")
Back to Archived Journals » Advances in Genomics and Genetics » Volume 5
Recent insights into the regulation of X-chromosome inactivation
Authors Valencia K, Wutz A
Received 3 February 2015
Accepted for publication 5 March 2015
Published 14 May 2015 Volume 2015:5 Pages 227—238
DOI https://doi.org/10.2147/AGG.S60399
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr John Martignetti
Karmele Valencia, Anton Wutz
Department of Biology, Institute of Molecular Health Sciences, Swiss Federal Institute of Technology Zurich, Zurich, Switzerland
Abstract: X-chromosome inactivation (XCI) is the mechanism by which mammals compensate gene dosage differences between males and females. XCI is required for female development and has implications for human disease. As a result, a single X chromosome is transcriptionally active in both male and female cells. Functional hemizygosity of the X chromosomes greatly predisposes to phenotypic consequences of mutations. In females, X chromosomes are randomly chosen to become inactivated leading to a mosaic pattern of cells expressing genes from either chromosome. This facilitates the masking of phenotypic consequences of heterozygous X-linked mutations. Skewing of XCI in favor of one chromosome can result in increased severity of disease symptoms, if the X chromosome with a gene mutation remains preferentially active. In addition, phenotypic masking of X-linked mutations is not always observed. Rett syndrome represents a paradigm of this statement. Dosage compensation can also mask some aspects of sex chromosome aneuploidies. X-chromosome aneuploidies include Klinefelter, Turner, and X-trisomy syndromes. In all these cases, a single active X chromosome is present. However, in those cases with two or more X chromosomes, some genes from the inactivated X chromosome escape from XCI becoming active. Therefore, dose imbalances of escape genes cause pathologies. Defects in the structure and silencing of the inactive X chromosome are further observed in human pluripotent stem cells and in certain tumors. Taken together, these findings suggest that aspects of XCI are relevant for a large number of human diseases. Here we review basic and clinical research on XCI with the aim of illustrating connections and highlighting opportunities for future investigation.
Keywords: XCI, X-linked diseases, sex chromosome aneuploidies, iPSCs, cancer
Recent basic XCI research: a brief sum up
X-chromosome inactivation (XCI) is the mechanism for dosage compensation in mammals. Gene dosage imbalances arising from the different numbers of X chromosomes between XX females and XY males are compensated by inactivation of one of the two X chromosomes in females. This process is specific for mammals and the mechanism is not naturally observed or easily recapitulated in other model species that are used in basic research laboratories today. Originally, the hypothesis of XCI was proposed by Mary Lyon in 1961, who was the first to recognize the silencing of one of the two X chromosomes in female cells.1 It is random chance that determines which of the two X chromosomes is inactivated and which will remain active leading to a mosaic of cells with opposite XCI patterns that can be observed in all female tissues.
For experimentally investigating the regulation and mechanism of XCI, a large number of studies have used mice as a simple mammalian model species. In mice, XCI is initiated during early development in two consecutive waves. The first one, around the four-cell embryo stage, is considered an imprinted X inactivation as the paternally inherited X (Xp) is exclusively selected. This imprinted XCI is thereafter maintained in extraembryonic lineages but lost in the inner cell mass of the blastocyst, where Xi will reactivate. A second wave then leads to random inactivation of the paternally or maternally inherited X in cells of the developing epiblast around embryonic day 6.5. Once XCI is initiated, the Xi is maintained through subsequent cell divisions resulting in a mosaic pattern in tissues. Although imprinted XCI is not observed in humans (and appears restricted to a minority of mammalian species2), other aspects of XCI appear well conserved throughout mammals suggesting that insights obtained from mouse models are useful for extrapolation to other mammals, although with caution.
Molecularly, the process of XCI involves several steps: counting the number of X chromosomes per nucleus, choice of the future Xi,3 initiation of chromosome-wide silencing, and maintenance of Xi repression in a stable manner. XCI is initiated by a specific control region, the X-inactivation center (Xic) that is located on the mouse and human X chromosomes at XD (46.12 cM) and Xq13.2, respectively (refer Augui et al4 and references therein). The Xic contains a long-noncoding RNA gene, which produces the X-inactive specific transcript (Xist) with remarkable properties. Xist RNA associates with the X chromosome from where it is expressed and mediates gene repression in a near–chromosome-wide manner. The mechanism of Xist function is not entirely clear. Potential RNA-binding proteins like YY15 or ATRX6 have been implicated in XCI. Repression of the Xi further involves a depletion of transcription initiation factors, RNA polymerase II and splicing factors from the Xist-covered chromatin domain. Recruitment of Polycomb repressive complexes (PcG) induces characteristic histone modifications that generally correlate with transcriptional silencing. Although much has been recently learned on PcG recruitment7 from studying XCI, the precise function of PcG complexes on the Xi remains to be established.
Xist is critical for the initiation of gene repression in the early embryo and also in embryonic stem cells (ESCs).8,9 In contrast, once cells have entered differentiation, gene silencing on the Xi becomes largely independent of Xist and is stabilized by other epigenetic mechanisms.10,11 Conversely, in differentiated cells, Xist also becomes insufficient to induce gene repression (reviewed in Lee12 and Wutz13). Maintenance of stable inactivation is generally thought to be Xist-independent but loss of Xist does seem to lead to impairment of the Xi silencing.14 A recent study showing that deletion of Xist induces cancer with high penetrance in mice suggests that Xist has an essential role in Xi maintenance.14 It is therefore conceivable that epigenetic factors might act in certain cells to promote differences to other somatic tissue cells. This idea could reconcile the different observations and could be useful for understanding the origin of tumor-initiating cells and the mechanism of X-chromosome reactivation.
To date, a large number of studies have elucidated aspects of the molecular mechanism of XCI and advanced the understanding of this complex process. Insights into developmental regulation have identified links between XCI with pluripotency (reviewed in Payer and Lee15), and studies of Xi chromatin organization have provided connections to three-dimensional conformation of chromosomes in the cell nucleus.16 Insights into aspects of XCI have thereby shed light on a broad range of phenomena and contributed greatly to advance our basic understanding of gene regulation in mammals. Notably, insights into the etiology of the natural process can also be useful for understanding pathologies that arise in the clinics due to aberrations in X-linked gene dosage and can be caused by several unrelated mechanisms (Figure 1).
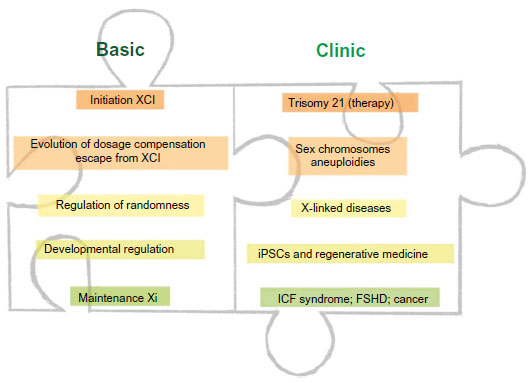 | Figure 1 Overview: XCI can be understood as a multistep process. |
When dosage compensation is compromised
X-linked gene dosage: escape from X inactivation and sex chromosome aneuploidies
Chromosomal aneuploidies are rarely compatible with embryonic development in mammals and in humans frequently cause death before birth or even before a pregnancy is detected. Exceptions are trisomy 21, 18, and 13 that occur in Down, Edwards, and Patau syndrome, respectively. X-chromosome aneuploidies are generally less detrimental than those of autosomes. The presence of extra sex chromosomes is largely compensated by XCI, which will inactivate all but one single X chromosome irrespective of the number of X chromosomes present in the diploid chromosome set. However, disorders do arise as a consequence of anomalous X-chromosome number.
Syndromes can arise due to the presence of extra copies of X chromosomes in males and females or a single X chromosome in females. These include Klinefelter syndrome, trisomy X, and Turner syndrome.17–19 Even when the dosage of gene expression is generally limited to one X chromosome in all instances, phenotypes in individuals with sex chromosome polyploidy result from an overexpression of genes that normally escape X inactivation.20,21 There are few genes known to escape XCI in mice and substantially more in humans. Escape genes are characterized with at least 10% of the expression level on the Xi compared to the level on the active X chromosome (Xa). From an evolutionary view, escapees are thought to have been added to the X chromosome in a relatively recent period so that they have not yet adapted to efficient XCI. They can be classified in two major groups: genes that lie within the pseudoautosomal regions of the X chromosome and have a homolog on the Y chromosome and escape genes located outside the pseudoautosomal regions. The first group will show similar expression levels in both sexes, whereas genes from the second group will be higher expressed in females than in males.22 Klinefelter patients (47, XXY) carry an additional X chromosome in their genome. Therefore, the expression of escape genes in the pseudoautosomal regions would potentially reach a triple rather than a normal double dose, although most of the supernumerary X-linked gene copies would be inactivated by XCI.23 In trisomy X patients (47, XXX), a triple dose of all escape genes can be expected. On the contrary, deficiency in escape genes is thought to play a major role in phenotypes observed in Turner patients (45, X), which carry a single X chromosome and therefore have a dosage reduction for all X-linked escape genes24 (Figure 2).
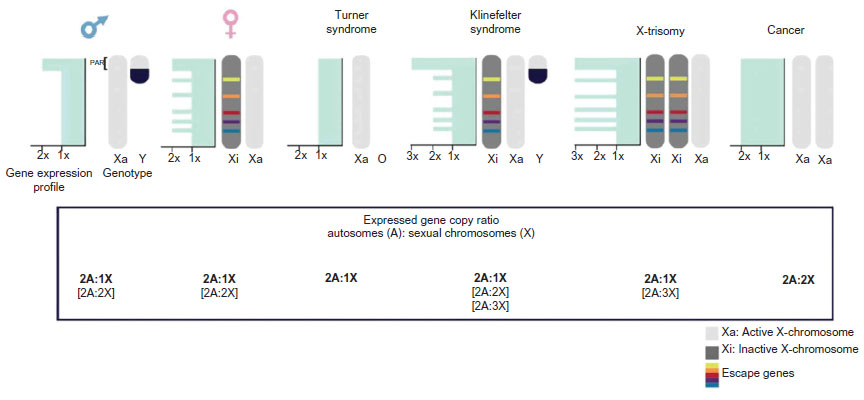 | Figure 2 X-linked gene dosage. |
Phenotypes of sex chromosome aneuploidies are generally milder in mouse models. Female mice carrying a single X chromosome (39, X) are near-normal and fertile, albeit they show reduced fertility when compared to normal female mice (40, XX).25 The reduced severity of X monosomy is consistent with the fact that fewer escape genes exist on the mouse Xi compared to the human Xi. Escapees account for 15% and 3% of X-linked genes in human and mouse, respectively.26 In addition, Klinefelter and X-trisomy mouse models produce pathologies that are corresponding to the human syndromes but have a moderate phenotype.27,28 Not only the number but also the distribution of escape genes differs between species. In mice, individual escape genes are embedded in regions of silenced chromatin and are distributed over the entire length of the X chromosome. In contrast, escape genes tend to cluster in humans. Of all escape genes, 80% reside on the short arm of the X chromosome, predominantly in the distal region that is furthest away from the centromere and the long arm that carries the Xic26 (Figure 3 and Table 1). This suggests that the position of genes on the chromosome could contribute to their susceptibility to be silenced. In addition, the spatial organization of chromatin has been suggested as a critical factor for the escape from X inactivation. Three-dimensional conformation assays suggest that escape genes localize preferentially in exterior positions of the Xi chromosome territory,29 which could reflect looping of specific chromatin domains from the condensed Xi heterochromatin compartment. Long-range associations between escape genes have been observed, but associations between escape genes and genes subject to XCI could not be detected,30 suggesting that different domains could coexist on the Xi. Recently, it has also been shown that Xist accumulates over the X chromosome by tracing the three-dimensional chromatin structure and subsequently inducing histone modifications and gene repression.16 Together, these findings suggest that the spatial organization of chromosomal DNA could be an important aspect for understanding chromosome-wide gene repression of the Xi. The position of the Xic relative to the centromere on the X chromosome might also influence the ability of spreading inactivation and underlie differences in escape genes between mouse and human. On the mouse X chromosome, the centromere is located at one extremity. Therefore, the centromere does not affect spreading of Xist along the long arm. On the human X chromosome, the centromere assumes a medial position and separates the short arm from the long arm, which carries the Xic region.26 In this case, the centromere might reduce the ability of Xist to spread and silence efficiently. This view is consistent with the fact that 80% of escape genes are found on the short arm of the X chromosome.
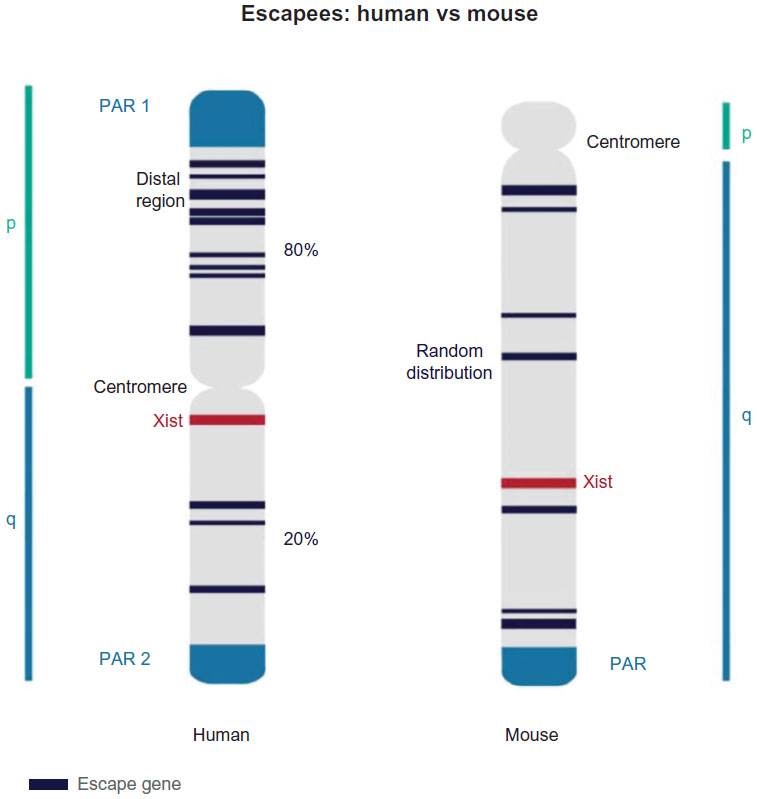 | Figure 3 Escape genes: differences between human and mouse. |
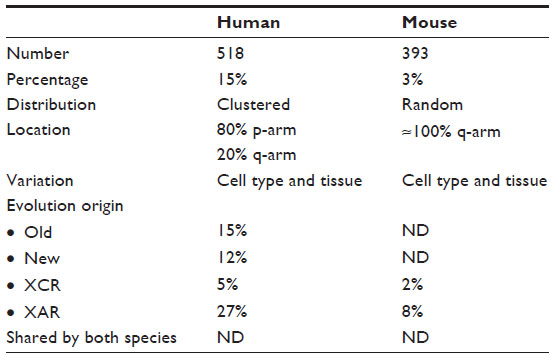 | Table 1 Escape gene features in human and mouse species |
In addition to the physical distribution of the genes, many studies point to a connection between the spatial location and protective regulatory elements for escape genes to resist XCI.31–33 For example, one of these regulatory elements, which is recognized by CCCTC-binding factor, might contribute to keep escape genes together and potentially facilitate expression through CCCTC-binding factor–RNA interactions.34 On the contrary, retrotransposons such as long interspersed nuclear elements are associated with the spreading of silencing.35,36 There is evidence that correlates fewer long interspersed nuclear element repeat elements with failed gene silencing.37–39 The promoters of escape genes maintain histone modifications associated with transcriptional activity, in contrast to the majority of genes on the Xi that are silenced.40 Other genomic repeat elements and sequence motifs including long terminal repeats or AT-rich motifs appear also depleted at escape genes.41–43 Several studies have investigated the mechanism by which genes escape from XCI. Escape of individual genes observed in mice during imprinted XCI could rely on local regulatory elements that lie in close proximity of the genes. However, mechanisms of escape may vary from gene to gene and will likely need to be understood on a gene-by-gene basis.44,45 The many factors influencing the escape of genes from XCI might also contribute to the variability of expression that specifically escape genes display between tissues and further also between individuals.46 Differences in escape from XCI between individuals could contribute to phenotypic variability in subtle ways and be potentially related to the evolution of sex differences of specific phenotypes.
Clinical studies have established that escape genes are important to brain development and function47 and X aneuploidy can be associated with certain disruptions in cognitive and emotional development.48 In addition, Klinefelter syndrome patients have higher risks of developing autoimmune diseases, diabetes, leg ulcers, osteopenia and osteoporosis, tumors of the breast and germ cells, systemic lupus erythematosus, rheumatoid arthritis, and Sjögren syndrome.23 Furthermore, gene expression from the Xi may also change over time during development into adulthood, and the Xi can be prone to partial reactivation during aging.49 Mutations of specific escape genes have been observed to contribute to a range of diseases including cancer. For example, the escape gene KDM6A is involved in medulloblastoma, prostate cancer, renal carcinoma development, and Kabuki syndrome, whereas KDM5C is important for neural development.50 Both, oncogenes and tumor suppressor genes are among the genes that escape XCI (reviewed in Spatz et al51). This suggests that understanding the mechanism and function of escape from XCI better could be relevant for a wide range of human diseases.
Regulation of randomness and X-linked diseases: mutations and skewing
The two X chromosomes in females normally have equal chances of becoming inactivated (Xi). Hence, the ratio of cells with the paternal X chromosome (Xp) inactivated to the cells having the maternal X (Xm) inactivated can be expected to be around 50%. Mosaicism caused by cells randomly inactivating Xm or Xp has been suggested to explain how some females can perceive more colors in comparison to males for expression of the two X-linked retinal photo pigment alleles.52 However, Xi choice can be skewed53 and the XCI ratios can deviate from random and also vary among tissues of the same individual.54 This fact generally protects the individual from developing a disease but in some cases preferential selection of the mutation renders symptoms with different degrees of severity and penetrance (Figure 4). Skewing has been classified in primary nonrandom pattern of XCI, which is established at the time when XCI is initiated, and secondary nonrandom XCI, which can arise as a consequence of cell selection for or against the expression of one of the two X chromosomes.55 The mechanisms that lead to skewing are likely diverse and remain to be resolved on a case by case basis.
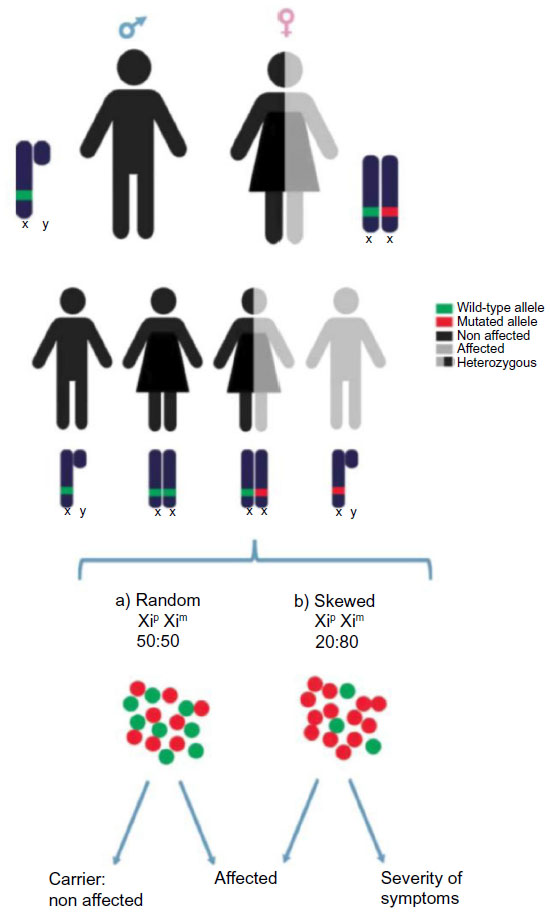 | Figure 4 X-linked disorders: skewing. |
In mouse, there is evidence that a genetic locus, the X controlling element (Xce), influences the choice of the future Xi.56 The Xce is defined as the cis element with four alternative alleles: Xcea, Xceb, Xcec, and Xced, with different tendency to remain active (Xcea > Xceb > Xcec > Xced). Xce is thought to lie in a region close to the Xic. If two X chromosomes with different Xce alleles are present, a primary nonrandom pattern of XCI will be observed. Recently, the Xce region has been reduced to a genomic interval of 176 kb that resides approximately 500 kb proximal (toward the X centromere) of the Xist gene.57 Other X chromosomal loci might also influence Xi choice and contribute to the Xce effect.58 Although the molecular mode of action of the Xce has not yet been identified, it is hypothesized that it serves as a binding site for trans-acting factors that exert an influence on other loci within the Xic including the Xist and Tsix genes. Tsix is a long-noncoding RNA that is transcribed in antisense orientation to Xist and acts as a repressor of Xist on the same X chromosome.59 Tsix is specific for the mouse Xic and does not appear to be functionally conserved in humans suggesting it could be related to the strong skewing characteristic of intercrosses between specific mouse strains carrying different Xce alleles. In humans, several etiologies for skewing have been considered including cis-regulatory elements within the Xic, stochasticity, and cell selection for X-linked gene mutations. However, there is no conclusive evidence for the presence of an Xce in humans (reviewed in Bolduc et al60 and references therein).
Skewing also influences the severity of disease symptoms when heterozygous mutations of X-linked genes are present. In the vast majority of the cases of random XCI, the mutated allele of an X-linked gene will reside on the inactive X chromosome in 50% of the cells. Therefore, half of the cells are essentially wild-type, which often will ameliorate disease phenotypes. Skewing in favor of expressing the mutated gene can cause a proportional increase in phenotypic severity that in the extreme case can approach the level of that observed in males where X-linked mutations are hemizygous. Rare cases of female hemophilia A61 and sideroblastic anemia have been described,62 in which the X chromosome carrying the intact factor VII and erythroid-specific 5-aminolevulinic acid synthase (ALAS2) gene is preferentially inactivated leaving a majority of cells expressing only the mutated allele.
The presence of 50% cells expressing the wild-type allele does not prevent the development of disease in all cases. A notable case is Rett syndrome (RTT). RTT is the second most common cause of X-linked mental retardation with an estimated prevalence of 1 in 10,000–15,000 girls (reviewed in Chahrour and Zoghbi63). In 1999, mutations in the X-linked gene methyl-CpG–binding protein 2 (MeCP2) were identified as disease-causing mutations in RTT patients.64 Female RTT patients carry MeCP2 heterozygous mutations, which leads to expression of the wild-type protein in only about half of their cells. Considering that MeCP2 mutations in males are in a hemizygous state and cause lethality, one can appreciate that also in this case random XCI alleviates much of the phenotypic consequences in females. However, in this case cells lacking MeCP2 cause a neurological phenotype. Dysregulation of MeCP2 expression is also correlated with other neurological diseases including Alzheimer and Huntington disease (refer Ausio et al65 and references therein). Although most cases of RTT develop from the loss of MeCP2 function, gain in MeCP2 dosage can also result in similar neurological disorders. Therefore, MeCP2 dosage appears critical for proper brain function, which is crucial for considering potential therapies. One approach that has been considered is the reactivation of the repressed but intact MeCP2 copy from the Xi. This idea was suggested in a study that could show a reversal of several symptoms after restoring MeCP2 function in a RTT mouse model, which was based on a conditional mutation of the mouse MeCP2 gene.66 Xi reactivation has been achieved in a small percentage of treated cells by a combination of inhibitors against DNA methylation and histone deacetylation. Xi reactivation appears to be incomplete and gene specific in most cases. Investigating the mechanism for maintenance of XCI hold promise to advance the possibilities for reactivating genes form the Xi. Recently, a screen has identified several genes that are involved in maintaining silencing on X known as trans-acting XCI factors. Interference with these factors either by RNA interference or in certain cases by chemical inhibitors can induce an upregulation of MeCP2 from Xi. In particular, inhibition of PDPK1 or PI3K function led to reactivation of MeCP2 in postmitotic mouse cortical neurons.67 These findings suggest that restoration of MeCP2 expression from the Xi in neurons of RTT patients might be a potential therapeutic opportunity. However, more research is needed before clinical application can be considered.
Developmental regulation of XCI: induced-pluripotent stem cells and regenerative medicine
Chromosome-wide Xi reactivation occurs normally in early mouse embryo development and also accompanies the reprogramming of somatic cells to a pluripotent state. Nuclear transfer cloning, cell fusion with pluripotent cells, and generation of induced-pluripotent stem cells (iPSCs) have all been used to demonstrate the reactivation of a somatic Xi in mouse cells. A recent report defines many sequential steps of reprogramming somatic mouse cells to induce pluripotency and links them to development.68 Reactivation in these cases is chromosome wide and complete. Human iPSCs are currently considered for modeling diseases and screening for drugs. In addition, they hold great promise for applications in regenerative medicine.69 Whereas Xi reactivation is observed during the reprogramming of mouse cells, reactivation is variable in human cell reprogramming. Also human ESCs show considerable variability regarding their XCI status (reviewed in Lessing et al70). The difference between mouse and human cells seems to reflect embryonic development of both species. In human, embryos X inactivation is not initiated until implantation and appears to involve a period of Xist expression from both X chromosomes that does, however, not lead to chromosomal inactivation.2 Recently, new culture conditions for human iPSCs have been introduced that facilitate a similar morphology and resemble mouse iPSCs to an even greater extent.71–73 In these cultures, Xi reactivation in human iPSCs has been demonstrated.74 The new type of naive human iPSCs provides significant technical advantages that could be important for regenerative medicine, but formally it remains to be shown if these cells represent authentic cell types of the human embryo or resemble a culture-induced cell fate.75 Differences between the mechanism of XCI in human and mouse embryos have been noted. In mice, the antisense regulator Tsix has a prominent role for preventing Xist upregulation from the Xa.59 However, Tsix function is not conserved in human cells. The human X chromosome encodes another long-noncoding RNA, XACT that appears to prevent inactivation of the chromosome and thereby counteract initiation of XCI. XACT is remarkable as it accumulates over the Xa in a manner that appears similar to Xist but XACT mediates activation rather than silencing.76 XACT is, thus far, only described in human cells and its function remains to be characterized. Species differences in the mechanism of XCI underscore limitations of model organisms and raise the necessity of working in human cells or embryos, which can add considerable difficulty to experimental exploration but is most relevant to the clinical situation.
Female human ESCs can be assigned into three classes based on their XCI status.70 Class I cells possess two active X chromosomes (XaXa) and are able to undergo XCI and upregulate Xist when induced to differentiate suggesting similarity to mouse ESCs; Class II cells have already initiated XCI and contain one Xi; Class III cell lines appear to contain an Xi that is partially repressed and do not express Xist. The latter state is interpreted as to reflect an erosion of XCI and might point to an epigenetic instability of the Xi. Loss of Xist expression in human cells is linked to an altered transcriptional profile, somatic mutations, copy number variations, and immunogenicity.77 Hall et al report that most cell lines in a panel of National Institutes of Health–approved female human ESCs showed anomalies in XCI initiation upon differentiation, suggesting that XCI aberration could be a prevalent feature of current female human ESCs cultures.78 Notably, these abnormalities can also include upregulation of cancer-associated genes.79,80 It is not clear at the moment if XCI-related changes will limit the use of some human pluripotent cells for regenerative medicine. In general, human iPSCs have teratocarcinogenic potential and can form tumors when injected into immunocompromised mice.81 Additional risk in iPSCs might come from the introduced reprogramming factors. For the safe use of stem cell–based treatments, several suggestions have been made.82 It would be interesting to also explore criteria based on XCI for assessing the quality of female human iPSCs and human ESCs.
Since a pioneering study by Rudolf Jaenisch’s group has illustrated the therapeutic use of iPSCs in a mouse model of sickle cell anemia,83 patient-specific human iPSCs have been generated for several diseases, including RTT (reviewed in Bellin et al84). In July 2013, a first clinical iPSCs trial for dry age-related macular degeneration has been launched in Japan with the first enrolled female patient. The iPSCs in this trial were assessed to be genetically stable and safe by a number of assays. It could be useful to include the XCI status as an additional criterion for characterization and safety.
Problems with Xi maintenance in cancer
XCI is a prerequisite for female mammal development, and in mice, the presence of two active X chromosomes causes early embryonic lethality.85 Yet, somatic cells can tolerate a limited increase in dosage of X-linked genes, which further can provide a selective advantage that contributes to tumor progression (reviewed in Spatz et al51). A number of observations point to a correlation between cancer and aberrations of the Xi.86 However, the molecular mechanism behind this link is largely unknown. It is accepted that cancer cells can be chromosomally instable. Kawakami et al87 observed the loss of the inactive X and potential subsequent gain of active X chromosomes in female-derived cancer cells (Figure 2). Loss of the Xi could be related to an asynchronous replication at a late time in S-phase of the cell cycle, which could make it more prone to subsequent missegregation.88 In addition, late replication has also been suggested to make the Xi susceptible to mutations consistent with the observation of hyper-mutated Xi in tumor cells.89
How gene repression on the Xi is maintained in differentiated cells has been investigated by a number of studies. Particularly, DNA methylation and hypomethylation of histone H4 have been correlated with gene repression. Structural maintenance of chromosomes flexible hinge domain-containing protein 1 (SmcHD1) was identified in a screen for epigenetic modifiers in mice and subsequently shown to be required for repression and DNA methylation of X-linked genes on the Xi.90 SmcHD1 is enriched on the Xi and could play a critical role in establishing DNA methylation over gene promoters. DNA methylation appears critical for maintaining repression but less important for the initiation of XCI in earlier embryonic stages. Mutations in SmcHD1 cause facioscapulohumeral muscular dystrophy type 2 in humans but a link to XCI has not been investigated in these cases to date.91 Furthermore, mice carrying mutations in the maintenance DNA methyltransferase gene Dnmt1 display a reactivation of gene expression from Xi in embryonic development.92 This suggests that DNA methylation could contribute to gene repression on Xi. This view is also consistent with the observation that in immunodeficiency, centromeric instability and facial anomalies (ICF) syndrome, a rare autosomal recessive syndrome caused by mutations in the DNA methyltransferase DNMT3B, repression on the Xi is destabilized. Hypomethylation on Xi of female ICF patients has been reported. ICF syndrome is also associated with loss of Polycomb protein binding and loss of histone H3 lysine 27 trimethylation.93 Taken together, data from ICF patients could indicate a role for DNMT3B in Xi maintenance and stability.
An elevation of X-linked genes has been described in a broad range of female and rare male tumors including male breast cancer, which accounts for less than 1% of all breast cancers.94 Klinefelter syndrome is associated with a 20-fold higher risk of developing breast cancer than the risk in normal males.95 Multiple X chromosomes can be observed in a small number of female breast cancer (reported as XXX or XXXX96), testicular germ cell tumors,97 and acute lymphoblastic leukemia in children.98 An increased X chromosome relative to autosome (X:A) gene dosage has further been correlated with the progression from chronic phase to blast crisis in chronic neutrophilic leukemia.99 It is clear that more research is needed to further confirm the significance of these observations but together the evidence suggest that in a subset of tumors elevation of X-linked gene expression could be a driver of disease progression. Further support for this idea comes also from the relative increase in X-linked gene dosage in strong hypoploidy in rare human tumors,100 where only a single set of autosomes is present, and therefore, autosomal expression levels equalize with the X chromosome (X:A =1).101 Although near-haploid tumors are infrequent, they still could represent a significant intermediate or transient stage in tumor development. Haploid cells can undergo subsequent amplification in genome copy number and become hyperdiploid, whereby they retain largely homozygosity for genetic markers.87 After genome amplification, haploid phases become difficult to detect, and thus, their occurrence could be underestimated at present. Interestingly loss of a fragment of the active X chromosome is also observed in a wide variety of cancers.102–105 This could best be explained by the loss of tumor suppressor genes localized therein and would be associated with the progression in the severity of the tumor phenotype.
Xist expression and a morphologically distinct Xi are also absent in some cases of BRCA1-deficient breast cancer.106 Albeit initial reports have connected BRCA1 to the mechanism of Xist localization and silencing, more recent studies suggest that BRCA1 status does not always correlate with Xist expression or localization.107,108 The association between loss of Xist and cancer is further strengthened by a recent study in mice showing that deletion of Xist in blood cells can cause leukemia.14
In addition, Xist function has been inferred from ectopic XCI initiation in transformed human cells when expressing Xist from transgenes.109,110 Also in a mouse T-cell lymphoma model, forced expression of Xist by an inducible system caused cell death and regression of tumors.111 Xist is also expressed in human testicular germ cell tumors with multiple inactive X chromosomes, and evidence for initiation of X inactivation in tumor cells has been reported.97,112,113 The epigenetic or cellular context that facilitates the initiation of XCI could further be important for cancer progression. The special AT-rich binding protein (SATB1) is thought to play an important role as a silencing factor for Xist in a mouse lymphoma model. SATB1 has also been shown to reprogram breast tumor cells to a metastatic phenotype and could collaborate with loss of ATM or p16 in cellular transformation.114,115 Several studies now link SATB1 expression to an aggressive and metastatic phenotype in breast, rectal, liver, prostate, gastric cancer, and melanoma. Consequently, interference with the DNA-binding ability of SATB1 by pharmacological inhibition has been considered as a potential therapeutic strategy (reviewed in Kohwi-Shigematsu et al116). Taken together, these observations suggest that the context for Xist-mediated silencing as well as aberrations of X-linked gene doses can contribute to tumorigenesis by different mechanisms. Basic research may suggest opportunities for diagnostic and therapeutic strategies. Several studies already report on correlations between different aspects of XCI and the progression of tumor development and response to therapy.112,117–120
Final remarks
It is widely assumed that sex chromosomes have evolved from an ancestral pair of autosomes on which a mutation introduced a sex-determining role.121 The XY sex chromosome system is estimated to have appeared about 210–180 million years ago, which would place its emergence shortly before the split of metatherians (marsupials) and eutherians (placental mammals).122,123 In placental mammals, it is the Y chromosome that specifies male fate through the presence of the SRY gene. The progressive dimorphism between the sex chromosomes X and Y might have induced the evolution of the dosage compensation mechanism (XCI) to compensate for the relative dosage difference of X-gene expression between XX and XY. The dosage compensation system presents considerable molecular differences between mammalian species and might be considered as still evolving in humans. Interestingly, it appears that XCI in mouse is much more complete that in humans with only a few X-linked genes escaping from silencing.26 Ongoing sequence diversification and in turn dosage adjustments second to gene loss and emergence of dosage compensation could potentially contribute to disease predisposition. As the molecular details of the mechanism of XCI become better understood, insights offer opportunities for the clinics for diagnosis and potentially treatment of a subset of diseases. The mechanism of XCI has also been exploited to generate tools for therapy. A recent study is noteworthy for its demonstration of eliminating expression from one chromosome 21 in human iPSCs derived from a Down syndrome patient using transgenic Xist expression.124 This chromosome therapy could be considered for correcting for the phenotypic consequences of trisomy 21 in culture before cell grafts for application in regenerative approaches are explored. It appears that basic and clinical research could mutually benefit from exchange to unravel the different aspects of the mammalian dosage compensation mechanism in a collaborative manner.
Acknowledgments
The authors thank A Monfort and T Beyer for critically reading the manuscript. This work was supported by the Swiss National Science Foundation (SNF grant 31003A_152814/1). KV is supported by “Fundación Alfonso Martín Escudero”.
Disclosure
The authors declare no conflict of interest.
References
Lyon MF. Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature. 1961;190:372–373. | |
Okamoto I, Patrat C, Thépot D, et al. Eutherian mammals use diverse strategies to initiate X-chromosome inactivation during development. Nature. 2011;472(7343):370–374. | |
Avner P, Heard E. X-chromosome inactivation: counting, choice and initiation. Nat Rev Genet. 2001;2(1):59–67. | |
Augui S, Nora EP, Heard E. Regulation of X-chromosome inactivation by the X-inactivation centre. Nat Rev Genet. 2011;12(6):429–442. | |
Jeon Y, Lee JT. YY1 tethers Xist RNA to the inactive X nucleation center. Cell. 2011;146(1):119–133. | |
Sarma K, Cifuentes-Rojas C, Ergun A, et al. ATRX directs binding of PRC2 to Xist RNA and polycomb targets. Cell. 2014;159(4):869–883. | |
da Rocha ST, Boeva V, Escamilla-Del-Arenal M, et al. Jarid2 is implicated in the initial xist-induced targeting of PRC2 to the inactive X chromosome. Mol Cell. 2014;53(2):301–316. | |
Marahrens Y, Panning B, Dausman J, Strauss W, Jaenisch R. Xist-deficient mice are defective in dosage compensation but not spermatogenesis. Genes Dev. 1997;11(2):156–166. | |
Penny GD, Kay GF, Sheardown SA, Rastan S, Brockdorff N. Requirement for Xist in X chromosome inactivation. Nature. 1996;379(6561):131–137. | |
Brown CJ, Willard HF. The human X-inactivation centre is not required for maintenance of X-chromosome inactivation. Nature. 1994;368(6467):154–156. | |
Csankovszki G, Panning B, Bates B, Pehrson JR, Jaenisch R. Conditional deletion of Xist disrupts histone macroH2A localization but not maintenance of X inactivation. Nat Genet. 1999;22(4):323–324. | |
Lee JT. Gracefully ageing at 50, X-chromosome inactivation becomes a paradigm for RNA and chromatin control. Nat Rev Mol Cell Biol. 2011;12(12):815–826. | |
Wutz A. Gene silencing in X-chromosome inactivation: advances in understanding facultative heterochromatin formation. Nat Rev Genet. 2011;12(8):542–553. | |
Yildirim E, Kirby JE, Brown DE, et al. Xist RNA is a potent suppressor of hematologic cancer in mice. Cell. 2013;152(4):727–742. | |
Payer B, Lee JT. Coupling of X-chromosome reactivation with the pluripotent stem cell state. RNA Biol. 2014;11(7):798–807. | |
Engreitz JM, Pandya-Jones A, McDonel P, et al. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science. 2013;341(6147):1237973. | |
Reindollar RH. Turner syndrome: contemporary thoughts and reproductive issues. Semin Reprod Med. 2011;29(4):342–352. | |
Tuttelmann F, Gromoll J. Novel genetic aspects of Klinefelter’s syndrome. Mol Hum Reprod. 2010;16(6):386–395. | |
Otter M, Schrander-Stumpel CT, Curfs LM. Triple X syndrome: a review of the literature. Eur J Hum Genet. 2010;18(3):265–271. | |
Heard E, Turner J. Function of the sex chromosomes in mammalian fertility. Cold Spring Harb Perspect Biol. 2011;3(10):a002675. | |
Linden MG, Bender BG, Robinson A. Sex chromosome tetrasomy and pentasomy. Pediatrics. 1995;96(4 pt 1):672–682. | |
Disteche CM, Filippova GN, Tsuchiya KD. Escape from X inactivation. Cytogenet Genome Res. 2002;99(1–4):36–43. | |
Tartaglia N, Cordeiro L, Howell S, Wilson R, Janusz J. The spectrum of the behavioral phenotype in boys and adolescents 47, XXY (Klinefelter syndrome). Pediatr Endocrinol Rev. 2010;8(Suppl 1):151–159. | |
Zinn AR, Ross JL. Molecular analysis of genes on Xp controlling Turner syndrome and premature ovarian failure (POF). Semin Reprod Med. 2001;19(2):141–146. | |
Burgoyne PS, Baker TG. Perinatal oocyte loss in XO mice and its implications for the aetiology of gonadal dysgenesis in XO women. J Reprod Fertil. 1985;75(2):633–645. | |
Berletch JB, Yang F, Disteche CM. Escape from X inactivation in mice and humans. Genome Biol. 2010;11(6):213. | |
Lue YH, Wang C, Liu PY, Erkilla K, Swerdloff RS. Insights into the pathogenesis of XXY phenotype from comparison of the clinical syndrome with an experimental XXY mouse model. Pediatr Endocrinol Rev. 2010;8(Suppl 1):140–144. | |
Endo A, Watanabe T. A case of X-trisomy in the mouse. Cytogenet Cell Genet. 1989;52(1–2):98–99. | |
Chaumeil J, Le Baccon P, Wutz A, Heard E. A novel role for Xist RNA in the formation of a repressive nuclear compartment into which genes are recruited when silenced. Genes Dev. 2006;20(16):2223–2237. | |
Splinter E, de Wit E, Nora EP, et al. The inactive X chromosome adopts a unique three-dimensional conformation that is dependent on Xist RNA. Genes Dev. 2011;25(13):1371–1383. | |
Filippova GN, Cheng MK, Moore JM, et al. Boundaries between chromosomal domains of X inactivation and escape bind CTCF and lack CpG methylation during early development. Dev Cell. 2005;8(1):31–42. | |
Horvath LM, Li N, Carrel L. Deletion of an X-inactivation boundary disrupts adjacent gene silencing. PLoS Genet. 2013;9(11):e1003952. | |
Simon MD, Pinter SF, Fang R, et al. High-resolution Xist binding maps reveal two-step spreading during X-chromosome inactivation. Nature. 2013;504(7480):465–469. | |
Kung JT, Kesner B, An JY, et al. Locus-specific targeting to the X chromosome revealed by the RNA interactome of CTCF. Mol Cell. 2015;57(2):361–375. | |
Chow J, Heard E. X inactivation and the complexities of silencing a sex chromosome. Curr Opin Cell Biol. 2009;21(3):359–366. | |
Chow JC, Ciaudo C, Fazzari MJ, et al. LINE-1 activity in facultative heterochromatin formation during X chromosome inactivation. Cell. 2010;141(6):956–969. | |
Bailey JA, Carrel L, Chakravarti A, Eichler EE. Molecular evidence for a relationship between LINE-1 elements and X chromosome inactivation: the Lyon repeat hypothesis. Proc Natl Acad Sci U S A. 2000;97(12):6634–6639. | |
Carrel L, Park C, Tyekucheva S, Dunn J, Chiaromonte F, Makova KD. Genomic environment predicts expression patterns on the human inactive X chromosome. PLoS Genet. 2006;2(9):e151. | |
Ross MT, Grafham DV, Coffey AJ, et al. The DNA sequence of the human X chromosome. Nature. 2005;434(7031):325–337. | |
Li N, Carrel L. Escape from X chromosome inactivation is an intrinsic property of the Jarid1c locus. Proc Natl Acad Sci U S A. 2008;105(44):17055–17060. | |
Tsuchiya KD, Greally JM, Yi Y, Noel KP, Truong JP, Disteche CM. Comparative sequence and x-inactivation analyses of a domain of escape in human xp11.2 and the conserved segment in mouse. Genome Res. 2004;14(7):1275–1284. | |
Wang Z, Willard HF, Mukherjee S, Furey TS. Evidence of influence of genomic DNA sequence on human X chromosome inactivation. PLoS Comput Biol. 2006;2(9):e113. | |
Nguyen DK, Yang F, Kaul R, et al. Clcn4-2 genomic structure differs between the X locus in Mus spretus and the autosomal locus in Mus musculus: AT motif enrichment on the X. Genome Res. 2011;21(3):402–409. | |
Mugford JW, Starmer J, Williams RL Jr, et al. Evidence for local regulatory control of escape from imprinted X chromosome inactivation. Genetics. 2014;197(2):715–723. | |
Yang F, Babak T, Shendure J, Disteche CM. Global survey of escape from X inactivation by RNA-sequencing in mouse. Genome Res. 2010; 20(5):614–622. | |
Carrel L, Willard HF. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature. 2005;434(7031):400–404. | |
Xu J, Disteche CM. Sex differences in brain expression of X- and Y-linked genes. Brain Res. 2006;1126(1):50–55. | |
Viana J, Pidsley R, Troakes C, et al. Epigenomic and transcriptomic signatures of a Klinefelter syndrome (47, XXY) karyotype in the brain. Epigenetics. 2014;9(4):587–599. | |
Berletch JB, Yang F, Xu J, Carrel L, Disteche CM. Genes that escape from X inactivation. Hum Genet. 2011;130(2):237–245. | |
Deng X, Berletch JB, Nguyen DK, Disteche CM. X chromosome regulation: diverse patterns in development, tissues and disease. Nat Rev Genet. 2014;15(6):367–378. | |
Spatz A, Borg C, Feunteun J. X-chromosome genetics and human cancer. Nat Rev Cancer. 2004;4(8):617–629. | |
Jameson KA, Highnote SM, Wasserman LM. Richer color experience in observers with multiple photopigment opsin genes. Psychon Bull Rev. 2001;8(2):244–261. | |
Migeon BR. Non-random X chromosome inactivation in mammalian cells. Cytogenet Cell Genet. 1998;80(1–4):142–148. | |
Wu H, Luo J, Yu H, et al. Cellular resolution maps of X chromosome inactivation: implications for neural development, function, and disease. Neuron. 2014;81(1):103–119. | |
Clerc P, Avner P. Random X-chromosome inactivation: skewing lessons for mice and men. Curr Opin Genet Dev. 2006;16(3):246–253. | |
Cattanach BM. Controlling elements in the mouse X-chromosome. 3. Influence upon both parts of an X divided by rearrangement. Genet Res. 1970;16(3):293–301. | |
Calaway JD, Lenarcic AB, Didion JP, et al. Genetic architecture of skewed X inactivation in the laboratory mouse. PLoS Genet. 2013;9(10):e1003853. | |
Thorvaldsen JL, Krapp C, Willard HF, Bartolomei MS. Nonrandom X chromosome inactivation is influenced by multiple regions on the murine X chromosome. Genetics. 2012;192(3):1095–1107. | |
Lee JT, Davidow LS, Warshawsky D. Tsix, a gene antisense to Xist at the X-inactivation centre. Nat Genet. 1999;21(4):400–404. | |
Bolduc V, Chagnon P, Provost S, et al. No evidence that skewing of X chromosome inactivation patterns is transmitted to offspring in humans. J Clin Invest. 2008;118(1):333–341. | |
Renault NK, Dyack S, Dobson MJ, Costa T, Lam WL, Greer WL. Heritable skewed X-chromosome inactivation leads to haemophilia A expression in heterozygous females. Eur J Hum Genet. 2007;15(6):628–637. | |
Cazzola M, May A, Bergamaschi G, Cerani P, Rosti V, Bishop DF. Familial-skewed X-chromosome inactivation as a predisposing factor for late-onset X-linked sideroblastic anemia in carrier females. Blood. 2000;96(13):4363–4365. | |
Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56(3):422–437. | |
Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23(2):185–188. | |
Ausio J, de Paz AM, Esteller M. MeCP2: the long trip from a chromatin protein to neurological disorders. Trends Mol Med. 2014;20(9):487–498. | |
Cobb S, Guy J, Bird A. Reversibility of functional deficits in experimental models of Rett syndrome. Biochem Soc Trans. 2010;38(2):498–506. | |
Bhatnagar S, Zhu X, Ou J, et al. Genetic and pharmacological reactivation of the mammalian inactive X chromosome. Proc Natl Acad Sci U S A. 2014;111(35):12591–12598. | |
Pasque V, Tchieu J, Karnik R, et al. X chromosome reactivation dynamics reveal stages of reprogramming to pluripotency. Cell. 2014;159(7):1681–1697. | |
Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–676. | |
Lessing D, Anguera MC, Lee JT. X chromosome inactivation and epigenetic responses to cellular reprogramming. Annu Rev Genomics Hum Genet. 2013;14:85–110. | |
Gafni O, Weinberger L, Mansour AA, et al. Derivation of novel human ground state naive pluripotent stem cells. Nature. 2013;504(7479):282–286. | |
Theunissen TW, Powell BE, Wang H, et al. Systematic identification of culture conditions for induction and maintenance of naive human pluripotency. Cell Stem Cell. 2014;15(4):471–487. | |
Takashima Y, Guo G, Loos R, et al. Resetting transcription factor control circuitry toward ground-state pluripotency in human. Cell. 2014;158(6):1254–1269. | |
Hanna J, Cheng AW, Saha K, et al. Human embryonic stem cells with biological and epigenetic characteristics similar to those of mouse ESCs. Proc Natl Acad Sci U S A. 2010;107(20):9222–9227. | |
Huang K, Maruyama T, Fan G. The naive state of human pluripotent stem cells: a synthesis of stem cell and preimplantation embryo transcriptome analyses. Cell Stem Cell. 2014;15(4):410–415. | |
Vallot C, Huret C, Lesecque Y, et al. XACT, a long noncoding transcript coating the active X chromosome in human pluripotent cells. Nat Genet. 2013;45(3):239–241. | |
Liang G, Zhang Y. Genetic and epigenetic variations in iPSCs: potential causes and implications for application. Cell Stem Cell. 2013;13(2):149–159. | |
Hall LL, Byron M, Butler J, et al. X-inactivation reveals epigenetic anomalies in most hESC but identifies sublines that initiate as expected. J Cell Physiol. 2008;216(2):445–452. | |
Bruck T, Yanuka O, Benvenisty N. Human pluripotent stem cells with distinct X inactivation status show molecular and cellular differences controlled by the X-Linked ELK-1 gene. Cell Rep. 2013;4(2):262–270. | |
Anguera MC, Sadreyev R, Zhang Z, et al. Molecular signatures of human induced pluripotent stem cells highlight sex differences and cancer genes. Cell Stem Cell. 2012;11(1):75–90. | |
Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448(7151):313–317. | |
World Health Organization. Recommendations for the evaluation of animal cell cultures as substrates for the manufacture of biological medicinal products and for the characterization of cell banks. WHO Technical Report Series No 978, Annex 3; 2013. Available from: http://www.who.int/biologicals/vaccines/TRS_978_Annex_3.pdf. | |
Hanna J, Wernig M, Markoulaki S, et al. Treatment of sickle cell anemia mouse model with iPS cells generated from autologous skin. Science. 2007;318(5858):1920–1923. | |
Bellin M, Marchetto MC, Gage FH, Mummery CL. Induced pluripotent stem cells: the new patient? Nat Rev Mol Cell Biol. 2012;13(11):713–726. | |
Schulz EG, Meisig J, Nakamura T, et al. The two active X chromosomes in female ESCs block exit from the pluripotent state by modulating the ESC signaling network. Cell Stem Cell. 2014;14(2):203–216. | |
Chaligné R, Popova T, Mendoza-Parra MA, et al. The inactive X chromosome is epigenetically unstable and transcriptionally labile in breast cancer. Genome Res. Epub 2015 Feb 4. | |
Kawakami T, Zhang C, Taniguchi T, et al. Characterization of loss-of-inactive X in Klinefelter syndrome and female-derived cancer cells. Oncogene. 2004;23(36):6163–6169. | |
Dutrillaux B, Muleris M, Seureau MG. Imbalance of sex chromosomes, with gain of early-replicating X, in human solid tumors. Int J Cancer. 1986;38(4):475–479. | |
Jäger N, Schlesner M, Jones DT, et al. Hypermutation of the inactive X chromosome is a frequent event in cancer. Cell. 2013;155(3):567–581. | |
Blewitt ME, Gendrel AV, Pang Z, et al. SmcHD1, containing a structural-maintenance-of-chromosomes hinge domain, has a critical role in X inactivation. Nat Genet. 2008;40(5):663–669. | |
Lemmers RJ, Tawil R, Petek LM, et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat Genet. 2012; 44(12):1370–1374. | |
Gribnau J, Hochedlinger K, Hata K, Li E, Jaenisch R. Asynchronous replication timing of imprinted loci is independent of DNA methylation, but consistent with differential subnuclear localization. Genes Dev. 2003;17(6):759–773. | |
Matarazzo MR, De Bonis ML, Vacca M, Della Ragione F, D’Esposito M. Lessons from two human chromatin diseases, ICF syndrome and Rett syndrome. Int J Biochem Cell Biol. 2009;41(1):117–126. | |
Rudas M, Schmidinger M, Wenzel C, et al. Karyotypic findings in two cases of male breast cancer. Cancer Genet Cytogenet. 2000;121(2):190–193. | |
Scheike O, Visfeldt J, Petersen B. Male breast cancer. 3. Breast carcinoma in association with the Klinefelter syndrome. Acta Pathol Microbiol Scand A. 1973;81(3):352–358. | |
Rooman RP, Van Driessche K, Du Caju MV. Growth and ovarian function in girls with 48, XXXX karyotype – patient report and review of the literature. J Pediatr Endocrinol Metab. 2002;15(7):1051–1055. | |
Kawakami T, Okamoto K, Sugihara H, et al. The roles of supernumerical X chromosomes and XIST expression in testicular germ cell tumors. J Urol. 2003;169(4):1546–1552. | |
Heinonen K, Mahlamaki E, Riikonen P, Meltoranta RL, Rahiala J, Perkkio M. Acquired X-chromosome aneuploidy in children with acute lymphoblastic leukemia. Med Pediatr Oncol. 1999;32(5):360–365. | |
Yamamoto K, Nagata K, Kida A, Hamaguchi H. Acquired gain of an X chromosome as the sole abnormality in the blast crisis of chronic neutrophilic leukemia. Cancer Genet Cytogenet. 2002;134(1):84–87. | |
Sukov WR, Ketterling RP, Wei S, et al. Nearly identical near-haploid karyotype in a peritoneal mesothelioma and a retroperitoneal malignant peripheral nerve sheath tumor. Cancer Genet Cytogenet. 2010;202(2):123–128. | |
Safavi S, Forestier E, Golovleva I, et al. Loss of chromosomes is the primary event in near-haploid and low-hypodiploid acute lymphoblastic leukemia. Leukemia. 2013;27(1):248–250. | |
Xu J, Meyers D, Freije D, et al. Evidence for a prostate cancer susceptibility locus on the X chromosome. Nat Genet. 1998;20(2):175–179. | |
Piao Z, Malkhosyan SR. Frequent loss Xq25 on the inactive X chromosome in primary breast carcinomas is associated with tumor grade and axillary lymph node metastasis. Genes Chromosomes Cancer. 2002; 33(3):262–269. | |
Choi C, Cho S, Horikawa I, et al. Loss of heterozygosity at chromosome segment Xq25-26.1 in advanced human ovarian carcinomas. Genes Chromosomes Cancer. 1997;20(3):234–242. | |
Mathur M, Das S, Samuels HH. PSF-TFE3 oncoprotein in papillary renal cell carcinoma inactivates TFE3 and p53 through cytoplasmic sequestration. Oncogene. 2003;22(32):5031–5044. | |
Ganesan S, Silver DP, Greenberg RA, et al. BRCA1 supports XIST RNA concentration on the inactive X chromosome. Cell. 2002;111(3):393–405. | |
Vincent-Salomon A, Ganem-Elbaz C, Manié E, et al. X inactive-specific transcript RNA coating and genetic instability of the X chromosome in BRCA1 breast tumors. Cancer Res. 2007;67(11):5134–5140. | |
Pageau GJ, Hall LL, Lawrence JB. BRCA1 does not paint the inactive X to localize XIST RNA but may contribute to broad changes in cancer that impact XIST and Xi heterochromatin. J Cell Biochem. 2007;100(4):835–850. | |
Chow JC, Hall LL, Baldry SE, Thorogood NP, Lawrence JB, Brown CJ. Inducible XIST-dependent X-chromosome inactivation in human somatic cells is reversible. Proc Natl Acad Sci U S A. 2007;104(24):10104–10109. | |
Hall LL, Byron M, Sakai K, Carrel L, Willard HF, Lawrence JB. An ectopic human XIST gene can induce chromosome inactivation in postdifferentiation human HT-1080 cells. Proc Natl Acad Sci U S A. 2002;99(13):8677–8682. | |
Agrelo R, Souabni A, Novatchkova M, et al. SATB1 defines the developmental context for gene silencing by Xist in lymphoma and embryonic cells. Dev Cell. 2009;16(4):507–516. | |
Kawakami T, Okamoto K, Ogawa O, Okada Y. XIST unmethylated DNA fragments in male-derived plasma as a tumour marker for testicular cancer. Lancet. 2004;363(9402):40–42. | |
Looijenga LH, Gillis AJ, van Gurp RJ, Verkerk AJ, Oosterhuis JW. X inactivation in human testicular tumors. XIST expression and androgen receptor methylation status. Am J Pathol. 1997;151(2):581–590. | |
Agrelo R, Kishimoto H, Novatchkova M, et al. SATB1 collaborates with loss of p16 in cellular transformation. Oncogene. 2013;32(48):5492–5500. | |
Ordinario E, Han HJ, Furuta S, et al. ATM suppresses SATB1-induced malignant progression in breast epithelial cells. PLoS One. 2012;7(12):e51786. | |
Kohwi-Shigematsu T, Poterlowicz K, Ordinario E, Han HJ, Botchkarev VA, Kohwi Y. Genome organizing function of SATB1 in tumor progression. Semin Cancer Biol. 2013;23(2):72–79. | |
Koivisto P, Hyytinen E, Palmberg C, et al. Analysis of genetic changes underlying local recurrence of prostate carcinoma during androgen deprivation therapy. Am J Pathol. 1995;147(6):1608–1614. | |
Thompson J, Hyytinen ER, Haapala K, et al. Androgen receptor mutations in high-grade prostate cancer before hormonal therapy. Lab Invest. 2003;83(12):1709–1713. | |
Piao Z, Lee KS, Kim H, Perucho M, Malkhosyan S. Identification of novel deletion regions on chromosome arms 2q and 6p in breast carcinomas by amplotype analysis. Genes Chromosomes Cancer. 2001;30(2):113–122. | |
Huang KC, Rao PH, Lau CC, et al. Relationship of XIST expression and responses of ovarian cancer to chemotherapy. Mol Cancer Ther. 2002;1(10):769–776. | |
Muller HJ. The relation of recombination to mutational advance. Mutat Res. 1964;106:2–9. | |
Luo ZX, Yuan CX, Meng QJ, Ji Q. A Jurassic eutherian mammal and divergence of marsupials and placentals. Nature. 2011;476(7361):442–445. | |
Marshall Graves JA. Weird animal genomes and the evolution of vertebrate sex and sex chromosomes. Annu Rev Genet. 2008;42:565–586. | |
Jiang J, Jing Y, Cost GJ, et al. Translating dosage compensation to trisomy 21. Nature. 2013;500(7462):296–300. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.