")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Receptor tyrosine kinase (c-Kit) inhibitors: a potential therapeutic target in cancer cells
Authors Abbaspour Babaei M, Kamalidehghan B, Saleem M, Zaman Huri H, Ahmadipour F
Received 23 May 2015
Accepted for publication 14 October 2015
Published 1 August 2016 Volume 2016:10 Pages 2443—2459
DOI https://doi.org/10.2147/DDDT.S89114
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Maryam Abbaspour Babaei,1 Behnam Kamalidehghan,2,3 Mohammad Saleem,4–6 Hasniza Zaman Huri,1,7 Fatemeh Ahmadipour1
1Department of Pharmacy, Faculty of Medicine, University of Malaya, Kuala Lumpur, Malaysia; 2Department of Medical Genetics, National Institute of Genetic Engineering and Biotechnology (NIGEB), Shahrak-e Pajoohesh, 3Medical Genetics Department, School of Medicine, Shahid Beheshti University of Medical Sciences, Tehran, Iran; 4Department of Urology, 5Department of Laboratory Medicine and Pathology, Masonic Cancer Center, University of Minnesota, 6Section of Molecular Therapeutics & Cancer Health Disparity, The Hormel Institute, Austin, MN, USA; 7Clinical Investigation Centre, University Malaya Medical Centre, Lembah Pantai, Kuala Lumpur, Malaysia
Abstract: c-Kit, a receptor tyrosine kinase, is involved in intracellular signaling, and the mutated form of c-Kit plays a crucial role in occurrence of some cancers. The function of c-Kit has led to the concept that inhibiting c-Kit kinase activity can be a target for cancer therapy. The promising results of inhibition of c-Kit for treatment of cancers have been observed in some cancers such as gastrointestinal stromal tumor, acute myeloid leukemia, melanoma, and other tumors, and these results have encouraged attempts toward improvement of using c-Kit as a capable target for cancer therapy. This paper presents the findings of previous studies regarding c-Kit as a receptor tyrosine kinase and an oncogene, as well as its gene targets and signaling pathways in normal and cancer cells. The c-Kit gene location, protein structure, and the role of c-Kit in normal cell have been discussed. Comprehending the molecular mechanism underlying c-Kit-mediated tumorogenesis is consequently essential and may lead to the identification of future novel drug targets. The potential mechanisms by which c-Kit induces cellular transformation have been described. This study aims to elucidate the function of c-Kit for future cancer therapy. In addition, it has c-Kit inhibitor drug properties and their functions have been listed in tables and demonstrated in schematic pictures. This review also has collected previous studies that targeted c-Kit as a novel strategy for cancer therapy. This paper further emphasizes the advantages of this approach, as well as the limitations that must be addressed in the future. Finally, although c-Kit is an attractive target for cancer therapy, based on the outcomes of treatment of patients with c-Kit inhibitors, it is unlikely that Kit inhibitors alone can lead to cure. It seems that c-Kit mutations alone are not sufficient for tumorogenesis, but do play a crucial role in cancer occurrence.
Keywords: c-Kit, cancer, oncogene, cancer therapy
Introduction
c-Kit, a type III receptor tyrosine kinase (RTK), plays a crucial role in cancer occurrence.1 Currently, c-Kit is mainly considered a stem cell factor (SCF), which participates in vital functions of the human body, such as fertility, homeostasis, and melanogenesis; nevertheless, early studies on c-Kit introduced it as an oncogene.2,3 Deregulation of c-Kit, including overexpression and gain of function mutations, has been detected in several human cancers. Leukemia is the first cancer linked to c-Kit activating mutation.4 Subsequent studies reported that c-Kit activating mutation is found in almost all cases of systemic mastocytosis and other hematopoietic cancers; these findings support the hypothesis that the c-Kit target is possibly located in the stem cell compartment.5 c-Kit has been reported to be mostly correlated with gastrointestinal stromal tumor (GIST), with 80% of all GIST cases involving c-Kit activating mutation. As such, the use of Kit inhibitors has provided novel insights for cancer treatment.6 In addition, Kit mutations have been detected in cancers such as leukemia,7 unilateral ovarian dysgerminoma,8–10 melanoma,11 and others.12–14 Evidence reveals that targeting c-Kit as an oncogene by using kinase inhibitor drugs such as imatinib is a promising approach for cancer treatment. However, several issues have been raised regarding this approach. For instance, resistance to imatinib, a famous c-Kit inhibitor drug, has been observed in several cases and is attributed to changes in c-Kit mutations; moreover, c-Kit is expressed in normal tissues such as breast epithelial, vascular endothelial, sweat glands, and retinal astrocytes.15 In this regard, c-Kit mutations cannot be considered a risk factor for cancer occurrence.16 Therefore, targeting c-Kit for cancer treatment is only feasible in cases where c-Kit is the “driver” of the cancer.
Gene and protein structures of c-Kit
c-Kit, a protooncogene in a region on the long arm of chromosome 4 (4q11–4q13), encodes the SCF receptor (CD117).17,18 c-Kit is the cellular equivalent of the v-kit oncogene, a transforming feline retrovirus, and a 145 kDa transmembrane glycoprotein, which belongs to class III of the RTK family. This family is categorized into three domains: a hydrophobic transmembrane, an extracellular ligand-binding domain, and a cytoplasmic domain with tyrosine kinase activity.19
Four c-Kit isomers caused by alternative RNA splicing have been found in humans.20 The presence of serine residues in the kinase insert region differentiates the two isoforms, though the function of a serine residue is still unknown. A stretch of four acids on the extracellular side also distinguishes the two other isoforms. At the molecular level, these isoforms differ in terms of ability to induce signal transduction and tumorigenic potential.21–26 The isoform without the tetrapeptide sequence is regarded as the strongest inducer and highest transformer.27
Another c-Kit isoform has been detected in murine testis; this isoform is truncated resulting from the controlled promoter element within intron 16, which contains 12 amino acids and a carboxyterminal tail without kinase activity.28 This isoform has also been found to be expressed in human prostate cancers.29 By contrast, one study reported that this isoform is mouse specific and cannot be found in humans.30
c-Kit in normal stem cells
c-Kit, an SCF receptor,1 plays an important role in stem cell maintenance and differentiation.31 c-Kit expression has been detected in various stem cells or cells with self-renewal potency and progenitor cells.32 Studies have also confirmed that c-Kit is expressed in different kinds of stem cells, especially hematopoietic cells.33,34 In several loss-of-function mutations of c-Kit, the mutated site has been linked to a wide range of defects, from minor defects in catalytic activity to critical flaws in the hematopoietic system in mice.35 c-Kit mutations has also been reported to significantly affect other systems such as the reproductive, pigmentation, and nervous system.36,37
Hematopoietic stem cells divide asymmetrically and can self-renew or differentiate into all hematopoietic cell lineages, including myeloid (monocytes and macrophages, neutrophils, basophils, eosinophils, erythrocytes, megakaryocytes/platelets, and dendritic cells) and lymphoid (T-cells, B-cells, and NK-cells) lineages.38 During differentiation, stemness and self-renewal are lessened, whereas cell proliferation is maintained; this phenomenon leads to increased cell numbers.39 Arguably, c-Kit, as an SCF receptor, is expressed in original hematopoietic cells, such as stem and progenitor cells. However, c-Kit expression vanishes during differentiation.40–43 Less than 1% of peripheral blood cells express c-Kit, which indicates the minimal role of c-Kit in differentiated hematopoietic cells.44–46 Furthermore, c-Kit is highly expressed in mast cells; as such, completely differentiated cells depend on c-Kit signal transduction for their survival, function, and growth.47 Finally, among lymphoid lineages, c-Kit is involved in immune system maintenance in adult animals.48
c-Kit affects germ cells in the fertility system, which are classified as progenitor cells.48 Previous studies showed that c-Kit mutations most likely and normally leads to the protection of germ cells from apoptosis and to the induction of cell migration and proliferation.49 The pathway necessary for apoptosis inhibition by c-Kit is the PI3 kinase/AKT pathway, which is essential in cellular processes such as protein synthesis, proliferation, and survival.50 Mutant mice overexpressing the c-Kit receptor cannot activate PI3 kinase, leading to infertility.51 c-Kit is also involved in germ cell biology, particularly during gametogenesis in male and female mice.
c-Kit exhibits other functions. Mutant mice underexpressing c-Kit present defects in pigmentation, which is related to the ability of SCF (a c-Kit cognate ligand) to induce proliferation and guide the migration of melanocytes from the neuronal crest to the dermis.52–54 Other studies revealed two tyrosine residues, namely, 568 and 570, which are necessary for normal pigmentation.55 The loss-of-function mutations of c-Kit has been detected in majority of humans with piebaldism syndrome, a rare autosomal dominant disorder of melanocyte development. This syndrome is characterized by hypopigmentation in the stomach, hair, and extremities as a result of melanocyte deficiency.56 Furthermore, c-Kit activity influences the digestive and nervous systems. c-kit is suggested to be involved in the interstitial cells of Cajal or (ICC) in sum. Loss of ICC is correlated with diseases such as slow transit constipation.57 Previous studies suggested the vital role of c-Kit in ICC development or function.58–60 In mice with c-Kit loss-of-function mutation, ICC is depleted.57 Several studies reported the role of c-Kit in the development and/or function of nervous system cells. c-Kit expression has also been detected in neuroproliferative cells in adult rat brains, in addition to neural cultures.61 Moreover c-Kit signaling is significant in migration of neural stem cells to injured areas of the brain.62 These studies suggest that c-Kit plays a crucial role in the stem and progenitor cells of different systems of the body and that loss-of-function mutations in c-Kit can lead to several defects.
c-Kit and cancers
In this section, we discuss the previous findings of c-Kit deregulations in several types of cancer. As will be shown, there are different type of deregulation of c-Kit, and each of them can result in tumorogenesis. Deregulation of c-Kit can result in cancer in different ways. This deregulation could occur in different ways such as gain of function, loss of function, overexpression, and point mutations.16 The role of c-Kit deregulation in cancer was first identified as a retroviral oncogene using mice as a reference. The role of c-Kit in cancer has not been completely uncovered. c-Kit is a marker for human acute myeloid leukemia (AML) and normal hemopoietic progenitor cells.1 Studies demonstrated the crucial function of c-Kit and its ligand in hematopoiesis,63 fertility,64,65 and melangenesis.66
In a number of cancers, c-Kit activation was detected through overexpression or mutations. Conversely, in other tumors, such as melanoma,67,68 thyroid carcinoma,69 and breast cancer,69 loss-of-function mutation of c-Kit was observed. Moreover, c-Kit gain-of-function mutation in metastatic melanoma induces apoptosis.70 By contrast, in uveal melanomas, c-Kit expression results in cell proliferation, for which treatment with kinase inhibitor drugs leads to apoptosis induction.71 The activating mutation of c-Kit, namely, L576P, has been reported in a small subset of highly metastatic melanomas.72 Thus, there are various c-Kit mutations involved in melanoma, so melanoma can be used as a model to clarify the complex roles of c-Kit in tumorigenesis.73 In certain cancer types, such as GIST, the main cause of molecular events in tumorogenesis is the activating mutations in c-Kit. In this case, targeting c-Kit with imatinib mesylate increases survival by approximately 70%–80% after 2 years compared with cancer treatment without c-Kit targeting drugs.74 In cancer cases where activating mutation in c-Kit is not the causative event, treatment targeting c-Kit results in poor treatment outcomes.
c-Kit mutations were first reported in human mast cell leukemia.4 These mutations have also been detected in GIST.12 Furthermore, c-Kit mutations have been found in most cases of systemic mastocytosis.5
The activation of c-Kit mutations is rarely detected in some cancer types. For instance, c-Kit activation mutations are uncommon in AML75 and rarely detected in other cancer types; for example, only 26% of germ cell cancers, or more specifically, testicular seminomas, have been associated with c-Kit mutations.8 These mutations have not been detected in urinary and ovarian cancers and only in 30% cases of urinal ovarian dysgerminoma.8 Moreover, a low frequency of activating c-Kit mutation and proliferation is found in melanoma.76
Different types of c-Kit mutations can cause tumors. The concurrent overexpression of c-Kit and its ligands occur in some types of tumors such as colorectal carcinoma, breast carcinoma, small-cell lung carcinoma, neuroblastoma, and gynecological tumor.77 Tumors resistant to chemotherapy, such as malignant mesothelioma, have been shown to be associated with simultaneous upregulated expression of c-Kit and its ligand.78
In several tumors, c-Kit overexpression is found without mutation and/or mutation in its ligand. In normal physiological circumstances, only a minority of hematopoietic cells express c-Kit. AML cells express c-Kit, which influences the malignant phenotype of this cancer.3,46 A previous study reported that c-Kit expression level is 7.4-fold higher in renal oncocytoma and chromophobe renal carcinoma than that in renal normal tissues.79 c-Kit overexpression has not been observed in other types of renal cancers.80
In cancers involving RTKs, gain-of-function mutations are the main events that lead to cancer progression.81 The first gain-of-function c-Kit mutation was identified in the human mast cell line HMC1. These mutations, known as D816V and V560G, are located in the juxtamembrane region and in the tyrosine kinase domain, respectively.4 Mutations in the juxtamembrane domain are disrupted by the interaction of this region with the kinase domain, which results in inhibited mutation. Furthermore, point mutations in this region can result in induced c-Kit dimerization.82 These events consequently induce the activation of kinase domains.
Another mutation hotspot is found in codon 816, located in the second part of the kinase domain, and leads to the activation of the domain. This mutation is considered as ligand-independent activation because of the switching of aspartic acid residues to asparagine, tyrosine, valine, or histidine residues.83 Nevertheless, other studies provide contradicting results, in which the kinase domain forms a dimer in the absence of SCF.82,84,85 Furthermore, studies showed that both mutants in the kinase domain (D814Y) and juxtamembrane domain (KΔ27) (in frame deletion at codons 547–555) lead to substrate alteration, which results in significant outcomes based on signaling pathway(s) activated by the mutant c-Kit.86,87
A broad range of juxtamembrane domain mutations have been detected in GIST; these mutations include duplications, point mutations, deletions, or their combinations. Mutations in this region have also been detected in approximately 30% of AML,88 sinonasal lymphoma,89 and rare cases of mastocytosis.90
A wide range of human cancers are caused by mutations in the c-Kit kinase domain; these cancers include different types of leukemia, such as acute myeloid, core-factor binding, and mast cell leukemia;7,91,92 testicular germ cell tumor; intracranial and ovarian dysgerminoma;9,93,94 mastocytosis;14–95 and papillary renal carcinomas.13
Hotspot region of Kit mutation
Numerous Kit mutation sites are found and vary in different cancer types,16 reflecting the effect of each mutation on downstream signaling pathways.12 Some “hotspots” in the Kit gene are regular in certain main domain structures.113 Mutations in domains, such as intracellular and extracellular juxtamembranes, located on exons 8, 9, and 11,12 as well as exon 17, which corresponds to the activation loop in the kinase domain,114 disrupt the autoinhibitory mechanisms of Kit.115,116
The importance of these two domains is reflected in their critical role in the Kit structure and function. The first group of extracellular juxtamembrane domains is responsible for the correct binding of the receptor monomer and the stabilization of dimers, which contain dimeric SCF.117 The mutations in this region, particularly in exons 8 and 9, are detected in AML and GIST, respectively.118,119 The most common mutations in c-Kit are summarized in Table 1 and are classified based on type of cancer, exon location, and the type of mutation.
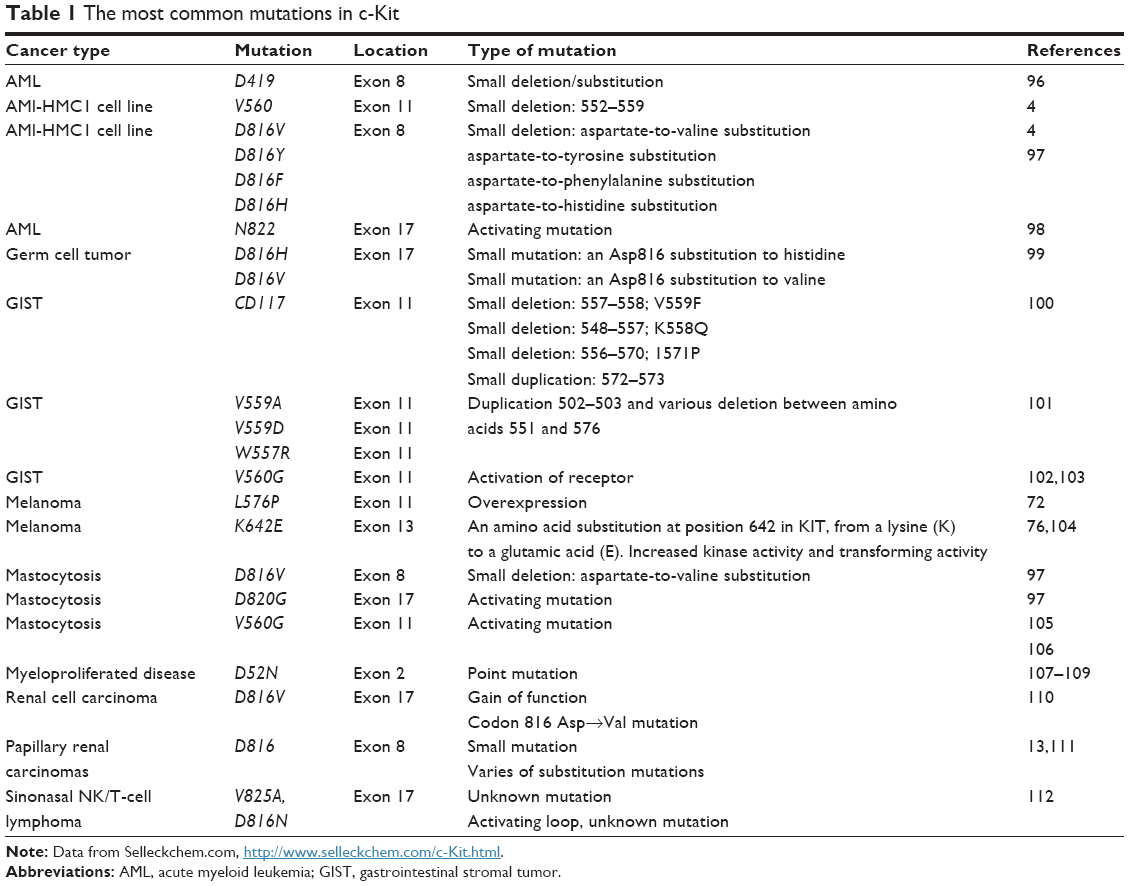 | Table 1 The most common mutations in c-Kit |
Kit mutations occur within exon 11 in almost 65% of all GIST cases. This exon encodes a key autoregulatory domain of the RTK, which is the intracellular juxtamembrane domain, and stabilizes the inactive conformation of the kinase domain.121 In addition, mutations in other exons, such as exons 11 and 17, have been identified in GIST122 and hematological cancers,123 respectively.
The most detected c-Kit mutations that lead to melanoma are located within exons 11 and 13, namely, L576P and K642E, respectively.76 Mutations in exon 17, which encodes the activation loop of the kinase domain, result in hemopoietic malignancies in germ cell tumors. After kinase activation, conformational shifts occur in this region.116 The KIT cDNA structure in different cancers and their respective mutations is illustrated in Figure 1. c-Kit mutation based on their gene location, corresponding cancer, and drug sensitivity are listed in Table 2.
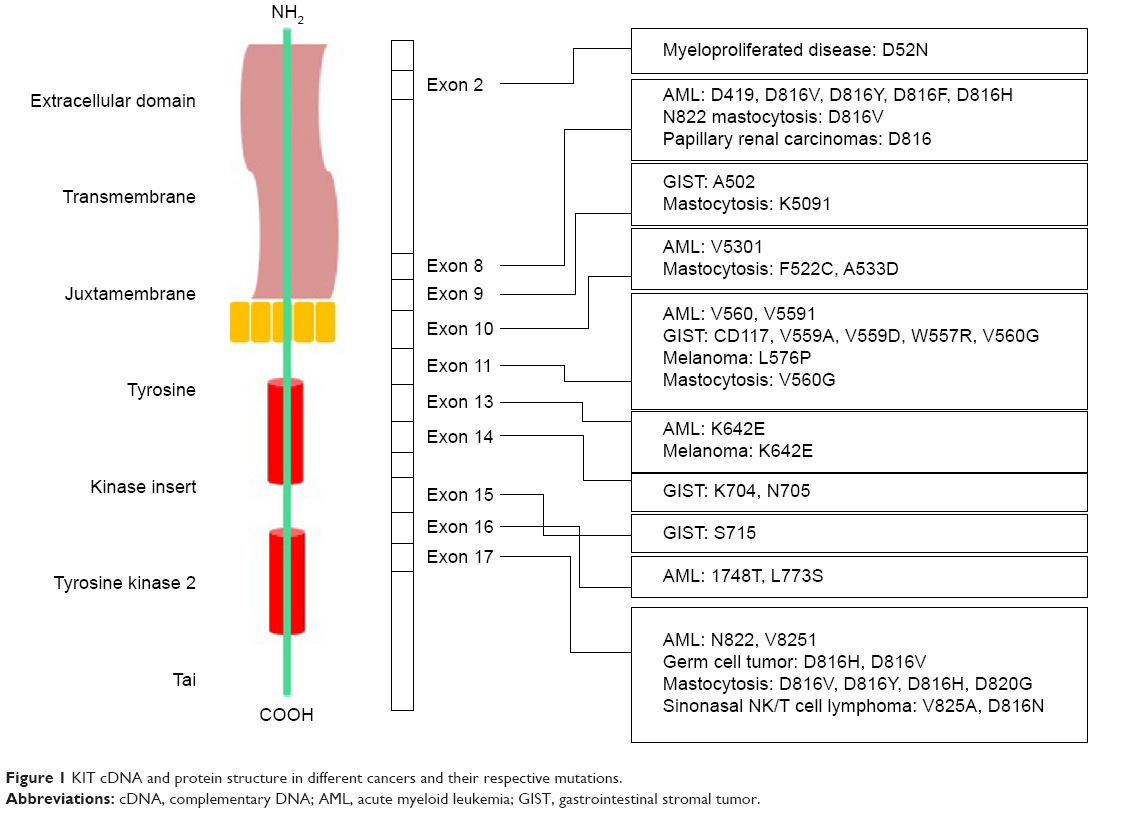 | Figure 1 KIT cDNA and protein structure in different cancers and their respective mutations. |
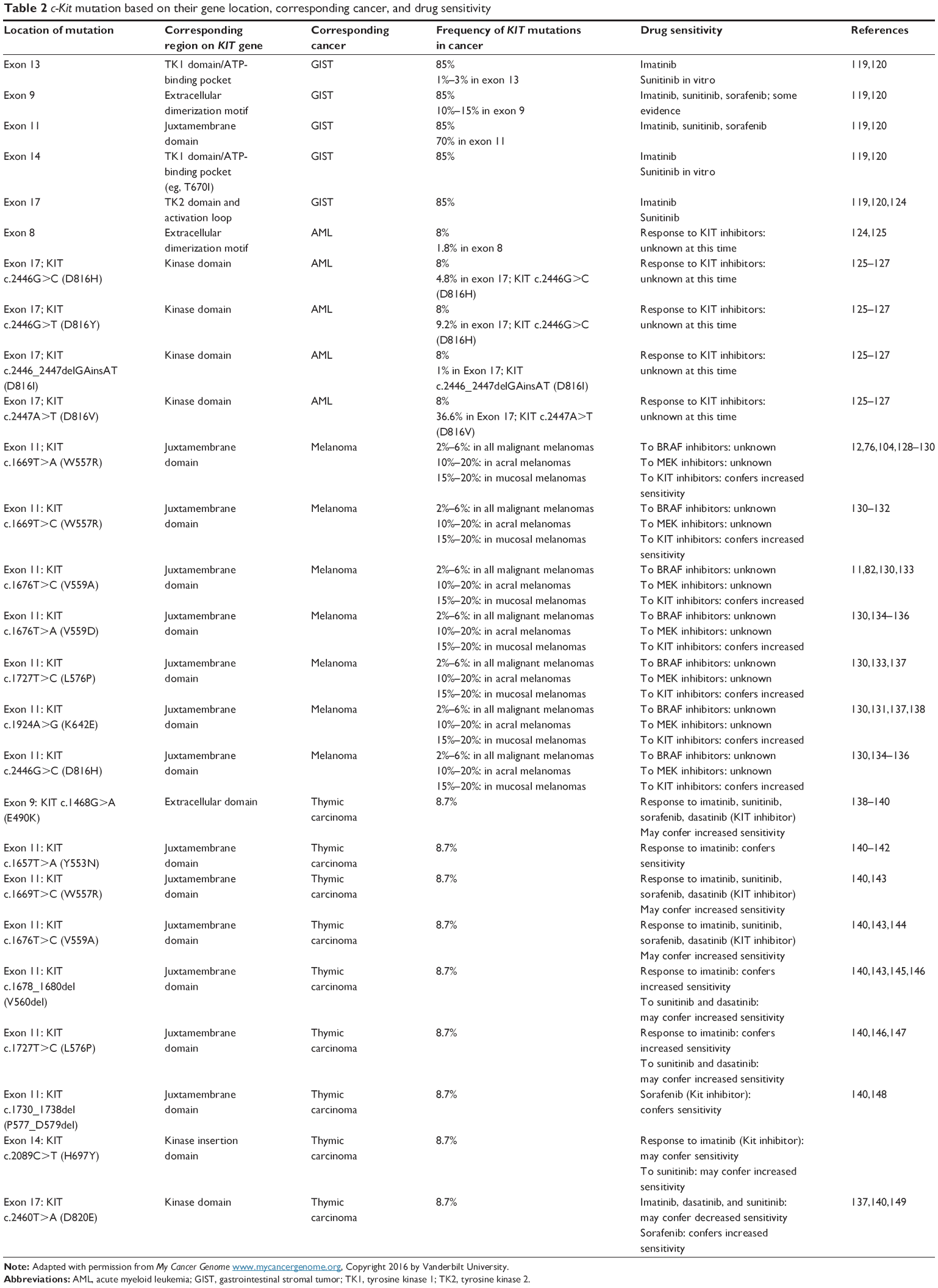 | Table 2 c-Kit mutation based on their gene location, corresponding cancer, and drug sensitivity |
c-Kit signal transduction
c-Kit is involved in several signaling pathways, as discussed.
PI3-kinase
PI3-kinase is the most studied pathways involving c-Kit. In this pathway, phosphatidylinositide 3′-kinase phosphorylates the 3′-hydroxyl group of the inositol ring of lipids in the cell membrane.79 As such, the negative electric potential across the lipid increases, which results in the interaction of PIP3 in the cell membrane with proteins comprising pleckstrin homology (PH) domains. Consequently, the PH domain transduces proteins from the cytoplasm to the plasma, thus activating AKT. This activation is crucial for the apoptosis ability of SCF. Serine/threonine kinase AKT is a vital PH domain that contains proteins in c-Kit signaling.34
Recent studies revealed that SCF interacts with the transcription factor FOXO3a, which leads to survival through AKT-mediated phosphorylation; this factor belongs to the O subclass of the forkhead family of transcription factors and is characterized by a diverse forkhead DNA-binding domain.34,81 As a result, the expression of proto-apoptotic protein Bim decreases and Mek-dependent phosphorylation is downregulated.80 In addition, PI3-kinase plays an important role in SCF-induced proliferation and regulation of the actin cytoskeleton and cell migration.83 Earlier studies confirmed the necessity of PI3-kinase in hematopoietic cell growth and tumorigenicity, which involves the active form of c-Kit in the genome.84,85
These studies focused on class I PI3-kinase, despite that class II PI3KC2β is significantly associated with c-Kit.86 Studies have also illustrated the interaction of C2 domains with phosphotyrosine residues,87 which can prove the probability of interaction between PI3K-C and c-Kit activation.
Src family kinase
The other signaling pathway that stimulates c-Kit activity is the Src family kinases (SFKs), which are cytoplasmic tyrosine kinases. This pathway is involved in several crucial biological functions such as survival, chemotaxis, and proliferation.88 Although studies prove that SFKs are activated by c-Kit, the function of this activation in the pathway remains unknown.89,90 In addition, the involvement of SFK in c-Kit internalization has been demonstrated at the cell biological level.91
The contribution of Lyn in c-Kit, STAT3, and JNK phosphorylation was determined using Lyn−/− bone marrow mast cells.92 PI3-kinase/AKT signaling is negatively regulated by Lyn, although the underlying mechanism remains unknown. SFK evidently plays a role in SCF-induced chemotaxis and proliferation of primary hematopoietic progenitor cells.91 In the study by Hong et al,93 it was demonstrated that SFK affects the downstream pathways of c-Kit, although other signaling molecules, such as APS, SHP1, and SHP2, also interact with the SFK-binding site.94,95
Phenotypic analysis on mutant c-Kit mice with inability to interact with SFK showed that SFK plays a role(s) in c-Kit signaling in lymphocytes.39,55 In addition to lymphocyte defects, severe problems in pigmentation, splenomegaly, and mast cell development have been observed in double c-Kity567/569f mutations. Thus, PI3-kinase and SFK are two important signaling pathways in most tissues, and any defect in these pathways can interrupt their interaction with c-Kit could lead to severe defects.150
Ras-Erk pathway
The next signaling pathway that stimulates the activity of c-Kit is the Ras-Erk pathway. The activation of MAP-kinases, namely, Erk1/2, plays a vital role in cell proliferation, differentiation, and survival. The main role of c-Kit in this signaling includes the recruitment of the guanine exchange factor Sos to its substrate, namely, the small GTPase Ras, which is located in the plasma membrane. By converting GTP to GDP, Sos provides the guanine nucleotide phosphate which binds to Ras, resulting in Ras activation. Active Ras consequently translocates Raf-1 to the plasma membrane. Mek, another kinase, and Erk are then activated. Finally, the activated Erk results in the alteration of protein activity and gene expression.97
In terms of the relevance of c-Kit to this pathway, a special region is found in c-Kit, namely, tyrosine residues 703 and 936, which directly interacts with the stable complex of Sos with the adaptor protein Grb2.98 Altogether, c-Kit is considered a target for cancer therapy because it mediates the activation of Erk1/2. In addition, in HL60 cells, Erk activation upregulates survivin expression by SCF mediation, which leads to resistance toward apoptosis induced by radiation.102
These signaling pathways are activated by Kit. Other important signaling pathways involving c-Kit include the JAK/STAT pathway and various signaling proteins, such as adaptor proteins (eg, Crk, Gab, APS, Gads, ShcA, Grb2, and Grb7), cytoplasmic tyrosine kinases, and protein tyrosine phosphatases.39,94,101,102,151 The role of c-Kit in the molecular function of the aforementioned signaling proteins is characterized to varied degrees. With respect to the diverse functions of c-Kit, large numbers of these proteins are affected in different ways. In addition to the direct or indirect effect of c-Kit on various signaling pathways or proteins, this kinase often functions in cooperation with other cytokines and growth factors.150,152 The summary of the signaling pathway with involvement of c-Kit in normal and cancer cells is demonstrated in Figures 2 and 3, respectively.
 | Figure 2 Signal transducer and activator of transcription tyrosine kinase domain c-Kit in normal cell. |
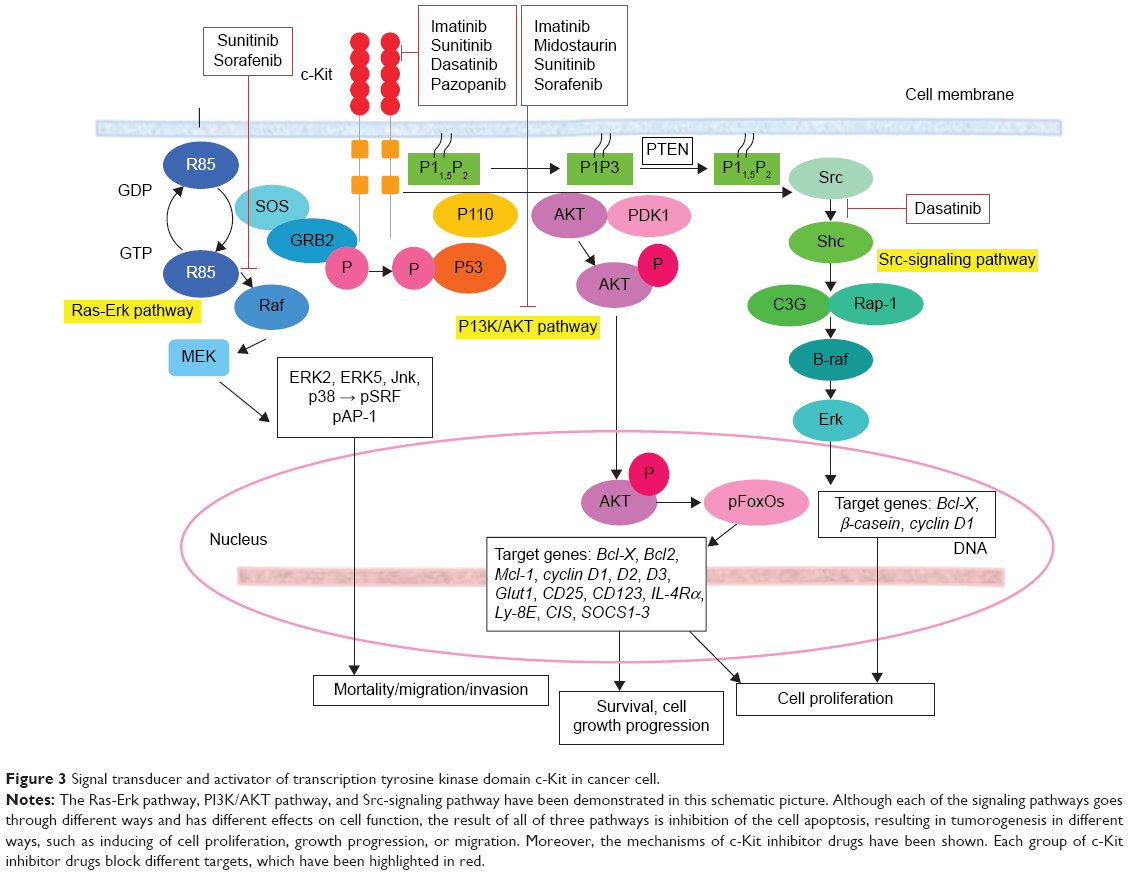 | Figure 3 Signal transducer and activator of transcription tyrosine kinase domain c-Kit in cancer cell. |
c-Kit: a potential target for cancer therapy
Kinase superfamily proteins are considered a main target for molecular cancer therapy. Numerous studies have uncovered the molecular chronicles that occur during cancer development. The gist of most researchers is that kinases are an essential factor for cancer progression and are overexpressed by tumors. As such, kinases should be targeted as a new method for cancer treatment. Different approaches have been considered for this strategy. The first approach uses antibodies against kinase proteins. For instance, trastuzumab (Herceptin®; Genentech Inc, South San Francisco, CA, USA) targets the extracellular domain of HER2. Other antibodies, such as low-molecular-weight kinase inhibitors, eg, gefitinib (Iressa®; AstraZeneca, London, UK), aim at enzymatic activity. The first group can only affect proteins with an extracellular domain, whereas the second group can target both transmembrane and intracellular proteins. The first group of proteins is very specific, whereas the specificity of the second group of proteins may be limited. Most kinase inhibitors target and bind to the enzymatic domain and compete with ATP; however, the specificity of these inhibitors are conserved because of unique binding patterns.101
The major concern in cancer management is the occurrence of resistance toward drugs. This resistance could be due to mutations in the target protein, which result in the reduction of the binding between the drug and the kinase. In addition, overexpression of transport proteins by cells may occur, which leads to decreased intracellular concentration of drugs. Additionally, some other oncogenes may substitute for or reimburse the inhibition of the drug target. For instance, for the drug used in chronic myelogenous leukemia (imatinib mesylate), drug resistance has been observed as a result of both the overexpression of Bcr-Abl and mutation in the kinase domain, which is specific for drug binding.104 c-Kit mutation based on their gene location, corresponding cancer, and drug sensitivity are listed in Table 2.
Low-molecular- weight inhibitors have been utilized for c-Kit targeting. For instance, imatinib mesylate is one of the c-Kit targeting drugs that inhibit both Abl and PDGFRs. List of the drugs that target c-Kit have been listed in Table 3, and are classified based on drug name, molecular formula, common and specific targets, structural formula, and general function. The list of c-Kit inhibitors with their complete list of their targets, besides c-Kit, with emphasize of their affectivity on c-Kit is listed in Table 4. In addition, c-Kit inhibitors classification based on their targets, chemical and structure formulae, and diseases they are tested on, have been listed in Table 5.
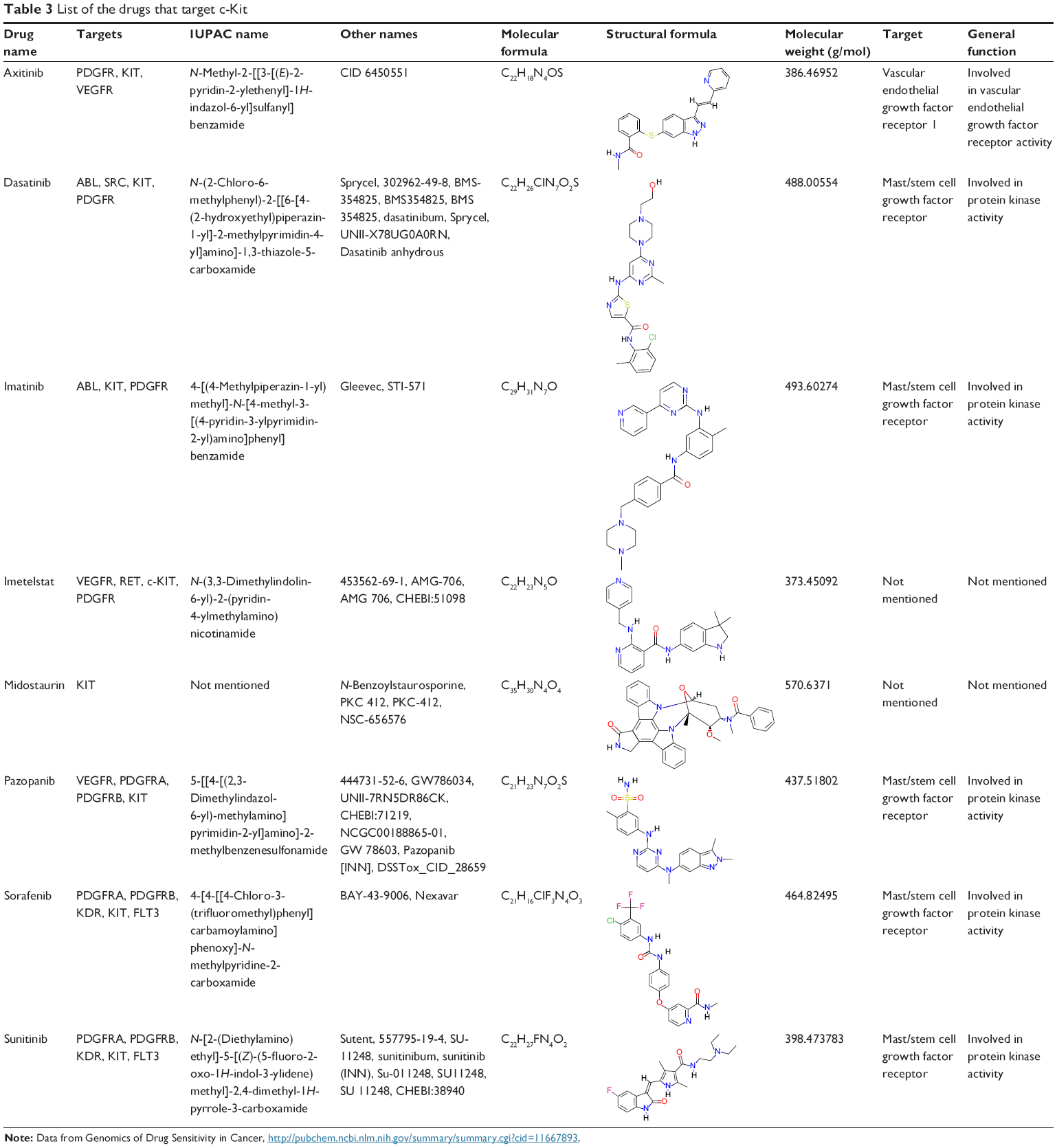 | Table 3 List of the drugs that target c-Kit |
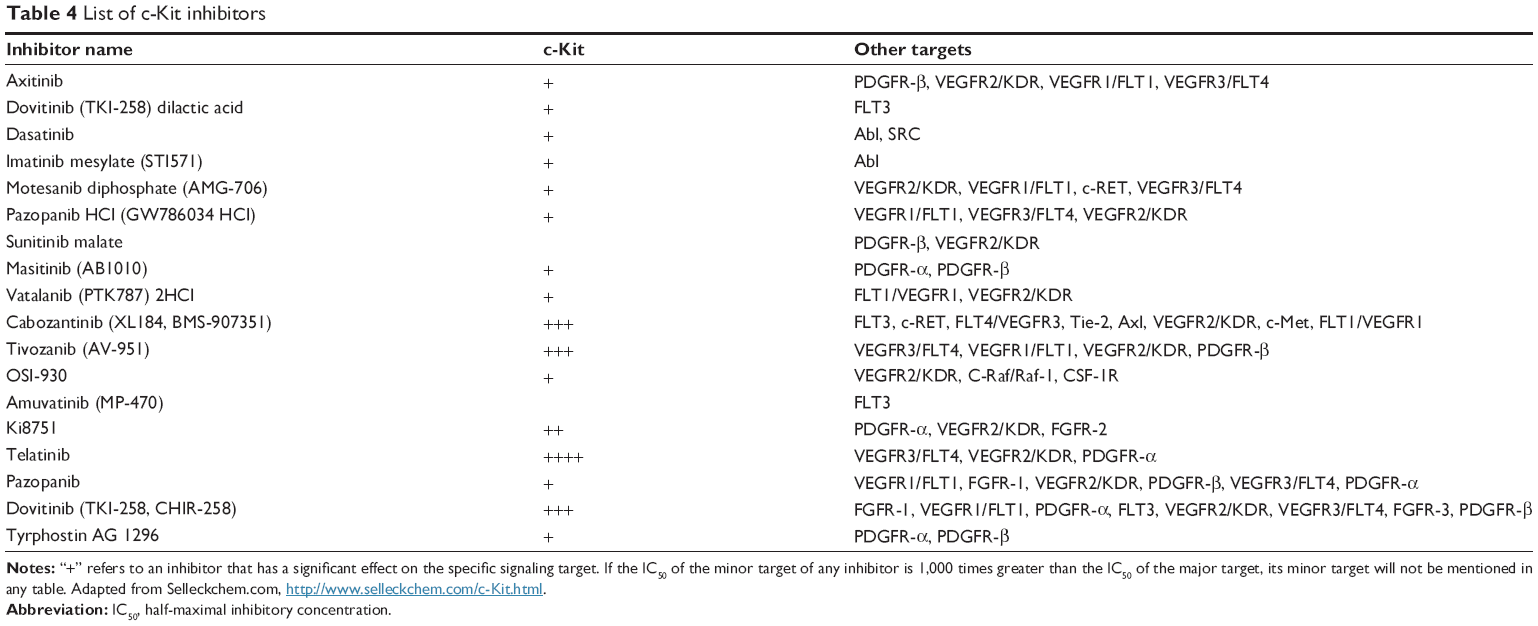 | Table 4 List of c-Kit inhibitors |
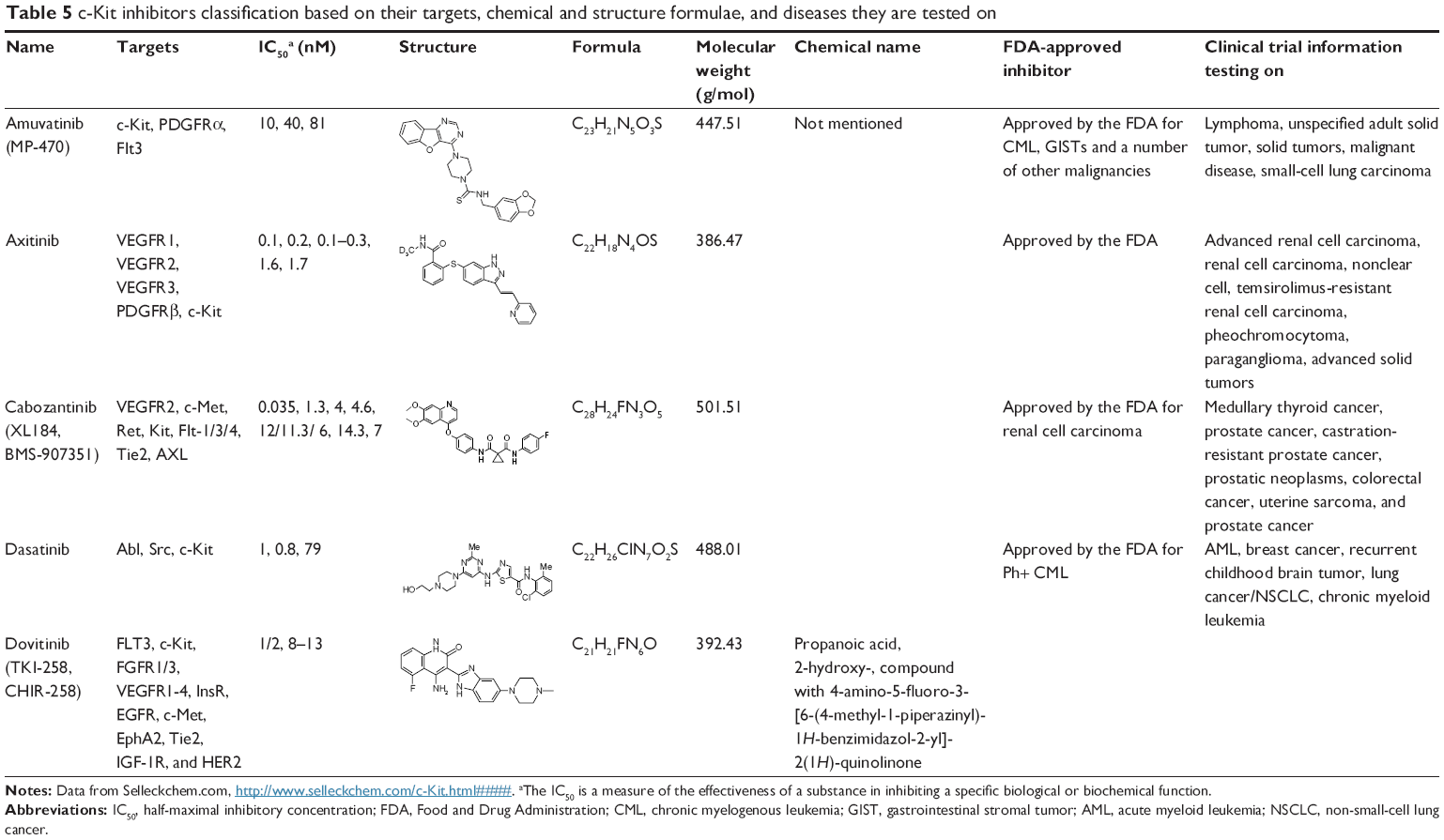 | Table 5 c-Kit inhibitors classification based on their targets, chemical and structure formulae, and diseases they are tested on |
Conclusion
Overall, particular mutations in c-Kit are accountable for cancer occurrence, such as GIST and SM (Systemic Mastocytosis) cases, as well as subsets of AML and melanoma. As the c-Kit mutations are the “drivers” in these cases, the use of kinase inhibitors, such as imatinib, could significantly improve cancer treatment. Nevertheless, the second mutation in c-Kit, which disturbs the biding region of kinase inhibitor, results in drug resistance. Thus far, no alternative solution for the current problem is established. This limitation could be addressed by elucidating the c-Kit pathway and its targeting genes. Cotargeting these pathways may also lead to tumor control. Finally, inhibition of cancer stem cells would be vital when considering the crucial role of cancer stem cells in drug resistance and cancer recurrence in cancer therapy.
Acknowledgments
The authors would like to express their utmost gratitude and appreciation to the University of Malaya Research Grant (RG084-13BIO), RP024A/14HTM UMRP grant, IPPP grant (PG082-2013B), the BKP grant (BK020-2012), and the Malaysian Ministry of Higher Education HIR Grants (UM.C/625/1/MOHE/MED/17 and UM.C/625/1/MOHE/MED/33) for providing financial support to conduct this study. The authors are thankful to Selleckchem.com for the reuse of their data.
Disclosure
The authors report no conflicts of interest in this work.
References
Ashman LK. The biology of stem cell factor and its receptor c-kit. Int J Biochem Cell Biol. 1999;31(10):1037–1051. | ||
Furitsu T, Tsujimura T, Tono T, et al. Identification of mutations in the coding sequence of the proto-oncogene c-kit in a human mast cell leukemia cell line causing ligand-independent activation of c-kit product. J Clin Invest. 1993;92(4):1736. | ||
Yavuz AS, Lipsky PE, Yavuz S, Metcalfe DD, Akin C. Evidence for the involvement of a hematopoietic progenitor cell in systemic mastocytosis from single-cell analysis of mutations in the c-kit gene. Blood. 2002;100(2):661–665. | ||
Corless CL, Fletcher JA, Heinrich MC. Biology of gastrointestinal stromal tumors. J Clin Oncol. 2004;22(18):3813–3825. | ||
Yarden Y, Kuang WJ, Yang-Feng T, et al. Human proto-oncogene c-kit: a new cell surface receptor tyrosine kinase for an unidentified ligand. EMBO J. 1987;6(11):3341. | ||
Giebel L, Strunk K, Holmes S, Spritz R. Organization and nucleotide sequence of the human KIT (mast/stem cell growth factor receptor) proto-oncogene. Oncogene. 1992;7(11):2207–2217. | ||
Ullrich A, Schlessinger J. Signal transduction by receptors with tyrosine kinase activity. Cell. 1990;61(2):203–212. | ||
Reith A, Ellis C, Lyman S, et al. Signal transduction by normal isoforms and W mutant variants of the Kit receptor tyrosine kinase. EMBO J. 1991;10(9):2451. | ||
Voytyuk O, Lennartsson J, Mogi A, et al. Src family kinases are involved in the differential signaling from two splice forms of c-Kit. J Biol Chem. 2003;278(11):9159–9166. | ||
Maulik G, Bharti A, Khan E, Broderick RJ, Kijima T, Salgia R. Modulation of c-Kit/SCF pathway leads to alterations in topoisomerase-I activity in small cell lung cancer. J Environ Pathol Toxicol Oncol. 2004;23(4):237–251. | ||
Pawson T. Protein modules and signalling networks. Nature. 1995;373(6515):573–580. | ||
Blume-Jensen P, Siegbahn A, Stabel S, Heldin CH, Rönnstrand L. Increased Kit/SCF receptor induced mitogenicity but abolished cell motility after inhibition of protein kinase C. EMBO J. 1993;12(11):4199. | ||
Zhu WM, Dong WF, Minden M. Alternate splicing creates two forms of the human kit protein. Leuk Lymphoma. 1994;12(5–6):441–447. | ||
Crosier P, Ricciardi ST, Hall LR, Vitas MR, Clark SC, Crosier KE. Expression of isoforms of the human receptor tyrosine kinase c-kit in leukemic cell lines and acute myeloid leukemia. Blood. 1993;82(4):1151–1158. | ||
Caruana G, Cambareri AC, Ashman LK. Isoforms of c-KIT differ in activation of signalling pathways and transformation of NIH3T3 fibroblasts. Oncogene. 1999;18:5573–5581. | ||
Huang E, Nocka K, Buck J, Besmer P. Differential expression and processing of two cell associated forms of the kit-ligand: KL-1 and KL-2. Mol Biol Cell. 1992;3(3):349–362. | ||
Albanesi C, Geremia R, Giorgio M, Dolci S, Sette C, Rossi P. A cell- and developmental stage-specific promoter drives the expression of a truncated c-kit protein during mouse spermatid elongation. Development. 1996;122(4):1291–1302. | ||
Paronetto MP, Farini D, Sammarco I, et al. Expression of a truncated form of the c-Kit tyrosine kinase receptor and activation of Src kinase in human prostatic cancer. Am J Pathol. 2004;164(4):1243–1251. | ||
Sakamoto A, Yoneda A, Terada K, et al. A functional truncated form of c-kit tyrosine kinase is produced specifically in the testis of the mouse but not the rat, pig, or human. Biochem Genet. 2004;42(11–12):441–451. | ||
DaSilva N, Hu Z, Ma W, Rosnet O, Birnbaum D, Drexler H. Expression of the FLT3 gene in human leukemia-lymphoma cell lines. Leukemia. 1994;8(5):885–888. | ||
Hu ZB, Ma W, Uphoff C, Quentmeier H, Drexler H. c-kit expression in human megakaryoblastic leukemia cell lines. Blood. 1994;83(8):2133–2144. | ||
Russell ES. Hereditary anemias of the mouse: a review for geneticists. Adv Genet. 1979;20:357. | ||
Keshet E, Lyman S, Williams D, et al. Embryonic RNA expression patterns of the c-kit receptor and its cognate ligand suggest multiple functional roles in mouse development. EMBO J. 1991;10(9):2425. | ||
Lev S, Blechman JM, Givol D, Yarden Y. Steel factor and c-kit protooncogene: genetic lessons in signal transduction. Crit Rev Oncog. 1994;5(2–3):141–168. | ||
Okada S, Nakauchi H, Nagayoshi K, Nishikawa S, Miura Y, Suda T. Enrichment and characterization of murine hematopoietic stem cells that express c-kit molecule. Blood. 1991;78(7):1706–1712. | ||
Broudy VC. Stem cell factor and hematopoiesis. Blood. 1997;90(4):1345–1364. | ||
Lyman SD, Jacobsen SEW. c-Kit ligand and Flt3 ligand: stem/progenitor cell factors with overlapping yet distinct activities. Blood. 1998;91(4):1101–1134. | ||
Ashman LK, Cambareri AC, To LB, Levinsky RJ, Juttner CA. Expression of the YB5. B8 antigen (c-kit proto-oncogene product) in normal human bone marrow. Blood. 1991;78(1):30–37. | ||
Papayannopoulou T, Brice M, Broudy VC, Zsebo KM. Isolation of c-kit receptor-expressing cells from bone marrow, peripheral blood, and fetal liver: functional properties and composite antigenic profile. Blood. 1991;78(6):1403–1412. | ||
Broudy VC, Smith F, Lin N, Zsebo K, Egrie J, Bernstein I. Blasts from patients with acute myelogenous leukemia express functional receptors for stem cell factor. Blood. 1992;80(1):60–67. | ||
Oliveira S, Lukacs N. Stem cell factor: a hemopoietic cytokine with important targets in asthma. Curr Drug Targets Inflamm Allergy. 2003;2(4):313–318. | ||
Waskow C, Paul S, Haller C, Gassmann M, Rodewald HR. Viable c-Kit W/W mutants reveal pivotal role for c-kit in the maintenance of lymphopoiesis. Immunity. 2002;17(3):277–288. | ||
Loveland K, Schlatt S. Stem cell factor and c-kit in the mammalian testis: lessons originating from Mother Nature’s gene knockouts. J Endocrinol. 1997;153(3):337–344. | ||
Blume-Jensen P, Janknecht R, Hunter T. The kit receptor promotes cell survival via activation of PI 3-kinase and subsequent Akt-mediated phosphorylation of Bad on Ser136. Curr Biol. 1998;8(13):779–785. | ||
Kissel H, Timokhina I, Hardy MP, et al. Point mutation in kit receptor tyrosine kinase reveals essential roles for kit signaling in spermatogenesis and oogenesis without affecting other kit responses. EMBO J. 2000;19(6):1312–1326. | ||
Scott G, Ewing J, Ryan D, Abboud C. Stem cell factor regulates human melanocyte-matrix interactions. Pigment Cell Res. 1994;7(1):44–51. | ||
Mackenzie MA, Jordan SA, Budd PS, Jackson IJ. Activation of the receptor tyrosine kinase Kit is required for the proliferation of melanoblasts in the mouse embryo. Dev Biol. 1997;192(1):99–107. | ||
Wehrle-Haller B. The role of Kit-ligand in melanocyte development and epidermal homeostasis. Pigment Cell Res. 2003;16(3):287–296. | ||
Kimura Y, Jones N, Klüppel M, et al. Targeted mutations of the juxtamembrane tyrosines in the Kit receptor tyrosine kinase selectively affect multiple cell lineages. Proc Natl Acad Sci U S A. 2004;101(16):6015–6020. | ||
Lyford G, He C, Soffer E, et al. Pan-colonic decrease in interstitial cells of Cajal in patients with slow transit constipation. Gut. 2002; 51(4):496–501. | ||
Huizinga JD, Thuneberg L, Kluppel M, Malysz J, Mikkelsen HB, Bernstein A. W/kit gene required for interstitial cells of Cajal and for intestinal pacemaker activity. Nature. 1995;373(6512):347–349. | ||
Ward SM, Burns AJ, Torihashi S, Harney SC, Sanders KM. Impaired development of interstitial cells and intestinal electrical rhythmicity in steel mutants. Am J Physiol. 1995;269(6):C1577–C1585. | ||
Ward SM, Burns AJ, Torihashi S, Sanders KM. Mutation of the proto-oncogene c-kit blocks development of interstitial cells and electrical rhythmicity in murine intestine. J Physiol. 1994;480(1):91–97. | ||
Jin K, Mao XO, Sun Y, Xie L, Greenberg DA. Stem cell factor stimulates neurogenesis in vitro and in vivo. J Clin Invest. 2002;110(3):311–319. | ||
Sun L, Lee J, Fine HA. Neuronally expressed stem cell factor induces neural stem cell migration to areas of brain injury. J Clin Invest. 2004;113(9):1364. | ||
Ashman LK, Griffith R. Therapeutic targeting of c-KIT in cancer. Expert Opin Investig Drugs. 2013;22(1):103–115. | ||
Lennartsson J, Jelacic T, Linnekin D, Shivakrupa R. Normal and oncogenic forms of the receptor tyrosine kinase kit. Stem Cells. 2005; 23(1):16–43. | ||
Huang S, Luca M, Gutman M, et al. Enforced c-KIT expression renders highly metastatic human melanoma cells susceptible to stem cell factor-induced apoptosis and inhibits their tumorigenic and metastatic potential. Oncogene. 1996;13(11):2339–2347. | ||
Willmore-Payne C, Holden JA, Tripp S, Layfield LJ. Human malignant melanoma: detection of BRAF-and c-kit–activating mutations by high-resolution amplicon melting analysis. Hum Pathol. 2005;36(5):486–493. | ||
D’Amato G, Steinert DM, McAuliffe JC, Trent JC. Update on the biology and therapy of gastrointestinal stromal tumors. Cancer Control. 2005;12(1):44–56. | ||
Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279(5350):577–580. | ||
Kelly LM, Gilliland DG. Genetics of myeloid leukemias. Annu Rev Genomics Hum Genet. 2002;3(1):179–198. | ||
Hoei-Hansen CE, Kraggerud SM, Abeler VM, Kærn J, Rajpert-De Meyts E, Lothe RA. Ovarian dysgerminomas are characterised by frequent KIT mutations and abundant expression of pluripotency markers. Mol Cancer. 2007;6(1):12. | ||
Beadling C, Jacobson-Dunlop E, Hodi FS, et al. KIT gene mutations and copy number in melanoma subtypes. Clin Cancer Res. 2008;14(21):6821–6828. | ||
Heinrich MC, Blanke CD, Druker BJ, Corless CL. Inhibition of KIT tyrosine kinase activity: a novel molecular approach to the treatment of KIT-positive malignancies. J Clin Oncol. 2002;20(6):1692–1703. | ||
Catalano A, Rodilossi S, Rippo MR, Caprari P, Procopio A. Induction of stem cell factor/c-Kit/slug signal transduction in multidrug-resistant malignant mesothelioma cells. J Biol Chem. 2004;279(45):46706–46714. | ||
Ikeda H, Kanakura Y, Tamaki T, et al. Expression and functional role of the proto-oncogene c-kit in acute myeloblastic leukemia cells. Blood. 1991;78(11):2962–2968. | ||
Huo L, Sugimura J, Tretiakova MS, et al. c-kit expression in renal oncocytomas and chromophobe renal cell carcinomas. Hum Pathol. 2005;36(3):262–268. | ||
Rodrigues GA, Park M. Oncogenic activation of tyrosine kinases. Curr Opin Genet Devel. 1994;4(1):15–24. | ||
Kitayama H, Kanakura Y, Furitsu T, et al. Constitutively activating mutations of c-kit receptor tyrosine kinase confer factor-independent growth and tumorigenicity of factor-dependent hematopoietic cell lines. Blood. 1995;85(3):790–798. | ||
Moriyama Y, Tsujimura T, Hashimoto K, et al. Role of aspartic acid 814 in the function and expression of c-kit receptor tyrosine kinase. J Biol Chem. 1996;271(7):3347–3350. | ||
Lam L, Chow R, Berger S. A transforming mutation enhances the activity of the c-Kit soluble tyrosine kinase domain. Biochem J. 1999;338:131–138. | ||
Tsujimura T, Hashimoto K, Kitayama H, et al. Activating mutation in the catalytic domain of c-kit elicits hematopoietic transformation by receptor self-association not at the ligand-induced dimerization site. Blood. 1999;93(4):1319–1329. | ||
Casteran N, De Sepulveda P, Beslu N, et al. Signal transduction by several KIT juxtamembrane domain mutations. Oncogene. 2003;22(30):4710–4722. | ||
Piao X, Paulson R, Van Der Geer P, Pawson T, Bernstein A. Oncogenic mutation in the Kit receptor tyrosine kinase alters substrate specificity and induces degradation of the protein tyrosine phosphatase SHP-1. Proc Natl Acad Sci U S A. 1996;93(25):14665–14669. | ||
Naoe T, Kiyoi H. Normal and oncogenic FLT3. Cell Mol Life Sci. 2004;61(23):2932–2938. | ||
Hongyo T, Li T, Syaifudin M, et al. Specific c-kit mutations in sinonasal natural killer/T-cell lymphoma in China and Japan. Cancer Res. 2000;60(9):2345–2347. | ||
Büttner C, Henz BM, Welker P, Sepp NT, Grabbe J. Identification of activating c-kit mutations in adult-, but not in childhood-onset indolent mastocytosis: a possible explanation for divergent clinical behavior. J Investig Dermatol. 1998;111(6):1227–1231. | ||
Ashman LK, Ferrao P, Cole SR, Cambareri AC. Effects of mutant c-kit in early myeloid cells. Leuk Lymphoma. 1999;34(5–6):451–461. | ||
Beghini A, Peterlongo P, Ripamonti CB, et al. c-kit mutations in core binding factor leukemias. Blood. 2000;95(2):726–728. | ||
Tian Q, Frierson HF, Krystal GW, Moskaluk CA. Activating c-kit gene mutations in human germ cell tumors. Am J Pathol. 1999;154(6):1643–1647. | ||
Pauls K, Wardelmann E, Merkelbach-Bruse S, Büttner R, Zhou H. c-KIT codon 816 mutation in a recurrent and metastatic dysgerminoma of a 14-year-old girl: case study. Virchows Arch. 2004;445(6):651–654. | ||
Sakuma Y, Sakurai S, Oguni S, Satoh M, Hironaka M, Saito K. c-Kit gene mutations in intracranial germinomas. Cancer Sci. 2004;95(9):716–720. | ||
Nagata H, Worobec AS, Oh CK, et al. Identification of a point mutation in the catalytic domain of the protooncogene c-kit in peripheral blood mononuclear cells of patients who have mastocytosis with an associated hematologic disorder. Proc Natl Acad Sci U S A. 1995;92(23):10560–10564. | ||
Longley BJ, Tyrrell L, Lu SZ, et al. Somatic c-KIT activating mutation in urticaria pigmentosa and aggressive mastocytosis: establishment of clonality in a human mast cell neoplasm. Nature Genet. 1996;12(3):312–314. | ||
Lin ZH, Han EM, Lee ES, et al. A distinct expression pattern and point mutation of c-kit in papillary renal cell carcinomas. Mod Pathol. 2004; 17(6):611–616. | ||
Hubbard SR. Juxtamembrane autoinhibition in receptor tyrosine kinases. Nat Rev Mol Cell Biol. 2004;5(6):464–471. | ||
DiNitto JP, Deshmukh GD, Zhang Y, et al. Function of activation loop tyrosine phosphorylation in the mechanism of c-Kit auto-activation and its implication in sunitinib resistance. J Biochem. 2010;147(4):601–609. | ||
Morgan SJ, Smith AD, Parker PJ. Purification and characterization of bovine brain type I phosphatidylinositol kinase. Eur J Biochem. 1990;191(3):761–767. | ||
Engström M, Karlsson R, Jönsson JI. Inactivation of the forkhead transcription factor FoxO3 is essential for PKB-mediated survival of hematopoietic progenitor cells by kit ligand. Exp Hematol. 2003;31(4):316–323. | ||
Möller C, Alfredsson J, Engström M, et al. Stem cell factor promotes mast cell survival via inactivation of FOXO3a-mediated transcriptional induction and MEK-regulated phosphorylation of the proapoptotic protein Bim. Blood. 2005;106(4):1330–1336. | ||
Möller C, Alfredsson J, Engström M, et al. Stem cell factor promotes mast cell survival via inactivation of FOXO3a-mediated transcriptional induction and MEK-regulated phosphorylation of the proapoptotic protein Bim. Blood. 2005;106(4):1330–1336. | ||
Vosseller K, Stella G, Yee N, Besmer P. c-Kit receptor signaling through its phosphatidylinositide-3′-kinase-binding site and protein kinase C: role in mast cell enhancement of degranulation, adhesion, and membrane ruffling. Mol Biol Cell. 1997;8(5):909–922. | ||
Hashimoto K, Matsumura I, Tsujimura T, et al. Necessity of tyrosine 719 and phosphatidylinositol 3′-kinase–mediated signal pathway in constitutive activation and oncogenic potential of c-kit receptor tyrosine kinase with the Asp814Val mutation. Blood. 2003;101(3):1094–1102. | ||
Shivakrupa R, Bernstein A, Watring N, Linnekin D. Phosphatidylinositol 3′-kinase is required for growth of mast cells expressing the kit catalytic domain mutant. Cancer Res. 2003;63(15):4412–4419. | ||
Arcaro A, Khanzada UK, Vanhaesebroeck B, Tetley TD, Waterfield MD, Seckl MJ. Two distinct phosphoinositide 3-kinases mediate polypeptide growth factor-stimulated PKB activation. EMBO J. 2002;21(19):5097–5108. | ||
Benes CH, Wu N, Elia AE, Dharia T, Cantley LC, Soltoff SP. The C2 domain of PKCδ is a phosphotyrosine binding domain. Cell. 2005;121(2):271–280. | ||
Bromann PA, Korkaya H, Courtneidge SA. The interplay between Src family kinases and receptor tyrosine kinases. Oncogene. 2004;23(48):7957–7968. | ||
Linnekin D, DeBerry CS, Mou S. Lyn associates with the juxtamembrane region of c-Kit and is activated by stem cell factor in hematopoietic cell lines and normal progenitor cells. J Biol Chem. 1997;272(43):27450–27455. | ||
Krystal GW, DeBerry CS, Linnekin D, Litz J. Lck associates with and is activated by Kit in a small cell lung cancer cell line: inhibition of SCF-mediated growth by the Src family kinase inhibitor PP1. Cancer Res. 1998;58(20):4660–4666. | ||
Broudy VC, Lin NL, Liles WC, et al. Signaling via Src family kinases is required for normal internalization of the receptor c-Kit. Blood. 1999;94(6):1979–1986. | ||
Shivakrupa R, Linnekin D. Lyn contributes to regulation of multiple Kit-dependent signaling pathways in murine bone marrow mast cells. Cell Signal. 2005;17(1):103–109. | ||
Hong L, Munugalavadla V, Kapur R. c-Kit-mediated overlapping and unique functional and biochemical outcomes via diverse signaling pathways. Mol Cell Biol. 2004;24(3):1401–1410. | ||
Wollberg P, Lennartsson J, Gottfridsson E, Yoshimura A, Ronnstrand L. The adapter protein APS associates with the multifunctional docking sites Tyr-568 and Tyr-936 in c-Kit. Biochem J. 2003;370: 1033–1038. | ||
Kozlowski M, Larose L, Lee F, Le DM, Rottapel R, Siminovitch KA. SHP-1 binds and negatively modulates the c-Kit receptor by interaction with tyrosine 569 in the c-Kit juxtamembrane domain. Mol Cell Biol. 1998;18(4):2089–2099. | ||
Agosti V, Corbacioglu S, Ehlers I, et al. Critical role for Kit-mediated Src kinase but not PI 3-kinase signaling in pro T and pro B cell development. J Exp Med. 2004;199(6):867–878. | ||
Murphy LO, Smith S, Chen RH, Fingar DC, Blenis J. Molecular interpretation of ERK signal duration by immediate early gene products. Nat Cell Biol. 2002;4(8):556–564. | ||
Thommes K, Lennartsson J, Carlberg M, Ronnstrand L. Identification of Tyr-703 and Tyr-936 as the primary association sites for Grb2 and Grb7 in the c-Kit/stem cell factor receptor. Biochem J. 1999;341:211–216. | ||
Hosseinimehr SJ, Inanami O, Hamasu T, et al. Activation of c-kit by stem cell factor induces radioresistance to apoptosis through ERK-dependent expression of survivin in HL60 cells. J Radiat Res. 2004;45(4):557–561. | ||
Lennartsson J, Wernstedt C, Engström U, Hellman U, Rönnstrand L. Identification of Tyr900 in the kinase domain of c-Kit as a Src-dependent phosphorylation site mediating interaction with c-Crk. Exp Cell Res. 2003;288(1):110–118. | ||
Brizzi MF, Zini MG, Aronica MG, Blechman JM, Yarden Y, Pegoraro L. Convergence of signaling by interleukin-3, granulocyte-macrophage colony-stimulating factor, and mast cell growth factor on JAK2 tyrosine kinase. J Biol Chem. 1994;269(50): 31680–31684. | ||
Pérez-Losada J, Sánchez-Martín M, Rodríguez-García A, et al. Zinc-finger transcription factor Slug contributes to the function of the stem cell factor c-kit signaling pathway. Blood. 2002;100(4): 1274–1286. | ||
Lennartsson J, Ronnstrand L. The stem cell factor receptor/c-Kit as a drug target in cancer. Curr Cancer Drug Targets. 2006;6(1):65–75. | ||
Deininger M, Buchdunger E, Druker BJ. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood. 2005;105(7):2640–2653. | ||
Wakita S, Yamaguchi H, Miyake K, et al. Importance of c-kit mutation detection method sensitivity in prognostic analyses of t (8; 21)(q22; q22) acute myeloid leukemia. Leukemia. 2011;25(9):1423–1432. | ||
Willmore-Payne C, Layfield LJ, Holden JA. c-Kit mutation analysis for diagnosis of gastrointestinal stromal tumors in fine needle aspiration specimens. Cancer Cytopathol. 2005;105(3):165–170. | ||
Frost MJ, Ferrao PT, Hughes TP, Ashman LK. Juxtamembrane mutant V560GKit is more sensitive to Imatinib (STI571) compared with wild-type c-Kit whereas the kinase domain mutant D816VKit is resistant. Mol Cancer Ther. 2002;1(12):1115–1124. | ||
Roberts KG, Odell AF, Byrnes EM, et al. Resistance to c-KIT kinase inhibitors conferred by V654A mutation. Mol Cancer Ther. 2007;6(3):1159–1166. | ||
Krüger S, Sotlar K, Kausch I, Horny HP. Expression of KIT (CD117) in renal cell carcinoma and renal oncocytoma. Oncology. 2005;68(2–3):269–275. | ||
Corbin AS, Demehri S, Griswold IJ, et al. In vitro and in vivo activity of ATP-based kinase inhibitors AP23464 and AP23848 against activation-loop mutants of Kit. Blood. 2005;106(1):227–234. | ||
Wang YY, Zhou GB, Yin T, et al. AML1-ETO and c-KIT mutation/overexpression in t (8; 21) leukemia: implication in stepwise leukemogenesis and response to Gleevec. Proc Natl Acad Sci U S A. 2005;102(4):1104–1109. | ||
Chan I, Kasprowicz S, Tharp M. Distinct signalling pathways for mutated KIT (V560G) and KIT (D816V) in mastocytosis. Clin Exp Dermatol. 2013;38(5):538–544. | ||
Lanternier F, Cohen-Akenine A, Palmerini F, et al. Phenotypic and genotypic characteristics of mastocytosis according to the age of onset. PLoS One. 2008;3(4):e1906. | ||
Schnittger S, Kohl TM, Haferlach T, et al. KIT-D816 mutations in AML1-ETO-positive AML are associated with impaired event-free and overall survival. Blood. 2006;107(5):1791–1799. | ||
Sakuma Y, Sakurai S, Oguni S, Hironaka M, Salto K. Alterations of the c-kit gene in testicular germ cell tumors. Cancer Sci. 2003;94(6):486–491. | ||
Orfao A, Garcia-Montero AC, Sanchez L, Escribano L. Recent advances in the understanding of mastocytosis: the role of KIT mutations. Br J Haematol. 2007;138(1):12–30. | ||
Nakata Y, Kimura A, Katoh O, et al. c-Kit point mutation of extracellular domain in patients with myeloproliferative disorders. Br J Haematol. 1995;91(3):661–663. | ||
Kimura A, Nakata Y, Katoh O, Hyodo H. c-Kit point mutation in patients with myeloproliferative disorders. Leuk Lymphoma. 1997;25(3–4):281–287. | ||
Scheijen B, Griffin JD. Tyrosine kinase oncogenes in normal hematopoiesis and hematological disease. Oncogene. 2002;21(21):3314–3333. | ||
Maki R, Keedy V. 2012. KIT Exon 14 Mutation in GIST [webpage on the Internet]. My Cancer Genome. Available from: https://www.mycancergenome.org/content/disease/gist/kit/50/. Accessed April 21, 2016. | ||
Heinrich MC, Griffith DJ, Druker BJ, Wait CL, Ott KA, Zigler AJ. Inhibition of c-kit receptor tyrosine kinase activity by STI 571, a selective tyrosine kinase inhibitor. Blood. 2000;96(3):925–932. | ||
Tuveson DA, Willis NA, Jacks T, et al. STI 571 inactivation of the gastrointestinal stromal tumor c-KIT oncoprotein: biological and clinical implications. Oncogene. 2001;20(36):5054–5058. | ||
Attoub S, Rivat C, Rodrigues S, et al. The c-kit tyrosine kinase inhibitor STI571 for colorectal cancer therapy. Cancer Res. 2002;62(17):4879–4883. | ||
Weisberg E, Boulton C, Kelly LM, et al. Inhibition of mutant FLT3 receptors in leukemia cells by the small molecule tyrosine kinase inhibitor PKC412. Cancer Cell. 2002;1(5):433–443. | ||
Wheeler S, Seegmiller A, Vnencak-Jones A. 2013. KIT c.2447A>T (D816V) Mutation in Acute Myeloid Leukemia [webpage on the Internet]. My Cancer Genome. Available from: https://www.mycancergenome.org/content/disease/acute-myeloid-leukemia/kit/275/. Accessed April 21, 2016. | ||
Yee KW, O’Farrell AM, Smolich BD, et al. SU5416 and SU5614 inhibit kinase activity of wild-type and mutant FLT3 receptor tyrosine kinase. Blood. 2002;100(8):2941–2949. | ||
Smolich BD, Yuen HA, West KA, Giles FJ, Albitar M, Cherrington JM. The antiangiogenic protein kinase inhibitors SU5416 and SU6668 inhibit the SCF receptor (c-kit) in a human myeloid leukemia cell line and in acute myeloid leukemia blasts. Blood. 2001;97(5):1413–1421. | ||
Kim DW, Jo YS, Jung HS, et al. An orally administered multitarget tyrosine kinase inhibitor, SU11248, is a novel potent inhibitor of thyroid oncogenic RET/papillary thyroid cancer kinases. J Clin Endocrinol Metabol. 2006;91(10):4070–4076. | ||
Von Bubnoff N, Gorantla S, Kancha R, Lordick F, Peschel C, Duyster J. Spotlight correspondence. Leukemia. 2005;19:1670–1671. | ||
Lovly C, Pao W, Sosman J. 2015. KIT c.2446G>C (D816H) Mutation in Melanoma [webpage on the Internet]. My Cancer Genome. Available from: https://www.mycancergenome.org/content/disease/melanoma/kit/84/. Accessed April 21, 2016. | ||
Sambol EB, Ambrosini G, Geha RC, et al. Flavopiridol targets c-KIT transcription and induces apoptosis in gastrointestinal stromal tumor cells. Cancer Res. 2006;66(11):5858–5866. | ||
Bellone G, Silvestri S, Artusio E, et al. Growth stimulation of colorectal carcinoma cells via the c-kit receptor is inhibited by TGF-β1. J Cell Physiol. 1997;172(1):1–11. | ||
Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol. 2006;24(26):4340–4346. | ||
Terheyden P, Houben R, Pajouh P, Thorns C, Zillikens D, Becker JC. Response to imatinib mesylate depends on the presence of the V559A-mutated KIT oncogene. J Investig Dermatol. 2010;130(1): 314–316. | ||
Woodman SE, Trent JC, Stemke-Hale K, et al. Activity of dasatinib against L576P KIT mutant melanoma: molecular, cellular, and clinical correlates. Mol Cancer Ther. 2009;8(8):2079–2085. | ||
Quintás-Cardama A, Lazar AJ, Woodman SE, Kim K, Ross M, Hwu P. Complete response of stage IV anal mucosal melanoma expressing KIT Val560Asp to the multikinase inhibitor sorafenib. Nat Clin Pract Oncol. 2008;5(12):737–740. | ||
Carvajal RD, Antonescu CR, Wolchok JD, et al. KIT as a therapeutic target in metastatic melanoma. JAMA. 2011;305(22):2327–2334. | ||
Harada H, Harada Y, Niimi H, Kyo T, Kimura A, Inaba T. High incidence of somatic mutations in the AML1/RUNX1 gene in myelodysplastic syndrome and low blast percentage myeloid leukemia with myelodysplasia. Blood. 2004;103(6):2316–2324. | ||
Guo J, Si L, Kong Y, et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol. 2011;29(21):2904–2909. | ||
Girard N. 2015. KIT c.2460T>A (D820E) Mutation in Thymic Carcinoma [webpage on the Internet]. My Cancer Genome. Available from: https://www.mycancergenome.org/content/disease/thymic-carcinoma/kit/120/. Accessed April 21, 2016. | ||
Buti S, Donini M, Sergio P, et al. Impressive response with imatinib in a heavily pretreated patient with metastatic c-KIT mutated thymic carcinoma. J Clin Oncol. 2011;29(33):e803–e805. | ||
Schirosi L, Nannini N, Nicoli D, et al. Activating c-KIT mutations in a subset of thymic carcinoma and response to different c-KIT inhibitors. Ann Oncol. 2012;23(9):2409–2414. | ||
Antonescu CR, Sommer G, Sarran L, et al. Association of KIT exon 9 mutations with nongastric primary site and aggressive behavior KIT mutation analysis and clinical correlates of 120 gastrointestinal stromal tumors. Clin Cancer Res. 2003;9(9):3329–3337. | ||
Rossi F, Yozgat Y, de Stanchina E, et al. Imatinib upregulates compensatory integrin signaling in a mouse model of gastrointestinal stromal tumor and is more effective when combined with dasatinib. Mol Cancer Res. 2010;8(9):1271–1283. | ||
Ströbel P, Hartmann M, Jakob A, et al. Thymic carcinoma with overexpression of mutated KIT and the response to imatinib. N Engl J Med. 2004;350(25):2625–2626. | ||
Antonescu CR, Besmer P, Guo T, et al. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res. 2005;11(11):4182–4190. | ||
Yoh K, Nishiwaki Y, Ishii G, et al. Mutational status of EGFR and KIT in thymoma and thymic carcinoma. Lung Cancer. 2008;62(3): 316–320. | ||
Dişel U, Öztuzcu S, Beşen AA, et al. Promising efficacy of sorafenib in a relapsed thymic carcinoma with c-KIT exon 11 deletion mutation. Lung Cancer. 2011;71(1):109–112. | ||
Guo T, Agaram NP, Wong GC, et al. Sorafenib inhibits the imatinib-resistant KITT670I gatekeeper mutation in gastrointestinal stromal tumor. Clin Cancer Res. 2007;13(16):4874–4881. | ||
Timokhina I, Kissel H, Stella G, Besmer P. Kit signaling through PI 3-kinase and Src kinase pathways: an essential role for Rac1 and JNK activation in mast cell proliferation. EMBO J. 1998;17(21):6250–6262. | ||
Goddard N, McIntyre A, Summersgill B, Gilbert D, Kitazawa S, Shipley J. KIT and RAS signalling pathways in testicular germ cell tumours: new data and a review of the literature. Int J Androl. 2007;30(4):337–349. | ||
Wandzioch E, Edling CE, Palmer RH, Carlsson L, Hallberg B. Activation of the MAP kinase pathway by c-Kit is PI-3 kinase dependent in hematopoietic progenitor/stem cell lines. Blood. 2004;104(1):51–57. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.