")
Back to Journals » Clinical and Experimental Gastroenterology » Volume 9
Recent insights into the molecular pathogenesis of Crohn's disease: a review of emerging therapeutic targets
Authors Manuc T, Manuc M, Diculescu M
Received 19 June 2015
Accepted for publication 21 September 2015
Published 15 March 2016 Volume 2016:9 Pages 59—70
DOI https://doi.org/10.2147/CEG.S53381
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Andreas M. Kaiser
Teodora-Ecaterina M Manuc,1 Mircea M Manuc,2 Mircea M Diculescu2
1Fundeni Clinical Institute, 2University of Medicine and Pharmacy "Carol Davila", Bucharest, Romania
Abstract: Chronic inflammatory bowel diseases (IBDs) are a subject of great interest in gastroenterology, due to a pathological mechanism that is difficult to explain and an optimal therapeutic approach still undiscovered. Crohn's disease (CD) is one of the main entities in IBD, characterized by clinical polymorphism and great variability in the treatment response. Modern theories on the pathogenesis of CD have proven that gut microbiome and environmental factors lead to an abnormal immune response in a genetically predisposed patient. Genome-wide association studies in patients with CD worldwide revealed several genetic mutations that increase the risk of IBD and that predispose to a more severe course of disease. Gut microbiota is considered a compulsory and an essential part in the pathogenesis of CD. Intestinal dysmicrobism with excessive amounts of different bacterial strains can be found in all patients with IBD. The discovery of Escherichia coli entero-invasive on resection pieces in patients with CD now increases the likelihood of antimicrobial or vaccine-type treatments. Recent studies targeting intestinal immunology and its molecular activation pathways provide new possibilities for therapeutics. In addition to antitumor necrosis factor molecules, which were a breakthrough in IBD, improving mucosal healing and resection-free survival rate, other classes of therapeutic agents come to focus. Leukocyte adhesion inhibitors block the leukocyte homing mechanism and prevent cellular immune response. In addition to anti-integrin antibodies, chemokine receptor antagonists and SMAD7 antisense oligonucleotides have shown encouraging results in clinical trials. Micro-RNAs have demonstrated their role as disease biomarkers but it could also become useful for the treatment of IBD. Moreover, cellular therapy is another therapeutic approach under development, aimed for severe refractory CD. Other experimental treatments include intravenous immunoglobulins, exclusive enteral nutrition, and granulocyte colony-stimulating factors.
Keywords: microbiota, GWAS, biologic therapy, micro-RNA, stem cell therapy
Introduction
Crohn’s disease (CD) is an immune-mediated inflammatory bowel disease (IBD), with a rising incidence during the past decade. It generally affects younger people with the age of onset between 15 and 40 years and has an incidence of 3.1 to 14.6 cases per 100,000 person-years in North America.1 Clinical, endoscopic, and pathological criteria for a CD diagnosis are highly variable due to the many possible disease patterns. Definitive characteristics for the CD are “skipping” intestinal lesions, presence of transmural inflammation, sometimes complicated by fistulae, and the presence of non-caseous granuloma on intestinal biopsies.2
Experimental medicine through murine models brought the opportunity of gene manipulation, thus revealing the importance of genetic determinants and triggering or protective factors of disease. Progress was also made in mucosal immunology, where recent evidence suggests that a defective innate immune response after disruption of the epithelial barrier is the primary defect in CD.3 Several mechanisms were incriminated in this process, such as increased intestinal permeability, alteration in mucus layer, bacterial aggression, chemical substances, and changes in the tight intercellular junctions.4–6
Activation of nuclear factor (NF)-kB pathway with the overproduction of tumor necrosis factor (TNF) alpha is already widely known as the central effector arm leading to inflammation.7 This common pathway bringing the anti-tumor necrosis factor (anti-TNF) to clinical practice was definitely a breakthrough in terms of disease management and outcome.8 However, almost 30% of patients do not respond to anti-TNF therapy or lose response in time,9 while adverse reactions such as systemic infections or malignancies are not negligible over the course of the treatment.10 In this context, the need for new, more potent, and targeted molecules is obvious.
This review will gather recent pathogenic factors incriminated, as well as the latest treatment options both available and under investigation, which might change the current management of the disease.
Methods
An electronic search of publications on PubMed database (through May 2015) was conducted. We also reviewed the reference lists of each included article and relevant review articles. The selection of studies presented was based on the most recently published trial results. Ongoing clinical trials were assessed from the pharmaceutical developers’ sites and from international IBD Foundations that are currently enrolling patients (http://www.clinicatrials.gov and http://www.clinicaltrialsregister.eu).
Pathogenic factors
The current pathogenic mechanism in CD is defined by an abnormal response of the intestinal immunity to innocuous luminal antigens in a genetically predisposed host, resulting in transmural, patchy inflammation of the intestinal wall. A first step in the process of the disease is the disruption of the intestinal barrier, permitting bacterial antigen passage into the submucosa and lamina propria.4 In patients with CD, the immune response is imbalanced through various factors, such as genetic predisposition, altered autophagy, and increased cytotoxicity of T cells. The defective immune response leads to increased cellular recruitment and overexpression of pro-inflammatory cytokines in association with diminished levels of immunosuppressive cytokines.
Genetic factors
The first signs for the genetic basis in CD started with monozygotic twin studies and other familial clusters of IBD.11 Lately, genome-wide association studies (GWAS) have identified more than 150 genetic risk loci for IBD, 70 of which may be associated with CD. There is also a great variation between European, American, and Asian populations with different gene mutations that can predispose to CD.12
The most important association is, by far, the nucleotide-binding oligomerization domain-containing protein 2 (NOD2)/caspase recruitment domain-containing protein 15 (CARD15) gene polymorphism, participating in the innate immune response. NOD2 is a protein in the NF-kB pathway, which acts as an intracellular sensor for muramyl dipeptide (bacterial wall component). Several experimental studies have shown that patients with NOD2 mutations have an inadequate antibacterial response, possibly through lower production of alpha defensins (antibacterial molecules produced by Paneth cells) or by impairment in the autophagy cascade, leading to increased levels of NF-kB.13 Clinically, variants of NOD2 are associated with ileal involvement, a stenosing or fistulizing pattern of disease and a higher risk of surgery.14 Nonetheless, GWAS studies in Korea found that NOD2 mutation is not present in their patients, even though clinical pattern is similar to Caucasians.15
Other genetic variants that may lead to an increased risk of CD are: Toll-like receptor 4 (TLR-4), CARD9, interleukin 23 receptor (IL-23R), signal transducer and activator of transcription 3 (STAT3) for innate immunity; human leukocyte antigen, tumor necrosis factor ligand superfamily 15 (TNFSF15), interferon regulatory factor 5 (IRF-5), protein tyrosine phosphatase non-receptor type 22 (PTPN-22) for adaptative immune system, etc.12
Due to the fact that GWAS results were consistent only for NOD2, larger sample sizes for gene susceptibility evaluation are required. Therefore, an International IBD Genetics Consortium was created. Until the present date, three GWAS have been published as a product of International IBD Genetics Consortium. These studies include more than 75,000 patients and evaluations for over 140 risk loci for CD.12,16
Genetic variations in the autophagic pathway can also increase the risk of CD. Autophagy is a lysosomal recycling mechanism of the cytoplasm that plays an important role in the innate immune response toward intracellular bacteria. Another pathway with close interaction to the autophagic pathway is the unfolded protein response induced by endoplasmic reticulum stress. Autophagy-related 16-like 1 gene (ATG16L1) and immunity-related guanosine triphosphatase gene (IRGM) have been linked to a higher susceptibility of CD.17
Individually, these mutations carry a moderate risk and need other associated factors like microbial interaction to trigger the disease. Future genetic research by fine-mapping and whole-genome sequencing studies, followed by statistical analysis of the population, will probably guide us toward new potential therapeutics.12
Micro-RNAs (miRNAs) are noncoding RNAs, which act like posttranscriptional factors in gene expression, altering gene transcription, and various mechanisms of cell signaling in innate and adaptive immunity. Over 5,400 miRNAs have been identified so far, each carrying possible implications in autoimmune-mediated diseases.18 In the context of IBD, correlations to the NOD-like receptors, TLRs and T-helper cells, especially Th17, were assessed in multiple investigational studies.19 The therapeutic application of miRNAs is not well established, but could comprise miRNA agonists and antagonists. The limitations of these molecules are represented by difficulty to target a specific organ and the associated systemic adverse reactions. However, the variability of miRNAs depending on the type of IBD, disease pattern, and response to treatment suggests that it could become a biomarker of disease.20
Intestinal barrier
The intestinal wall has four layers: mucosa, submucosa, muscular layer, and serosa. Epithelial cells in the mucosa are a dynamic interface and form a physical, chemical, and immune barrier. In normal people, a mucus layer protects the epithelium and the layers below from luminal bacteria and allows interactions mainly through the Peyer’s patches. The diminished mucosal protection may lead to an increased bacterial adhesion and invasion, with a final inflammatory process. Disruption in the mucosal barrier is now thought to be a key element in the onset of disease and in the frequent relapses.4 Alteration in the small vasculature of the mucosal layer followed by the appearance of aphthous ulcers is the earliest pathological and endoscopic step in the course of the disease.21 Some mechanisms incriminated are a low production of antimicrobial proteins by Paneth cells (alpha-defensin in ileal CD and beta-defensins, LL-37, SLPI, elafin in colonic CD), impaired autophagy, and increased permeability of the intestinal epithelium (genetic, environmental, and lifestyle factors are implicated).4,22
Microbial factors
The hypothesis of a microbial involvement in CD can be supported by the location of the lesions with aphthous ulcers appearing in Peyer’s patches, by the predominance of lesions where the bacterial concentration is the highest and also by the favorable therapeutic effects of temporary transit derivation and antibiotics in moderate flares.23
Gut microbiota is the subject of an unprecedented interest in all the fields of gastroenterology. All recent studies confirm the existence of a dysmicrobiosis phenomenon with low levels of Firmicutes, Bifidobacteria, and Lactobacilli, while higher numbers of Escherichia coli and other strains of Enterobacteriae.6,24,25
Adherent-invasive E. coli (AIEC) is a new bacterial entity emerging from observational studies proving that E. coli in CD patients has a greater pathogenic effect than those in control subjects.24 In a study on CD intestinal tissue, Darfeuille-Michaud found this type of E. coli in 65% of intestinal chronic lesions and in 100% of early postoperative recurrence lesions.26 In CD patients, invasive bacteria can enter the macrophages and stimulate the TNF-alpha production, while possibly promoting granuloma formation (in vitro finding). Other in vitro findings relate AIEC to a lower expression of miRNA leading to a reduced autophagic process.27
In animals, Mycobacterium avium paratuberculosis (MAP) is an infection that causes a CD-like disease with fibrosing and stenosing patterns and a predisposition to granuloma formation. Human exposure to this agent is achieved via environmental factors (water, meat, milk, and breast milk), and it can be isolated in blood cultures in patients with IBD and in control subjects.28 Its influence in IBD and CD is highly debatable with several counter-arguments, such as the higher prevalence of MAP DNA in healthy subjects compared to the patients with IBD (47%–16%), the failure of antibiotic therapy, and the effect of immunosuppressive therapy, which could worsen the MAP infection.28–30 Other findings supporting a possible implication of MAP were the presence of antibodies against Saccharomyces cerevisiae (ASCA) in the plasma of patients with CD (MAP is a source of ASCA-epitope) and the in vitro studies showing that MAP impairs E. coli phagocytosis by the macrophages.30 Genetic research found that SLC11A1 gene polymorphism was associated with CD and MAP infection.31 Therapeutic implications of MAP are currently under development.
The involvement of Helicobacter pylori (HP) infection in CD pathogenesis has been highly debated with discordant results between researchers. Epidemiological studies showed that HP infection is inversely correlated with the IBD demonstrating CD onset shortly after HP eradication with case reports.32 Another retrospective study from People’s Republic of China found that decreased HP infection was parallel to increased CD severity.33
Clostridium difficile infection is a risk factor for flare-ups in patients with IBD probably due to a disrupted microbiotic environment and frequent antibiotic treatment. Even though it can complicate the course of disease, no correlation with the pathogenesis of IBD has been proved.34 Campylobacter and Salmonella infections were associated with a higher risk of IBD in the first year after a positive stool test. However, in Jess et al study, patients with a negative stool test also had a high risk for IBD within the first year.35
Environmental components
There are many theories that correlate possible environmental factors with the higher incidence of CD in some specific populations. The frequency of IBD in developed countries is definitely higher than in the poorer countries, suggesting that low exposure to pathogenic infections could disturb the mucosal immune balance, thus increasing the risk of IBD.36 Another aspect is the increased rate of parasitic colonization in underdeveloped countries. Helminths elicit a type-2 T-helper cell response (Th2), which lowers the Th1 response commonly increased in CD.37
Lifestyle choices, such as previous or present smoking, have been typically associated with a higher risk of CD and an increased number of flares in those patients.38 The underlying mechanism can be related to an increased epithelial cell apoptosis, a higher intestinal permeability, and also to changes in the mucosal immune response without a clearly proven correlation. Nicotine can also influence the intestinal immune balance by lowering T-cell proliferation and altering macrophagic response.39
Diet is considered a pathological trigger in some cases, as nutritional principles can affect the intestinal permeability and the clearance of bacterial antigens, consequently influencing the immune system.40 Extrapolating from the effectiveness of exclusive enteral nutrition in pediatric CD,41 there have been many recent studies targeting the dietary components that increase the risk of CD.42 In a recent study, Kawaguchi et al showed that food antigens can trigger CD4+ T cell activation in mice affected by CD and are associated with high IgG plasma levels.43 With the help of GWAS correlated with nutrigenetic and nutrigenomic research, a new, more personalized approach to the patient with IBD is expected.43,44
Another important aspect when discussing dietary habits is their influence on intestinal microbiota. Colonic bacteria can metabolize a large spectrum of complex carbohydrates and other fermentation products resulting in short chain fatty acids, acetate, butyrate, propionate, which in turn can influence colonic pH, intestinal permeability, and normal gut microbiota, possibly facilitating the development of IBD.45 One small study (eight participants) researching the influence of diet in altering fecal microbiota of the patients with CD noted that after a specific carbohydrate diet, there was an increased representation of Faecalibacterium prausnitzii, an anti-inflammatory commensal. In contrast, a low residue diet led to a decrease in microbiome diversity.46 One major study involving dietary therapy and gut microbiome in patients with IBD is currently recruiting participants.47
Immuno-inflammatory factors
In IBD, various aspects of the mucosal immune system are modified. The implicated cells include intestinal epithelia, innate lymphoid cells, macrophages, dendritic cells, B cells, and T cells. The interaction of the antigen-presenting cells (APCs) with the bacterial antigens leads to differentiation of naïve T-cells into effector T-helper cells, which occurs mainly in Peyer’s patches and lymphoid tissue.
The CD is considered a Th1 inflammatory disease with an overexpression of cell-mediated immune response, and therefore a high concentration of IL-12 produced by APCs and macrophages. Th2 cells are part of an immune-mediated process and respond by producing IL-4, IL-5, IL-6, IL-10, and IL-13. The communication between the two CD 4+ subsets is also cytokine mediated, with IFN-gamma (produced by Th1) inhibiting Th2 and IL-4, IL-10 and IL-13 inhibiting the Th1 response.48
In IBD, cytokine imbalance leads to a chronic intestinal inflammation. On one hand, the pro-inflammatory Th1 lymphocytes increase the levels of TNF, IL-6, IL-12, IL-17, IL-23. On the other hand, inhibition of intermediate molecular pathways leads to increased plasma levels of immunosuppressive cytokines (such as TGF-beta and IL-10) that have limited activity. For example, SMAD7 pathway was shown to relate a blockage in TGF-beta signaling by binding to its receptor.49 Other pro-inflammatory pathways, such as PI3K/AKT/PTEN pathway, increase reactive oxygen species and lead to NF-kB activation via IB kinase, as final mechanism.50
IL-12/IL-23 is a pro-inflammatory axis with a role in the differentiation of Th17 lymphocytes, a new subtype of T-lymphocytes implicated in many chronic autoimmune- mediated diseases. IL-12 and IL-23 share a common p40 subunit. While IL-12 participates in the Th1 response, IL-23 is oriented toward stimulating the Th17 arm. The latter has a key position in the adaptive immune system against extracellular bacteria and fungi not well covered by Th1 and Th2, while also being responsible for producing the IL-17 family of cytokines (IL-17A, IL-17B, IL-17C, IL-17D, IL-17E [IL-25], and IL-17F).48
Other pro-inflammatory cells stimulated by IL-23 are the innate lymphoid cells, which mirror the Th cytokine secretion and which could provide both pro-inflammatory and protective/regulatory responses toward the commensal bacteria. Thus, the manipulation of those cells could also provide future treatment targets for IBD.51
Therapeutic targets
The therapeutic goal for CD has been the induction and maintenance of remission, with an emerging emphasis on mucosal healing52 and, more recently, on histological remission.53 CD treatment guidelines include salicylate compounds, antibiotics, immunosuppressive, and immune-modulating drugs.
The discovery of biologic agents, especially anti-TNF, has revolutionized the medical care by improving the resection-free survival rate and the morbidity in patients with moderate to severe CD, finally changing the natural course of the disease.54 However, there are still severe cases in which neither of the current medical tools can provide a good clinical response, prevent the frequent relapses after induction, maintain the remission, and avoid the associated adverse reactions.55
Therefore, the development of new drugs for an efficient treatment of the disease is mandatory, and comprehensive research in mucosal immunology brings many new potential molecular targets into the light.
Microbial modifiers
Gut microbiota is considered an essential trigger in chronic IBD. Bacterial overgrowth treatment with intraluminal antibiotics (Rifaximin) induced remission in patients with moderately active CD.56 The discovery of AIEC and MAP as etiopathogenic factors promotes the hypothesis of antibacterial drugs as therapeutics.
QBECO Site Specific Immunomodulator is a vaccine derived from inactivated E. coli bacteria, which has shown excellent results in the induction of clinical response and remission in subjects with moderate to severe CD (Phase I/II trials placebo-controlled).57 The basis of this therapy is to stimulate the innate immune system to remove the underlying trigger. During the trial, patients followed their normal therapeutic regimen for the CD.
MAP is the subject of the study MAP US Phase III, a randomized double-blind study aimed to investigate RHB-104 antibiotic (combining clarithromycin, rifabutin, and clofazimine) efficacy in inducing remission in patients with moderately to severely active CD.58 One previous study with the same antibiotic combination administered for 2 years had positive results at 18 weeks, but no final beneficial outcome.59
Prebiotics and probiotics could change intestinal microbiota and elicit an anti-inflammatory response. Although for ulcerative colitis (UC), prebiotics such as oligofructose-enriched inulin have shown some benefit in maintaining remission, for CD the results were not consistent. Probiotic strains from the Lactobacillus, Bifidobacterium, and Saccharomyces families were attempted for active CD and for the maintenance of remission in CD. The results were not conclusive, with some researchers stating that Lactobacillus GG might actually increase relapse rate.60 Currently, probiotics are not recommended in CD, whereas in UC there is an evidence supporting the effectiveness in induction and maintenance of remission.61
Adhesion molecules and leukocyte recruitment
A completely different approach to IBD treatment is the inhibition of the lymphocyte gut-homing mechanism. This can be done by blocking adhesion molecules, which permit leukocyte migration or by inhibiting different receptors on the surface of CD4(+) T cells. Such a process prevents the activation of an inflammatory cascade, minimizing the intestinal damage.62
Natalizumab is a humanized IgG4 monoclonal antibody (mAb) that inhibits α4β7 integrin/mucosal addressin cell adhesion molecule-1 (MAdCAM-1) interaction and α4β1/VCAM-1 binding.63 Unfortunately, due to severe adverse reactions, like multifocal leukoencephalopathy, occurring from its systemic passage, it was withdrawn from the European market.64
Vedolizumab (VDZ) is a selective inhibitor of the α4β7 integrin/MAdCAM-1 interaction, which provides a low systemic effect and therefore, a limited therapeutic effect on the intestinal layers. Its clinical efficacy has been established by three Phase III double-blind vs placebo studies (GEMINI I, II, III) and is now approved for the treatment of moderate-severe CD.65–69
GEMINI II65 enrolled 368 patients with moderate to severe CD, 51% having previous anti-TNF exposure. These patients received VDZ 300 mg IV at week 0 and 2 with the first evaluation at 6 weeks. The primary end point was met, with CDAI response 31.4% vs 25.7% in placebo and noted a lower C-reactive protein (CRP) in 19.9% vs 21.1%. This trial had modest efficacy results primarily due to the late onset of therapeutic effect and motivated the following study.
GEMINI III68 enrolled 315 patients with moderate, severe CD, and all were subjected to previous anti-TNF therapy. It also introduced another end point. Results showed that, by 10 weeks, clinical remission was achieved in 26.6% of the patients with VDZ vs 12.1% in the placebo group. In terms of steroid-free remission and durable remission, after 52 weeks, VDZ every 8 weeks had 45% and 31.7%, compared with 30.1% and 15.9% in the placebo group.
In conclusion, VDZ has a gut-specific mechanism that lowers the infectious risk present in classical anti-TNF therapy, and can be used in patients who do not tolerate or have lost response to Infliximab (IFX). The only criterion for such treatment selection is that patients afford to wait 10 weeks for the onset of the pharmacological effect.67,70
AMG-181, Abrilumab, is a fully human anti-α4β7 antibody heterodimer under development. Phase II trials are enrolling patients with moderate to severe CD, but results are expected to be similar to VDZ, since they have similar mechanisms of action.71 Two other anti-integrins, Firategrast and TRK-170, are also in Phase II trials, without any results published so far.72
PF 00547,659 is a MAdCAM-1 inhibitor, subject of the OPERA study, which was tested for active refractory CD. Despite preliminary results having a high placebo rate of response, further analysis showed good response and remission in patients with CRP levels above 18, in a dose-dependent manner.73
Other biologic molecules targeting integrin ligands and receptors include intercellular adhesion molecule-1 inhibitors, anti-NKG2D mAb, anti-CXCL-10 mAb, and chemokine receptor type 9 (CCR9).74–76 Vercirnon is a selective CCR9 antagonist under research. CCR9 is expressed on CD4(+) T cells and binds to CCL25, a small intestinal chemoattractant, facilitating lymphocyte gut homing. In addition to this, CCR9 signaling can inhibit Treg cell development in mouse models. Phase II trials – PROTECT77 and the last Phase III trial – SHIELD 478 showed efficacy at inducing and maintaining remission in moderate-severe CD, even though previous Phase III studies failed. Possible explanations for divergent results include a dose-dependent efficacy and a different study design with different inclusion criteria and a higher rate of discontinuation.79 CCX507 is another CCR9 antagonist in Phase I that has had good results in preclinical studies when combined with anti-α4β7 antibodies.80
BL-7040 is an oligonucleotide acting like a TLR-9 agonist, which suppresses acetylcholinesterase and which indirectly has macrophagic immune-modulating properties. It has been tested in a Phase II trial in patients with IBD with good results achieving also a significant reduction in neutrophil cell levels and in IL-6 levels.81
Anti-cytokines
Anti-TNF and TNF-inhibiting molecules
Anti-TNF molecules revolutionized the medical treatment for patients with IBD in the past decade. The molecules specifically target the TNF, either soluble or transmembrane, and prove highly effective in the management of moderate to severe disease. They act by blocking the final inflammatory mediator. There are four main compounds approved in IBD: IFX, Adalimumab (ADA), Certolizumab pegol (CTZ), and Golimumab.54
IFX is a mAb that binds to transmembrane-TNF in macrophages and activated T-cells, thus inducing apoptosis. Both efficacy and safety have been proven in multiple clinical studies for all phenotypes of the disease.54 Since it is derived from animal antibodies, its administration comes with a higher risk of allergic reaction55 and a loss of response due to anti-IFX antibodies, which may lead to treatment discontinuation. 82
ADA is similar to IFX but uses humanized mAbs, which theoretically have a lower immunogenic response. Current indications are similar to IFX as most clinical studies have shown comparable results, with some studies suggesting IFX as a better option in induction of remission in UC.83 For patients who have experienced a loss of clinical response to anti-TNF, dose intensification can be an effective measure in regaining remission. IBD studies comparing IFX/ADA dose-escalation revealed that 25% of patients with CD require dose escalation within the 1 year of treatment, independent of the therapeutic agent used. Statistically, more patients with UC on IFX treatment needed intensification of treatment (72% vs 25%).84
CTZ is also a humanized mAb with the Fab segment linked to polyethylene glycol that neutralizes both transmembrane and soluble TNF, unlike the previous two, it does not have an Fc-portion and does not activate the complement cascade thus having no apoptotic effect.85 In a 2014 systematic review, the ADA had a superior effect in induction and the maintenance of disease remission compared with CTZ.86
Golimumab is the latest anti-TNF agent that has been approved for the treatment of IBD, especially for UC, but more recently also in refractory CD.76 It is a fully human mAb against soluble and transmembrane TNF-alpha with a less pronounced apoptotic effect compared with IFX or ADA.86,87
Etanercept and onercept are soluble TNF receptors (inhibiting p75 and p55 units) that were suggested as potential therapeutics, but pilot and small double-blind studies (43 patients for etanercept) proved them ineffective.88,89 Etanercept did not receive market approval in spite of the authors’ hypothesis that higher doses may be required for clinical response.90
Other anti-TNF molecules, called TNF kinoids, were the subject of Phase I/II trials in 2010 and 2012 with inconclusive results.91 These molecules intend to induce an immune response and generate polyclonal antibodies that bind different epitopes of TNF-alpha, thus minimizing antibody-mediated resistance. Further clinical trials are warranted.
Thalidomide has marked anti-angiogenic and anti-inflammatory properties, by means of inhibiting the production of TNF-alpha, IL-6, IL-10, and IL-12, while modulating natural killer cell activity and stimulating Th2 cytokines (IL-2, IL-4, IL-5).92 While its precise mechanism of action remains unknown, multiple clinical essays show a benefit in introducing it in refractory and pediatric cases of CD.93,94 A retrospective study by Gerich et al95, in which 47 patients were treated for a median of 4.4 months, obtained clinical responses in 54% of cases and clinical remission in 19% of patients. Steroids could have been discontinued in 40% of patients. This study also showed that response is dose dependent, since for a dose of less than 50 mg, the rates of response reached 40%, while a dose higher than 50 mg had a response of 80%.95
Other studies, which used thalidomide in patients with previous treatment with anti-TNF or in the fistulizing pattern of disease, had encouraging results, worth future investigations.94 Combinations of anti-TNF or cyclophosphamide with thalidomide have shown promising effects in Chinese studies.96 However, thalidomide comes with frequent side effects, which limits its use in current medical practice. Changes in thalidomide drug delivery system by artificial alginate-poly-l-lysine-alginate microcapsules had good results in the context of a more targeted, intraepithelial limited effect.97
Anti-IL-12 and IL-23 antibodies
Anti-IL-12 and IL-23 antibodies (briakinumab) were tested but failed to produce significant clinical responses.98 An oral inhibitor for IL-12 and IL-23 was researched (apilimod mesylate) but results in the treatment of rheumatoid arthritis were superior. These molecules are no longer being studied in CD at the present moment.99
Inhibitors for the p40 subunit have been considered as an important therapeutic target in autoimmune diseases, such as psoriasis, because blocking IL-12 and IL-23 would prevent activation of Th1, Th17, natural killer, and APC. Ustekinumab is a p40 inhibitor, which has been successfully used in the treatment of CD when anti-TNF therapy failed.100 A French multicenter GETAID study including 97 patients who did not respond to anti-TNF therapy and immune-suppressants showed a clinical benefit in 71% of cases at 3 months and 86% at 12 months. Eight out of nine patients with perianal disease have shown clinical improvement at the end of the study.101,102
MEDI2070/AMG-139 is another IL-23 inhibitor that binds to the p19 subunit while leaving the IL-12 pathway unblocked.103 This compound is in its Phase II trials with preliminary reports showing good clinical response vs placebo at 8-week end point in a cohort of 119 patients, 90% of whom previously received anti-TNF therapy.104
IL-6 is another pro-inflammatory cytokine implicated in Th17 activation. Tocilizumab is a human mAb binding IL-6, which has proven clinical efficacy in rheumatoid arthritis and in active CD, but has been ineffective regarding endoscopic and histological aspects in CD.105 The ANDANTE study is a Phase II trial for PF-04236921, another anti-IL-6 mAb. Primary pilot studies for this compound had good results in patients with active CD.106
Anti-IL-13 mAb Dectrekumab (QAX576) is of interest in CD with perianal fistulas according to the ongoing Phase II clinical studies. Primary concept studies aimed for complete fistula closure for more than 4-weeks during the trial.107 Two other compounds, tralokimumab and anrunkinzumab, were investigated in CD and UC but failed to meet primary end points.108,109
Signaling pathways mediated by cytokines
Janus kinase (JAK) inhibitors represent a possible molecular target due to their implication in signal transduction between IL-2R, IL-6R, IL-12, and IL-23. Tofacitinib is an oral JAK inhibitor that has shown effectiveness in rheumatoid arthritis and in some UC studies.110 The CD results of a Phase II showed that 10 and 15 mg doses twice a day over 4 weeks had no effect in induction or remission. However, at the dose of 15 mg, both CRP levels and fecal calprotectin were significantly decreased, justifying a second trial with stricter inclusion criteria.111 Other JAK inhibitors are being tested in CD (GLPG0634) and in UC (Peficitinib).112
Laquinimod is a small, oral molecule that has a modulatory effect on APC, directing T-cells toward an anti-inflammatory phenotype and lowering inflammatory cytokines. Two double-blind Phase II trials demonstrated that at low doses of 0.5 mg it could induce clinical response and remission, while decreasing fecal calprotectin.113 Further research is required.
SMAD7 antisense oligonucleotides are designed to lower intestinal SMAD7 and prevent the blocking of TGF-β1 receptor, thus permitting its immunosuppressive effect. Mongersen (GED0301) is a synthetic single-stranded modified DNA oligonucleotide that in experimental colitis has shown an effect in reducing SMAD7 in the ileal and colonic mucosa. It can also interfere with the T-cell gut-homing mechanism. A first double-blind study on patients with active CD showed high rates of clinical response (reduction of a minimum of 100 CDAI points on day 28) after receiving a 2-week regimen on a daily basis, 10 mg (37%), 40 mg (58%), or 160 mg (72%) vs placebo (17%). Such a promising new molecule definitely needs further study to extend its use as an objective inflammatory biomarker.114
Table 1 summarizes current and future biological therapies, their stage of development and mechanism of action.
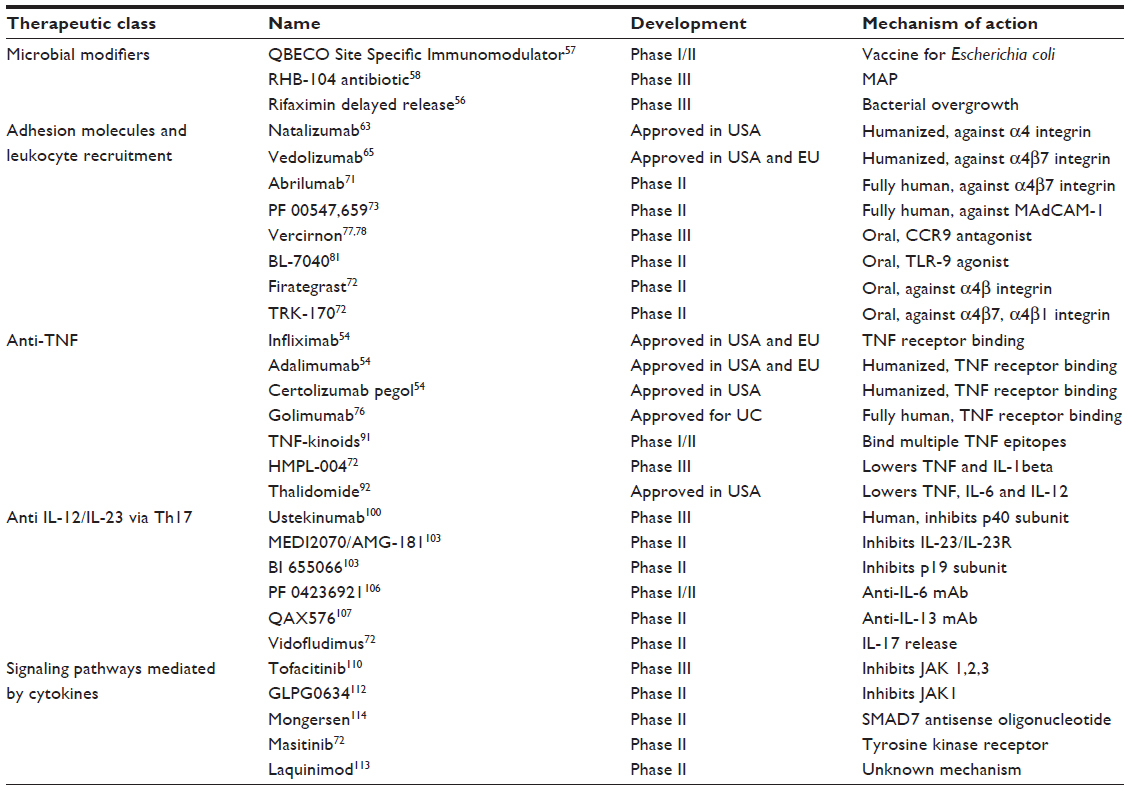 | Table 1 Therapeutical targets for Crohn’s disease |
Immune reconstitution – cellular therapy
There is a growing interest in using stem cell therapy in immune-mediated diseases, where immune reconstitution is needed. Both hematopoietic and mesenchymal stem cells (MSCs) have shown good results when they induced a long-lasting clinical remission in severe cases of CD.115
Hematopoietic stem cell transplantation (HSCT) was successfully used in refractory cases or when concomitant diseases occurred (lymphoma). The theory was that Allo-HSCT might change the genetic predisposition, whereas auto-HSCT eliminated committed lymphocytes. Attempts of both types of transplantation were made in severe, complicated cases with remission rates ranging from 80% to 91%.116 In 2010, a prospective study was published, featuring 24 patients with anti-TNF refractory disease during a 5-year follow-up. The results were 91% remission at 1 year, 57% at 3 years, and 19% at 5 years, after the initial conditioning by cyclophosphamide and granulocyte colony-stimulating factor.117 Researchers hypothesize that a mutation in the IL10RB gene probably constitutes the genetic factor in these cases and that HSCT (allogenic or autogenic) proves beneficial in highly selected cases.118
MSCs were tested, especially for fistulizing pattern diseases. The differentiation capacity of these cells facilitates the closure of the fistulas. Moreover, the immunosuppressive effect they have could also help in active luminal disease. Phase I studies evaluating autologous bone marrow derived MSC transfusion in a single dose are currently being conducted.119
Intravenous infusion of human placenta-derived cells (PDA001) has an immunosuppressive potential as shown by studies on humans and mice. PDA001 is a compound of mesenchymal-like adherent cells from postpartum placenta, which is currently used in clinical trials for the treatment-resistant patients with CD. Preliminary results are comparable to other cellular therapies with CDAI response and remission induction.120
Intraperitoneal administration of autologus tolerogenic dendritic cells (tolDC’s) exhibits better results in experimental mice colitis. A Phase I pilot-study of Jauregui-Amezaga including only nine patients with refractory CD had modest, but positive results. Endoscopic findings improved markedly in three patients at week 12 and remission was achieved in one patient. Further analysis is required.121
Other possible therapies
Helminths therapies could have an impact through inducing an intense immune regulatory circuit. Experimental infections have shown that nematode infections increase the number of dendritic cells, while rising IL-10 level in the intestine and can also shift the immune response toward a Th2 phenotype. In vitro and animal studies showed promising results but future testing is needed considering the disadvantages of living with a parasitic infection.122
Granulocyte colony-stimulating factor promotes granulocytic and macrophagic differentiation from bone marrow precursor cells contributing to an immunoregulatory mechanism. In spite of the in vitro studies showing good results on tapering intestinal inflammation and promoting a normal intestinal epithelium, human studies failed to achieve their end points. Three randomized studies on 537 patients concluded that there was no significant difference in terms of clinical remission or 100-point clinical response.123
Immunoglobulin therapy (intravenous/subcutaneous) has shown effectiveness in some isolated refractory patients with CD, even though controlled trials are not available. Historically, intravenous immunoglobulins in high doses were used for cortico-resistant CD before the biologic therapies. Other advantages of this therapy include a very good safety profile, a clinical application for some extraintestinal manifestations, and the possibility of subcutaneous usage.124
Conclusion
The CD is a multifactorial disease in which genetic, environmental, immunological, and microbial components shape its pathogenesis, without revealing the exact mechanism of onset. The genetic susceptibility is thought to interact with bacterial antigens, nutrients in the diet, and other types of epithelial injuries by triggering an abnormal immune response. Microbial agents have emerged as compulsory factors in the pathogenesis of CD, suggesting that vaccine-type treatments and antibiotics would constitute a good therapeutic approach.
Current interest in CD management is guided toward eliminating inflammation and achieving endoscopic remission rather than focusing on clinical remission. Anti-TNF agents represent the most explored therapeutic line for almost 20 years of clinical practice, leading to persistent remission in patients with CD. However, the failure of those molecules in certain cases, as well as the need for a lower infection risk, leaves room for the development of new therapeutic targets. Anti-adhesion molecules, such as VDZ, offer the advantage of a gut-specific immune-modulation and improve the safety profile. Other biologic agents, such as ustekinumab, have proven clinical benefits, especially in patients who do not respond to anti-TNF therapy. Regarding the upcoming new molecules, anti-MAdCAM, anti-IL23 antibodies, and SMAD7 antisense oligonucleotides should be mentioned as promising. Nevertheless, clinical testing is compulsory since neither theoretical nor animal models may predict the human response, as shown in the previous studies.
For severe refractory CD, a new approach is cellular therapy, with good clinical results in a limited number of cases. Given the extensive research focusing on the immunological and molecular biology of the CD, the future medical approach is expected to change dramatically.
Disclosure
The authors report no conflicts of interest in this work.
References
Centers for Disease Control and Prevention. CDC—Epidemiology of the IBD – Inflammatory Bowel Disease. 2015. Atlanta, GA: Centers for Disease Control and Prevention. Available from: http://www.cdc.gov/ibd/ibd-epidemiology.htm. Accessed April 15, 2015. | |
Magro F, Langner C, Driessen A, et al. European consensus on the histopathology of inflammatory bowel disease. J Crohns Colitis. 2013;7(10):827–851. | |
Coulombe F, Behr MA. Crohn’s disease as an immune deficiency? Lancet. 374(9692):769–770. | |
Antoni L, Nuding S, Wehkamp J, Stange EF. Intestinal barrier in inflammatory bowel disease. World J Gastroenterol. 2014;20(5):1165–1179. | |
Merga Y, Campbell BJ, Rhodes JM. Mucosal barrier, bacteria and inflammatory bowel disease: possibilities for therapy. Dig Dis. 2014;32(4):475–483. | |
Fava F, Danese S. Crohn’s disease: bacterial clearance in Crohn’s disease pathogenesis. Nat Rev Gastroenterol Hepatol. 2010;7(3):126–128. | |
Mantzaris GJ. When can we cure Crohn’s? Best Pract Res Clin Gastroenterol. 28(3):519–529. | |
Baumgart DC, Sandborn WJ. Crohn’s disease. Lancet. 2012; 380(9853):1590–1605. | |
Ben-Horin S, Chowers Y. Review article: loss of response to anti-TNF treatments in Crohn’s disease. Alimen Pharmacol Ther. 2015; 33(9):987–995. | |
Ding T, Deighton C. Complications of anti-TNF therapies. Fut Rheumatol. 2007;2(6):587–597. | |
Van Limbergen J, Wilson DC, Satsangi J. The genetics of Crohn’s disease. Annu Rev Genomics Hum Genet. 2009;10:89–116. | |
Liu JZ, Anderson CA. Genetic studies of Crohn’s disease: past, present and future. Best Pract Res Clin Gastroenterol. 2014;28(3):373–386. | |
Strober W, Asano N, Fuss I, Kitani A, Watanabe T. Cellular and molecular mechanisms underlying NOD2 risk-associated polymorphisms in Crohn’s disease. Immunol Rev. 2014;260(1):249–260. | |
Adler J, Rangwalla SC, Dwamena BA, Higgins PD. The prognostic power of the NOD2 genotype for complicated Crohn’s disease: a meta-analysis. Am J Gastroenterol. 2011;106(4):699–712. | |
Lee KM, Lee JM. Crohn’s disease in Korea: past, present, and future. Korean J Intern Med. 2014;29(5):558–570. | |
Lee JC, Parkes M. Genome-wide association studies and Crohn’s disease. BriefFunct Genomics. 2011;10(2):71–76. | |
Fritz T, Niederreiter L, Adolph T, Blumberg RS, Kaser A. Crohn’s disease: NOD2, autophagy and ER stress converge. Gut. 2011; 60(11):1580–1588. | |
Bai J, Li Y, Shao T, et al. Integrating analysis reveals microRNA-mediated pathway crosstalk among Crohn’s disease, ulcerative colitis and colorectal cancer. Mol BioSyst. 2014;10(9):2317–2328. | |
Archanioti P, Gazouli M, Theodoropoulos G, Vaiopoulou A, Nikiteas N. Micro-RNAs as regulators and possible diagnostic bio-markers in inflammatory bowel disease. J Crohns Colitis. 2011;5(6):520–524. | |
Chapman CG, Pekow J. The emerging role of miRNAs in inflammatory bowel disease: a review. Therap Adv Gastroenterol. 2015;8(1):4–22. | |
Sankey EA, Dhillon AP, Anthony A, et al. Early mucosal changes in Crohn’s disease. Gut. 1993;34(3):375–381. | |
Teshima CW, Dieleman LA, Meddings JB. Abnormal intestinal permeability in Crohn’s disease pathogenesis. Ann N Y Acad Sci. 2012;1258:159–165. | |
Scribano ML, Prantera C. Use of antibiotics in the treatment of Crohn’s disease. World J Gastroenterol. 2013;19(5):648–653. | |
Bosca-Watts MM, Tosca J, Anton R, Mora M, Minguez M, Mora F. Pathogenesis of Crohn’s disease: bug or no bug. World J Gastrointest Pathophysiol. 2015;6(1):1–12. | |
Juste C, Kreil DP, Beauvallet C, et al. Bacterial protein signals are associated with Crohn’s disease. Gut. 2014;63(10):1566–1577. | |
Barnich Nicolas, D-MA. Adherent-invasive Escherichia coli and Crohns disease. Curr Opin Gastroenterol. 2007;23(1):16–20. | |
Nguyen HT, Dalmasso G, Seibold F, Müller S, Darfeuille-Michaud A. 436e Crohn’s disease-associated adherent-invasive Escherichia coli suppress autophagy response to replicate intracellularly by regulating host microRNA expression. Gastroenterology. 2013;144(5):S–82. | |
Hermon-Taylor J. Mycobacterium avium subspecies paratuberculosis, Crohn’s disease and the Doomsday scenario. Gut Pathog. 2009; 1(1):15. | |
Parrish NM, Radcliff RP, Brey BJ, et al. Absence of Mycobacterium avium subsp. paratuberculosis in Crohn’s patients. Inflamm Bowel Dis. 2008;15(4):558–565. | |
Bach H. What role does Mycobacterium avium subsp. paratuberculosis play in Crohn’s disease? Curr Infect Dis Rep. 2015;17(2):463. | |
Stewart LC, Day AS, Pearson J, et al. SLC11A1 polymorphisms in inflammatory bowel disease and Mycobacterium avium subspecies paratuberculosis status. World J Gastroenterol. 2010;16(45):5727–5731. | |
Papamichael K, Konstantopoulos P, Mantzaris GJ. Helicobacter pylori infection and inflammatory bowel disease: is there a link? World J Gastroenterol. 2014;20(21):6374–6385. | |
Xiang Z, Chen YP, Ye YF, et al. Helicobacter pylori and Crohn’s disease: a retrospective single-center study from China. World J Gastroenterol. 2013;19(28):4576–4581. | |
Navaneethan U, Venkatesh PG, Shen B. Clostridium difficile infection and inflammatory bowel disease: understanding the evolving relationship. World J Gastroenterol. 2010;16(39):4892–4904. | |
Jess T, Simonsen J, Nielsen NM, et al. Enteric Salmonella or Campylobacter infections and the risk of inflammatory bowel disease. Gut. 2011;60(3):318–324. | |
Sartor RB. Mechanisms of disease: pathogenesis of Crohn’s disease and ulcerative colitis. Nat Clin Pract Gastroenterol Hepatol. 2006;3(7):390–407. | |
Elliott DE, Urban J Jr, Argo CK, Weinstock JV. Does the failure to acquire helminthic parasites predispose to Crohn’s disease? FASEB J. 2000;14(12):1848–1855. | |
Slattery E, Mitchell P, Mulcahy HE. Cigarette smoking in Crohns disease: can we do more? J Crohns Colitis. 2011;5(5):505–505. | |
Graham C. Understanding smoking and nicotine effects on the gastrointestinal tract. Gastrointest Nurs. 2013;11(1):44. | |
Pfeffer-Gik T, Levine A. Dietary clues to the pathogenesis of Crohn’s disease. Dig Dis. 2014;32(4):389–394. | |
Schulman J, Shaoul R. P435 Maintenance of remission with partial enteral nutrition therapy in pediatric Crohn’s disease: a retrospective study. J Crohns Colitis. 2015;9(Suppl 1):S298. | |
Caprilli R, Lapaquette P, Darfeuille-Michaud A. Eating the enemy in Crohn’s disease: an old theory revisited. J Crohns Colitis. 2010;4(4):377–383. | |
Kawaguchi T, Mori M, Saito K, et al. Food antigen-induced immune responses in Crohn’s disease patients and experimental colitis mice. J Gastroenterol. 2014;50(4):394–405. | |
Ferguson LR. Potential value of nutrigenomics in Crohn’s disease. Nat Rev Gastroenterol Hepatol. 2012;9(5):260–270. | |
Scott KP, Gratz SW, Sheridan PO, Flint HJ, Duncan SH. The influence of diet on the gut microbiota. Pharm Res. 2013;69(1):52–60. | |
Walters SS, Quiros A, Rolston M, Grishina I, Li J. Analysis of gut microbiome and diet modification in patients with Crohn’s disease. SOJ Microbiol Infect Dis. 2014;2(3):1–13. | |
ClinicalTrials.gov. Dietary therapy and gut microbiome in Crohn’s disease and ulcerative colitis. Available from: https://clinicaltrials.gov/ct2/show/NCT02412553. NLM identifier: NCT02412553. Accessed May 1, 2015. | |
Wallace KL, Zheng LB, Kanazawa Y, Shih DQ. Immunopathology of inflammatory bowel disease. World J Gastroenterol. 2014;20(1):6–21. | |
Monteleone G, Kumberova A, Croft NM, McKenzie C, Steer HW, MacDonald TT. Blocking Smad7 restores TGF-β1 signaling in chronic inflammatory bowel disease. J Clin Invest. 1082001:601–609. | |
Tokuhira N, Kitagishi Y, Suzuki M, et al. PI3K/AKT/PTEN pathway as a target for Crohn’s disease therapy (Review). Int J Mol Med. 2014; 35(1):10–16. | |
Geremia A, Arancibia C, Gwela A, Chapman RW, Travis SPL, Powrie F. OP001. Innate lymphoid cells accumulate in IBD. J Crohns Colitis. 2014;8(Suppl 1):S1. | |
De Cruz P, Kamm MA, Prideaux L, Allen PB, Moore G. Mucosal healing in Crohn’s disease: a systematic review. Inflamm Bowel Dis. 2012;19(2):429–444. | |
Bryant RV, Winer S, Travis SP, Riddell RH. Systematic review: histological remission in inflammatory bowel disease. Is ‘complete’ remission the new treatment paradigm? an IOIBD initiative. J Crohns Colitis. 2014;8(12):1582–1597. | |
Kawalec P, Mikrut A, Wisniewska N, Pilc A. Tumor necrosis factor-alpha antibodies (infliximab, adalimumab and certolizumab) in Crohn’s disease: systematic review and meta-analysis. Arch Med Sci. 2013; 9(5):765–779. | |
Ali T, Kaitha S, Mahmood S, Ftesi A, Stone J, Bronze MS. Clinical use of anti-TNF therapy and increased risk of infections. Drug Healthc Patient Saf. 2013;5:79–99. | |
Prantera C, Lochs H, Grimaldi M, Danese S, Scribano ML, Gionchetti P. Rifaximin-extended intestinal release induces remission in patients with moderately active Crohn’s disease. Gastroenterology. 2011;142(3):473–481.e4. | |
SSI Efficacy Crohn’s Disease Remission Compassionate Use Results. Vancouver: Qu Biologics Inc. 2015. Available from: http://www.qucrohnstrial.com/ssi-intro/qbeco-ssi-efficacy-and-safety/. Accessed February 10, 2015. | |
ClinicalTrials.gov. Efficacy and safety of anti-MAP therapy in adult Crohn’s disease. Available from: https://clinicaltrials.gov/ct2/show/NCT01951326. NLM identifier: NCT01951326. Accessed April 15, 2015. | |
RedHill Biopharma Ltd. RedHill Biopharma Provides Update on Progress With RHB-104 Ongoing Phase III Program for Crohn’s Disease Following FDA Meeting. Tel Aviv: RedHill Biopharma Ltd; 2015. Available from: http://globenewswire.com/news-release/2015/01/29/701089/10117655/en/RedHill-Biopharma-Provides-Update-on-Progress-With-RHB-104-Ongoing-Phase-III-Program-for-Crohn-s-Disease-Following-FDA-Meeting.html. Accessed January 29, 2015. | |
Orel R, Kamhi Trop T. Intestinal microbiota, probiotics and prebiotics in inflammatory bowel disease. World J Gastroenterol. 2014;20(33):11505–11524. | |
Ghouri YA, Richards DM, Rahimi EF, Krill JT, Jelinek KA, DuPont AW. Systematic review of randomized controlled trials of probiotics, prebiotics, and synbiotics in inflammatory bowel disease. Clin Exp Gastroenterol. 2014;7:473–487. | |
Saruta M, Papadakis KA. Lymphocyte homing antagonists in the treatment of inflammatory bowel diseases. Gastroenterol Clin North Am. 2014;43(3):581–601. | |
Yu Y, Schurpf T, Springer TA. How natalizumab binds and antagonizes alpha4 integrins. J Biol Chem. 2013;288(45):32314–32325. | |
Juillerat P, Wasan SK, Fowler SA, et al. Efficacy and safety of natalizumab in Crohn’s disease patients treated at 6 Boston academic hospitals. Inflamm Bowel Dis. 2013;19(11):2457–2463. | |
Hanauer SB, Feagan BG, Macintosh DG, et al. Tu1138 Efficacy of Vedolizumab in Crohn’s disease by prior treatment failure in Gemini II, a randomized, placebo-controlled, double-blind, multicenter study. Gastroenterology. 144(5):S–772. | |
Mosli MH, Feagan BG. Vedolizumab for Crohn’s disease. Expert Opin Biol Ther. 2013;13(3):455–463. | |
Raine T. Vedolizumab for inflammatory bowel disease: changing the game, or more of the same? United European Gastroenterol J. 2014;2(5):333–344. | |
Sands B, Feagan B, Rutgeerts P, et al. Vedolizumab induction therapy for patients with Crohn’s disease and prior anti-TNF antagonist failure: a randomized, placebo-controlled, double-blind, multicenter trial: P-26. Inflamm Bowel Dis. 2012;18:S24–S25. | |
Poole RM. Vedolizumab: first global approval. Drugs. 2014;74(11):1293–1303. | |
Wang MC, Zhang LY, Han W, et al. PRISMA – efficacy and safety of vedolizumab for inflammatory bowel diseases: a systematic review and meta-analysis of randomized controlled trials. Medicine (Baltimore). 2014;93(28):e326. | |
Pan WJ, Hsu H, Rees WA, et al. Pharmacology of AMG 181, a human anti-α(4)β(7) antibody that specifically alters trafficking of gut-homing T cells. Br J Pharmacol. 2013;169(1):51–68. | |
Amiot A, Peyrin-Biroulet L. Current, new and future biological agents on the horizon for the treatment of inflammatory bowel diseases. Therap Adv Gastroenterol. 2015;8(2):66–82. | |
OP022. Anti-MAdCAM-1 Antibody (PF-00547659) for active refractory Crohn’s disease: results of the OPERA study. J Crohn’s Colitis. 2015;9(Suppl 1):S14. | |
Allez M, Petryka R, Skolnick BE, Wisniewska-Jarosinska MA. Mo1213 efficacy and safety of NNC0142-0002, a novel human monoclonal antibody targeting NKG2D: a randomized, double-blind, single-dose phase 2 trial in patients with Crohn’s disease. Gastroenterology. 146(5):S–587. | |
D’Haens GR, Sartor RB, Silverberg MS, Petersson J, Rutgeerts P. Future directions in inflammatory bowel disease management. J Crohns Colitis. 2014;8(8):726–734. | |
Yacyshyn B, Chey WY, Wedel MK, Yu RZ, Paul D, Chuang E. A randomized, double-masked, placebo-controlled study of alicaforsen, an antisense inhibitor of intercellular adhesion molecule 1, for the treatment of subjects with active Crohn’s disease. Clin Gastroenterol Hepatol. 2007;5(2):215–220. | |
Arseneau KO, Cominelli F. Vercirnon for the treatment of Crohn’s disease. Expert Opin Investig Drugs. 2013;22(7):907–913. | |
ChemoCentryx, Inc. ChemoCentryx releases Phase III SHIELD 4 clinical results in patients with Crohn’s disease (NASDAQ:CCXI). Presented at: 79th Annual Scientific Meeting of the American College of Gastroenterology; October 17–22; 2015; Philadelphia, USA. | |
Wendt E, Keshav S. CCR9 antagonism: potential in the treatment of inflammatory bowel disease. Clin Exp Gastroenterol. 2015;8:119–130. | |
ChemoCentryx, Inc. ChemoCentryx’s orally administered CCR9 inhibitor, CCX507, shown to be well tolerated and effective in CCR9 blockade in Phase I study (NASDAQ:CCXI). Presented at: 79th Annual Scientific Meeting of the American College of Gastroenterology; October 21; 2014; Philadelphia, USA. | |
BL-7040 Overview Inflammatory Bowel Disease. 2015. Jerusalem: BioLineRx Ltd. Available from: http://www.biolinerx.com/default.asp?pageid=13&itemid=23. Accessed March 10, 2015. | |
Gisbert JP, Arredondo M, Chaparro M. et al. Drug survival and reasons for discontinuation of anti-TNF therapy in inflammatory bowel disease (IBD) in clinical practice. J Crohns Colitis. 2014;8:S27. | |
Thorlund K, Druyts E, Mills EJ, Fedorak RN, Marshall JK. Adalimumab versus infliximab for the treatment of moderate to severe ulcerative colitis in adult patients naive to anti-TNF therapy: an indirect treatment comparison meta-analysis. J Crohns Colitis. 2014;8(7):571–581. | |
P408. Differences in biologic efficacy and dose-escalation among anti-TNF agents in Crohn´s disease and ulcerative colitis. J Crohns Colitis. 2015;9(Suppl 1):S283. | |
Rutgeerts MF, Séverine V, Paul. Certolizumab pegol in the treatment of Crohn’s disease. Expert Opin Biol Ther. 2013;13(4):595–605. | |
Singh S, Garg SK, Pardi DS, Wang Z, Murad MH, Loftus EV Jr. Comparative efficacy of biologic therapy in biologic-naive patients with Crohn disease: a systematic review and network meta-analysis. Mayo Clin Proc. 2014;89(12):1621–1635. | |
Bohm M, El Chafic AH, Fischer M. Golimumab rescue for active Crohn’s disease refractory to other therapies. Am J Gastroenterol. 2014;109:S514–S515. | |
Rutgeerts P, Sandborn WJ, Fedorak RN, et al. Onercept for moderate-to-severe Crohn’s disease: a randomized, double-blind, placebo-controlled trial. Clin Gastroenterol Hepatol. 2006;4(7):888–893. | |
Mozaffari S, Nikfar S, Abdolghaffari AH, Abdollahi M. New biologic therapeutics for ulcerative colitis and Crohn’s disease. Expert Opin Biol Ther. 2014;14(5):583–600. | |
Sandborn WJ, Hanauer SB, Katz S, et al. Etanercept for active Crohn’s disease: a randomized, double-blind, placebo-controlled trial. Gastroenterology. 2001;121(5):1088–1094. | |
Neovacs presents successful results of Crohn’s disease drug trial at European Crohn’s & Colitis Congress [webpage on the Internet]. Drugs.com; 2011 [cited February 2011]. Available from: http://www.drugs.com/clinical_trials/neovacs-presents-successful-results-crohn-s-trial-european-crohn-s-amp-colitis-congress-11235.html. Accessed January 21, 2016. | |
De Sanctis JB, Mijares M, Suárez A, Compagnone R, Garmendia J, Moreno D, et al, Pharmacological properties of thalidomide and its analogues. Recent Pat Inflamm Allergy Drug Discov. 2010; 4(2):144–148. | |
Qian J, Li Y. Su1235 efficacy and safety of thalidomide in chinese patients with refractory Crohn’s disease. Gastroenterology.144(5):S–435. | |
Felipez LM, Gokhale R, Tierney MP, Kirschner BS. Thalidomide use and outcomes in pediatric patients with Crohn disease refractory to infliximab and adalimumab. J Pediatr Gastroenterol Nutr. 2011; 54(1):28–33. | |
Gerich ME, Yoon JL, Targan SR, Ippoliti AF, Vasiliauskas EA. Long-term outcomes of thalidomide in refractory Crohn’s disease. Aliment Pharmacol Ther. 2014;41(5):429–437. | |
Tang J, Gao X, Zhi M, et al. P436 Intravenous cyclophosphamide combined with thalidomide has a promising effect in refractory Crohn’s disease. J Crohns Colitis. 2014;8(Suppl 1): S247–S248. | |
Fakhoury M, Coussa-Charley M, Al-Salami H, et al. Anti-inflammatory potential of artificial microcapsules containing thalidomide for use in treating Crohn’s disease. J Drug Deliv Therap. 2013;3(5):11–17. | |
Bankhead C. ACG: Mixed Results With Biologic for Crohn’s. 2010. Available from: http://www.medpagetoday.com/MeetingCoverage/ACG/22913. Accessed March 15, 2015. | |
Sands BE, Jacobson EW, Sylwestrowicz T, et al. Randomized, double-blind, placebo-controlled trial of the oral IL-12/23 inhibitor apilimod mesylate for treatment of active Crohn’s disease. Inflamm Bowel Dis. 2009;16(7):1209–1218. | |
Settesoldi A, Coppola M, Rogai F, Annese V. Ustekinumab: moving the target from psoriasis to Crohn’s disease. Expert Rev Gastroenterol Hepatol. 2014;8(1):5–13. | |
DOP029. Ustekinumab efficacy and safety in Crohn’s disease patients refractory to conventional and anti-TNF therapy: a multicenter retrospective experience. J Crohns Colitis. 2015;9(Suppl 1):S37–S39. | |
P368. Ustekinumab in super-refractory Crohn’s disease patients. J Crohns and Colitis. 2015;9(Suppl 1):S262–S263. | |
Goldhill J. Advances in Drug Discovery: Promising Crohn Disease Data Presented at ECCO for the IL-23 mAB, MEDI2070/AMG-139. 2015. Available from: http://leaddiscovery.blogspot.ro/2015/02/updatesplus-ibd-alert-promising-crohn.html. Accessed March 5, 2015. | |
OP025. A randomized, double-blind placebo-controlled phase 2a induction study of MEDI2070 (anti-p19 antibody) in patients with active Crohn’s disease who have failed anti-TNF antibody therapy. J Crohns Colitis. 2015;9(Suppl 1):S15–S16. | |
Ito H, Takazoe M, Fukuda Y, et al. A pilot randomized trial of a human anti- IL-6 receptor monoclonal antibody in active Crohn’s disease. Gastroenterology. 2004;126(4):989–996; discussion 947. | |
Pfizer’s Immunology Pipeline Has Few Bright Spots Besides Xeljanz. 2015. Available from: http://seekingalpha.com/article/2778925-pfizers-immunology-pipeline-has-few-bright-spots-besides-xeljanz. Accessed March 15, 2015. | |
Adis Insight. A multi-center, randomized, double-blind, active controlled study to assess efficacy, safety and tolerability of the anti-IL13 monoclonal antibody QAX576 in the treatment of perianal fistulas in patients suffering from Crohn’s disease. Available from: http://adisinsight.springer.com/trials/700196337. Accessed January 21, 2016. | |
Schmid B. OP011. Tralokinumab (CAT-354), an interleukin 13 antibody, in moderate to severe ulcerative colitis: a phase 2 randomized placebo-controlled study. Paper presented at: 9th ECCO Congress; Febuary 20–22, 2014; Copenhagen. | |
Ferner C. United European Gastroenterology: document detail. United European Gastroenterol J. 2013;1(Suppl 1):A79. | |
Panés J, Su C, Bushmakin AG, Cappelleri JC, Mamolo C, Healey P. Randomized trial of tofacitinib in active ulcerative colitis: analysis of efficacy based on patient-reported outcomes. BMC Gastroenterol. 2015;15(1):14. | |
Sandborn WJ, Ghosh S, Panes J, Vranic I, Wang W, Niezychowski W. A phase 2 study of tofacitinib, an oral Janus kinase inhibitor, in patients with Crohn’s disease. Clin Gastroenterol Hepatol. 2014;12(9):1485–1493. e1482. | |
Buchert M, Burns CJ, Ernst M. Targeting JAK kinase in solid tumors: emerging opportunities and challenges. Oncogene. 2015. | |
D’Haens G, Sandborn WJ, Colombel JF, et al. A phase II study of laquinimod in Crohn’s disease. Gut. 2015;64(8):1227–1235. | |
Monteleone G, Caruso R, Pallone F. Role of Smad7 in inflammatory bowel diseases. World J Gastroenterol. 2012;18(40):5664–5668. | |
Knyazev OV, Parfenov AI, Shcherbakov PL, Ruchkina IN, Konoplyannikov AG. Cell therapy of refractory Crohn’s disease. Bull Exp Biol Med. 2013;156(1):139–145. | |
Ricart JP, Ingrid O, Elena R. Stem cell treatment for Crohn’s disease. Exp Rev Clin Immunol. 2014;6:597–605. | |
Burt RK, Craig RM, Milanetti F, et al. Autologous nonmyeloablative hematopoietic stem cell transplantation in patients with severe anti-TNF refractory Crohn disease: long-term follow-up. Blood. 2010;116(26):6123–6132. | |
Al-toma A, Nijeboer P, Bouma G, Visser O, Mulder CJ. Hematopoietic stem cell transplantation for non-malignant gastrointestinal diseases. World J Gastroenterol. 2014;20(46):17368–17375. | |
CCFA: A Phase I Study Evaluating Autologous Bone Marrow Derived Mesenchymal Stromal for Crohn’s Disease. 2015. Available from: http://www.ccfa.org/research/participate-in-research/find-studies-and-clinical-trials/emory-msc/emory-msc-1.html?referrer=https://www.google.ro/. Accessed February 15, 2015. | |
Mayer L, Pandak WM, Melmed GY, et al. Safety and tolerability of human placenta-derived cells (PDA001) in treatment-resistant crohn’s disease: a phase 1 study. Inflamm Bowel Dis. 2013;19(4):754–760. | |
Jauregui-Amezaga A, Cabezón R, Ramírez-Morros A, et al. Intraperitoneal administration of autologous tolerogenic dendritic cells for refractory Crohn’s disease: a Phase I study. J Crohns Colitis. 2015;9(12):1071–1078. | |
Taghipour N, Aghdaei HA, Haghighi A, Mossafa N, Tabaei SJ, Rostami-Nejad M. Potential treatment of inflammatory bowel disease: a review of helminths therapy. Gastroenterol Hepatol Bed Bench. 2014;7(1):9–16. | |
Roth L, MacDonald JK, McDonald JW, Chande N. Sargramostim (GM-CSF) for induction of remission in Crohn’s disease: a cochrane inflammatory bowel disease and functional bowel disorders systematic review of randomized trials. Inflamm Bowel Dis. 2012;18(7):1333–1339. | |
Shah S, Terdiman J, Gundling K, Mahadevan U. Immunoglobulin therapy for refractory Crohn’s disease. Therap Adv Gastroenterol. 2014;7(2):99–102. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.