulation,59 which may also be the reason why the Tmax of the drug is about 2.5 h.
For IVA-suspension, the significant inter-individual variability was seen in the standard deviations of the major pharmacokinetic parameters, the mean (±SD) for area under the concentration-time curve (AUC (0–24 h)) and maximum concentration (Cmax) were 49.368 ± 14.654 μg·h/mL, 6.895 ± 1.682 μg/mL (Fed) and 11.547 ± 3.876 μg·h/mL, 1.963 ± 0.548 μg/mL (Fasted), respectively. Further, after oral administration of IVA-suspension in the fed state, pharmacokinetic parameters of AUC (0–24 h) and Cmax were 4.3-fold and 3.5-fold higher than those in the fasted state, respectively (Table 6). These results are consistent with earlier literature that showed the exposure of market product IVA increased approximately 2.5- to 4-fold when given with food that contains fat in healthy volunteers.13,14
In contrast to the significant effects of food on absorption of IVA-suspension or marketed product, the food has no significant effect on the pharmacokinetics of IVA with IVA-SNEDDS formulation (Table 6), as the relative bioavailability (IVA-suspension (Fed) as reference) of IVA-SNEDDS in the Fed and Fasted state were 149.89% and 153.63%, respectively. Besides that, IVA-SNEDDS formulation showed reduced inter-individual variability with the standard deviations of the major pharmacokinetic parameters after oral administration of the IVA-SNEDDS formulation was decreased. This may be due to the fact that after oral administration of IVA-SNEDDS, IVA-SNEDDS can form a nanoemulsion under the slight peristalsis of the gastrointestinal tract, and the nanoemulsion increases the absorption area of IVA in the gastrointestinal tract.60 In addition, the drug is dissolved in the oil phase and is located in the core of the nanoemulsion, which is not affected by environmental changes such as pH, ions, and food in the gastrointestinal tract, and can also reduce the degradation effect of enzymes on it.61
For water-insoluble drugs such as IVA, the ability to dissolve in the gastrointestinal tract is a prerequisite for drug absorption. After oral administration of drugs, due to individual differences, the gastrointestinal environment varies greatly, which has a great impact on the dissolution and absorption of drugs. Even due to the different eating habits of each person, the environment of the gastrointestinal tract before and after meals will also undergo dramatic changes, which will also lead to changes in drug absorption.62 IVA-SNEDDS can rapidly form a nanoemulsion under peristalsis of the gastrointestinal tract, and the solubility of the drug wrapped in SNEDDS is not only unaffected by food, pH and bile in the gastrointestinal tract but it also protects the drug from enzymatic degradation. Therefore, SNEDDS could be a promising alternative for delivery of IVA, by improving oral bioavailability and reducing the food effect on drug absorption. Nevertheless, further studies are needed to recommend the optimal IVA-SNEDDS formulation for use in clinical applications.
Conclusion
In this paper, a newly developed lipid substance DHML was used to construct an IVA-SNEDDS which showed solubility enhancement and no food effect of IVA. Due to the diversity of structural properties of DHML, it showed it greatly improves the solubility of IVA compared with other oils. The IVA-SNEDDS formulation consisting of DHML, Tween 80, and Transcutol HP spontaneously formed uniform nanoemulsions in different pH media with slight stirring and the mean globule size was less than 75 nm regardless of dilution ratio and pH conditions. In addition, IVA-SNEDDS showed good stability at the end of six months under 40 ± 2℃ and 65 ± 5% relative humidity (RH) storage conditions. The in vitro drug release showed complete and fast release of above 95% within 15 min. The results of the in vivo pharmacokinetic study show that the prepared IVA-SNEDDS using DHML can enhance the solubility of IVA as well as increase the oral absorption in the fasted state, thereby reducing the food effect on the absorption of IVA. In conclusion, this paper indicated that SNEDDS prepared by using DHML as oil phase can increase the drug loading of insoluble drugs in SNEDDS. These data provide proof of concept that, when IVA-SNEDDS is used in clinical practice, oral bioavailability of IVA will be increased together with a reduction in inter-individual absorption variability and the impact of food on drug absorption.
Acknowledgments
The authors are grateful to Taian Rutocel co., Ltd (Tai’an, Shandong, China) for providing DHML as a gift sample. This research was funded by Taishan University “1958 Talent Project”, grant numbers 2019-48.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Alqahtani MS, Kazi M, Alsenaidy MA, Ahmad MZ. Advances in oral drug delivery. Front Pharmacol. 2021;12:618411. doi:10.3389/fphar.2021.618411
2. Murdande SB, Pikal MJ, Shanker RM, Bogner RH. Solubility advantage of amorphous pharmaceuticals: I. A thermodynamic analysis. J Pharm Sci. 2010;99(3):1254–1264. doi:10.1002/jps.21903
3. Morishita M, Peppas NA. Advances in oral drug delivery: improved bioavailability of poorly absorbed drugs by tissue and cellular optimization. Preface Adv Drug Deliv Rev. 2012;64(6):479. doi:10.1016/j.addr.2012.02.008
4. Meola TR, Joyce P, Wignall A, Bremmell KE, Prestidge CA. Harnessing the potential of nanostructured formulations to mimic the food effect of lurasidone. Int J Pharm. 2021;608:121098. doi:10.1016/j.ijpharm.2021.121098
5. Nasir S, Hussain A, Abbas N, Bukhari NI, Hussain F, Arshad MS. Improved bioavailability of oxcarbazepine, a BCS class II drug by centrifugal melt spinning: in-vitro and in-vivo implications. Int J Pharm. 2021;604:120775. doi:10.1016/j.ijpharm.2021.120775
6. Schultz HB, Meola TR, Thomas N, Prestidge CA. Oral formulation strategies to improve the bioavailability and mitigate the food effect of Abiraterone acetate. Int J Pharm. 2020;577:119069. doi:10.1016/j.ijpharm.2020.119069
7. Sager M, Grimm M, Aude P, et al. In vivo characterization of enTRinsic™ drug delivery technology capsule after intake in fed state: a cross-validation approach using salivary tracer technique in comparison to MRI. J Control Release. 2019;313:24–32. doi:10.1016/j.jconrel.2019.10.023
8. Frost C, Wang J, Nepal S, et al. Apixaban, an oral, direct factor Xa inhibitor: single dose safety, pharmacokinetics, pharmacodynamics and food effect in healthy subjects. Br J Clin Pharmacol. 2013;75(2):476–487. doi:10.1111/j.1365-2125.2012.04369.x
9. Dening TJ, Rao S, Thomas N, Prestidge CA. Silica encapsulated lipid-based drug delivery systems for reducing the fed/fasted variations of ziprasidone in vitro. Eur J Pharm Biopharm. 2016;101:33–42. doi:10.1016/j.ejpb.2016.01.010
10. Darade A, Pathak S, Sharma S, Patravale V. Atovaquone oral bioavailability enhancement using electrospraying technology. Eur J Pharm Sci. 2018;111:195–204. doi:10.1016/j.ejps.2017.09.051
11. Davis PB, Yasothan U, Kirkpatrick P. Ivacaftor. Nat Rev Drug Discov. 2012;11(5):349–350. doi:10.1038/nrd3723
12. Wainwright CE, Elborn JS, Ramsey BW. Lumacaftor-Ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N Engl J Med. 2015;373(18):1783–1784. doi:10.1056/NEJMoa1409547
13. Liddy AM, McLaughlin G, Schmitz S, D’Arcy DM, Barry MG. The pharmacokinetic interaction between ivacaftor and ritonavir in healthy volunteers. Br J Clin Pharmacol. 2017;83(10):2235–2241. doi:10.1111/bcp.13324
14. Wainwright CE. Ivacaftor for patients with cystic fibrosis. Expert Rev Respir Med. 2014;8:533–538. doi:10.1586/17476348.2014.951333
15. Welshman IR, Sisson TA, Jungbluth GL, Stalker DJ, Hopkins NK. Linezolid absolute bioavailability and the effect of food on oral bioavailability. Biopharm Drug Dispos. 2001;22(3):91–97. doi:10.1002/bdd.255
16. Kaur S, Jena SK, Samal SK, Saini V, Sangamwar AT. Freeze dried solid dispersion of exemestane: a way to negate an aqueous solubility and oral bioavailability problems. Eur J Pharm Sci. 2017;107:54–61. doi:10.1016/j.ejps.2017.06.032
17. Smeets A, Koekoekx R, Clasen C, Van den Mooter G. Amorphous solid dispersions of darunavir: comparison between spray drying and electrospraying. Eur J Pharm Biopharm. 2018;130:96–107. doi:10.1016/j.ejpb.2018.06.021
18. Patel V, Lalani R, Bardoliwala D, Ghosh S, Misra A. Lipid-based oral formulation strategies for lipophilic drugs. AAPS Pharm Sci Tech. 2018;19(8):3609–3630. doi:10.1208/s12249-018-1188-8
19. Inugala S, Eedara BB, Sunkavalli S, et al. Solid self-nanoemulsifying drug delivery system (S-SNEDDS) of darunavir for improved dissolution and oral bioavailability: in vitro and in vivo evaluation. Eur J Pharm Sci. 2015;74:1–10. doi:10.1016/j.ejps.2015.03.024
20. Singh G, Pai RS, Atazanavir-loaded Eudragit RL. 100 nanoparticles to improve oral bioavailability: optimization and in vitro/in vivo appraisal. Drug Deliv. 2016;23(2):532–539. doi:10.3109/10717544.2014.930760
21. Meka AK, Jenkins LJ, Dàvalos-Salas M, et al. Enhanced solubility, permeability and anticancer activity of vorinostat using tailored mesoporous silica nanoparticles. Pharmaceutics. 2018;10(4):283. doi:10.3390/pharmaceutics10040283
22. Elsayad MK, Mowafy HA, Zaky AA, Samy AM. Chitosan caged liposomes for improving oral bioavailability of rivaroxaban: in vitro and in vivo evaluation. Pharm Dev Technol. 2021;26(3):316–327. doi:10.1080/10837450.2020.1870237
23. Shah S, Parmar B, Soniwala M, Chavda J. Design, optimization, and evaluation of lurasidone hydrochloride nanocrystals. AAPS Pharm Sci Tech. 2016;17(5):1150–1158. doi:10.1208/s12249-015-0449-z
24. Vaishali YL, Aishwarya BD, Sarita RS, Yogesh AK. Lurasidone-β-cyclodextrin complexes: physicochemical characterization and comparison of their antidepressant, antipsychotic activities against that of self microemulsifying formulation. J Mol Struc. 2018;1157:395–400. doi:10.1016/j.molstruc.2017.12.042
25. Date AA, Nagarsenker MS. Design and evaluation of self-nanoemulsifying drug delivery systems (SNEDDS) for cefpodoxime proxetil. Int J Pharm. 2007;329(1–2):166–172. doi:10.1016/j.ijpharm.2006.08.038
26. Bernkop-Schnürch A, Jalil A. Do drug release studies from SEDDS make any sense? J Control Release. 2018;271:55–59. doi:10.1016/j.jconrel.2017.12.027
27. Ameeduzzafar E-BI, Alruwaili NK, Elkomy MH, et al. Development of novel dapagliflozin loaded solid self-nanoemulsifying oral delivery system: physiochemical characterization and in vivo antidiabetic activity. J Drug Deliv Sci Technol. 2019;54:101279. doi:10.1016/j.jddst.2019.101279
28. Kim JS, Din FU, Lee SM, et al. Comparison of three different aqueous microenvironments for enhancing oral bioavailability of sildenafil: solid self-nanoemulsifying drug delivery system, amorphous microspheres and crystalline microspheres. Int J Nanomedicine. 2021;16:5797–5810. doi:10.2147/IJN.S324206
29. Panigrahi KC, Patra CN, Rao MEB. Quality by design enabled development of oral self-nanoemulsifying drug delivery system of a novel calcimimetic cinacalcet HCl using a porous carrier: in vitro and in vivo characterisation. AAPS Pharm Sci Tech. 2019;20(5):216. doi:10.1208/s12249-019-1411-2
30. Thomas N, Holm R, Müllertz A, Rades T. In vitro and in vivo performance of novel supersaturated self-nanoemulsifying drug delivery systems (super-SNEDDS). J Control Release. 2012;160(1):25–32. doi:10.1016/j.jconrel.2012.02.027
31. Miao Y, Chen G, Ren L, Pingkai O. Characterization and evaluation of self-nanoemulsifying sustained-release pellet formulation of ziprasidone with enhanced bioavailability and no food effect. Drug Deliv. 2016;23(7):2163–2172. doi:10.3109/10717544.2014.950768
32. Woo JS, Song YK, Hong JY, Lim SJ, Kim CK. Reduced food-effect and enhanced bioavailability of a self-microemulsifying formulation of itraconazole in healthy volunteers. Eur J Pharm Sci. 2008;33(2):159–165. doi:10.1016/j.ejps.2007.11.001
33. Perlman ME, Murdande SB, Gumkowski MJ, et al. Development of a self-emulsifying formulation that reduces the food effect for torcetrapib. Int J Pharm. 2008;351(1–2):15–22. doi:10.1016/j.ijpharm.2007.09.015
34. Zhang Y, Bai Y, Chen H, Huang Y, Yuan P, Zhang L. Preparation of a colon-specific sustained-release capsule with curcumin-loaded SMEDDS alginate beads. Rsc Adv. 2017;7:22280–22285. doi:10.1039/C6RA27693H
35. Ishak RA, Osman R. Lecithin/TPGS-based spray-dried self-microemulsifying drug delivery systems: in vitro pulmonary deposition and cytotoxicity. Int J Pharm. 2015;485(1–2):249–260. doi:10.1016/j.ijpharm.2015.03.019
36. Chaudhari KS, Akamanchi KG. Novel bicephalous heterolipid based self-microemulsifying drug delivery system for solubility and bioavailability enhancement of efavirenz. Int J Pharm. 2019;560:205–218. doi:10.1016/j.ijpharm.2019.01.065
37. Constantinides PP. Lipid microemulsions for improving drug dissolution and oral absorption: physical and biopharmaceutical aspects. Pharm Res. 1995;12(11):1561–1572. doi:10.1023/A:1016268311867
38. Zafar A, Imam SS, Alruwaili NK, et al. Development of piperine-loaded solid self-nanoemulsifying drug delivery system: optimization, in-vitro, ex-vivo, and in-vivo evaluation. Nanomaterials. 2021;11(11):2920. doi:10.3390/nano11112920
39. Rashid R, Kim DW, Yousaf AM, et al. Comparative study on solid self-nanoemulsifying drug delivery and solid dispersion system for enhanced solubility and bioavailability of ezetimibe. Int J Nanomedicine. 2015;10:6147–6159. doi:10.2147/IJN.S91216
40. Lefebvre G, Riou J, Bastiat G, et al. Spontaneous nano-emulsification: process optimization and modeling for the prediction of the nanoemulsion’s size and polydispersity. Int J Pharm. 2017;534(1–2):220–228. doi:10.1016/j.ijpharm.2017.10.017
41. Müller RH, Jacobs C, Kayser O. Nanosuspensions as particulate drug formulations in therapy. Rationale for development and what we can expect for the future. Adv Drug Deliv Rev. 2001;47(1):3–19. doi:10.1016/S0169-409X(00)00118-6
42. Bhagwat DA, Swami PA, Nadaf SJ, et al. Capsaicin loaded solid SNEDDS for enhanced bioavailability and anticancer activity: in-vitro, in-silico, and in-vivo characterization. J Pharm Sci. 2021;110(1):280–291. doi:10.1016/j.xphs.2020.10.020
43. Alothaid H, Aldughaim MS, Yusuf AO, et al. A comprehensive study of the basic formulation of supersaturated self-nanoemulsifying drug delivery systems (SNEDDS) of albendazolum. Drug Deliv. 2021;28(1):2119–2126. doi:10.1080/10717544.2021.1986601
44. Annisa R, Yuwono M, Hendradi E. Formulation and characterization of Eleutherine palmifolia extract-loaded self-nanoemulsifying drug delivery system (SNEDDS). J Basic Clin Physiol Pharmacol. 2021;32(4):859–865. doi:10.1515/jbcpp-2020-0400
45. Anne ML, Gavin M, Susanne S, Deirdre MD, Michael GB. Stability indicating RP-HPLC method development and validation for the determination of ivacaftor in bulk and its pharmaceutical formulation. J Int Pharm Res. 2021;13:14–21.
46. Patel MH, Sawant KK. Self microemulsifying drug delivery system of lurasidone hydrochloride for enhanced oral bioavailability by lymphatic targeting: in vitro, Caco-2 cell line and in vivo evaluation. Eur J Pharm Sci. 2019;138:105027. doi:10.1016/j.ejps.2019.105027
47. Singh N, Bansal P, Maithani M, Chauhan Y. Development and validation of a novel stability-indicating RP-HPLC method for simultaneous determination of tezacaftor and ivacaftor in fixed dose combination. J Chromatogr Sci. 2020;58(4):346–354. doi:10.1093/chromsci/bmz120
48. Patel MR, Patel MH, Patel RB. Preparation and in vitro/ex vivo evaluation of nanoemulsion for transnasal delivery of paliperidone. Appl Nanosc. 2016;6:1095–1104. doi:10.1007/s13204-016-0527-x
49. O’Shea JP, Holm R, O’Driscoll CM, Griffin BT. Food for thought: formulating away the food effect - a PEARRL review. J Pharm Pharmacol. 2019;71(4):510–535. doi:10.1111/jphp.12957
50. Ameeduzzafar AJ, Bhatnagar A, Kumar N, Ali A. Chitosan nanoparticles amplify the ocular hypotensive effect of cateolol in rabbits. Int J Biol Macromol. 2014;65:479–491. doi:10.1016/j.ijbiomac.2014.02.002
51. Rashid R, Kim DW, Din FU, et al. Effect of hydroxypropylcellulose and Tween 80 on physicochemical properties and bioavailability of ezetimibe-loaded solid dispersion. Carbohydr Polym. 2015;130:26–31. doi:10.1016/j.carbpol.2015.04.071
52. Zeng L, Xin X, Zhang Y. Development and characterization of promising Cremophor EL-stabilized o/w nanoemulsions containing short-chain alcohols as a cosurfactant. RSC Adv. 2017;7(32):19815–19827. doi:10.1039/C6RA27096D
53. Craig DQ, Barker SA, Banning D, Booth SW. An investigation into the mechanisms of self-emulsification using particle size analysis and low frequency dielectric spectroscopy. Int J Pharm. 1995;114:103–110.
54. Balakumar K, Raghavan CV, Abdu S. Self nanoemulsifying drug delivery system (SNEDDS) of rosuvastatin calcium: design, formulation, bioavailability and pharmacokinetic evaluation. Colloids Surf B Biointerfaces. 2013;112:337–343. doi:10.1016/j.colsurfb.2013.08.025
55. Beg S, Jena SS, Patra C, et al. Development of solid self-nanoemulsifying granules (SSNEGs) of ondansetron hydrochloride with enhanced bioavailability potential. Colloids Surf B Biointerfaces. 2013;101:414–423. doi:10.1016/j.colsurfb.2012.06.031
56. Koziolek M, Alcaro S, Augustijns P, et al. The mechanisms of pharmacokinetic food-drug interactions - A perspective from the UNGAP group. Eur J Pharm Sci. 2019;134:31–59. doi:10.1016/j.ejps.2019.04.003
57. Koziolek M, Carrière F, Porter CJH. Lipids in the stomach - implications for the evaluation of food effects on oral drug absorption. Pharm Res. 2018;35(3):55. doi:10.1007/s11095-017-2289-x
58. Berthelsen R, Klitgaard M, Rades T, Müllertz A. In vitro digestion models to evaluate lipid based drug delivery systems; present status and current trends. Adv Drug Deliv Rev. 2019;142:35–49. doi:10.1016/j.addr.2019.06.010
59. Deng J, Zhu X, Chen Z, et al. A review of food-drug interactions on oral drug absorption. Drugs. 2017;77(17):1833–1855. doi:10.1007/s40265-017-0832-z
60. Zafar A. Development of oral lipid based nano-formulation of dapagliflozin: optimization, in vitro characterization and ex vivo intestinal permeation study. J Oleo Sci. 2020;69(11):1389–1401. doi:10.5650/jos.ess20162
61. Smith L, Serrano DR, Mauger M, Bolás-Fernández F, Dea-Ayuela MA, Lalatsa A. Orally bioavailable and effective buparvaquone lipid-based nanomedicines for visceral leishmaniasis. Mol Pharm. 2018;15(7):2570–2583. doi:10.1021/acs.molpharmaceut.8b00097
62. Kim JS, Ud Din F, Lee SM, et al. Comparative study between high-pressure homogenisation and Shirasu porous glass membrane technique in sildenafil base-loaded solid SNEDDS: effects on physicochemical properties and in vivo characteristics. Int J Pharm. 2021;592:120039. doi:10.1016/j.ijpharm.2020.120039
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.