")
Back to Journals » OncoTargets and Therapy » Volume 10
Recent progress on the effects of microRNAs and natural products on tumor epithelial–mesenchymal transition
Authors He SJ , Xiang CQ, Zhang Y , Lu XT , Chen HW, Xiong LX
Received 13 April 2017
Accepted for publication 27 May 2017
Published 12 July 2017 Volume 2017:10 Pages 3435—3451
DOI https://doi.org/10.2147/OTT.S139546
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr XuYu Yang
Shu-Jin He,1,2,* Chu-Qi Xiang,1,3,* Yu Zhang,3 Xiang-Tong Lu,1 Hou-Wen Chen,1,4 Li-Xia Xiong1,4
1Department of Pathophysiology, Medical College, Nanchang University, 2Second Clinical Medical College, Nanchang University, 3First Clinical Medical College, Nanchang University, 4Jiangxi Province Key Laboratory of Tumor Pathogenesis and Molecular Pathology, Nanchang, People’s Republic of China
*These authors contributed equally to this work
Abstract: Epithelial–mesenchymal transition (EMT) is a biological process of phenotypic transition of epithelial cells that can promote physiological development as well as tissue healing and repair. In recent years, cancer researchers have noted that EMT is closely related to the occurrence and development of tumors. When tumor cells undergo EMT, they can develop enhanced migration and local tissue invasion abilities, which can lead to metastatic growth. Nevertheless, two researches in NATURE deny its necessity in specific tumors and that is discussed in this review. The degree of EMT and the detection of EMT-associated marker molecules can also be used to judge the risk of metastasis and to evaluate patients’ prognosis. MicroRNAs (miRNAs) are noncoding small RNAs, which can inhibit gene expression and protein translation through specific binding with the 3' untranslated region of mRNA. In this review, we summarize the miRNAs that are reported to influence EMT through transcription factors such as ZEB, SNAIL, and TWIST, as well as some natural products that regulate EMT in tumors. Moreover, mutual inhibition occurs between some transcription factors and miRNAs, and these effects appear to occur in a complex regulatory network. Thus, understanding the role of miRNAs in EMT and tumor growth may lead to new treatments for malignancies. Natural products can also be combined with conventional chemotherapy to enhance curative effects.
Keywords: epithelial–mesenchymal transition, miRNA, tumor, natural products, ZEB, SNAIL
Introduction
As a biological process, epithelial–mesenchymal transition (EMT) plays important roles in tissue growth, remodeling, and repair of various pathological injuries. EMT is also a significant factor in determining the malignancy of a tumor1 for an important consideration given that metastatic growth is the direct cause of death in many cancer patients. Consequently, the relationship between EMT and tumors is a significant focus in cancer research. The three main features of EMT on which research is focused include 1) intracellular cell protein levels, including decreased expression of epithelial marker proteins such as E-cadherin (E-CAD), β-catenin, and cytokeratin; it also focuses on the increased expression of mesenchymal marker proteins such as N-cadherin (N-CAD) and vimentin (VIM). 2) Cell phenotypes and biological behaviors, such that epithelial cells lose their polarity and connection with the basal membrane, and then transform into mesenchymal phenotypes with higher migration and invasive tendencies, antiapoptotic features, and production of an extracellular matrix. When EMT occurs in tumors, it tends to increase the migratory ability of the cells and give rise to malignancies. 3) In epigenetics, the occurrence of EMT is associated with epigenetic reprogramming of the genome, involving DNA methylation and post-translational modifications of histone.2 These changes can promote the expression of stromal genes or inhibit the expression of genes related to the epithelial phenotype.
The microRNAs (miRNAs) are 22–24-nucleotides (nts) endogenous, noncoding small RNAs that play a variety of regulatory roles in cells. Most miRNAs regulate target mRNA through base-pairing between seed sequences, which range from nt positions 2–8, located at the 5′ end of the miRNA and target the mRNA 3′ untranslated region (UTR).3 Because they have a smaller number of nts in a complementary sequence and relatively low requirements for matching, a single miRNA can target a variety of mRNAs, with many individual mRNA sequences being regulated by various miRNAs.4 In recent years, many miRNAs that play a regulatory role in EMT have been identified, and most of those that inhibit EMT have a common feature: their expression is decreased in tumor cells, thus promoting occurrence of EMT. However, miRNA re-expression will inhibit that process, mainly by reducing levels of EMT-related transcription factors such as ZEB, SNAIL, TWIST, and SLUG. However, some miRNAs promote EMT through other mechanisms. In addition to miRNAs, some natural products are currently known to regulate EMT or, alternatively, tumor inhibition. The structural diversity and highly selective mechanisms of action of these natural products suggest that they may have great value for the development of antineoplastic drugs.5
In this paper, we address the role of EMT in neoplasms, and summarize the effect of miRNAs in the regulation of EMT. We will discuss the function and mechanism of miRNAs and natural products in detail.
EMT and tumor metastasis
In the traditional view, tumors were considered to be a homogeneous proliferation of malignant cells, but more recent studies have shown that solid neoplasms are complex tissues composed of multiple cell types, with considerable intratumoral heterogeneity. The difference between cancer stem cells and non-stem cells is a major source of this heterogeneity. Apart from showing remarkable genetic and phenotypic variabilities, cancer stem cells maintain plasticity between epithelial and mesenchymal cell types under the regulation of their microenvironment, which influences tumor metastasis.6 EMT has been identified and described as the underlying process for tumor metastasis in the last two decades.7 Metastases occur through a series of steps: local infiltration, entry of cancer cells into blood vessels, transport in the circulatory system, and finally, exuding from blood vessels, and achieving colonization.8 The neoplastic cells then disseminate from the primary site and form secondary tumors at other distant sites. Metastasis is a significant hallmark of malignancy, and it seriously affects the patient’s prognosis, treatment plan, and quality of life.
EMT and local infiltration of tumor
EMT is the first step in the cascade of steps involved in tumor invasion and metastasis. The original malignant epithelial cells lose intercellular junctions and lumen–basement polarity and thus develop the capacity to migrate, cross basement membranes, and invade blood vessels. After entering their local blood vessels, these cells can give rise to clusters of circulating tumor cells (CTCs) in the bloodstream. At some point, they can leave the blood vessels, escape the circulation, and form micrometastases at distant organs.9 For example, for tissue invasion and metastasis, colorectal cancer cells (CRCs) at the tumor–normal tissue interface frequently undergo EMT, losing epithelial marker proteins and acquiring mesenchymal ones. Then, they lose cell-to-cell adhesion, and finally become invasive. We have known that the progression of colorectal cancer could be promoted by the pro-inflammatory factor interleukin 6 (IL-6) in the tumor microenvironment. Liu et al10 suggested that an aberrant IL-6/STAT3/Fra-1 signaling pathway promoted cellular invasion and metastasis in colorectal cancer patients through EMT. They found that the ectopic expression of Fra-1 significantly reversed the STAT3-knockdown effect, and enhanced CRC cell aggressiveness, by promoting the expression of EMT-related factors ZEB1, SNAIL, SLUG, matrix metalloproteinases-2 (MMP-2), and MMP-9. Furthermore, it has been reported that breast cancer cell line py2t from the MMTV-PyMT transgenic mouse can be induced to undergo reversible EMT by cell transforming growth factor-β (TGF-β) treatment in cell model systems, accompanied by downregulation of E-CAD, the gain of stromal cell markers, and expression of EMT transcription factors. At the same time, the motility and aggressiveness of the single cancer cells were elevated.11
The relationship between EMT and tumor cells exuding from vessel lumens
EMT is often accompanied with the detyrosination of certain proteins like α-tubulin. This mediates the formation of tubulin microantenna, which induces CTCs of mesenchymal phenotypes to attach to the endothelial layer, and further migrate from blood vessels to colonize distant loci. Furthermore, GLu microprotein (a kind of α-tubulin) increases the life span of tumor cells in blood vessels and promotes cells exuding from blood vessels.
EMT and tumor metastasis
In the process of tumor colonization, cancer cells usually regain epithelial features via reversing EMT (or mesenchymal–epithelial transformation – [MET]) to form a secondary tumor and large patchy metastases, resulting in the completion of the cascade reaction of invasion and metastasis. Although metastatic tumors show the typical characteristics of EMT in both their original tumor cells and the CTC clusters, the distant metastases of most epithelial carcinomas usually have epithelial features. MET plays an important role in assisting disseminated tumor cells to form large metastatic foci in distant organs.6 It has been reported that the invasive ability and transepithelial migration of E-CAD-negative prostate cancer cells is greater than that of E-CAD-positive prostate cancer cells.12
EMT score and tumor evaluation
Interestingly, it has been reported that conversion between epithelial and mesenchymal phenotypes is not necessarily complete. Neoplastic cells can acquire a mixed epithelial–mesenchymal phenotype that is called incomplete or partial EMT.9 The level of EMT in ovarian cancer, breast cancer, and colorectal cancer has been assessed by measuring the transcription level of tumor-specific EMT markers, and an EMT score was evaluated and used to analyze the interaction between EMT and tumor progression. The results showed that there was a good correlation between the EMT score and previously identified tumor-specific EMT markers in ovarian cancer and colorectal cancer, but not in breast cancer.13 The EMT score can be used to diagnose and evaluate the development of tumors as well as their drug resistance.
Unconventional discovery: EMT is not required for tumor metastasis
It is worth noting that a study in NATURE has put forward different ideas and challenged the existing hypotheses about the role of EMT in tumor metastasis. They invented a mesenchymal-specific, Cre-mediated fluorescent marker switch strategy and established a triple transgenic mouse model (Tri-PyMT, MMTV-PyMT/Rosa26-RFP-GFP/Fsp1-Cre), which facilitated breast-to-lung metastasis. As a result, they found that tumor cells maintained their original epithelial phenotype in the primary tumor and they did not activate the mesenchymal-specific FSP1 promoter, but retained their epithelial phenotype during metastasis. More importantly, tumor cells in metastatic lesions were not manifested to have experienced EMT process during metastasis either. Then they injected miR-200-overexpressing Tri-PyMT cells in vivo. To much depression, inhibition of EMT by miR-200 overexpression did not impair the ability of tumor cells to form distant lung metastases. Therefore, they believe that EMT is not required for lung metastasis in breast cancer.14 At the same time, there is another study in NATURE that points out that EMT is not essential in the metastasis of pancreatic cancer.15 Different from the above article, they confirmed their conclusion in the reverse perspective. They would like to check whether EMT inhibition will repress tumor metastasis. Finally, it was found that knocking out twist, snail genes did not inhibit tumor metastasis in vivo at all.
Both research methods are new and reasonable, and they put forward different conclusions from past views. There may be two reasons accounting for this disparity. First, most of the studies about EMT are in vitro experiments, while there is a gap between them and the actual situation of tumor development in vivo. In addition, what both studies point out is that the tumor does not undergo EMT during metastasis, which is not contradictory to previous verdict that EMT promotes tumor metastasis. The EMT process leads to a decrease in cell adhesion, endowing tumor cells features contributing to easier migration. There is no doubt that EMT promotes tumor metastasis, but whether it happens during the complicated process in all metastatic situations is uncertain. That is understandable. After all, tumor metastasis involves so many known and undiscovered mechanisms. What is more, these two researches are studied in mouse, remaining the argument of the potential role of EMT in human cancer metastasis.
Regulation of miRNA on EMT in tumors
Inhibition of EMT via miRNA/ZEB pathway
Oba et al16 revealed 82 candidate miRNAs that have possible interaction with ZEB-2 through analyzing complementary sequences of miRNA and ZEB-2 mRNA 3′ UTR. Then, each miRNA and a luciferase reporter vector containing the 3′ UTR region of the ZEB-2 mRNA were cotransfected into NIH3T3 cells. They found that miR-200, miR-666-5p, and miR-183 had significant effects, decreasing the activity of ZEB-2 more than 20% compared to the control group. The ZEB-2 protein inhibits E-CAD expression, allowing these miRNAs to inhibit tumor EMT. This has been confirmed in their experiments. There are three kinds of stable state of miR-200/ZEB: high miR-200/low ZEB; low miR-200/high ZEB; and moderate miR-200/ZEB, which represent the epithelial, mesenchymal, and mixed states, respectively.17
p53/miRNA/ZEB
It is summarized in a review that miRNA molecules regulated by P-53 include miR-192 family (miR-192, miR-194, miR-215), miR-107, miR-145, miR-200s, and miR-34s (+7).18 Through a comparative study of (CD24−CD44+, stem) HMECS and (non-CD24−CD44+, non-stem) MCF12A cells, it was found that miR-183 and miR-200c were significantly downregulated in stem cells (>2 times). Further studies have revealed that P53 can act on the upstream transcription initiation region of both miR-183 and miR-200c. Chromatin immunoprecipitation analysis confirmed that P53 could bind specifically with their promoter-specific fragment and promote their expression. Besides, P53 was able to regulate EMT in MCF12A cells through miR-200c. P53 knockdown decreased the expression of E-CAD, while N-cadherin, VIM, and ZEB1 were increased. The exogenous expression of P53 gene could reverse the TGF-β-induced EMT.19 This study links the most important genetic alteration in human cancer to the emerging fields of EMT and cancer stem cells, which are of importance for metastasis.20 In ovarian cancer cells, the luciferase reporter assay revealed that mutations in P53 binding region in the miR-200b/a/429 and miR-200c/141 promoters reduced luciferase activity by 40% (P<0.003, t-test).21 Another relevant research indicated overexpression of P53 upregulated miR-145 and inhibited EMT process, with ZEB2, fibronectin, and VIM expression increased but E-CAD decreased in prostate cancer, and inhibit bone metastasis in vivo.22 In prostate cancer, double-negative feedback loop between ZEB2 and miR-145 contributes to PCa progression and metastasis, and might have therapeutic relevance.23
The expression of miR-192 family is also related to upregulation of P53 protein expression. P53-regulated miR-192 family members also repress ZEB2 expression.24 Overexpression of these molecules can inhibit the expression of MDM2 (murine double minute 2) at mRNA and protein levels, thus reducing the inhibitory effect of MDM2 on P53 gene. This autoregulatory loop is a positive feedback loop with the feature of double-negative regulation.25
The role of miRNA/ZEB in head and neck squamous cell carcinoma
EMT in head and neck squamous cell carcinoma (HNSCC) can be inhibited by miR-138, which reduces local invasion and metastases. It mainly involves three mechanisms: 1) direct targeting of VIM inhibits mRNA translation, 2) targeting of ZEB2 and SNAI2 genes to regulate the E-CAD transcription, and 3) directly acting on histone lysine methyltransferase EZH2 (or enhancer of zeste homolog 2), which can inhibit the transcription of downstream E-CAD genes. These points help to promote the inhibitory effect of miR-138 on EMT.26
The role of miRNA/ZEB in breast cancer
MiR-200b-3p/5p was found to upregulate MET-related genes and epithelial-cadherin gene (Cadherin-1) and attenuate cell migration in triple-negative breast cancer (TNBC). Interestingly, there was no obvious decrease of ZEB, and it decreased neoplastic invasion by inhibiting the Rho signaling pathway rather than the classical pathway, miR-200/ZEB/EMT, in this process.27 Kong et al28 proposed that 53BP1 was a tumor suppressor gene in breast cancer, and that its overexpression in MDA-MB-231 cells increased E-CAD content, accompanied by a significant decrease in ZEB1 and increased expression of miR-429 and miR-200b. Therefore, 53BP1 may inhibit EMT of breast cancer cells and metastasis in vitro and in vivo and through the miR-200/ZEB/EMT pathway. Nevertheless, some studies have put forward opposite conclusion. Low level of miR-200 expression was detected in claudin-low breast cancer (CLBC). Re-expression of miR-200c in vitro did not change the expression of ZEB1/2 significantly. Invasion chamber assay showed little diminishment in tumor cell invasion either.20 What is more, another study has shown that miR-141/200 re-expression increased the ability to invade and migrate in human CLBC cell line MDA-MB-231. So the universality of miRNA200s function remains to be further studied.29
In animal experiments, MDA-MB-231 cells (human breast cancer cells) were implanted into the left ventricles of mice,30 with bone metastases occurring 8–12 weeks later and bone metastasis lesions formed. The differential expression of 118 kinds of miRNA (>2 times) was found by analyzing primary tumor cells (231-P) and metastatic tumor cells (231-B). Sixteen kinds of miRNA expression were upregulated; conversely, another 102 kinds of miRNA were downregulated. Specifically, miR-429 was significantly downregulated in 231-B cells. That was consistent with the results of previous studies: miR-429 is known to inhibit the invasion and metastasis of various tumors. Further studies found that overexpression of miR-429 in 231-B cells could significantly reduce their invasive and metastatic activity. Western blot studies showed that ZEB1 and Crk-like protein were significantly reduced by miR-429, and EMT was repressed as well. In addition, miR-486-5P was also found to be significantly downregulated in the 231-B cells, which has been reported recently to inhibit lung cancer metastasis. Also, miR-147 has also been shown to inhibit EMT induced by TGF-β in A549 lung cancer cells via acting on ZEB.31
The role of miRNA/ZEB in lung cancer
A new study discovered Foxf2 (forkhead box F2, a member of transcriptional regulators) as the new targeting molecule of miR-200b/c, which mediated EMT and promoted invasion and migration of lung cancer cells in non-small-cell lung cancer (NSCLC).32 The miR-155 and miR-200c expressions were downregulated in gemcitabine-resistant (GR) cells (HCC827GR), with their target molecules Smad and ZEB1 increased at the same time. EMT process was detected as well.33 Decitabine is able to reverse TGF-β-mediated EMT through inhibition of miR-200 promoter methylation and repression of miR-200/ZEB axis in NSCLC.34 Chen et al35 concluded that GSK-3 was involved in EMT through snail inhibition. In docetaxel-resistant lung adenocarcinoma cell model, miR-451 re-expression inhibited extracellular-signal-regulated kinase (ERK)-dependent GSK-3 phosphorylation, which represented its inactivation via acting on C-MYC, so as to repress snail-mediated EMT.
The role of miRNA/ZEB in pancreatic cancer
In the mechanisms of EMT, ZEB interacts with the C-terminal-binding protein (CtBP) to inhibit expression of miR-200s. Both CtBP and ZEB have interaction sites on the miR-200 family (miR-200a, miR-200b, miR-200c, miR-141, and miR-429). Conversely, miR-200s regulates EMT through ZEB inhibition. Sass et al36 found that overexpression of miR-141 and miR-200c led to a reduced expression of CtBP and ZEB1 in pancreatic cancer cells, and then inhibited the occurrence of EMT. Thus, miR-200a plays the same role in pancreatic cancer cells through ZEB1.37 Moreover, miR-655 can also inhibit invasion and migration of pancreatic and breast cancer cells by acting on ZEB.38
The role of miRNA/ZEB in bladder cancer
It was found in two bladder cancer cell lines that invasion by CRL1749 cells was inhibited by enhancing the expression of miR-141 or miR-200b, but opposite results occurred with HTB9 cells after downregulating miR-141 or miR-200c. Moreover, an increase of miRNA expression in the CRL1749 cells was accompanied by a significant decrease in the activity of MMP-2 and MMP-9, while the opposite was found in HTB9 cells. Because MMPs could promote tumor invasion and metastasis, we anticipated that both MMP and EMT inhibition were significant mechanisms by which miR-200s repressed tumor metastasis.39
Inhibiting EMT via miRNA/SNAIL pathway
The SNAIL/miR-34 family (miR-34a, miR-34b, and miR-34c) forms another system for epithelial and mesenchyme morphogenesis.40 The miR-34 inhibits their translation through base-pairing in conserved region of SNAIL1, SNAIL2, and ZEB1 3′ UTR, and ultimately inhibits the occurrence of EMT. Conversely, SNAIL can directly inhibit the miR-34 promoter to reduce the expression of miR-34. Actually, miR-34 expression is usually downregulated while SNAIL expression increased in tumor cells, which further inhibits the expression of miR-34. Such bidirectional negative regulation mechanism, making each molecule linked more closely and more efficient, has better regulating effect on tumor cell EMT. Noticeably, miR-34 may repress EMT via other mechanism instead of SNAIL. It was confirmed that miR-34a acted on SNAIL to regulate EMT in breast cancer and lung cancer cells, but there was no such regulation in prostate cancer PCa cells, replaced by significant downregulation of lymphoid enhancer-binding factor 1 (LEF-1). Then the expression of E-CAD was increased, while N-CAD expression decreased.41 Therefore, regulating the expression or activity of miR-34a or LEF-1 in prostate cancer cells may play a positive role in cancer treatment.
p53/miRNA/SNAIL
There was a study pointing out that when P53 gene got mutation or lost its function, the miRNA-34 level was reduced. It could not inhibit SNAIL1 at protein level as usual, which promoted tumor cell EMT. Although P53 gene could directly affect cell-cycle regulation, cell apoptosis, and DNA repair pathways, the EMT and metastasis process induced by P53 inactivation completely depended on the expression of SNAIL1, which is common and important in terminal cancer.42
miRNA/SNAIL/ZNF281
There is a molecule closely related to SNAIL/miR-34/EMT regulatory system – ZNF281. ZNF281 is a transcription factor that mediates transcriptional activation or inhibition, regulating tumor cells proliferation, differentiation, and apoptosis. A study about colorectal cancer showed that SNAIL could promote the expression of ZNF281, which is positively correlated with mesenchymal markers such as VIM in breast cancer SKBR3 cells, pancreatic cancer MiaPaCa2 cells, and colorectal carcinoma DLD-1 cells. When they used small interfering RNA (siRNA) to interfere with ZNF281 expression, the EMT induced by SNAIL was significantly weakened.43 C-MYC inducing the occurrence of EMT also depends on ZNF281, and C-MYC cannot bind with ZNF281 promoter, but activates downstream EMT via promoting the expression of SNAIL. It maintains mesenchyme phenotype in CRC cell lines, and promotes tumor invasion and metastasis.43
miRNA/SNAIL/G9a
G9a, as a kind of histone H3 lysine 9 methyltransferase, is a downstream molecule of SNAIL, which can form a complex with DNA methyltransferase under the recruitment of SNAIL, and bring methylation of E-CAD promoter to inhibit its expression. G9a knockdown reduced cell invasion and metastasis in vitro and lung metastasis in vivo of CLBC.44 Liu et al45 have also confirmed that G9a promotes EMT and lymph node metastasis in head and neck cancer cells through such mechanism.
The role of miRNA/SNAIL in liver cancer
The expression of miR-451 is downregulated in liver cancer cells, which mediates the overexpression of C-MYC, thus activating the Erk1/2 signaling pathway. Then this pathway regulates the GSK-3β/SNAIL/E-CAD cascade reaction to promote EMT. In addition, miR-451 also inhibits cellular growth and promotes apoptosis. Therefore, it is likely that a new method for treatment of metastatic liver cancer can be developed by regulating the miR-451/C-MYC/Erk1 and 2 axis.46 Injecting miR-449a into hepatocellular carcinoma (HCC) cells can inhibit its formation of foci of invasion and metastasis in vitro.47 It combined with the 3′ UTR of FOS (FBJ murine osteosarcoma viral oncogene homolog) and MET (pro-oncogene, receptor tyrosine kinase) to reduce their expression, thereby inhibiting the downstream signaling pathways, including Protein Kinase B (AKT) phosphorylation and the GSK-3α/β activity. Then it decreased nuclear SNAIL accumulation and raised the E-CAD level. While the miR-29b/SNAIL axis inhibited EMT in prostate cancer, Yan et al48 found that miR-29b inhibited MMP-2 expression, which contributed to EMT as well in HCC. It is also recognized that fucoidan upregulated miR-29b expression in HCC, in addition to TGF-β receptors and SMAD signaling inhibition. All of these effects contributed to cooperative inhibition of the occurrence of EMT in HCC.
The LET-7 family consists of 12 miRNAs, which are also regulators of cell differentiation, directly taking effect on transcription factors related to oncogene activity or SNAIL expression to inhibit tumor growth. LET-7 expression was decreased in various tumors including lung cancer, colon cancer, and ovarian cancer.40 Chen et al49 found that LET-7g expression was the lowest, and transfection of the LET-7g gene in vitro significantly inhibited the malignant invasion of HCC cells. Mechanistic studies showed that LET-7g re-expression inhibited EMT in HCC cells through blocking the K-Ras/HMGA2A/SNAIL axis. Liver cancer cell line HCCLM3 was transplanted into nude mice to form a tumor. Then LET-7g plasmids were injected into the tumor, after which its volume was significantly reduced. Therefore, LET-7g may serve as an ideal treatment for early HCC patients. In NSCLC and liver cancer, miR-30a promoted E-CAD expression by acting on SNAIL1.50 However, miR-30c was directly targeted to VIM and fibronectin to inhibit tumor metastasis in lung cancer and hepatoma. The fragile histidine triad gene product (FHIT) could upregulate miR-30c, and thus FHIT may provide an effective intervention for early lung cancer and hepatoma.51
The role of miRNA/SNAIL in gastrointestinal tumor
The expression of miR-22 was significantly reduced in clinical gastrointestinal tumors and was tightly associated with poor prognosis. The introduction of miR-22 significantly inhibited gastroenteric cancer cell growth, migration, and invasion in vitro. It targeted mRNA of SNAIL and MMP14 molecules to inhibit tumor progression; meanwhile, experiments in vivo have confirmed that overexpression of miR-22 inhibited tumor growth, peritoneal dissemination, and lung metastasis.52
The role of miRNA/SNAIL in prostatic cancer
The miR-29b and miR-30 could also inhibit the expression of SNAIL. In prostate cancer cells, overexpression of miR-29 induced complete mesenchymal epithelial transition, which greatly depressed the invasiveness of prostate cancer cells.53
Inhibition of EMT via the miRNA/TWIST pathway
The role of miRNA/TWIST in breast cancer
MiR-300 was an obvious type of miRNA that was downregulated after HNSCC cells, and breast cancer cells had undergone EMT. Transducing miR-300 inhibitor in low-metastatic HN-4 and MCF-7 cells could induce EMT, but miR-300 could reduce TWIST expression in highly metastatic HN-12 and MDA-MB-231 cells. Moreover, overexpression of TWIST could weaken the inhibition of miR-300 on EMT, and the complementary region of miR-300 and TWIST 3′ UTR further indicated that miR-300 directly targets TWIST to suppress EMT.54
Similarly, miR-129-5p can also inhibit the expression of EMT in breast cancer cells. But miR-129-5p is also one of the transcriptional regulatory targets of TWIST and SNAIL. Gene chip detection using the luciferase reporter gene assay showed that overexpression of TWIST1 or SNAIL decreased miR-129-5p promoter activity, but knockdown of TWIST1 or SNAIL increased it. Therefore, there are inhibitory effects between miR-129-5p and SNAIL. Downregulation of miR-129-5p in tumor cells indicates an increased expression of TWIST and occurrence of EMT, which results in poor prognosis in patients with breast cancer.55
The expression of miR-33b in breast cancer cells is also decreased, and its ectopic expression could inhibit the proliferation, invasion, and metastasis of tumor stem cells.56 Results of initial screening of miR-33b target genes using online databases, quantitative reverse transcription polymerase chain reaction, and luciferase detection indicate that TWIST1, HMGA2, and SNAIL are downstream targets of miR-33b. MiR-155 can downregulate TWIST1, TCF4 (transcription factor 4), VIM-1, and ZEB2 in mouse breast cancer cells. Experiments in vivo show that it does not affect the growth of primary tumor cells, but inhibits EMT.57
The role of miRNA/TWIST in liver cancer
PDGF-D (platelet-derived growth factor D) plays an important role in EMT and drug resistance through the PDGF-D/miR-106a/TWIST pathway in HCCs. Introducing the miR-106a analog into HCC cells decreases TWIST expression and suppresses invasion and metastasis by GR HCC cells. But PDGF-D mainly inhibits the expression of miR-106a. In contrast to prostate cancer cells, which develop EMT with inhibition by miR-200b, there are no changes of miR-200b level in the HCC cells.58 Inactivation of the PDGF-D/miR-106a/TWIST pathway or direct activation of miR-106a expression may be a novel strategy for the treatment of HCC.
The role of miRNA/TWIST in prostatic cancer
In in vitro experiments, miR-186-TWIST axis inhibited EMT initiation in prostate cancer (M12 cell line), accompanied by decreased cell invasion and migration abilities. Moreover, Kaplan–Meier survival analysis revealed that patients with high level of miR-186 had a higher survival rate than those with low level of miR-186 (P=0.028).59
In summary, in different types of tumors, there may be different miRNA molecules that inhibit EMT, and these miRNAs can directly or indirectly regulate multiple target molecules; multiple miRNAs can regulate EMT by the same molecular pathway. The list of miRNAs and their target molecules and the tumor types that they inhibit to affect the occurrence of EMT, as elucidated in recent years, are provided in Table 1.
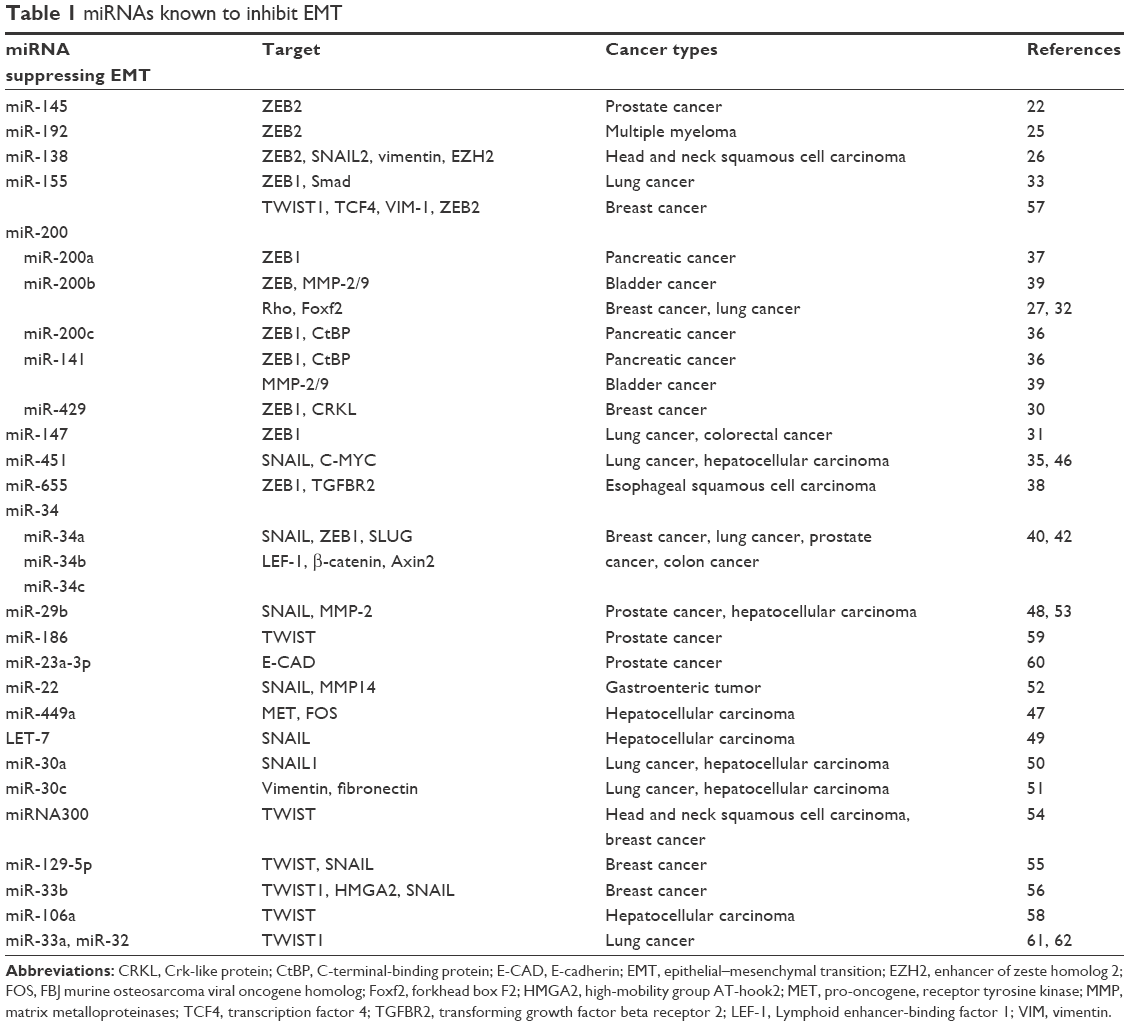 | Table 1 miRNAs known to inhibit EMT |
miRNA promoting EMT
Although most of the miRNA molecules such as miR-200 and miR-34 inhibit invasion and metastatic process of neoplastic cells by depressing EMT-related transcription factors, there are still some miRNAs that exert the opposite effect.
miR-9 promoting EMT
Song et al63 reported that miR-9 could bind with E-CAD mRNA 3′ UTR. A stable transfection of miR-9 on esophageal adenocarcinoma cell lines HKESC1 and KYSE410 decreased E-CAD expression at both RNA and protein levels. It promoted EMT in colon cancer cells as well, and the mechanism is related to the PROX1 (Prospero homeobox 1) gene.64 In liver cancer, miR-9 promoted EMT invasion and metastasis through KLF17 (Krüppel-like factor 17).65
miR-27a promoting EMT
Xu et al66 found that SPFFbxo45 (the atypical E3 ubiquitin ligase complex SKp1-Pam-Fbxo45) could downregulate core transcription factors of EMT, such as ZEB1/2, SNAIL, and TWIST1, through the SPRY domain (ZEB2) and F-box domain (SNAIL1/2, TWIST2), respectively. Additionally, miR-27a could inhibit the expression of Fbxo45 through direct interaction, to maintain the phenotype of EMT. Therefore, inhibiting miR-27a or raising E3 activity may provide new methods to treat cancer and other diseases.66
miR-221/miR-373 promoting EMT in breast cancer
MiR-373 targets the 3′ UTR of TXNIP (thioredoxin-interacting protein) mRNA to reduce the expression of TXNIP, and then increases ROS (reactive oxygen species) and activates the HIF-1α (hypoxia-inducible factor)-TWIST signal axis, which will promote EMT in the end. The TWIST binding with miR-373 promoter sequences could stimulate miR-373 expression and form a positive feedback loop to strengthen these effects. Finally, the presence of miR-373 worsens prognosis in breast cancer patients.67 Overexpression of miR-221 in tumor cells also promotes EMT.68 As a proto-oncogene-related miRNA, miR-221 plays an important role in the proliferation and migration of pancreatic cancer, mainly via inhibiting PTEN, P27kip1, P57kip2, PUMA, and other tumor suppressors.69 Peptide nucleic acid (PNA) is an analog of DNA, and the sugar-phosphate backbone is replaced by N-(2-aminoethyl) glycine. Piva et al70 found that experimental units with invasive effects on PNA targeting miR-221 could significantly inhibit the invasive ability of breast cancer cell lines in vitro. In vivo experiments also verified that PNA-anti-miR-21 significantly inhibited tumor growth.
miR-214/miR-346 promoting EMT in lung cancer
In A549 and NCI-H1650 cell lines, ectopic expression of miR-214 promoted the development of EMT in a lung adenocarcinoma cell line.71 The XPC (xeroderma pigmentosum complementation group C) expression is positively correlated with the outcome of NSCLC patients. However, it was found that miR-346 promoted the invasion and migration of NSCLC cells, which directly targeted XPC and then activated the ERK/SNAIL pathway, resulting in decreased expression of E-CAD.72
miR-21/miR-181a promoting EMT in liver cancer
In liver cancer, miR-21 inhibited PTEN and human sulfate-1 (sulfatase-1, hSulf-1) expression, resulting in activation of the AKT/ERK pathway and;73 a similar effect was seen in cholangiocarcinoma,74 and clear cell renal carcinoma.75 In mice with experimental NSCLC, treatment for 35 days with oral silibinin significantly reversed high miR-21/low miR-200 levels in the neoplastic cells, thereby inhibiting EMT via decreasing downstream ZEB and N-CAD expression.76 Applying bioinformatic analysis, Chang et al found 34 miRNAs expression upregulated in HCC, with miR-181a the most marked. To make a further study, 55 HCC tumor tissue samples and the adjacent normal tissues were selected. The qPCR assay verified increased miR-181a expression in tumor tissues, and the higher the stage of tumor, node, and metastasis, the higher level of miR-181a. MiR-181a knockdown reduced SNAIL, N-CAD, and MMP2/9, while increased E-CAD, accompanied by weaker invasiveness. It was proved that miR-181a promoted the occurrence of tumor EMT and promoted the invasion of HCC cells. In addition, they observed a decrease in P-AKT levels during the process. Therefore, it can be inferred that miR-181a regulates EMT process through phosphoinositide 3-kinase (PI3K)/AKT. Double luciferase assay revealed that PTEN was the target molecule of miR-181a.77
In conclusion, miRNAs promoting EMT significantly contribute to the invasiveness and metastatic tendency of neoplasms. They are potential targets of tumor therapy (Table 2).
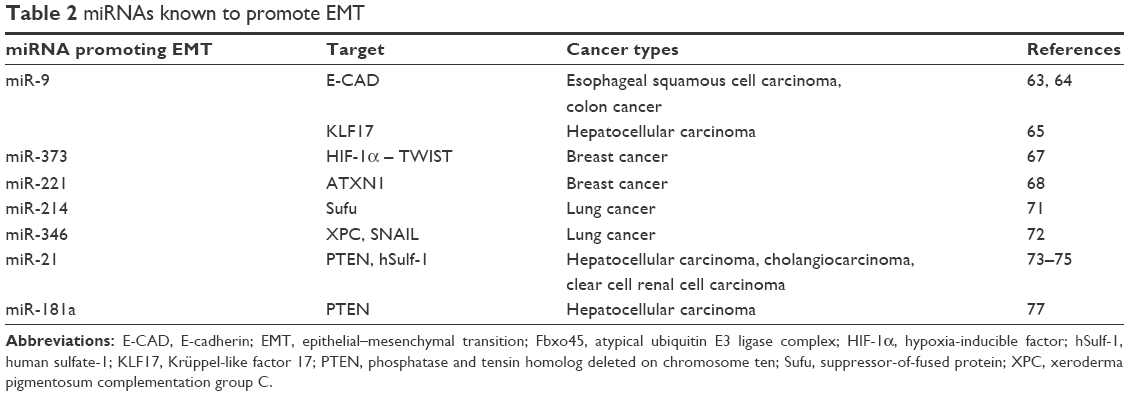 | Table 2 miRNAs known to promote EMT |
Effects of several common natural products on tumor EMT
The general mechanisms of natural products in regulating EMT in tumor cell
There are some general mechanisms of natural drugs in regulating EMT in tumor cells. In the analysis of biopsies from the tumors of 178 gastric cancer patients, Yadav et al78 found that TGF-β1 expression was negatively correlated with E-CAD (r=−0.452, P<0.001), and the Kaplan–Meier survival curves showed that prognosis of patients with low expression of E-CAD was poorer overall (P<0.001). Notch, Wnt, Hedgehog (Hh), and TGF-β are common signal pathways that can activate TWIST, SNAIL, SLUG, ZEB, and other EMT-related transcription factors, mediating the conversion of epithelial cells into mobile mesenchymal cells. This is related to tumor cell invasion and migration.79 In addition, the Notch pathway interacts with the Hh pathway closely. Specifically, the Notch pathway can inhibit Hh through Hes-1-mediated Gli-1 transcription inhibition. Abnormal activation of Notch increased the protein level of Gli2, and the Hh signaling pathway’s role in promoting monocilia choroid plexus glial cells depends on the function of the Notch signaling pathway. Hh pathway inactivation also can inhibit the Notch pathway, and both mediate mesenchymal epithelial transition.80–84 The nuclear factor kappa B (NF-kB) signaling pathway is also involved in EMT, by activating SNAIL, SLUG, ZEB1/2, and TWIST, and upregulating MMPs, fibronectin, VIM, and other mediators in mesenchymal tissue.85 In addition, the PI3K/Akt/mammalian target of rapamycin (mTOR) pathway is an important mechanism of tumor EMT, which can be regulated by botanicals (plant-derived medicinal agents). On the other hand, some botanical drugs directly regulate the EMT-related miRNAs to inhibit tumor invasion and migration. Recent researches have revealed Fra-1(a member of activating protein-1 transcription factor complex) as a target of various natural products and it can be activated by STAT3. Both of them can be regulated by natural products such as curcumin, resveratrol, and luteolin in tumor EMT progression.86
Curcumin
Curcumin inhibits hepatocyte growth factor-induced EMT in lung cancer cells (A549, PC9), restoring E-CAD and restraining N-CAD expression in a concentration-dependent manner, mainly through regulating the c-MET/PI3K/Akt/mTOR pathway. Thus, curcumin is thought to inhibit invasion and migration of lung cancer cells. Experiments in vivo also confirmed that injection of exogenous curcumin can repress neoplastic growth in tumor-bearing mice. Immunohistochemistry showed increased expression of E-CAD, while VIM, vascular endothelial growth factor (VEGF), and CD34 expressions were decreased, suggesting that curcumin may inhibit EMT and VEGF-mediated angiogenesis.87
Curcumin can inhibit migration of breast cancer stem cells in vitro through inhibiting the translocation of β-catenin into the nucleus as well as the EMT-related transcription factor SLUG.88 PAC (u4-hydroxy-3-methoxybenzylidene-N-methyl-4-piperidone) is a synthetic curcumin analog, with higher bioavailability. It can inhibit Erk1, AKT, and TWIST in breast cancer cells to control EMT in vitro and in vivo, thus inhibiting cell migration and invasion. Notably, its effect in estrogen receptor (ER)-negative cells (MDA-MB-231) is stronger than in ER-positive cells (MCF-7), which is of great significance for improving chemotherapeutic efficacy and prognosis of TNBC cells.89 Bakiri et al90 proposed that ectopic expression of Fra-1 induced mesenchymal phenotype transformation in completely polarized nontumorigenic mouse mammary epithelial EpH4 cells, with increased expression of TGF-β1, ZEB1, ZEB2, and slug. At the genetic level, Fra-1 could combine with the promoter of TGF-β1, ZEB2, and the conserved region in the first intron of ZEB1. In their experiments, they found that Fra-1-activating P3 fragment in ZEB2 promoter depended on three AP-1 binding sites, suggesting that Fra-1/AP-1 can regulate EMT-related transcription factor ZEB1 and ZEB2 expression through binding with their genome regulatory region. Previously, two Fra-1/AP-1 binding regions have been reported in human breast and CRC ZEB1 gene.91 And curcumin could inhibit Fra-1/AP-1-mediated gene expression by inhibiting the binding of AP-1 to its cognate motif.92 Liu et al93 also found that curcumin could effectively reduce the low concentration of benzidine-induced EMT in human immortalized urothelial cell (SV-HUC-1 cell line) by inhibiting the ERK5/AP-1 pathway. Therefore, curcumin may inhibit the occurrence of EMT by repressing the expression of EMT-related transcription factors mediated by AP-1.
In cholangiocarcinoma, curcumin (EF24) inhibited NF-kB transcriptional activity in a concentration- and time-dependent manner. Moreover, it inhibited EMT, broke the cytoskeleton, and weakened the invasiveness and migration of tumor cells.94 It was also found that diphenyl difluoroketone (EF24), a curcumin analog, inhibited EMT by upregulating miR-33b in cultured melanoma cells (in both the Lu1205 and A375 cell lines). The luciferase reporter gene showed that miR-33b could bind with HMG 3′ UTR to repress its expression, and also regulated the formation of HMGA-2-dependent dynamic fibers and the activation of FAK, Src, and RhoA. All these effects contributed to inhibiting tumor metastasis.95 A recent study indicated that EF24 inhibited phosphorylation of Src in HCC cells, then repressed tumor invasion and metastasis.96
Curcumin can also reverse EMT in the pancreatic cancer cell line PANC-1 and inhibit the invasion and migration of tumor cells. This may be related to the inhibition of the Shh (sonic hedgehog)-GLI1 signaling pathway.97 It was also found to block EMT progression in PANC-1 cell by inhibiting the PI3K/Akt/NF-kB pathway.98 The antitumor mechanisms of curcumin in pancreatic cancer included upregulation of tumor suppressors, such as miR-26a, miR-101, miR-146a, miR-200b, miR-200c, and the LET-7 family, as well as downregulation of miR-21, which could contribute to EMT. It could induce cell apoptosis and inhibit cell proliferation, migration, and invasion.99–103 The same effects were confirmed in vivo.97,100 Isoflavone mixtures (G2535), synthetic 3,3′-bis (indolyl) methanes, or synthetic curcumin analogs all can inhibit miR-221-related proliferation and migration of pancreatic tumor cells.69
Curcumin may inhibit the Wnt signaling pathway and upregulate NKD2 expression to suppress EMT in colon cancer. In the detection of Wnt- and EMT-related molecular events, it was found that curcumin could inhibit TCF and β-catenin expression in the Wnt signaling pathway and promote Axin (the Wnt negative regulator) expression in a concentration-dependent manner. In addition, it has been shown that transfection of CXCR4 plasmid or NKD2 siRNA to SW620 cells reversed the inhibitory effects of curcumin on the Wnt signaling pathway and EMT, indicating that the mechanism of curcumin here was related to these two molecules as well.104 Toden et al105 found the same results and revealed that curcumin could downregulate the expression of ZEB1 and increase EMT inhibitory miR-200b, C, 141, 429, 101. More importantly, curcumin can induce epithelial expression in 5-fluoride-resistant cells to inhibit EMT, so as to ameliorate the drug resistance. This is of great value for improved therapeutic efficiency by curcumin. Montgomery et al106 reported that curcumin and silymarin, two plant-derived agents used in colon cancer, can coordinate the antitumor effect. Treatment with curcumin alone can make the activity of cysteinyl aspartic enzyme 3/7 increase by a factor of 3, whereas in combination therapy with silymarin, the activity can be increased by a factor of 5.
Genistein
Genistein is an isoflavone angiogenesis inhibitor and a phytoestrogen. In laryngeal carcinoma HEP-2 cells, genistein showed inhibitory effects on EMT, and significantly reduced cell invasion and migration in vitro.107 The genistein treatment regimen produced the same findings in ASPC-1 pancreatic cancer cells, and mainly promoted miR-200b expression inhibited by the Notch-1 signaling pathway.108 Nevertheless, it can downregulate miR-223 expression in GR pancreatic cancer cells and this process can reverse EMT in GR cells, sharing the same effect with miR-223 inhibitor. Both of them can inhibit invasion and metastasis of GR cells and restore their sensitivity to gemcitabine.109 Prostate cancer cells (ARCaP-E/ARCaP-M cells) are also sensitive to genistein.110 It raised tumor suppressors such as miR-145 and miR-29a through methylation of the promoter, while miR-222, miR-221, and miR-151 (which stimulate tumor growth) were inhibited.111 The inhibition of EMT was attributable to downregulation of the TGF-β pathway in ovarian cancer.112 GEN-27, a new synthetic isoflavone derived from genistein, can inhibit the nuclear translocation of NF-kB in THP-1 and HCT116 colon cancer cell lines. Wang et al113 found that 100 nM Guqu trichostatin A combined with genistein significantly enhanced the antitumor effect of genistein; the potential mechanism may be related to AKT pathway inhibition. This kind of combined chemotherapy is an effective strategy for cancer treatment.107
Resveratrol
Resveratrol (3,5,4′-trihydroxy-trans-stilbene) is isolated from red grape skins and can inhibit ZEB2 expression directly through epigenetic regulation in oral cancer.114
Lipopolysaccharide (LPS)-mediated EMT is closely related to inflammation-induced melanoma metastasis; Chen et al115 found that resveratrol could inhibit LPS-induced tumor cell migration, accompanied by decreased EMT markers. The cellular mechanisms lay in weakened NF-kB nuclear localization and transcriptional activity.
Breast cancer cell EMT in MDA-MB-231 cells and MCF-7 cells is significantly inhibited by 3,6-dihydroxyflavone, by downregulating TWIST, SLUG, SNAIL, and N-CAD expression while decreasing E-CAD. As a result, tumor invasion, migration, and stem cell formation were inhibited. Further studies showed that Notch1 inhibition and miR-34a upregulation were involved.116 Nevertheless, in tamoxifen-resistant MCF-7TR breast cancer cells, resveratrol reversed EMT by reducing endogenous TGF-β and inhibiting SMAD phosphorylation.117
Oxidative stress and ROS induced by Klebsiella pneumoniae infection can lead to EMT in A549 cells and increase the expression of transcription factors, including TWIST, SNAIL, and ZEB (P<0.01). Nevertheless, the antioxidant effects of resveratrol inhibited these processes, and reduced tumor cell invasion and migration.118 In osteosarcoma cells, resveratrol reduced hypoxia-induced EMT through downregulation of HIF-1 protein expression.119
Resveratrol in colon cancer LoVo cells mainly suppressed SNAIL expression mediated by the TGF-β/SMAD signaling pathway.120 On the other hand, Buhrmann et al121 found that resveratrol downregulated nuclear localization, phosphorylation, and acetylation of NF-kB in colon cancer HCT cells and showed epithelium induction in colon cancer HCT116 cells. These findings point to important mechanisms by which resveratrol can inhibit tumor invasion and migration.
The findings for prostatic carcinoma seem different to some degree, in that in these studies resveratrol directly inhibited the level of ERK-1/2, AKT protein, androgen, and insulin-like growth factor-1 receptors, thus increasing apoptosis and reducing proliferation and migration. In addition, resveratrol reduced the levels of several oncogene miRNAs including miR-17, miR-18, miR-20a, miR-20b, miR-926, miR-106b, miR-106a, and miR-7. In contrast, resveratrol increased tumor suppressors including miR-149, miR-150, miR-152, and miR-654-5p.122 Treatment with 100 μM resveratrol in cervical adenocarcinoma Hela cells could inhibit STAT3, Notch, and Wnt activation.123 Kotha et al124 first identified Src-STAT3 signaling as a target of resveratrol in malignant cells. As stated above, STAT3-Fra1 inhibition can exert a negative effect on EMT, which may be another mechanism of resveratrol’s function.
Catechin
Green tea catechins are water-soluble polyphenols, the most potent and abundant of which is EGCG (epigallocatechin-3-gallate). Catechins have been studied for possible use in cancer prevention and treatment. In oral squamous cell carcinomas (ALDH1+ stem cells), decreased miR-204 expression correlated with the stem characteristics and lymph node metastasis. However, EGCG treatment could improve the expression of miR-204 and inhibit tumor-related EMT processes. It was also shown to restrict tumor growth in vivo.125 Through its regulation on EMT and anti-angiogenesis effects, EGCG inhibited the invasion and migration of breast cancer cells.126 In addition, EGCG competed for binding sites for ATP in the PI3K/mTOR active region and then blocked the PI3K/AKT/mTOR pathway.127 Another study showed that EGCG mediated HMG-box transcription factor 1 (HBP1), which is an inhibitory factor, blocking the Wnt/β-catenin signaling pathway, and inhibiting the proliferation and migration of breast cancer cells.128 TGF-β/Smad pathway plays an important role in EMT process. Ko et al claimed that EGCG inhibited acetylation of Smad2 and Smad3 by p300/CBP during TGF-β1-induced EMT and reversed the upregulation of EMT-associated genes.129 Activation of the MAPK family member Erk5 was involved in gastric cancer cell EMT mediated by tobacco. Lu et al fed 50 or 100 ng/mL EGCG to mice that were exposed to cigarette smoke, and found EGCG treatment reversed EMT via inhibiting Erk5.130 In HCT116 colon cancer cells, EGCG inhibited stem cell mass formation. Moreover, EGCG in 5FUR HCT116 and SW480 cells also inhibited the Notch pathway, which was closely related to tumor cell self-renewal, and downregulated CD44, CD133, Bmi1, and Notch1, which represented tumor stem characteristics. In addition, EGCG treatment increased miR-34a, miR-145, and miR-200c in colon cancer cells. These tumor-suppressing molecules may be one mechanism by which EGCG inhibits colon cancer EMT processes, invasion, migration, and chemoresistance.131
Luteolin
Luteolin is a natural flavonoid compound with anticancer properties. Besides TGF-β1/Smad pathway, Chen et al132 have verified that TGF-β induced EMT via PI3K/Akt/NF-κB/SNAIL pathway in lung cancer A549 cells, and luteolin could inhibit this pathway and then repressed migration of A549 cells. Hypoxia can significantly reduce the expression of E-CAD but increase N-CAD, which indicates an EMT progression. However, luteolin treatment inhibited this hypoxia-inducible EMT through β-3 integrin/FAK signaling depression in malignant melanoma and NSCLC in vivo and in vitro.133,134 Huang et al found that luteolin inhibited the expression of MMP2, MMP-7, and MMP-9 in PANC-1 and SW1990 cell lines in a concentration-dependent manner. IL-6 treatment increased the activity of STAT3, which could be blocked by luteolin.135
Others
Cucurbitacin B reduced Wnt/beta-catenin levels, inhibited neoplastic cell migration, and angiogenesis, and reversed TGF-β-induced EMT in NSCLC.136 It also reduced mesenchymal molecular expression in a concentration-dependent manner.136 When tested in human HCC cells PLC and Bel7402, apigenin inhibited EMT and increased cell adhesion by reducing NF-kB and SNAIL expression, and then inhibited the proliferation, invasion, and migration of tumor cells in a concentration-dependent manner.137
Mangostin-encapsulated PLGA nanoparticles also inhibited EMT in pancreatic cancer cell lines AsPC-1 and PANC-1, manifesting decreased expression of N-CAD and SLUG, as well as increased E-CAD expression. Simultaneously, cellular proliferation and migration were attenuated. The underlying mechanism was mainly attributed to inhibition of the Shh pathway.138 It was shown that tripterine in ulcerative-colitis-associated colon cancer downregulated SNAIL, VIM, and N-CAD, while raising E-CAD expression. At the same time, NF-kB activity was inhibited, which may be one of the mechanisms of EMT inhibition.139
Osthole is a bioactive coumarin extracted from medicinal plants. It can block EMT in androgen-independent prostate cancers and inhibits tumor invasion and metastasis. On the one hand, it reduces SNAIL production and targets the E-CAD promoter to inhibit EMT. On the other hand, it can significantly inhibit the production of miR-23a-3p, which is able to combine with E-CAD 3′ UTR. Therefore, osthole inhibits invasion and metastasis by prostate cancer.60
From what we have stated above, we summarize the common natural products and their functions in detail as in Table 3.
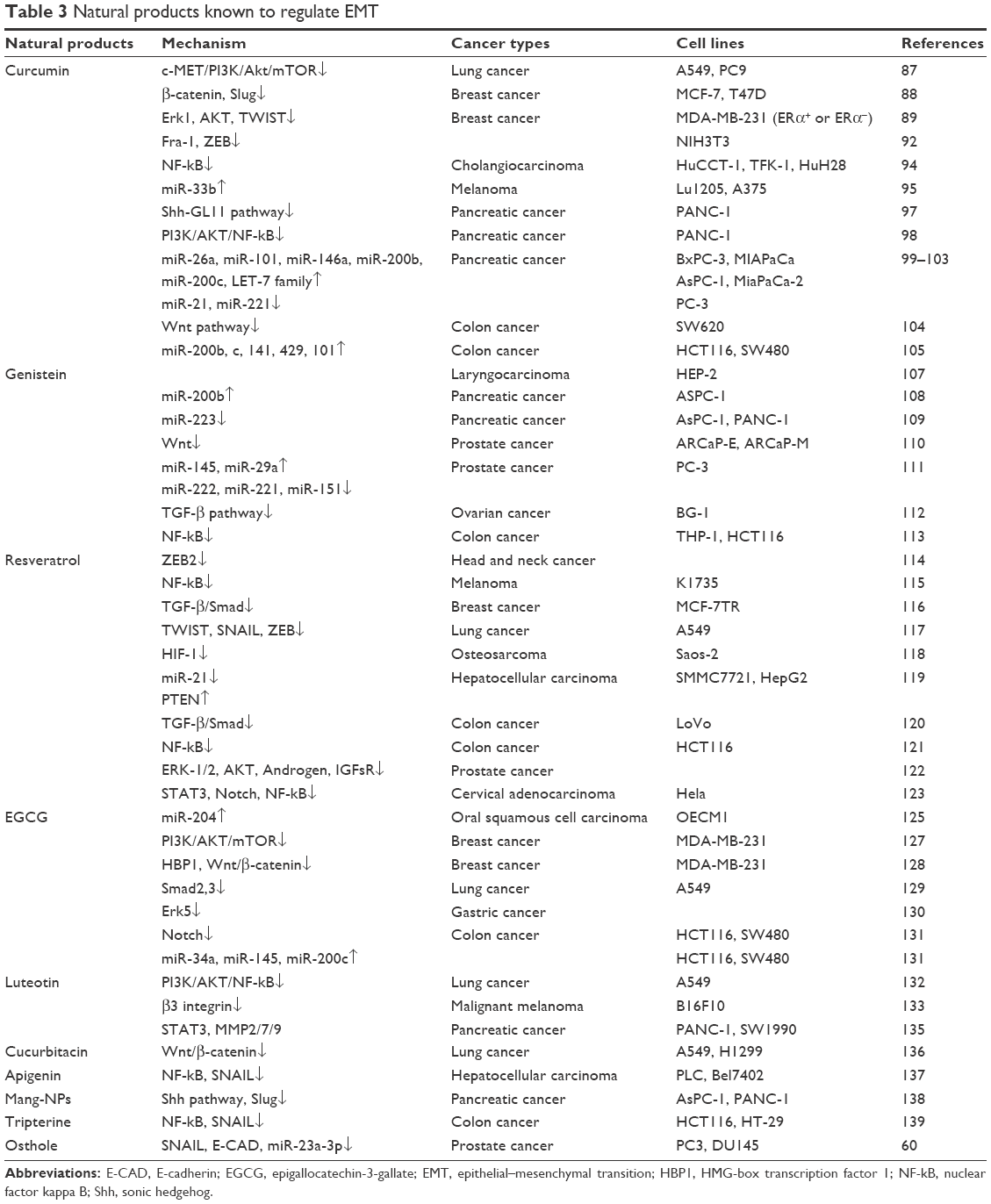 | Table 3 Natural products known to regulate EMT |
Conclusions and perspectives
An increasing number of studies into EMT have revealed its association with tumor invasion, metastasis, and drug resistance. Accordingly, some new strategies for tumor treatment aimed at EMT are also being developed. This may prove to be an important new direction for further research. Studies on miRNA have gradually revealed the key functions of noncoding RNAs on regulating human gene expression. Abnormal expression of these factors is intimately related to tumor development. EMT-inhibiting miRNAs often have the effect of inhibiting tumor invasion and metastasis, but are generally downregulated in tumors. Conversely, EMT-inducing miRNAs exhibit the opposite effect. Although a small proportion of miRNAs have been identified through biochemical or bioinformatics methodologies, the current state of knowledge about these agents is far from satisfactory. Further studies are needed to explain various biological functions of miRNAs in more detail. It would be good to develop a comprehensive knowledge of this system, which is of significance for tumor diagnosis and treatment, and of the appropriate biomarkers. In addition, natural products have long been recognized as valuable resources and starting compounds for the development of novel drugs, following the model of the many existing agents. 3-Hydroxy-3-methylglutaryl coenzyme A reductase inhibitors (statins) and paclitaxel are excellent examples of medications derived from natural products. More and more research has revealed that tumor EMT can be regulated by natural products. Moreover, it has been shown that combined use of these natural products with chemotherapy significantly improves the therapeutic effects of the treatments. Isolating and purifying specific pharmacological components from the known assortment of traditional medicinal and natural products is still the focus of relevant research. There is little doubt that this strategy is of great clinical relevance, suggesting that we can invest more effort in this work to achieve breakthroughs in tumor treatment.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No 81200069, No 31660287); the National natural science foundation of Jiangxi province of China (No 20161BAB205204); Science and technology plan of education department of Jiangxi province of China (No 150217); the postgraduates innovation special fund project of Nanchang University of China (No cx2016356).The first four items belong to Li-Xia Xiong and the last one belongs to Xiang-Tong Lu.
Disclosure
The authors report no conflicts of interest in this work.
References
Wendt MK, Allington TM, Schiemann WP. Mechanisms of the epithelial-mesenchymal transition by TGF-beta. Future Oncol. 2009;5(8):1145–1168. | ||
Bedi U, Mishra VK, Wasilewski D, Scheel C, Johnsen SA. Epigenetic plasticity: a central regulator of epithelial-to-mesenchymal transition in cancer. Oncotarget. 2014;5(8):2016–2029. | ||
Subramanyam D, Blelloch R. From microRNAs to targets: pathway discovery in cell fate transitions. Curr Opin Genet Dev. 2011;21(4):498–503. | ||
Lamouille S, Subramanyam D, Blelloch R, Derynck R. Regulation of epithelial-mesenchymal and mesenchymal-epithelial transitions by microRNAs. Curr Opin Cell Biol. 2013;25(2):200–207. | ||
Cragg GM, Newman DJ. Natural products: a continuing source of novel drug leads. Biochim Biophys Acta. 2013;1830(6):3670–3695. | ||
Luo M, Brooks M, Wicha MS. Epithelial-mesenchymal plasticity of breast cancer stem cells: implications for metastasis and therapeutic resistance. Curr Pharm Des. 2015;21(10):1301–1310. | ||
Nantajit D, Lin D, Li JJ. The network of epithelial-mesenchymal transition: potential new targets for tumor resistance. J Cancer Res Clin Oncol. 2015;141(10):1697–1713. | ||
Tsai JH, Yang J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013;27(20):2192–2206. | ||
Jolly MK, Boareto M, Huang B, et al. Implications of the hybrid epithelial/mesenchymal phenotype in metastasis. Front Oncol. 2015;5:155. | ||
Liu H, Ren G, Wang T, et al. Aberrantly expressed Fra-1 by IL-6/STAT3 transactivation promotes colorectal cancer aggressiveness through epithelial-mesenchymal transition. Carcinogenesis. 2015;36(4):459–468. | ||
Waldmeier L, Meyer-Schaller N, Diepenbruck M, Christofori G. Py2T murine breast cancer cells, a versatile model of TGFbeta-induced EMT in vitro and in vivo. PLoS One. 2012;7(11):e48651. | ||
Drake JM, Strohbehn G, Bair TB, Moreland JG, Henry MD. ZEB1 enhances transendothelial migration and represses the epithelial phenotype of prostate cancer cells. Mol Biol Cell. 2009;20(8):2207–2217. | ||
Tan TZ, Miow QH, Miki Y, et al. Epithelial-mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO Mol Med. 2014;6(10):1279–1293. | ||
Fischer KR, Durrans A, Lee S, et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. 2015;527(7579):472–476. | ||
Zheng X, Carstens JL, Kim J, et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527(7579):525–530. | ||
Oba S, Mizutani T, Suzuki E, et al. A useful method of identifying of miRNAs which can down-regulate Zeb-2. BMC Res Notes. 2013;6:470. | ||
Lu M, Jolly MK, Levine H, Onuchic JN, Ben-Jacob E. MicroRNA-based regulation of epithelial-hybrid-mesenchymal fate determination. Proc Natl Acad Sci U S A. 2013;110(45):18144–18149. | ||
Lin CP, Choi YJ, Hicks GG, He L. The emerging functions of the p53-miRNA network in stem cell biology. Cell Cycle. 2012;11(11):2063–2072. | ||
Chang CJ, Chao CH, Xia W, et al. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat Cell Biol. 2011;13(3):317–323. | ||
Jones R, Watson K, Bruce A, Nersesian S, Kitz J, Moorehead R. Re-expression of miR-200c suppresses proliferation, colony formation and in vivo tumor growth of murine claudin-low mammary tumor cells. Oncotarget. 2017;8(14):23727–23749. | ||
Knouf EC, Garg K, Arroyo JD, et al. An integrative genomic approach identifies p73 and p63 as activators of miR-200 microRNA family transcription. Nucleic Acids Res. 2012;40(2):499–510. | ||
Suh SO, Chen Y, Zaman MS, et al. MicroRNA-145 is regulated by DNA methylation and p53 gene mutation in prostate cancer. Carcinogenesis. 2011;32(5):772–778. | ||
Sekhon K, Bucay N, Majid S, Dahiya R, Saini S. MicroRNAs and epithelial-mesenchymal transition in prostate cancer. Oncotarget. 2016;7(41):67597–67611. | ||
Kim T, Veronese A, Pichiorri F, et al. p53 regulates epithelial-mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J Exp Med. 2011;208(5):875–883. | ||
Pichiorri F, Suh SS, Rocci A, et al. Downregulation of p53-inducible microRNAs 192, 194, and 215 impairs the p53/MDM2 autoregulatory loop in multiple myeloma development. Cancer Cell. 2010;18(4):367–381. | ||
Jin Y, Chen D, Cabay RJ, Wang A, Crowe DL, Zhou X. Role of microRNA-138 as a potential tumor suppressor in head and neck squamous cell carcinoma. Int Rev Cell Mol Biol. 2013;303:357–385. | ||
Rhodes LV, Martin EC, Segar HC, et al. Dual regulation by microRNA-200b-3p and microRNA-200b-5p in the inhibition of epithelial-to-mesenchymal transition in triple-negative breast cancer. Oncotarget. 2015;6(18):16638–16652. | ||
Kong X, Ding X, Li X, Gao S, Yang Q. 53BP1 suppresses epithelial-mesenchymal transition by downregulating ZEB1 through microRNA-200b/429 in breast cancer. Cancer Sci. 2015;106(8):982–989. | ||
Choi SK, Kim HS, Jin T, Hwang EH, Jung M, Moon WK. Overexpression of the miR-141/200c cluster promotes the migratory and invasive ability of triple-negative breast cancer cells through the activation of the FAK and PI3K/AKT signaling pathways by secreting VEGF-A. BMC Cancer. 2016;16:570. | ||
Ye ZB, Ma G, Zhao YH, et al. miR-429 inhibits migration and invasion of breast cancer cells in vitro. Int J Oncol. 2015;46(2):531–538. | ||
Lee CG, McCarthy S, Gruidl M, Timme C, Yeatman TJ. MicroRNA-147 induces a mesenchymal-to-epithelial transition (MET) and reverses EGFR inhibitor resistance. PLoS One. 2014;9(1):e84597. | ||
Kundu ST, Byers LA, Peng DH, et al. The miR-200 family and the miR-183~96~182 cluster target Foxf2 to inhibit invasion and metastasis in lung cancers. Oncogene. 2016;35(2):173–186. | ||
Narita M, Shimura E, Nagasawa A, et al. Chronic treatment of non-small-cell lung cancer cells with gefitinib leads to an epigenetic loss of epithelial properties associated with reductions in microRNA-155 and -200c. PLoS One. 2017;12(2):e0172115. | ||
Zhang N, Liu Y, Wang Y, Zhao M, Tu L, Luo F. Decitabine reverses TGF-beta1-induced epithelial-mesenchymal transition in non-small-cell lung cancer by regulating miR-200/ZEB axis. Drug Des Devel Ther. 2017;11:969–983. | ||
Chen D, Huang J, Zhang K, et al. MicroRNA-451 induces epithelial-mesenchymal transition in docetaxel-resistant lung adenocarcinoma cells by targeting proto-oncogene c-Myc. Eur J Cancer. 2014;50(17):3050–3067. | ||
Sass S, Dietmann S, Burk UC, et al. MicroRNAs coordinately regulate protein complexes. BMC Syst Biol. 2011;5:136. | ||
Lu Y, Lu J, Li X, et al. MiR-200a inhibits epithelial-mesenchymal transition of pancreatic cancer stem cell. BMC Cancer. 2014;14:85. | ||
Harazono Y, Muramatsu T, Endo H, et al. miR-655 is an EMT-suppressive microRNA targeting ZEB1 and TGFBR2. PLoS One. 2013;8(5):e62757. | ||
Liu W, Qi L, Lv H, et al. MiRNA-141 and miRNA-200b are closely related to invasive ability and considered as decision-making biomarkers for the extent of PLND during cystectomy. BMC Cancer. 2015;15:92. | ||
Diaz-Lopez A, Moreno-Bueno G, Cano A. Role of microRNA in epithelial to mesenchymal transition and metastasis and clinical perspectives. Cancer Manag Res. 2014;6:205–216. | ||
Liang J, Li Y, Daniels G, et al. LEF1 targeting EMT in prostate cancer invasion is regulated by miR-34a. Mol Cancer Res. 2015;13(4):681–688. | ||
Kim NH, Kim HS, Li XY, et al. A p53/miRNA-34 axis regulates Snail1-dependent cancer cell epithelial-mesenchymal transition. J Cell Biol. 2011;195(3):417–433. | ||
Hahn S, Jackstadt R, Siemens H, Hunten S, Hermeking H. SNAIL and miR-34a feed-forward regulation of ZNF281/ZBP99 promotes epithelial-mesenchymal transition. EMBO J. 2013;32(23):3079–3095. | ||
Dong C, Wu Y, Yao J, et al. G9a interacts with Snail and is critical for Snail-mediated E-cadherin repression in human breast cancer. J Clin Invest. 2012;122(4):1469–1486. | ||
Liu S, Ye D, Guo W, et al. G9a is essential for EMT-mediated metastasis and maintenance of cancer stem cell-like characters in head and neck squamous cell carcinoma. Oncotarget. 2015;6(9):6887–6901. | ||
Huang JY, Zhang K, Chen DQ, et al. MicroRNA-451: epithelial-mesenchymal transition inhibitor and prognostic biomarker of hepatocelluar carcinoma. Oncotarget. 2015;6(21):18613–18630. | ||
Chen SP, Liu BX, Xu J, et al. MiR-449a suppresses the epithelial-mesenchymal transition and metastasis of hepatocellular carcinoma by multiple targets. BMC Cancer. 2015;15:706. | ||
Yan MD, Yao CJ, Chow JM, et al. Fucoidan elevates microRNA-29b to regulate DNMT3B-MTSS1 axis and inhibit EMT in human hepatocellular carcinoma cells. Mar Drugs. 2015;13(10):6099–6116. | ||
Chen KJ, Hou Y, Wang K, et al. Reexpression of Let-7g microRNA inhibits the proliferation and migration via K-Ras/HMGA2/snail axis in hepatocellular carcinoma. Biomed Res Int. 2014;2014:742417. | ||
Chan SH, Wang LH. Regulation of cancer metastasis by microRNAs. J Biomed Sci. 2015;22:9. | ||
Suh SS, Yoo JY, Cui R, et al. FHIT suppresses epithelial-mesenchymal transition (EMT) and metastasis in lung cancer through modulation of microRNAs. PLoS Genet. 2014;10(10):e1004652. | ||
Zuo QF, Cao LY, Yu T, et al. MicroRNA-22 inhibits tumor growth and metastasis in gastric cancer by directly targeting MMP14 and Snail. Cell Death Dis. 2015;6:e2000. | ||
Ru P, Steele R, Newhall P, Phillips NJ, Toth K, Ray RB. miRNA-29b suppresses prostate cancer metastasis by regulating epithelial-mesenchymal transition signaling. Mol Cancer Ther. 2012;11(5):1166–1173. | ||
Yu J, Xie F, Bao X, Chen W, Xu Q. miR-300 inhibits epithelial to mesenchymal transition and metastasis by targeting Twist in human epithelial cancer. Mol Cancer. 2014;13:121. | ||
Yu Y, Zhao Y, Sun XH, et al. Down-regulation of miR-129-5p via the Twist1-Snail feedback loop stimulates the epithelial-mesenchymal transition and is associated with poor prognosis in breast cancer. Oncotarget. 2015;6(33):34423–34436. | ||
Lin Y, Liu AY, Fan C, et al. MicroRNA-33b inhibits breast cancer metastasis by targeting HMGA2, SALL4 and Twist1. Sci Rep. 2015;5:9995. | ||
Xiang X, Zhuang X, Ju S, et al. miR-155 promotes macroscopic tumor formation yet inhibits tumor dissemination from mammary fat pads to the lung by preventing EMT. Oncogene. 2011;30(31):3440–3453. | ||
Wang R, Li Y, Hou Y, et al. The PDGF-D/miR-106a/Twist1 pathway orchestrates epithelial-mesenchymal transition in gemcitabine resistance hepatoma cells. Oncotarget. 2015;6(9):7000–7010. | ||
Zhao X, Wang Y, Deng R, et al. miR186 suppresses prostate cancer progression by targeting Twist1. Oncotarget. 2016;7(22):33136–33151. | ||
Wen YC, Lee WJ, Tan P, et al. By inhibiting snail signaling and miR-23a-3p, osthole suppresses the EMT-mediated metastatic ability in prostate cancer. Oncotarget. 2015;6(25):21120–21136. | ||
Li L, Wu D. miR-32 inhibits proliferation, epithelial-mesenchymal transition, and metastasis by targeting TWIST1 in non-small-cell lung cancer cells. Onco Targets Ther. 2016;9:1489–1498. | ||
Yang L, Yang J, Li J, et al. MircoRNA-33a inhibits epithelial-to-mesenchymal transition and metastasis and could be a prognostic marker in non-small cell lung cancer. Sci Rep. 2015;5:13677. | ||
Song Y, Li J, Zhu Y, et al. MicroRNA-9 promotes tumor metastasis via repressing E-cadherin in esophageal squamous cell carcinoma. Oncotarget. 2014;5(22):11669–11680. | ||
Lu MH, Huang CC, Pan MR, Chen HH, Hung WC. Prospero homeobox 1 promotes epithelial-mesenchymal transition in colon cancer cells by inhibiting E-cadherin via miR-9. Clin Cancer Res. 2012;18(23):6416–6425. | ||
Sun Z, Han Q, Zhou N, et al. MicroRNA-9 enhances migration and invasion through KLF17 in hepatocellular carcinoma. Mol Oncol. 2013;7(5):884–894. | ||
Xu M, Zhu C, Zhao X, et al. Atypical ubiquitin E3 ligase complex Skp1-Pam-Fbxo45 controls the core epithelial-to-mesenchymal transition-inducing transcription factors. Oncotarget. 2015;6(2):979–994. | ||
Chen D, Dang BL, Huang JZ, et al. MiR-373 drives the epithelial-to-mesenchymal transition and metastasis via the miR-373-TXNIP-HIF1alpha-TWIST signaling axis in breast cancer. Oncotarget. 2015;6(32):32701–32712. | ||
Ke J, Zhao Z, Hong SH, et al. Role of microRNA221 in regulating normal mammary epithelial hierarchy and breast cancer stem-like cells. Oncotarget. 2015;6(6):3709–3721. | ||
Sarkar S, Dubaybo H, Ali S, et al. Down-regulation of miR-221 inhibits proliferation of pancreatic cancer cells through up-regulation of PTEN, p27(kip1), p57(kip2), and PUMA. Am J Cancer Res. 2013;3(5):465–477. | ||
Piva R, Spandidos DA, Gambari R. From microRNA functions to microRNA therapeutics: novel targets and novel drugs in breast cancer research and treatment (Review). Int J Oncol. 2013;43(4):985–994. | ||
Long H, Wang Z, Chen J, et al. microRNA-214 promotes epithelial-mesenchymal transition and metastasis in lung adenocarcinoma by targeting the suppressor-of-fused protein (Sufu). Oncotarget. 2015;6(36):38705–38718. | ||
Sun CC, Li SJ, Yuan ZP, Li DJ. MicroRNA-346 facilitates cell growth and metastasis, and suppresses cell apoptosis in human non-small cell lung cancer by regulation of XPC/ERK/Snail/E-cadherin pathway. Aging (Albany NY). 2016;8(10):2509–2524. | ||
Bao L, Yan Y, Xu C, et al. MicroRNA-21 suppresses PTEN and hSulf-1 expression and promotes hepatocellular carcinoma progression through AKT/ERK pathways. Cancer Lett. 2013;337(2):226–236. | ||
Liu Z, Jin ZY, Liu CH, Xie F, Lin XS, Huang Q. MicroRNA-21 regulates biological behavior by inducing EMT in human cholangiocarcinoma. Int J Clin Exp Pathol. 2015;8(5):4684–4694. | ||
Cao J, Liu J, Xu R, Zhu X, Liu L, Zhao X. MicroRNA-21 stimulates epithelial-to-mesenchymal transition and tumorigenesis in clear cell renal cells. Mol Med Rep. 2016;13(1):75–82. | ||
Cufi S, Bonavia R, Vazquez-Martin A, et al. Silibinin suppresses EMT-driven erlotinib resistance by reversing the high miR-21/low miR-200c signature in vivo. Sci Rep. 2013;3:2459. | ||
Chang S, Chen B, Wang X, Wu K, Sun Y. Long non-coding RNA XIST regulates PTEN expression by sponging miR-181a and promotes hepatocellular carcinoma progression. BMC Cancer. 2017;17(1):248. | ||
Yadav A, Kumar B, Datta J, Teknos TN, Kumar P. IL-6 promotes head and neck tumor metastasis by inducing epithelial-mesenchymal transition via the JAK-STAT3-SNAIL signaling pathway. Mol Cancer Res. 2011;9(12):1658–1667. | ||
Kim HA, Koo BK, Cho JH, et al. Notch1 counteracts WNT/beta-catenin signaling through chromatin modification in colorectal cancer. J Clin Invest. 2012;122(9):3248–3259. | ||
Xie G, Karaca G, Swiderska-Syn M, et al. Cross-talk between Notch and Hedgehog regulates hepatic stellate cell fate in mice. Hepatology. 2013;58(5):1801–1813. | ||
Doyle AJ, Redmond EM, Gillespie DL, et al. Differential expression of Hedgehog/Notch and transforming growth factor-beta in human abdominal aortic aneurysms. J Vasc Surg. 2015;62(2):464–470. | ||
Li L, Grausam KB, Wang J, et al. Sonic Hedgehog promotes proliferation of Notch-dependent monociliated choroid plexus tumour cells. Nat Cell Biol. 2016;18(4):418–430. | ||
Schreck KC, Taylor P, Marchionni L, et al. The Notch target Hes1 directly modulates Gli1 expression and Hedgehog signaling: a potential mechanism of therapeutic resistance. Clin Cancer Res. 2010;16(24):6060–6070. | ||
Ringuette R, Atkins M, Lagali PS, et al. A Notch-Gli2 axis sustains Hedgehog responsiveness of neural progenitors and Muller glia. Dev Biol. 2016;411(1):85–100. | ||
Min C, Eddy SF, Sherr DH, Sonenshein GE. NF-kappaB and epithelial to mesenchymal transition of cancer. J Cell Biochem. 2008;104(3):733–744. | ||
Carpenter RL, Lo HW. STAT3 target genes relevant to human cancers. Cancers (Basel). 2014;6(2):897–925. | ||
Jiao D, Wang J, Lu W, et al. Curcumin inhibited HGF-induced EMT and angiogenesis through regulating c-Met dependent PI3K/Akt/mTOR signaling pathways in lung cancer. Mol Ther Oncolytics. 2016;3:16018. | ||
Mukherjee S, Mazumdar M, Chakraborty S, et al. Curcumin inhibits breast cancer stem cell migration by amplifying the E-cadherin/beta-catenin negative feedback loop. Stem Cell Res Ther. 2014;5(5):116. | ||
Al-Howail HA, Hakami HA, Al-Otaibi B, et al. PAC down-regulates estrogen receptor alpha and suppresses epithelial-to-mesenchymal transition in breast cancer cells. BMC Cancer. 2016;16:540. | ||
Bakiri L, Macho-Maschler S, Custic I, et al. Fra-1/AP-1 induces EMT in mammary epithelial cells by modulating Zeb1/2 and TGFbeta expression. Cell Death Differ. 2015;22(2):336–350. | ||
Yang S, Du J, Wang Z, et al. Dual mechanism of deltaEF1 expression regulated by bone morphogenetic protein-6 in breast cancer. Int J Biochem Cell Biol. 2009;41(4):853–861. | ||
Huang TS, Lee SC, Lin JK. Suppression of c-Jun/AP-1 activation by an inhibitor of tumor promotion in mouse fibroblast cells. Proc Natl Acad Sci U S A. 1991;88(12):5292–5296. | ||
Liu Z, Liu J, Zhao L, et al. Curcumin reverses benzidine-induced epithelial-mesenchymal transition via suppression of ERK5/AP-1 in SV-40 immortalized human urothelial cells. Int J Oncol. 2017;50(4):1321–1329. | ||
Yin DL, Liang YJ, Zheng TS, et al. EF24 inhibits tumor growth and metastasis via suppressing NF-kappaB dependent pathways in human cholangiocarcinoma. Sci Rep. 2016;6:32167. | ||
Zhang P, Bai H, Liu G, et al. MicroRNA-33b, upregulated by EF24, a curcumin analog, suppresses the epithelial-to-mesenchymal transition (EMT) and migratory potential of melanoma cells by targeting HMGA2. Toxicol Lett. 2015;234(3):151–161. | ||
Zhao R, Tin L, Zhang Y, et al. EF24 suppresses invasion and migration of hepatocellular carcinoma cells in vitro via inhibiting the phosphorylation of Src. Biomed Res Int. 2016;2016:8569684. | ||
Sun XD, Liu XE, Huang DS. Curcumin reverses the epithelial-mesenchymal transition of pancreatic cancer cells by inhibiting the Hedgehog signaling pathway. Oncol Rep. 2013;29(6):2401–2407. | ||
Li W, Ma J, Ma Q, et al. Resveratrol inhibits the epithelial-mesenchymal transition of pancreatic cancer cells via suppression of the PI-3K/Akt/NF-kappaB pathway. Curr Med Chem. 2013;20(33):4185–4194. | ||
Ali S, Ahmad A, Banerjee S, et al. Gemcitabine sensitivity can be induced in pancreatic cancer cells through modulation of miR-200 and miR-21 expression by curcumin or its analogue CDF. Cancer Res. 2010;70(9):3606–3617. | ||
Bao B, Ali S, Banerjee S, et al. Curcumin analogue CDF inhibits pancreatic tumor growth by switching on suppressor microRNAs and attenuating EZH2 expression. Cancer Res. 2012;72(1):335–345. | ||
Bao B, Ahmad A, Kong D, et al. Hypoxia induced aggressiveness of prostate cancer cells is linked with deregulated expression of VEGF, IL-6 and miRNAs that are attenuated by CDF. PLoS One. 2012;7(8):e43726. | ||
Bao B, Ali S, Kong D, et al. Anti-tumor activity of a novel compound-CDF is mediated by regulating miR-21, miR-200, and PTEN in pancreatic cancer. PLoS One. 2011;6(3):e17850. | ||
Ali S, Ahmad A, Aboukameel A, et al. Increased Ras GTPase activity is regulated by miRNAs that can be attenuated by CDF treatment in pancreatic cancer cells. Cancer Lett. 2012;319(2):173–181. | ||
Zhang Z, Chen H, Xu C, et al. Curcumin inhibits tumor epithelial-mesenchymal transition by downregulating the Wnt signaling pathway and upregulating NKD2 expression in colon cancer cells. Oncol Rep. 2016;35(5):2615–2623. | ||
Toden S, Okugawa Y, Jascur T, et al. Curcumin mediates chemosensitization to 5-fluorouracil through miRNA-induced suppression of epithelial-to-mesenchymal transition in chemoresistant colorectal cancer. Carcinogenesis. 2015;36(3):355–367. | ||
Montgomery A, Adeyeni T, San K, Heuertz RM, Ezekiel UR. Curcumin sensitizes silymarin to exert synergistic anticancer activity in colon cancer cells. J Cancer. 2016;7(10):1250–1257. | ||
Du R, Liu Z, Hou X, Fu G, An N, Wang L. Trichostatin A potentiates genistein-induced apoptosis and reverses EMT in HEp2 cells. Mol Med Rep. 2016;13(6):5045–5052. | ||
Bao B, Wang Z, Ali S, et al. Notch-1 induces epithelial-mesenchymal transition consistent with cancer stem cell phenotype in pancreatic cancer cells. Cancer Lett. 2011;307(1):26–36. | ||
Ma J, Fang B, Zeng F, et al. Down-regulation of miR-223 reverses epithelial-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Oncotarget. 2015;6(3):1740–1749. | ||
Phillip CJ, Giardina CK, Bilir B, et al. Genistein cooperates with the histone deacetylase inhibitor vorinostat to induce cell death in prostate cancer cells. BMC Cancer. 2012;12:145. | ||
Li Y, Kong D, Ahmad A, Bao B, Dyson G, Sarkar FH. Epigenetic deregulation of miR-29a and miR-1256 by isoflavone contributes to the inhibition of prostate cancer cell growth and invasion. Epigenetics. 2012;7(8):940–949. | ||
Kim YS, Choi KC, Hwang KA. Genistein suppressed epithelial-mesenchymal transition and migration efficacies of BG-1 ovarian cancer cells activated by estrogenic chemicals via estrogen receptor pathway and downregulation of TGF-beta signaling pathway. Phytomedicine. 2015;22(11):993–999. | ||
Wang Y, Lu P, Zhang W, et al. GEN-27, a newly synthetic isoflavonoid, inhibits the proliferation of colon cancer cells in inflammation microenvironment by suppressing NF-kappaB pathway. Mediators Inflamm. 2016;2016:2853040. | ||
Chang YC, Lin CW, Yu CC, et al. Resveratrol suppresses myofibroblast activity of human buccal mucosal fibroblasts through the epigenetic inhibition of ZEB1 expression. Oncotarget. 2016;7(11):12137–12149. | ||
Chen MC, Chang WW, Kuan YD, Lin ST, Hsu HC, Lee CH. Resveratrol inhibits LPS-induced epithelial-mesenchymal transition in mouse melanoma model. Innate Immun. 2012;18(5):685–693. | ||
Chen J, Chang H, Peng X, et al. 3,6-dihydroxyflavone suppresses the epithelial-mesenchymal transition in breast cancer cells by inhibiting the Notch signaling pathway. Sci Rep. 2016;6:28858. | ||
Vergara D, Valente CM, Tinelli A, et al. Resveratrol inhibits the epidermal growth factor-induced epithelial mesenchymal transition in MCF-7 cells. Cancer Lett. 2011;310(1):1–8. | ||
Leone L, Mazzetta F, Martinelli D, et al. Klebsiella pneumoniae is able to trigger epithelial-mesenchymal transition process in cultured airway epithelial cells. PLoS One. 2016;11(1):e0146365. | ||
Harris DM, Besselink E, Henning SM, Go VL, Heber D. Phytoestrogens induce differential estrogen receptor alpha- or Beta-mediated responses in transfected breast cancer cells. Exp Biol Med (Maywood). 2005;230(8):558–568. | ||
Ji Q, Liu X, Han Z, et al. Resveratrol suppresses epithelial-to-mesenchymal transition in colorectal cancer through TGF-beta1/Smads signaling pathway mediated Snail/E-cadherin expression. BMC Cancer. 2015;15:97. | ||
Buhrmann C, Shayan P, Popper B, Goel A, Shakibaei M. Sirt1 is required for resveratrol-mediated chemopreventive effects in colorectal cancer cells. Nutrients. 2016;8(3):145. | ||
Bosutti A, Zanconati F, Grassi G, Dapas B, Passamonti S, Scaggiante B. Epigenetic and miRNAs dysregulation in prostate cancer: the role of nutraceuticals. Anticancer Agents Med Chem. 2016;16(11):1385–1402. | ||
Zhang P, Li H, Yang B, et al. Biological significance and therapeutic implication of resveratrol-inhibited Wnt, Notch and STAT3 signaling in cervical cancer cells. Genes Cancer. 2014;5(5–6):154–164. | ||
Kotha A, Sekharam M, Cilenti L, et al. Resveratrol inhibits Src and Stat3 signaling and induces the apoptosis of malignant cells containing activated Stat3 protein. Mol Cancer Ther. 2006;5(3):621–629. | ||
Ying Z, Li Y, Wu J, et al. Loss of miR-204 expression enhances glioma migration and stem cell-like phenotype. Cancer Res. 2013;73(2):990–999. | ||
Xiang LP, Wang A, Ye JH, et al. Suppressive effects of tea catechins on breast cancer. Nutrients. 2016;8(8). pii: E458. | ||
Oliveira MR, Nabavi SF, Daglia M, Rastrelli L, Nabavi SM. Epigallocatechin gallate and mitochondria-A story of life and death. Pharmacol Res. 2016;104:70–85. | ||
Kim J, Zhang X, Rieger-Christ KM, et al. Suppression of Wnt signaling by the green tea compound (-)-epigallocatechin 3-gallate (EGCG) in invasive breast cancer cells. Requirement of the transcriptional repressor HBP1. J Biol Chem. 2006;281(16):10865–10875. | ||
Ko H, So Y, Jeon H, et al. TGF-beta1-induced epithelial-mesenchymal transition and acetylation of Smad2 and Smad3 are negatively regulated by EGCG in human A549 lung cancer cells. Cancer Lett. 2013;335(1):205–213. | ||
Lu L, Chen J, Tang H, et al. EGCG suppresses ERK5 activation to reverse tobacco smoke-triggered gastric epithelial-mesenchymal transition in BALB/c mice. Nutrients. 2016;8(7):130. | ||
Toden S, Tran HM, Tovar-Camargo OA, Okugawa Y, Goel A. Epigallocatechin-3-gallate targets cancer stem-like cells and enhances 5-fluorouracil chemosensitivity in colorectal cancer. Oncotarget. 2016;7(13):16158–16171. | ||
Chen KC, Chen CY, Lin CR, et al. Luteolin attenuates TGF-beta1-induced epithelial-mesenchymal transition of lung cancer cells by interfering in the PI3K/Akt-NF-kappaB-Snail pathway. Life Sci. 2013;93(24):924–933. | ||
Ruan JS, Liu YP, Zhang L, et al. Luteolin reduces the invasive potential of malignant melanoma cells by targeting beta3 integrin and the epithelial-mesenchymal transition. Acta Pharmacol Sin. 2012;33(10):1325–1331. | ||
Ruan J, Zhang L, Yan L, et al. Inhibition of hypoxia-induced epithelial mesenchymal transition by luteolin in non-small cell lung cancer cells. Mol Med Rep. 2012;6(1):232–238. | ||
Huang X, Dai S, Dai J, et al. Luteolin decreases invasiveness, deactivates STAT3 signaling, and reverses interleukin-6 induced epithelial-mesenchymal transition and matrix metalloproteinase secretion of pancreatic cancer cells. Onco Targets Ther. 2015;8:2989–3001. | ||
Shukla S, Sinha S, Khan S, et al. Cucurbitacin B inhibits the stemness and metastatic abilities of NSCLC via downregulation of canonical Wnt/beta-catenin signaling axis. Sci Rep. 2016;6:21860. | ||
Qin Y, Zhao D, Zhou HG, et al. Apigenin inhibits NF-kappaB and snail signaling, EMT and metastasis in human hepatocellular carcinoma. Oncotarget. 2016;7(27):41421–41431. | ||
Verma RK, Yu W, Shrivastava A, Shankar S, Srivastava RK. α-Mangostin-encapsulated PLGA nanoparticles inhibit pancreatic carcinogenesis by targeting cancer stem cells in human, and transgenic (Kras(G12D), and Kras(G12D)/tp53R270H) mice. Sci Rep. 2016;6:32743. | ||
Lin L, Sun Y, Wang D, Zheng S, Zhang J, Zheng C. Celastrol ameliorates ulcerative colitis-related colorectal cancer in mice via suppressing inflammatory responses and epithelial-mesenchymal transition. Front Pharmacol. 2015;6:320. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.