")
Back to Journals » Journal of Pain Research » Volume 13
Rediscovery of Ceruletide, a CCK Agonist, as an Analgesic Drug
Authors Keppel Hesselink JM
Received 27 September 2019
Accepted for publication 24 December 2019
Published 15 January 2020 Volume 2020:13 Pages 123—130
DOI https://doi.org/10.2147/JPR.S232714
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Michael A Ueberall
Jan M Keppel Hesselink 1, 2
1Faculty of Health, University of Witten/Herdecke, Witten, Germany; 2Department Research and Development, Institute for Neuropathic Pain, Bosch en Duin, The Netherlands
Correspondence: Jan M Keppel Hesselink Tel +31651700527
Email [email protected]
Abstract: Ceruletide (CRL) is a decapeptide, originating from the skin of a tropical frog, and is many times more potent that cholecystokinin (CCK) in a number of assays. The compound was first isolated and characterized around 50 years ago, and its analgesic properties were subsequently identified. Since the 1980s it has been available in the clinic as a parenteral solution and is used as a diagnostic tool to characterize pancreas and gall bladder malfunctions. Its analgesic properties were evaluated in a number of indications: cancer pain, burns, colic pains and migraine. Preclinically, CRL reduces pain in low microgram dose range and promotes clear and long-lasting analgesic activity in nanograms when applied centrally. CCK is amongst the most widely expressed neuropeptides in the brain. CCK-induced analgesic effects in response to persistent and inflammatory pain have recently been associated with CCK2 receptor signaling. CRL, a potent CCK agonist, might be worthwhile to rediscover as a putative analgesic drug and could represent a potential analgesic intrathecal strategy to patients with cancer-related pain.
Keywords: caerulein, neuropeptide, pain, cancer, Kambo
Introduction
In light of our interest in the repositioning of old drugs in new indications, we explored in some detail the cases of dermorphin and phenytoin for the treatment of chronic pain, in the light of the importance of identifying novel non-opioid medication for pain control, especially related to cancer. We found that the clinical development of dermorphin as an analgesic stagnated in the 1980s without any good reason. New formulations for dermorphin could represent a useful intrathecal strategy for cancer-induced pain control.1 The prototype of all anti-epileptic drugs, the broad acting sodium channel blocker phenytoin, has already been repositioned by our group at the Institute of Neuropathic Pain in the Netherlands, to treat peripheral neuropathic pain. Painful diabetic neuropathy and chemotherapy-induced neuropathic pain, as well as pain in small fiber neuropathies seem to be the spectrum of indications where the repositioning of phenytoin makes sense.2–4 We tested our assumption that the further development of a topical phenytoin formulation in a simple Phase III program would be feasible during a consultation with the Dutch Medicines Evaluation Board (MEB). The MEB agreed that no further preclinical data, nor extensive clinical data were required, and that two clinical studies based on an enriched population would suffice to obtain registration. We are now in the last phase of designing such studies and a number of Dutch hospitals, including two academic ones, plan on joining our group in bringing this investigator-driven development plan to a good end. This case clearly demonstrates that investigators are able to develop drugs and that many millions of Euros are not required to support the notion that the rediscovery and repositioning of old drugs are possible and can be cheaper compared to industry-developed novel drugs.5
In this paper, we explore the neuropharmacological literature related to potential leads for the rediscovery of analgesic drugs, based on what endogenous tribes in the Amazonian forest call “Kambo”.6 Kambo is a dermal secretion of a specific Amazon frog (Phyllomedusa bicolor) that contains many bioactive peptides and was first explored in the early 1990s by the Italian research group of Professor Vittorio Erspamer (1909–1999). These tribes have known for centuries about the remarkable properties of this excretion, which is administered to fresh burn wounds after the blistered skin is scraped away.7 The bioactive compounds quickly enter the lymphatic system and subsequently the blood. Its effects are immediate and amongst others consist of hypotension, tachycardia, nausea, vomiting, facial edema and sweating. These short-time effects are seen by users as the detoxification of the body, and members of certain Amazonian tribes use it to improve their hunting skills (especially the Katukina, Kaxinawá, Matsés, Mayoruna and Yawanawá tribes). After an administration of Kambo, the reported long-term effects apparently last for days and result in the hunters developing sharper sight, smell and the ability to foresee game. The hunters claim they could freely move in the jungle for long distances without any effort and they felt more powerful. As a result, hunting was very successful. These long-term effects could very well have been caused by some of the bioactive peptides with high affinity for the opiate receptor systems, dermorphin, deltorphin or by the cholecystokinin (CCK) agonist CRL; all of these peptides are known for having additional analgesic effects.
In this review, we present and discuss relevant preclinical and clinical studies that demonstrate general and analgesic effects of ceruletide and suggest a rediscovery of this molecule as a non-opioid strategy for the control of cancer-related pain. Ceruletide is one of the several molecules that are not completely explored in drug development. The information we disclose might be of value in determining a possible different strategy for the management of cancer pain, relevant given the recent problems that countries, especially the US, are experiencing with opioid discriminate overuse.
Ceruletide: Introduction
CRL (cerulein or caerulein) is a bioactive peptide that stimulates smooth muscle, increases digestive secretions and demonstrates analgesic properties. Interest from the scientific community in this peptide peaked in 1985, with 100 papers indexed in PubMed. Since then, however, interest has declined (see Figure 1) with an absence of clear reasons. Its analgesic effects in humans were first mentioned at the beginning of the 1980s8–11 and these effects have been further evaluated in a number of academic clinical trials. We will review these studies and analyze the analgesic potential of CRL in order to be able to recommend the further rediscovery of CRL as a treatment for cancer pain. This analysis will also enable us to outline some weak points and flaws in the scientific exploration of this neuroactive peptide.
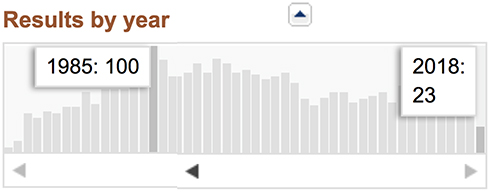 |
Figure 1 PubMed indexed papers on CRL – search results 12-12-2018. |
Professor Vittorio Erspamer (1909–1999), who was twice nominated for a Nobel Prize, and was supported by the Nobel Laureate Rite Levi-Montalcini, first reported a new bioactive peptide isolated from the skin of a frog, the Hyla caerulea (new nomenclature Litoria caerulea), in 1966.12 Erspamer referred to this peptide as caerulein, due to the frog’s green color and its origin in the Hyla caerulea; the compound was characterized as a decapeptide. Later on, the peptide was found to be present in large amounts in the skins of a number of different species of frogs. In 1966, the initial pharmacological actions were on reducing blood pressure, ‘conspicuous effects on some external secretions’, and a spasmogenic action on extravascular smooth muscles. In 1976, together with scientists from the Italian company Farmitalia, the decapeptide was further characterized as Pyr-Gln-Asp-Tyr(SO3H)-Thr-Gly-Trp-Met-Asp-Phe-NH2, and the similarity to cholecystokinin (CCK) and gastrin structures was noted, all sharing the common C-terminal amino acid sequence: Gly-Trp-Met-Asp-Phe-NH2.13 The decapeptide CRL has great similarities to cholecystokinin octapeptide (CCK8): Asp-Tyr(SO3H)-Met-Gly-Trp-Met-Asp-Phe-NH2.
In the first series of pharmacological experiments conducted by Erspamer et al (1967), CRL potently induced gallbladder contractions in an in-situ model of gallbladder which 1 mcg was equipotent to 7–15 mg of pure CCK.14 Using a different gallbladder model, the threshold dose of CRL was around ng/kg in dogs. Emesis and diarrhea were also monitored as GI effects, and in an intact conscious dog, the threshold emetic dose was approximately 0.5 mcg and 2 mcg/kg BW (kilogram-bodyweight), given via IV and SC route, respectively. The first human application of CRL was reported in Erspamer’s 1967 study.14 The peptide was administered via intradermal injection of an unspecified dose into the forearm, and an increased capillary permeability was reported, approximately half as intense as the effect caused by bradykinin. In addition, CRL produced gallbladder contractions at IV doses as low as 1–2 ng/kg BW. Its effects on the stomach, pancreas and gallbladder secretion were also noted, and these effects were suggested to mimic many of the effects of bradykinin, gastrin and cholecystokinin-pancreozymin. Several synthetic CRL-like polypeptides at that time (1967) were synthesized. The peptide and a number of its analogues were subsequently taken up for further exploration by the Italian company Farmitalia Carlo Erba.15 CRL soon became available under the brand names Takus and Ceosunin and was initially distributed in the USA by Adria Laboratories Inc. As early as 1980, various producers marketed CRL-me-too’s lacking parts of the original molecule and with suboptimal biological effects, which might have created some confusion in relation to the biological activity of the molecule.15
CRL – an Analgesic Peptide Not Explored to Its Full Potential
CRL still clinically available in and is registered by the FDA as CRL diethylamide injectable, injection (Pharmacia and Upjohn) 0.02 mg/mL N018296 001, under the brand name Tymtran.16 It is also available in Europe, sold for instance as Cerulex in France and as Takus in Germany, formulation: CRL diethylamine 0.02 mg/mL.17 In Germany 1 mL vials containing 5 µg CRL are available, commercialized by Pharmacia. The recommended dose is 0.050 µg/kg BW.18 In Italy, the current producer is PFIZER Italia S.r.l.
See Figure 2 for its chemical structure. The diethylamine salt of CRL is commercially available in an aqueous solution containing an antioxidant (sodium thiomalate) and its pH is adjusted to 7.0. It is still used in the gastroenterological clinic as its biological action is more pronounced in comparison to CCK. When assessed in a gallbladder bio-assay, CRL was 10 times more active than CCK on a molar basis and 47 times more potent on a weight basis.19
Preclinically, CRL produced an analgesic effect in different animal models of nociception. CRL was 114 and 15 times more potent than morphine on a hot plate and writhing test, respectively, while cholecystokinin was 3–10 times less active than CRL.20 In a series of elegant animal experiments, Barber et al (1989) concluded that on a molar basis CRL is much more potent than CCK and morphine, that the analgesic effect of the peptides is probably mediated centrally, and that although specific opioid receptors may be involved it is unlikely that there is a direct interaction of CRL with known opioid receptors.21
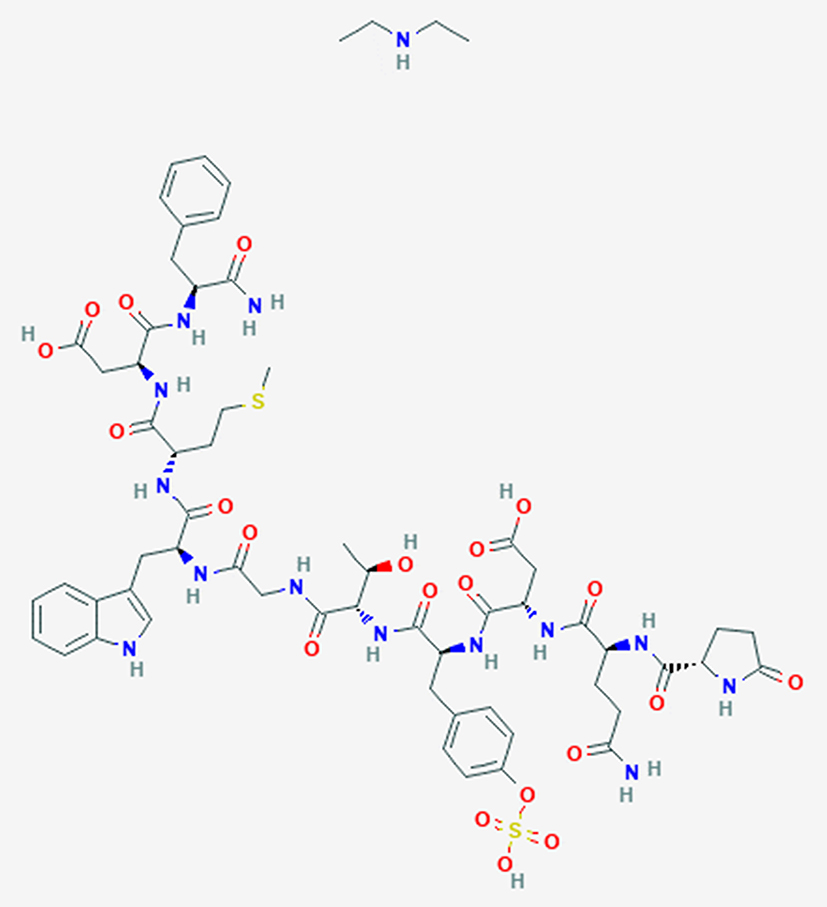 |
Figure 2 Structure of the decapeptide CRL diethylamine (PubChem, open chemistry database). |
Stacher et al (1982) from the University of Vienna, Austria, tested CRL in an acute pain test paradigm in men, based on electrically thermally induced cutaneous pain.22 In the first series of experiments on 24 volunteers, IV infusions of 60 and 120 ng/kg BW per hour of CRL produced potent analgesic effects which differed significantly from those of placebo infusions. In a follow-up study published in the same paper, CRL in a low dose of 6 and 60 ng/kg BW per hour were compared to those of a clinically effective dose of pentazocine. The low dose was slightly effective, and pentazocine was more effective compared to the 60 ng CRL group. Naloxone could reverse the analgesic effect of pentazocine, but it had no effect after the two doses of CRL. Both doses of CRL caused a slight fall in blood pressure, whereas a marked rise of systolic as well as of diastolic pressure occurred after treatment with pentazocine. CRL had no systematic influence on the respiratory rate while pentazocine caused a marked depression, which could be blocked by the administration of naloxone.
In random order and in a double-blind study, Basso et al (1982) first infused either a placebo or an (IV) CRL in doses of 1, 2 or 4 ng/kg BW to 8 claudicatio patients with rest pain once daily for 1 min.23 In this experiment rest pain was totally abolished, the analgesic effects lasting for around 6 hrs following the 1 ng dose, 9 hrs based on the 2 ng dose of CRL, and 7 hrs following the 4 ng dose. The placebo did not alter the rest pain. In a follow-up study presented in the same paper, and to unravel its mechanism, IV placebo or naloxone HC1 (0.8 mg) was administered 15 mins prior to a CRL administration of 2 ng/kg BW. Naloxone could totally abolish the analgesic effect, and beta-endorphin levels in the blood were raised significantly up to 5 hrs after infusion (doubled compared to baseline) of 2 ng/kg BW.
In 1983 the analgesic effects of CRL (single-blind) were evaluated in 45 patients suffering from pain in malignancies (n=8), burns (n=22) and cardiac infarctions (n=15); there were 15 control patients and 4 epileptic patients entered as extra control patients.24 The authors first reviewed the ‘quite pronounced analgesic effects, in biliary colics’ (p.41) of CRL based on earlier material (Santamaria et al, 1979; Saggario, 1979), as well as its analgesic effects in malignancies (Piazza, 1981), burns and myocardial infarction (Dolocek et al, 1982).25–28 The dose administered in this case-collection was 5 mcg infused in 30 mins, twice a day in most patients, and in some patients the same amount was administered as an IM injection. A complete disappearance of pain was reported in 24 patients, marked decreases in pain in 12 patients, and no additional analgesics were needed with any patient. Nine patients reported a slight decrease of pain and in 6 patients no analgesic effect was established. In many cases, the analgesic effects lasted 8–12 hrs after administration. With the severely burned patients, an increasing analgesic effect of CRL became apparent after several days of administration, and the analgesia was accompanied in half of all cases by a generally improved mood and even euphoria. It was noted that the infusion should start 1 hr before wound-dress changes, in order to reduce pain sufficiently during the wound care as well as for the rest of the day. Side effects were rare, only some minor ones were reported – headache, nausea and abdominal colic, mostly when the infusion rate was too high.
Malfertheiner (1983) reported three patients with refractory migraine attacks, treated with an IV infusion of CRL 2 ng/kg/min, and in all cases, a substantial pain decline was reported.29
Pardo et al (1984) reported the results of a randomized double-blind study in 60 patients suffering from moderate to severe biliary colic pain, based on the clinical picture and stones in situ and on radiological or echographical evidence.30 Thirty subjects received CRL 1 ng/kg IV over a period of 1 min and 30 received only a saline solution. A second dose of CRL or saline solution was injected 20 mins thereafter to those patients who did not respond to the first injection. Intensity of pain before receiving the injection was scored on the basis of a 3-point scale (0 = absent; 1 = mild; 2 = severe) in rest and during the Murphy provocation. CRL was more effective than a placebo in reducing the intensity of pain: only 4 (13.3%) subjects needed to be re-medicated in the group that received CRL, compared to 24 (80%) in the group that received placebo (P < 0.01). There were no reported serious side effects, two subjects had slight vertigo (one also had itching) and another had diarrhea.
Stacher et al (1984) administered 5, 10, or 20 mcg of CRL intramuscularly or placebo to 16 healthy men in an electrical and mechanical pain paradigm.31 All doses were significantly analgesic compared to the placebo in both paradigms, but the highest dose had the strongest effect. The compound also was assessed for its effect on jejunal mobility, and its effect resembled post-prandial activity. They identified a dose-response curve. The authors stressed the potentially beneficial effects of CRL in the postoperative phase, where both intestinal atonia and pain needs to be treated.
In a placebo-controlled double-blind RCT, 24 patients with biliary colic pain were treated with 1 ng/kg BW/min infused IV over 90 s.32 The analgesic effect was significantly higher than a placebo (p < 0.001) and could not be reversed by naloxone. The mechanism of action was probably mechanical, related to the reduction of distending pressure within the biliary tree.
In 1988, German investigators analyzed the safety and efficacy profile of the intramuscular (IM) administration of 5 mcg CRL in comparison to 10 mcg of morphine given one time to cancer patients.33 However, these data were never referenced in any other clinical article, and this line of exploration died without any firm scientific reason. In a randomized clinical trial, CRL and morphine were tested in 36 patients suffering from cancer pain. Pain scores were documented via a 100 mm VAS scale, and those patients had a pain score of at least 50 and had not received any analgesics for at least 6 hrs before the experimental treatment. Pain scores were assessed after 15, 30 and 60 mins, and 2, 3, 4 and 6 hrs after the IM administration. A 20 mm reduction on the VAS was defined as a meaningful clinical response. There was no statistical difference between the clinical responses in both groups. Pain reduction started around 30 mins and lasted between 4 and 5 hrs. However, patients treated with morphine reported several side effects: 56% of all patients treated with morphine suffered from nausea, vomiting, dizziness, sweating, dry mouth and cognitive impairment, whilst only 11% of all patients treated with CRL suffered from nausea and cognitive impairment. The rationale for selecting the CRL dose of 5 mcg in this study was missing, and given the benign side effect profile it might very well be that a higher dose could have been superior compared to morphine. In 1996, a last clinical study on the analgesic effects of CRL was published using a challenge paradigm comparable to that used by Stacher et al in 1982 and 1984. This study was inconsistent in reporting a robust analgesic effect on various pain modalities in healthy males.34
Gullo (1988) assessed the maximal effective dose of CRL infusions relating to the pancreatic function.35 He assessed the effect of infusions of 50 and 100 ng/kg BW/hour and found a clear dose-response, 100 ng/kg BW/hour being the more effective dose. Gullo also presented data from a German group, dosing 20 ng/kg BW as an IV bolus; adverse events were not reported for any of the doses explored. In Table 1 we summarized the details of all studies reviewed above.
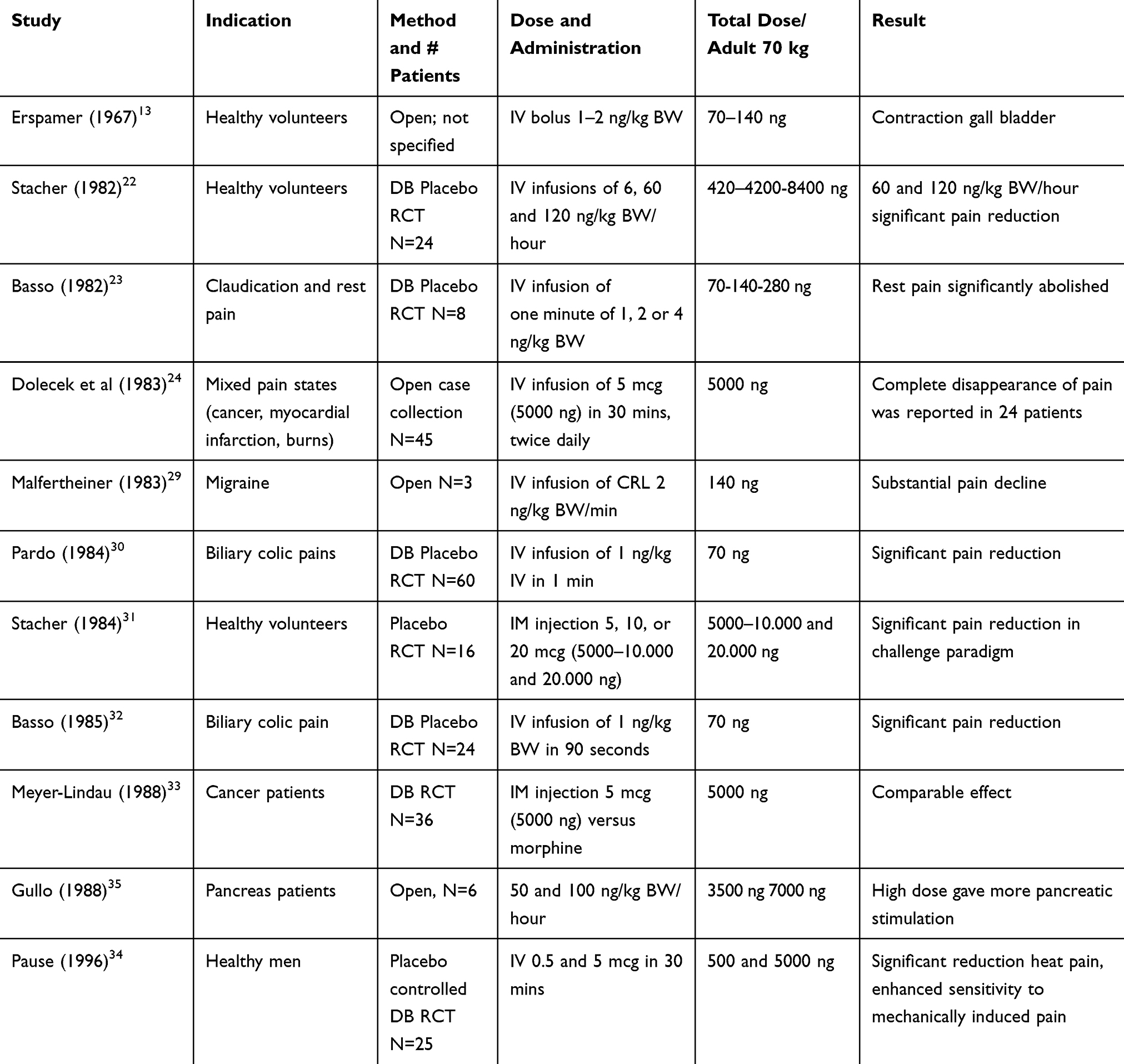 |
Table 1 Summarized Details of All Studies Reviewed |
Very different doses were administered, either as an IV bolus, an IV drip or an IM injection. The total dose calculated for an adult of 70 kg varied between 70 ng and 8400 ng. Standardized dose-finding for CRL is still lacking, although it seems both at the low end of a total dose of 70 ng and at the high-end dose range at 8400 (patients) and 20.000 ng (healthy volunteers) biological relevant responses were seen without dose-limiting adverse events, which is quite reassuring for future studies. Plasma levels have not been assessed in any of the studies conducted with CRL in pain due to the complexity of measuring such peptides.
Mechanism of Action
Clearly, many of the CRL effects on both smooth muscle and secretion are mediated by peripheral mechanisms of action. Subcutaneous administered CRL led to behavioral changes, catalepsia, ptosis and reduction of locomotor activity (sedation), suggesting a central mechanism of action.20 The analgesic effect of CRL and CCK-8 was the sensitivity towards naloxone: already a low dose of naloxone could completely block the analgesic effect of these peptides in the hot plate test.20 This suggested that CRL has an indirect mechanism of action via centrally located receptors, e.g. the CCK receptors. Zetler (1982) pointed out that sedation and naloxone-resistant ptosis would not support the notion that centrally located opioid receptors or released endogenous opioid peptides are involved. Moreover, peripherally administered CRL could inhibit convulsions triggered by the drug harman.36 Other centrally mediated symptoms, such as tremors induced by harmaline and ibogaine could be reversed by CRL sc.37 Systemically administered CRL could enhance the dopamine turnover in the striatum.38 Matsumoto et al (1990) administered a single dose of CRL at 0.8 mcg/kg BW intramuscularly every 6–8 days to 11 patients suffering from extrapyramidal choreatic disorders.38 EMG documented extrapyramidal movements as well as scored movements were clearly and significantly reduced following the administration. In some patients, the authors pointed out that involuntary movements were dramatically reduced in all parts of the body, and the orofacial movements in particular almost disappeared. The hypothesis of the mechanism of action was either via peripheral receptors on the vagal nerve or via dopamine neurons in the brain.
Roca-Lapirot et al (2018) recently discussed CRL related to the analgesic properties by the cholecystokinin (CCK)/CCK2 receptor system, and concluded that CRL and CCK-induced analgesic effects in response to persistent pain are associated with plasticity of the CCK/CCK receptor system in amongst others the central amygdala, based on CCK2 receptor signaling. The presence of inflammation further triggers changes in the system, whereby the analgesic activity of CRL might increase them. These recent findings also support further exploration of CRL as an analgesic compound.39 Further studies might be warranted to confirm the mechanisms and underlie the intracellular pathways involved with CRL and CCK effects.
Discussion
The above analysis demonstrates that drug development is a complicated matter, and this case serves to show that a potential analgesic drug-like CRL has not been sufficiently explored in order to decide whether it is of value or not, for instance in the treatment of cancer pain. A rationale for the selected dose was not given in any of the studied clinical papers. The dose-range studied in various indications is quite wide (70–20.000 ng) and only a few studies have systematically explored a part of this dose-range. Moreover, the route of administration varied between an IM injection, an IV bolus and an IV drip during 1 hr, giving rise to considerable variation in blood levels. Blood levels have rarely been explored, due to the complexity of assessing peptide levels. The exact mechanism of action of CRL is still vague, but what is clear is its high potency related to CCK. The debate of the analgesic mechanism of action of CRL via central or peripheral mechanisms still needs to be addressed, although there are studies that suggest that both mechanisms could contribute to analgesia, and the central role of the CCK system situated in the amygdala has recently been highlighted.39
Given the poverty of our therapeutic armamentarium for the treatment of chronic cancer pain, it seems to us that the absence of further interest in the use of CRL in this indication could be a missed opportunity. As CRL is a non-opioid peptide and does not act directly via the opiate receptors, a role as an intrathecal infusion against cancer pain in palliation seems worthwhile to explore, especially since some of the complications of intrathecal morphine delivery might be avoided such as tip of catheter granuloma and pH issues. As there is currently a high pressure to wean patients off opioids, we might want to consider to rediscover CRL for the treatment of cancer pains, perhaps as an intrathecal formulation, as we discussed previously for dermorphin.1 As CRL has no direct opiate-receptor activity, many of the clinical problems related to intrathecal use of morphine will not limit its use. Additional long-term studies with CRL could offer some information regarding the development of tolerance an adverse effect commonly reported to long- term use of opioids. This could offer an additional advantage in comparison to opioids. There are further advantages of bringing back studies and the use of CRL for pain control. This non-opioid strategy could also be relevant to reduce many of the deaths related to the misuse or overuse of opioid in several countries, especially the US.
Disclosure
The author is patent holder of two patents related to the topical formulations of phenytoin in the treatment of pain: 1) Topical phenytoin for the use in the treatment of peripheral neuropathic pain; and 2) Topical pharmaceutical composition containing phenytoin and a (co-)analgesic for the treatment of chronic pain; and reports no other conflicts of interest in this work.
References
1. Keppel Hesselink JM, Schatman ME. Rediscovery of old drugs: the forgotten case of dermorphin for postoperative pain and palliation. J Pain Res. 2018;11:2991–2995. doi:10.2147/JPR.S186082
2. Keppel Hesselink JM. The quest for better analgesics for the treatment of peripheral neuropathic pain: navigating the sodium channels. Trends Gen Pract. 2018;2:2–3.
3. Keppel Hesselink JM, Notermans NC. Topical phenytoin formulations for pain in small fiber neuropathy, a pathogenetic approach. Gen Int Med Clin Innov. 2018;3:2–4. doi:10.15761/GIMCI
4. Kopsky DJ, Keppel Hesselink JM. Single-blind placebo-controlled response test with phenytoin 10% cream in neuropathic pain patients. Pharmaceuticals. 2018;11:122. doi:10.3390/ph11040122
5. Keppel Hesselink JM, Mulder JK, Kopsky DJ. Rationale for the repositioning of phenytoin as a topical analgesic for peripheral neuropathic pain and a personalized and investigator-driven development plan. Ann Pharmacol Pharm. 2018;3:1144.
6. Keppel Hesselink JM. Kambo: A ritualistic healing substance from an amazonian frog and a source of new treatments. Open J Pain Med. 2018;2:004–006. doi:10.17352/ojpm.000007
7. Keppel Hesselink JM. Kambô: a shamanistic ritual arriving in the West - description, risks and perception by the users. Int J Psychol Psychoanal. 2018;4:034.
8. Basso N, Materia A, Fiocca F, et al. Treatment of biliary colic with ceruletide. Gastroenterology. 1980;78:1137.
9. Basso N, Materia A, Fiocca F, et al. Behandlung der gallenkolik mit ceruletid. Therapiewoche. 1981;31:2784–2786.
10. Basso N, Bagarani M, Gizzonio D, et al. Analgesic effect of ceruletide (CRL) in biliary and renal colic. Gastroenterology. 1981;80:1165.
11. Basso N, Materia A, D’Intinosante V, et al. Effect of ceruletide on pituitary-hypothalamic peptides and on emotion in man. Peptides. 1981;2(Suppl 2):71–75. doi:10.1016/0196-9781(81)90014-0
12. Erspamer V, Roseghini M, Endean R, et al. Biogenic amines and active polypeptides in the skin of Australian amphibians. Nature. 1966;212:204. doi:10.1038/212204a0
13. Anastasi A, Erspamer V, Endean R. Isolation and structure of caerulein, an active decapeptide from the skin of Hyla caerulea. Experientia. 1967;23:699–700. doi:10.1007/BF02154119
14. Erspamer V, Bertaccini G, De Caro G, et al. Pharmacological actions of caerulein. Experientia. 1967;23:702–703. doi:10.1007/BF02154121
15. De Castiglione R, Di Salle E. Is commercial “cerulein” always true cerulein? Gastroenterology. 1980;78:1113–1114. doi:10.1016/0016-5085(80)90820-3
16. Approved Drug Products with Therapeutics Equivalence Evaluations (Orange Book).
17. CERULETIDE USES. Available from: https://www.ndrugs.com/?s=ceruletide.
18. Takus 5 µg. Available from: https://medikamio.com/de-de/medikamente/takus-5-ug/pil.
19. Yau W, Maklouf G, Edwards L, Farrar JT. Mode of action of cholecystokinin and related peptides on gallbladder muscle. Gastroenterology. 1973;65:451–456. doi:10.1016/S0016-5085(19)33077-X
20. Zetler G. Analgesia and ptosis caused by caerulein and cholecystokinin octapeptide (CCK-8). Neuropharmacology. 1980;19:415–422. doi:10.1016/0028-3908(80)90047-7
21. Baber NS, Dourish CT, Hill DR. The role of CCK, caerulein, and CCK antagonists in nociception. Pain. 1989;39:307–328. doi:10.1016/0304-3959(89)90045-6
22. Stacher G, Steinringer H, Schmierer G, et al. Ceruletide increases threshold and tolerance to experimentally induced pain in healthy man. Peptides. 1982;3:955–962. doi:10.1016/0196-9781(82)90064-X
23. Basso N, Pona V, Dintinosante V, et al. Effect of ceruletide on rest pain in patients with arterial insufficiency of the lower extremity. Eur J Clin Pharmacol. 1982;22:531–533. doi:10.1007/BF00609626
24. Dolecek R, Jezek M, Adámková M, et al. Endorphin releasers: a new possible approach to the treatment of pain after burns–a preliminary report. Burns Incl Therm Inj. 1983;10:41–44. doi:10.1016/0305-4179(83)90126-2
25. Santamaria A, Lucani G, Montorsi M, et al. Action de la ceruleine sur la colique hepatique. Nouv Presse Med. 1979;8:2482.
26. Saggioro A. Wirkung verschiedener dosen von ceruletide auf die schmerzen bei gallenkolik. Therapiewoche. 1981;31:2787.
27. Piazza E, Brambilla M, Cattaneo MT, et al.
28. Dolecek R, Kubis M, Sajnar J, et al. Endorphin releasers-a new method of pain management? Cas Lek Cesk. 1982;121:944.
29. Malfertheiner P. Successful alleviation of migraine pain with short ceruletide infusion. Anaesthesist. 1983;32–1:33–35.
30. Pardo A, Celotti F, De Paolis C. Ceruletide analgesia in biliary colic. Clin Pharmacol Ther. 1984;36:510–514. doi:10.1038/clpt.1984.211
31. Stacher G, Steinringer H, Schmierer G, et al. Ceruletide increases dose dependently both jejunal motor activity and threshold and tolerance to experimentally induced pain in healthy man. Gut. 1984;25:513–519. doi:10.1136/gut.25.5.513
32. Basso N, Bagarani M, Materia A, et al. Effect of caerulein in patients with biliary colic pain. Gastroenterology. 1985;89:605–609. doi:10.1016/0016-5085(85)90457-3
33. Meyer-Lindau F, Pfister E, Gyr N, et al. Randomized double-blind study of the analgesic effect of caerulein and morphine in chronic tumor pain. Onkologie. 1988;11:77–80. doi:10.1159/000216492
34. Pause BM, Drews C, Scherhag C, et al. Analgesic effect of ceruletide in men is limited to specific pain qualities. Physiol Behav. 1996;59:1025–1031. doi:10.1016/0031-9384(95)02230-9
35. Gullo L. Maximal effective dose of cerulein in the secretin-cerulein test. Digestion. 1988;39:225–229. doi:10.1159/000199630
36. Zetler G. Ceruletide, ceruletide analogues and cholecystokinin octapeptide (CCK-8): effects on motor behaviour, hexobarbital-induced sleep and harman-induced convulsions. Peptides. 1982;3:701–704. doi:10.1016/0196-9781(82)90174-7
37. Zetler G. Cholecystokinin octapeptide (CCK-8), ceruletide and analogues of ceruletide: effects on tremors induced by oxotremorine, harmine and ibogaine. A comparison with prolyl-leucylglycine amide (MIF), anti-Parkinsonian drugs and clonazepam. Neuropharmacology. 1983;22:757–766. doi:10.1016/0028-3908(83)90100-4
38. Matsumoto T, Nakahara T, Ushimura H, et al. Effect of systematically administered caerulein on dopamine metabolism in rat brain. Brain Res. 1984;325:195–199. doi:10.1016/0006-8993(84)90643-7
39. Roca-Lapirot O, Fossat P, Ma S, et al. Acquisition of analgesic properties by the cholecystokinin (CCK)/CCK2 receptor system within the amygdala in a persistent inflammatory pain condition. Pain. 2018.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.