")
Back to Journals » International Journal of Nanomedicine » Volume 15
Recent Clinical Developments of Nanomediated Drug Delivery Systems of Taxanes for the Treatment of Cancer
Authors van Eerden RAG, Mathijssen RHJ, Koolen SLW
Received 17 July 2020
Accepted for publication 15 September 2020
Published 22 October 2020 Volume 2020:15 Pages 8151—8166
DOI https://doi.org/10.2147/IJN.S272529
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Ruben AG van Eerden,1 Ron HJ Mathijssen,1 Stijn LW Koolen1,2
1Department of Medical Oncology, Erasmus MC Cancer Institute, Rotterdam, the Netherlands; 2Department of Hospital Pharmacy, Erasmus MC University Medical Center, Rotterdam, the Netherlands
Correspondence: Ruben AG van Eerden
Department of Medical Oncology, Erasmus MC Cancer Institute, Erasmus University Medical Center, PO Box 2040, Rotterdam 3000 CA, the Netherlands
Tel +31 10 7039640
Email [email protected]
Abstract: Conventional taxanes are used as cornerstone of the chemotherapeutical treatment for a variety of malignancies. Nevertheless, a large proportion of patients do not benefit from their treatment while they do suffer from severe adverse events related to the solvent or to the active compound. Cremophor EL and polysorbate 80 free formulations, conjugates, oral formulations and different types of drug delivery systems are some examples of the several attempts to improve the treatment with taxanes. In this review article, we discuss recent clinical developments of nanomediated drug delivery systems of taxanes for the treatment of cancer. Targeting mechanisms of drug delivery systems and characteristics of the most commonly used taxane-containing drug delivery systems in the clinical setting will be discussed in this review.
Keywords: nanomedicine, nanoparticle, drug delivery systems, taxane, cancer
Introduction
Taxanes are widely used anti-mitotic drugs in the treatment of a variety of tumor types. Within this class of drugs, paclitaxel, docetaxel and cabazitaxel are approved by the FDA.1–3
In the early 1960s, paclitaxel was the first to be discovered. Paclitaxel is a natural taxoid extracted from the North American pacific yew tree, taxus brevifolia.4 Later, in the 1980s, docetaxel, a semisynthetic taxoid derived from the inactive compound 10-deacetylbaccatin III – which is found in the needles of the European yew tree, taxus baccata – was discovered.5 Most recently, in 2010, cabazitaxel, which is also a semisynthetic taxoid derived from 10-deacetylbaccatin III, was approved by the FDA to treat prostate cancer.3,6
The target of taxanes is free tubulin which is found intracellularly and plays an important role during mitosis. Binding of taxanes leads to unusually stable microtubules, thereby blocking DNA segregation and thus cell division eventually leading to cell death.7
The application of taxanes in the clinical setting is characterized by severe side effects as (potentially life-threatening) hypersensitivity reactions (ie, respiratory distress, hypotension, angioedema, urticaria and rash), peripheral neuropathy and hematological toxicity while a substantial part of patients receiving taxanes unfortunately do not benefit from their treatment.8–10 Ineffectiveness might (partly) be caused by insensitivity of the tumor or ineffective concentrations at the target site.11
Docetaxel and paclitaxel are pharmacologically characterized by their hydrophobicity with an aqueous solubility of 3 µg/mL and <10 µg/mL, respectively.12,13 Because of their poor solubility, they have to be dissolved in a solvent with a surfactant. In the current formulation of docetaxel, polysorbate 80 (PS80) is used to increase the solubility while in the case of paclitaxel, Cremophor EL (polyethoxylated castor oil; CrEL) is used for this purpose. Despite the wide use of the current formulations, serious problems are ascribed to the used surfactants. PS80 and CrEL are suggested to contribute to a variety of adverse events related to the infusion of docetaxel and paclitaxel, including peripheral neuropathy, fluid retention, taxane related cardiovascular adverse events, and severe hypersensitivity reaction during intravenous infusions.14–18 Corticosteroids are prescribed as premedication prior to the infusion of taxanes to reduce the incidence of allergic reactions. In the case of paclitaxel, histamine is also added to the prescribed premedication for the same purpose. This premedication could potentially cause side-effects as hyperglycemia and increases the risk of peptic ulcers.19 Despite the administration of premedication, a part of the patients receiving paclitaxel still experience a hypersensitivity reaction.9
In addition, also the pharmacokinetic profile is influenced by these solvents causing non-linear pharmacokinetics and changes in drug distribution.16,20,21 At higher doses, CrEL entraps paclitaxel in micelles which makes paclitaxel less available for metabolic pathways and tumor distribution, which results in a higher systemic exposure and therefore higher probability of severe toxicity.20 PS80 is associated with a reduced clearance of unbound docetaxel, which leads to increased unbound docetaxel systemic exposure and eventually possibly toxicity.22 Moreover, CrEL and PS80 inhibit OATP1B-type transporters which are involved in the uptake of taxanes into cells.23 OATP1B transporters are widely expressed by a variety of tumor types and therefore inhibition of these transporters by these solvents could lead to a reduced tumor exposure to taxanes.23
Because of all the drawbacks of conventional taxane formulations, there is an increasing interest in novel formulations. These new formulations aim to improve drug efficacy and try to deliver the drug more selectively to the tumor while reducing the incidence and severity of adverse events. Several approaches have been explored, for example, surfactant-free intravenous formulations, oral formulations and drug delivery systems.24–26 Examples of these drug delivery systems are liposomes, micelles, polymeric nanoparticles, and other nanoparticles.
In this review, we aim to discuss the recent clinical developments and global characteristics of nanomediated drug delivery systems of taxanes for the treatment of cancer. Furthermore, we will discuss if the proposed approaches and expectations raised in preclinical studies result in the desired reduced toxicity, increased efficacy, optimized pharmacokinetic profile and selective delivery of taxanes in clinical practice.
Targeting
Targeting the tumor site by changing the chemical properties and thereby the pharmacokinetic profile of the drug is a relatively new strategy to improve the risk–benefit ratio. Two ways of tumor targeting are known to selectively target the tumor and deliver the drug at that specific location; active and passive targeting, respectively.
An active targeting nanoparticle uses tumor cell-specific characteristics to selectively attack the tumor. All (malignant) cells are covered with markers on the surface which are essential for a wide variety of processes. The profile of these markers varies between different types of tissue, and these differences in expression of surface markers offer a chance for nanoparticles to actively and specifically target the tumor (Figure 1).27 Coupling a cytostatic drug with an antibody, ligand, or small molecule offers a possibility for a drug delivery system to selectively bind to tumor cells and direct their payload towards the site of action (Figure 1).28–30 These conjugated antibodies can target a marker such as HER2, EGFR and CD-19, which are expressed on the surface of the cell.30,31 Ligand conjugated drug delivery systems are using receptors on the surface of a cancer cell to target the tumor via their ligand. Examples of ligands used are folate which targets the folate receptor, and transferrin which targets the transferrin receptor.31 Another possible approach is targeting the tumor via small molecules such as PSMA.32
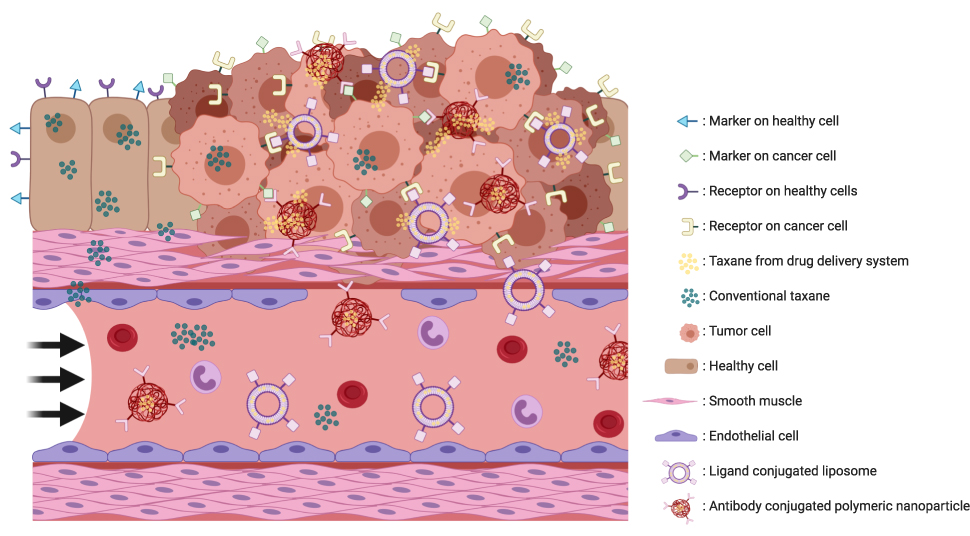 |
Figure 1 Schematic impression of active targeting drug delivery systems. Surface markers varies between healthy and tumor tissue. An targeting drug delivery system has one specific target which causes a release of the payload if the drug delivery system is bound to the target. When the target is not expressed or not bound the drug delivery system will not release its payload and will be transported to others locations. This figure was created with BioRender.com. |
BIND-014 is an example of a polymeric nanoparticle, which encapsulates docetaxel and actively targets the tumor via PSMA.32 It is 100 nm in size and consists of a hydrophobic polylactic acid polymeric core and a hydrophilic polyethylene glycol shell covered with Prostate-Specific Membrane Antigen (PMSA) targeting ligands.29 PSMA is expressed by prostate cancer cells, as well as on the neovascularization of solid tumors, while very little types of healthy tissue express PSMA.27,33 In a preclinical trial, BIND-014 was found to have a different pharmacokinetic profile compared to conventional docetaxel.32 The in vivo systemic exposure (area under the curve; AUC) to docetaxel was 46- to 670-fold higher after administration of the nanoparticle.32 After up to 50 hours, almost all of the docetaxel measured in plasma was still encapsulated by the particle.32 The nanoparticle prevented rapid distribution of docetaxel from the systemic compartment which resulted in a lower distribution volume compared to conventional docetaxel.32 The intratumoral concentration of docetaxel after the administration of the nanoparticle was 7-fold higher with a significantly improved efficacy compared to conventional docetaxel in mice.32 The administration of the nanoparticle was well tolerated in vivo and no hypersensitivity reactions were found.32 Recently, the results of a Phase II study investigating BIND-014 dosed at 60 mg/m2 every three weeks (without premedication), combined with prednisone 5 mg twice daily in metastatic castration-resistant prostate cancer were published.28 The overall response rate at this dosing regimen is 32% with a progression-free survival of 9.9 months.28 Neutropenia, fatigue, neuropathy, diarrhea, nausea and mucositis are the most commonly reported clinical adverse events of BIND-014.28,29 Contrary to the improved toxicity profile in preclinical studies, the toxicity profile of BIND-014 in clinical practice seems to be similar to conventional docetaxel. The observed pharmacokinetic characteristics in the clinical trials showed similarities with the preclinical data; eg, higher AUC, lower distribution volume and high stability.28,29,32
In contrast to active targeting, passive targeting uses characteristics of the microenvironment in the tumor to deliver the payload of a nanoparticle at the aimed site of action. The “enhanced permeability and retention” (EPR) effect is a generally accepted concept, explaining the passive targeting of nanoparticles based on a combination of the tissue and location characteristics (Figure 2).34 The tumor angiogenesis and vasculature substantially differ compared to vasculature in healthy organs (Figure 2).35,36 Most malignant cells divide at a higher rate, and in a more chaotic manner compared to healthy cells. To maintain the growth rate, tumor cells require nutrients which are supplied by the vascular circulation. Aggregates of tumor cells which are 150–200 µm (or larger) also need neovascularization to acquire enough blood supply for their maintenance.37 The angiogenesis rate, with elevated VEGF expression, is therefore high.38 Leaky vessels are formed due to the high rate of neovascularization (Figure 2).39 The tumor vasculature is characterized by poorly aligned endothelial cells resulting in wide fenestrations, lack of a smooth muscle layer, loss of auto-regulated homeostatic blood flow control and loss of angiotensin II receptors.34,35,38,39 Moreover, elevated levels of bradykinin, nitric oxide, peroxynitrite and vascular permeability factor are found in solid tumors which results in an even higher permeability of the local tumor vessels.34 A reduced intratumoral lymphatic drainage also contributes to the EPR effect.38 As a result of the increased permeability and lack of lymphatic drainage, drug delivery systems – ranging in size from 50 to 500 nm – can cross the endothelial barrier of the vasculature and be retained in the tumor (Figure 2).37,38,40 Nevertheless, in clinical practice, a wide heterogeneity in EPR effect exists. Various tumor characteristics such as oxygenation, necrosis, fibrosis, levels of EPR related effectors or enzymes, tumor size and vasculature all contribute to the variability of the effect.41 It is widely known that the vascularization differs between tumor types. The heterogeneity in EPR effect could be reduced by coadministration of drugs modulating the levels of the enzymes or effectors related to the EPR effect such as bradykinin and nitric oxide.41
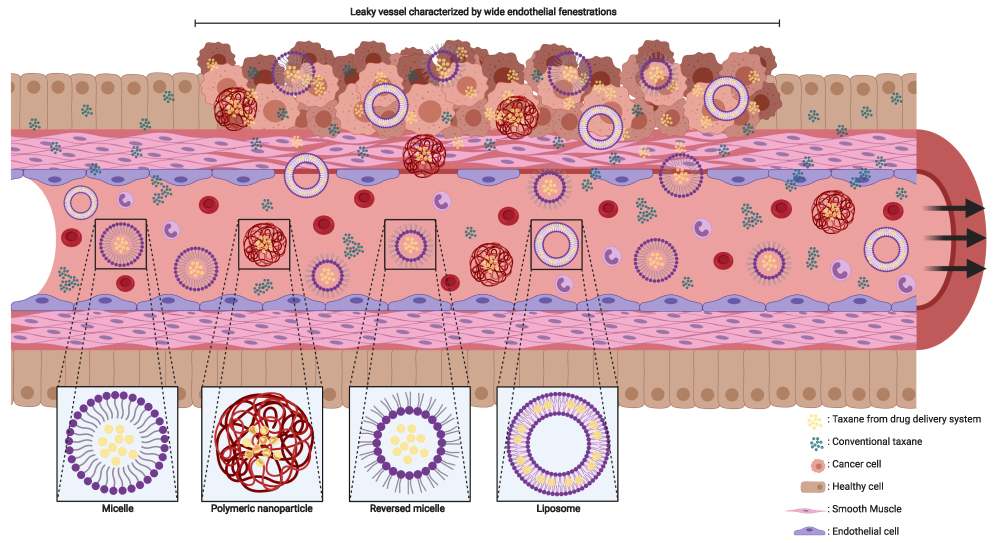 |
Figure 2 Schematic impression of EPR effect and nanoparticles. The fenestrae in the vascular wall of the tumor are wider than in normal tissue and the smooth muscle cells in the vascular wall are arranged in a chaotic manner compared to healthy tissue. Nanoparticles and conventional taxanes can easily pass the vascular wall inside the tumor. In contrast, the endothelial cells and smooth muscle cells are good aligned in the vascular wall of healthy tissue, which makes it hard for nanoparticle to cross the wall while conventional taxanes can still penetrate inside healthy tissue. This figure was created with BioRender.com. |
Targeting the tumor passively can also be achieved by using small differences in pH or temperature between tumor and healthy tissue.42 Nanoparticles such as liposomes, micelles and polymeric nanoparticles can be modified to be sensitive to a certain pH or temperature.43,44 If the pH or temperature-sensitive nanoparticles reach the target site by passive diffusion, the equilibrium of the nanoparticle with the encapsulated drug may shift and the drug may be released.42
Drug Delivery Systems
Each type of drug delivery system has its own characteristics in composition, structure, size and surface characteristics (Table 1). All these factors affect the stability, solubility, drug distribution, delivery mechanism, drug release, efficacy, toxicity profile and clearance of the nanoparticle. Drug delivery systems containing taxanes can be found in Table 2.
 |
Table 1 Types of Most Used Drug Delivery Systems Containing Taxanes and Their Characteristics |
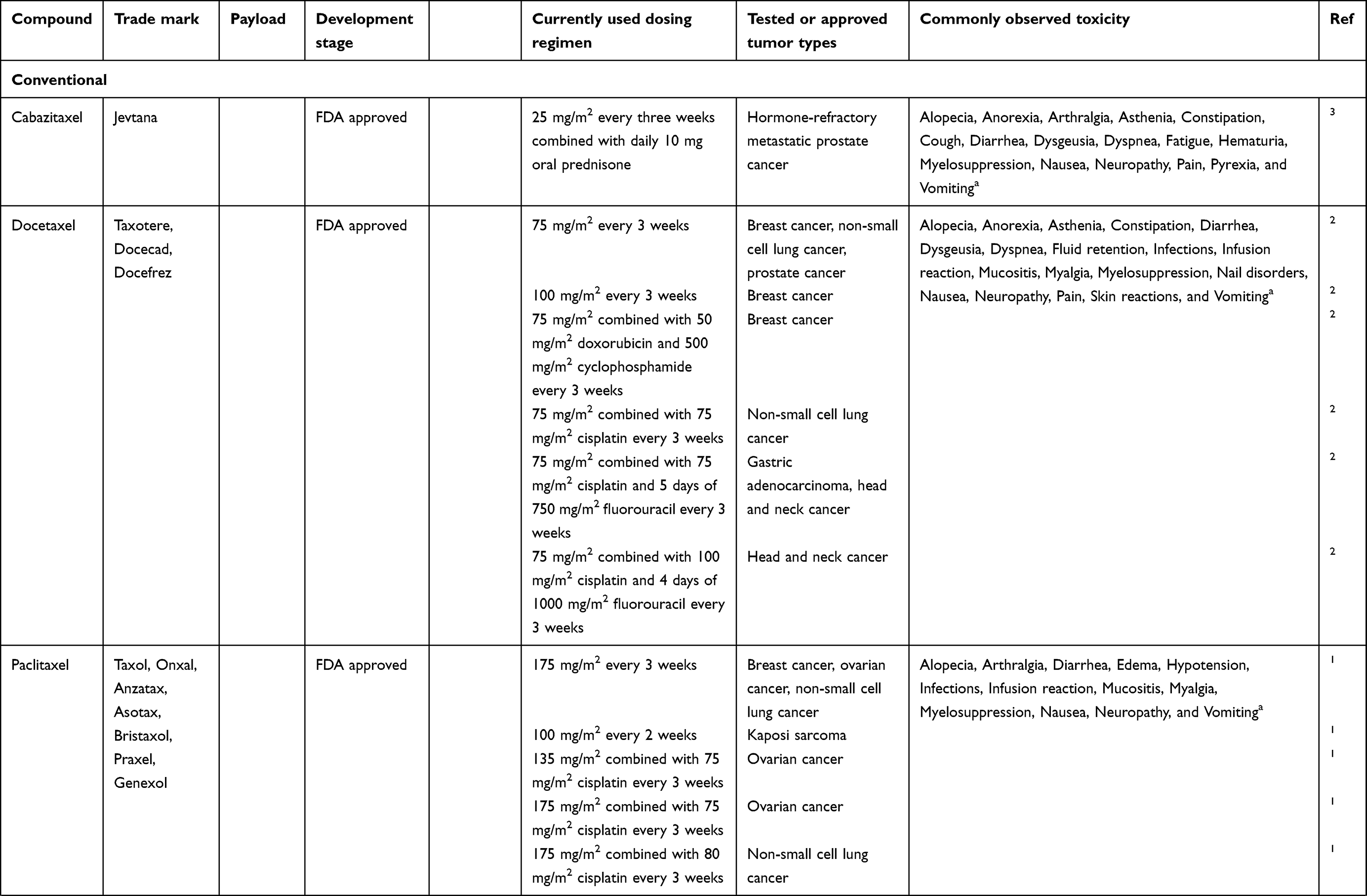 | 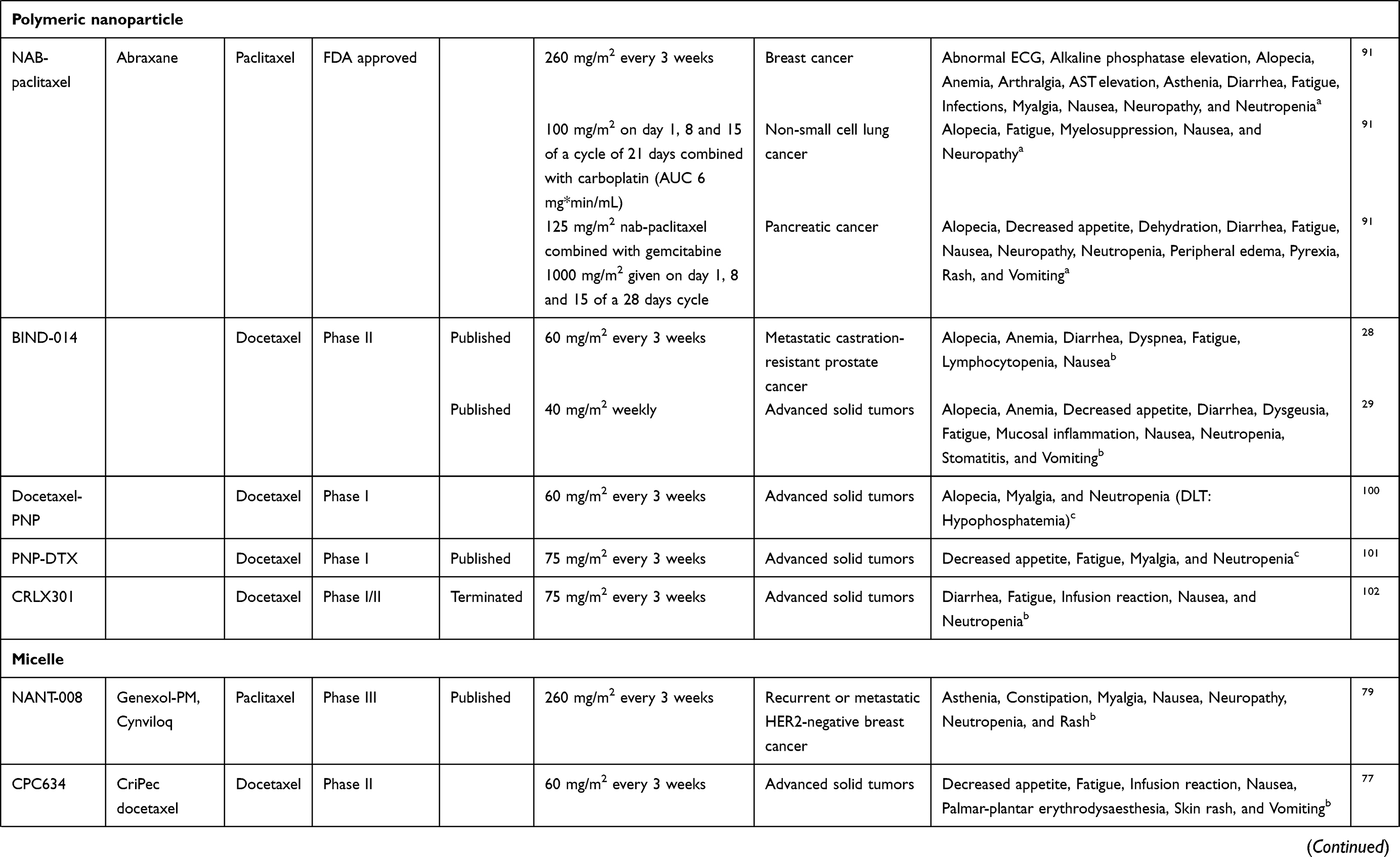 | 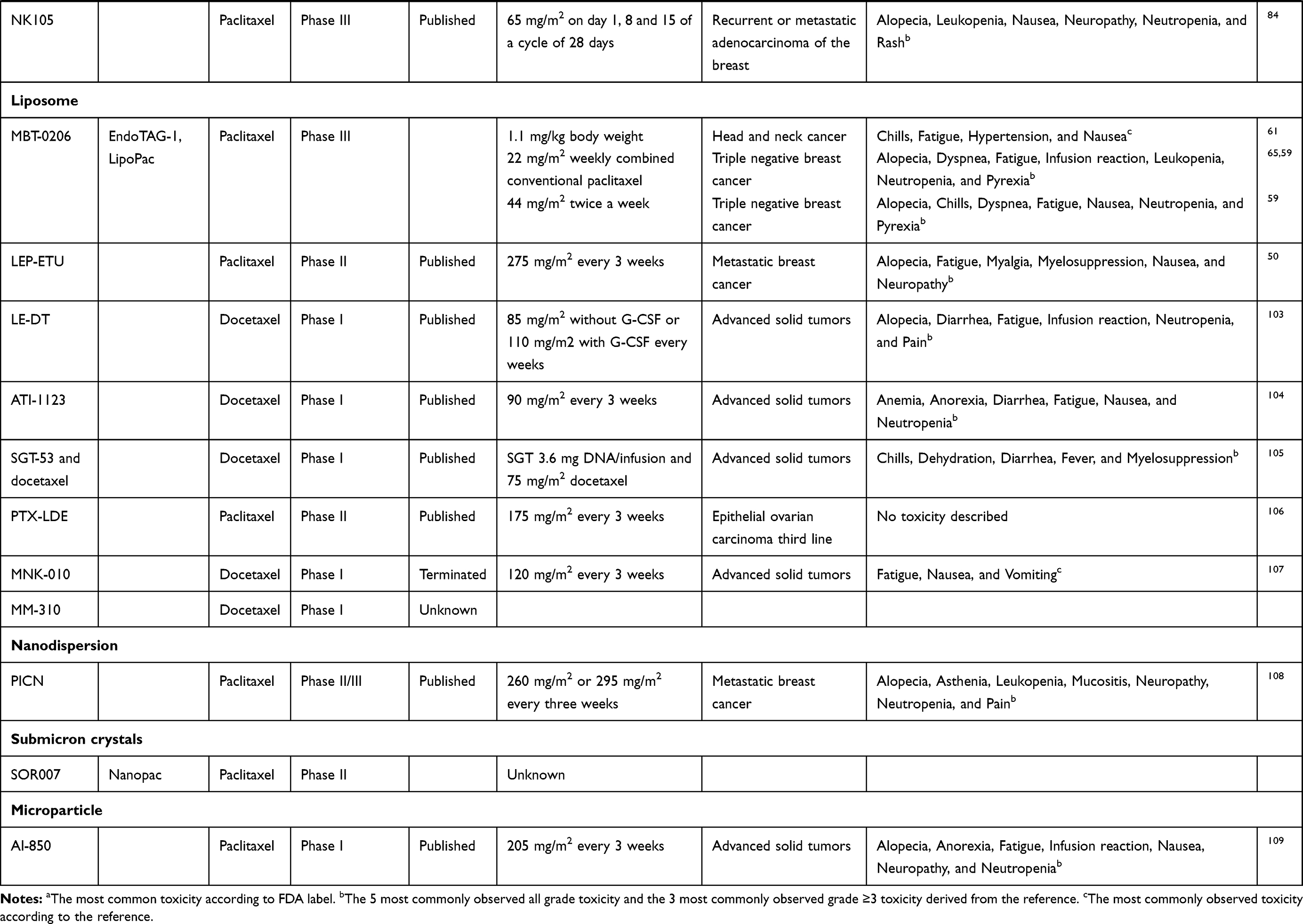 |
Table 2 Clinical Tested Drug Delivery Systems Containing Taxanes and Their Stage of Development and Dosing Schedules |
Liposomes
Liposomes as drug delivery systems have been investigated for several drugs. For some of these drugs, liposomal formulations are even already approved for clinical use.45,46
Liposomes are spherical-formed vesicles which consist of one (unilamellar) or multiple (multilamellar) phospholipid bilayer(s) and mainly use the EPR effect to increase intratumoral exposure (Figure 2).12,30,47–49 Cholesterol and phosphatidylcholine are often used lipids for the phospholipid bilayer.30,47 In the core of this bilayer, an aqueous compartment is formed which can encapsulate hydrophilic drugs (Figure 2).47,49 Hydrophobic substances such as paclitaxel and docetaxel can be incorporated in the phospholipid bilayers (Figure 2).47 This gives liposomes an amphipathic (ie, having hydrophobic and hydrophilic groups) character. A wide range of sizes is described for liposomes; ranging between 50–400 nm.47 The ability to modify the surface of the liposome is very important. Conventional liposomes are rapidly cleared from the bloodstream.50 This rapid clearance is caused by the reticuloendothelial system (RES) in the systemic circulation.51 This drawback can be overcome by coating highly hydrophilic chains such as PEG to the surface of liposomes.42,50 Pegylated liposomes are also called “stealth” (sterically stabilized) liposomes.12,42 These stealth liposomes are characterized by a lower accumulation in healthy tissue, preferential uptake by the tumor and favorable pharmacokinetics, caused by a reduced uptake by the reticuloendothelial system and a smaller distribution volume leading to a slower clearance.12,30,52,53 Nevertheless, earlier research also described an “accelerated blood clearance” (ABC effect) during repeated infusions of stealth liposomes, which might be caused by serum IgM activating the complement system.54 This could eventually lead to increased liver uptake and clearance of the liposomes which possibly results in liver toxicity and decrease of efficacy.54 Liposomes could also be coated with monoclonal antibodies which permits them to actively and selectively target a certain type of cells.50,55 Moreover, components of liposomes are described to modify multidrug resistance caused by P-glycoprotein.56 Consequently, liposomes might increase drug concentrations inside the tumor and increase the permeability of the blood–brain barrier.56 On the other hand, potential drawbacks of liposomes are batch-to-batch variation, poor stability, insufficient drug loading, immune reactions and accelerated blood clearance.12,13,30,50 During clinical trials, immune reactions were observed which needed to be treated with routine administration of antihistamines and steroids as premedication.57,58 EndoTAG-1 is one of the taxane-containing liposomes in which a large proportion of the treated patients required premedication.59 1,2 dioleoyl-3-trimethylammonium-propane (DOTAP) and 1,2 dioleoyl-sn-glycero-3-phosphocholine (DOPC) are used to construct EndoTAG-1 which encapsulates paclitaxel.60,61 The size of the EndoTAG-1 typically ranges between 180 and 200 nm.60 Cationic liposomes have been proven to selectively accumulate within the tumor vascularization.62 In line with this, EndoTAG-1 showed a 4-fold selectivity compared with surrounding healthy tissue in vivo.63 A preclinical study with EndoTAG-1 demonstrated that it impairs the vascularization of the tumor by reducing vessel diameter and functional vessel density.60 EndoTAG-1 reduced the tumor growth significantly compared to conventional paclitaxel in vivo.63,64 Several treatment regimens have been tested clinically in Phase I and II trials (Table 2).59,61,65,66 When premedication was not routinely given to patients still approximately 60% of the included patients required systemic corticosteroids.59 The most common toxicities were hypersensitivity, fatigue and chills.59,66 Other commonly reported adverse events were nausea, vomiting, abdominal pain and dyspnea.59,66 The combination of EndoTAG-1 plus paclitaxel led to a higher incidence of neutropenia but a lower incidence of neuropathy compared to conventional paclitaxel monotherapy.59 In the phase II trial, the response rate of first-line EndoTAG-1 monotherapy observed in triple-negative breast cancer was 25% with a median PFS of 3.0 months and OS of 11.9 months which was lower than the response rate and PFS of paclitaxel monotherapy.59 The combination of EndoTAG-1 plus paclitaxel led to a response rate of 45% with a median PFS of 3.7 months and OS of 13.0 months.59 Currently, two Phase III trials are ongoing; one is investigating the combination of EndoTAG-1, gemcitabine and paclitaxel in triple-negative breast cancer with visceral metastasis (trial number: NCT03002103), the other investigates EndoTAG-1 and gemcitabine in patients with locally advanced or metastatic adenocarcinoma of the pancreas who failed on FOLFIRINOX (trial number: NCT03126435). The pharmacokinetic profile of EndoTAG-1 is characterized by a Cmax at the end of the infusion, low intravascular stability of the particle, rapid clearance from the plasma compartment, and a low distribution volume compared to conventional paclitaxel.66
Another liposome containing a taxane is LEP-ETU, which is an acronym of “Liposome Encapsulating Paclitaxel – Easy To Use”. LEP-ETU consists of 1.2‐dioleoyl‐sn‐glycero‐3‐phosphocholine (DOPC), cholesterol, and cardiolipin, and it is 150 nm in diameter with a drug-entrapment efficiency of >90%.67 In vitro, less than 6% of the payload was released during the first 120 hours at physiologic temperatures.67 The in vivo Cmax and AUC of LEP were, respectively, 1.4- to 3-fold and 2-fold higher compared to conventional paclitaxel.68 The distribution volume was equal between the two formulations.68 The pharmacokinetic profile of LEP was linear. In vivo, no significant differences in efficacy were observed between LEP and conventional paclitaxel.68 The maximum tolerable dose of LEP-ETU in patients is 150 mg/m2 weekly or 3-weekly 325 mg/m2, both without premedication.58,69,70 In clinical trials, the most reported adverse events were myelosuppression, fatigue, alopecia, myalgia and neuropathy.69,70 However, the neuropathy observed was not worse than the neuropathy caused by conventional paclitaxel.69 The pharmacokinetic profile of LEP-ETU and conventional paclitaxel were bioequivalent.58,70 Therefore, the overall pharmacological advantages and increased tolerability of the liposome seem to be marginal, which is contrary to the suggested improved pharmacokinetic profile and toxicological characteristics of LEP in the preclinical study. No phase II trial has been published yet publicly.
Micelles
Micelles are spherically formed with a hydrophobic core and a hydrophilic shell (also called corona) (Figure 2).47 A less commonly used formation is the reverse micelle (Figure 2).71 The hydrophilic head of the polymers forms the core, while the hydrophobic nonpolar tail forms the outside of the shell of the reverse micelle.44,47,48 The shell of a micelle consists of polymeric molecules such as amphiphilic polymers (eg, block-polymers) or surfactants.44,47,48 The most widely used hydrophilic polymer (to create the shell of the micelle) is PEG.72 PLA and PCL are commonly used as hydrophobic block.30 The sizes of micelles range between 10 and 100 nm which is relatively small.44,47 The advantages of micelles include increased solubility, protection of the payload against degradation, ability to inhibit drug efflux transporters, highly permeable shell, relatively small uptake by the RES system, ability to modify the composition (ie, surface and size), use without premedication and enhanced use of the EPR effect.44,47,48,73 Micelles are also described to have a favorable drug release. Nevertheless, avoiding premature (burst) drug release, controlling the amount of drug released over time and tumor selectivity, remains challenging.49,74 Several methods have been attempted to address this among which controlling the surface charge and covalently binding of the nanoparticle shell and core.24,75 CPC634 is a docetaxel encapsulating polymeric micelle which uses the technique of covalently binding. The polymeric micelle is 65 nm in size and consists of monomethoxy poly(ethylene glycol) (mPEG) and N−2-hydroxypropyl methacrylamide mono- or dilactate (HPMAmLacn).76,77 Covalently binding of the payload to the micelle resulted in vivo in a decrease of premature drug release and a sustained drug release over time. In vivo CPC634 administration resulted in a significantly higher intratumoral docetaxel concentration at different time-points up to 4 days.76 Moreover, in vivo, the docetaxel remained entrapped in the particle for several days in the bloodstream.76 CPC634 revealed to be superior compared to a conventional formulation of docetaxel in efficacy as well as in tolerability in rats.76 The recommended phase II dose of CPC634 is 60 mg/m2 three weekly, and dexamethasone is given as premedication.77 The toxicity profile observed in patients mainly consists of rash, stomatitis, fatigue and neuropathy.24,77 Neuropathy was seen in more cases after CPC634 administration than after conventional docetaxel.24 However, the incidence of neutropenia during CPC634 administration is lower.24 Our research group performed an extensive intrapatient head-to-head pharmacokinetic analysis, comparing CPC634 to conventional docetaxel.24 The Cmax and AUC of total docetaxel –which is docetaxel in the nanoparticle plus unbound docetaxel– and released docetaxel (unbound docetaxel) were significantly higher after CPC634 infusion, compared to conventional docetaxel.24 In addition, the intrapatient intratumoral exposure was compared between CPC634 and conventional docetaxel.24 CPC634 resulted in an equal intratumoral released docetaxel concentration, while the exposure to total docetaxel was 4-fold higher after CPC634 administration, compared to conventional docetaxel.24 Currently, a phase II study is ongoing, evaluating the response rates of platinum-resistant ovarian cancer (trial number: NCT03742713).
Recently two other taxane containing polymeric micelles were investigated in phase III trials. The first one is NANT-008 which consists of a shell formed by monomethoxy poly-(ethylene glycol) (mPEG) and a core consisting of poly-(D,L-lactic acid) (PDLLA).78 The in vivo concentrations of paclitaxel in tissues were 2- to 3-fold higher for NANT-008 in comparison with conventional paclitaxel.78 Compared to conventional paclitaxel, the plasma Cmax and AUC of NANT-008 were lower while the distribution volume of NANT-008 was higher in mice.78 In vivo data indicated that NANT-008 was well tolerated and the MTD was 3-fold higher for NANT-008 with an improved antitumor efficacy.78 During the phase III clinical trial in patients diagnosed with recurrent or metastatic HER2-negative breast cancer,79 NANT-008 was administered at 260 mg/m2 every three weeks.79 Premedication was not obliged.79 However, during the study hypersensitivity reactions were frequently observed which resulted in 83% of the patients receiving premedication.79 Frequently observed adverse events were neuropathy, neutropenia, anorexia and myalgia.80–83 However, no differences in the incidence of neuropathy and myalgia were observed between the micelle and the conventional formulation.79 The overall response rate was 39% with a progression-free survival of 8.0 months and an overall survival of 28.8 months compared to a response rate of 24% for conventional paclitaxel.79 Survival (PFS and OS) was not significantly different between the micelle and conventional formulation.79 The AUC and Cmax of NANT-008 are lower compared to conventional paclitaxel, which is probably a result of a rapid distribution.83
The second polymeric micelle tested in phase III also encapsulates paclitaxel, and is called NK105.84 The hydrophilic part of the micellar shell consists of PEG.85 The hydrophobic part of the particle consists of modified polyaspartate.85 NK105 is approximately 85 nm in diameter. In vivo, the systemic exposure was 50- to 86-fold higher than the exposure to conventional paclitaxel.85 The distribution volume was approximately 15 times smaller.85 The intratumoral Cmax and AUC in mice were, respectively, 3 and 25 times higher, compared to conventional paclitaxel.85 The intratumoral concentrations accumulated up to 72h.85 The in vivo antitumor efficacy was significantly better for NK105 compared to conventional paclitaxel.85 The peripheral nerves of the mice were less affected by NK105 than by conventional paclitaxel.85 During the clinical phase III trial, including patients with metastatic or recurrent breast cancer, weekly 65 mg/m2 NK105 (administration on days 1, 8 and 15 of a cycle of 28 days) was given, while NK105 was dosed at 150 mg/m2 every 3 weeks in a phase II trial in gastric cancer.84,86 Only 22% of the patients in the phase III trial needed premedication with a hypersensitivity incidence of 4%.84 The safety profile of NK105 was roughly the same compared to conventional paclitaxel.86,87 The incidence of neuropathy was significantly lower after NK105 administration, while no differences were seen in hematological toxicity.84 The observed response rate was 32% with a PFS of 8.4 months and OS of 31.2 months, respectively, while conventional paclitaxel resulted in a response rate of 39% with a PFS of 8.5 months and an OS of 36.2 months.84 The pharmacokinetic profile was remarkably different compared to the pharmacokinetic profile of conventional paclitaxel. NK105 displayed a linear pharmacokinetic profile with a 7-fold higher Cmax, 9- to 50-fold higher AUC and 10- to 25-fold lower distribution volume.86–88 Of the total paclitaxel exposure, 5% was released from the micelle.88
Polymeric Nanoparticles
Polymeric nanoparticles are solid particles which encapsulate drugs in a polymeric matrix with a size ranging between 10 and 1000 nm (Figure 2).30,48 Because of the variety of polymers and copolymers that can be used to composite the particle, structure and characteristics of polymeric nanoparticles are highly variable. Poly(lactic-co-glycolic acid) (PLGA) is the most widely used polymer because of its biodegradability.30 Nevertheless, other natural (ie, albumin, chitosan, etc.) and synthetic (ie, PEG, PLA, PGA, etc.) polymers are also widely used.30 It is even possible to blend different polymers and create a double-walled particle which results in a lower initial release of the payload or the ability to store different types of payload in the particle.30,89 Polymeric nanoparticles are commonly coated with nonionic surfactants to reduce opsonization by the RES system, hydrophobic interactions and hydrogen bonding.90 The payload inside a polymeric nanoparticle could be released by diffusion through the matrix of polymers, erosion of the particle or as a response to the target location.30,48 The advantages of this type of drug delivery system are their high stability during storage, protection of the payload against degradation, controlled drug release, multiple administration routes and generally low toxicity profile and systemic clearance.48
To date, nanoparticle-albumin-bound paclitaxel (nab-paclitaxel or Abraxane®) is the only FDA approved example of a taxane-loaded polymeric nanoparticle.91 Nab-paclitaxel is 130 nm in diameter and consists of particles formulated of human serum albumin loaded with paclitaxel.92 Paclitaxel is not covalently bound to the albumin. In vivo, differences in pharmacokinetic profiles exist between nab-paclitaxel and conventional paclitaxel. The distribution volume of nab-paclitaxel was approximately 50% higher.92,93 Nab-paclitaxel is hypothesized to lead to a higher intratumoral paclitaxel exposure by enhancing albumin mediated transcytosis (ie, transcellular transport) and the EPR effect.92 In vivo, Nab-paclitaxel was rapidly partitioned into the tissue with an absorption constant which was 3.3-fold higher.92 The endothelial binding and crossing through the endothelial monolayer of paclitaxel were, respectively, 10-fold and 4-fold increased in vitro after administration of nab-paclitaxel, while the in vivo intratumoral paclitaxel exposure was 33% higher after nab-paclitaxel administration.92 Nab-paclitaxel was less toxic in vivo, and still, the efficacy was improved compared to conventional paclitaxel.92 The recommended dose for metastatic breast cancer is 260 mg/m2 every three weeks.91 Nab-paclitaxel is dosed at 100 mg/m2 for non-small cell lung cancer (NSCLC) and 125 mg/m2 for pancreatic carcinoma (regimen showed in Table 2).91 No premedication is needed.91 The most commonly seen adverse events of nab-paclitaxel are myelosuppression, alopecia, sensory neuropathy, fatigue, myalgia, nausea and diarrhea. In NSCLC patients, significantly more thrombocytopenia and anemia were observed after nab-paclitaxel administration, while grade ≥3 sensory neuropathy, neutropenia, arthralgia and myalgia were observed less.94 In contrast, in breast cancer patients, grade ≥3 fatigue was more described after nab-paclitaxel while the incidence of severe neutropenia and sensory neuropathy was equal between the groups.95 Response rates in NSCLC patients were significantly higher for nab-paclitaxel compared to conventional paclitaxel, while survival did not significantly differ.94 According to a meta-analysis, the overall response rate and progression-free survival in the metastatic breast cancer group were significantly higher for nab-paclitaxel compared to conventional paclitaxel.95 However, overall survival was not significantly different.95 In the pharmacokinetic profiles, some differences exist between the two formulations. The fraction of unbound/free paclitaxel was significantly higher for nab-paclitaxel compared to conventional paclitaxel. The Cmax of unbound paclitaxel is 10-fold higher and Cmax of total paclitaxel 3.8- to 6.5-fold higher after nab-paclitaxel.93,96 The AUC of unbound paclitaxel is 2.7-fold higher while the AUC is equal between the formulations.93,96 The distribution volume of nab-paclitaxel is significantly higher for total paclitaxel.93 During the infusion of nab-paclitaxel, nanoparticles dissolve and albumin-paclitaxel complexes are formed.97 Following this, paclitaxel can bind and unbind albumin in the bloodstream.96 Because of this transient systemic binding and unbinding of paclitaxel with albumin, it can be questioned whether nab-paclitaxel is a true nanoparticle. Despite that, a paclitaxel formulation without CrEL is a huge advantage.
Conclusions
Over the last years, several new drug delivery systems containing taxanes have been developed to improve the tolerability and the efficacy of the treatment with taxanes. Based on the EPR effect, drug delivery systems should lead to a higher drug exposure in the tumor which could possibly increase efficacy. The tumor selectivity of drug delivery systems (by passive or active targeting the tumor) should lead to an increased tolerability profile compared to the conventional formulation. An important additional advantage of the nanoparticle formulation is that they do not require toxic surfactants such as CrEL or PS80. In preclinical studies, drug delivery systems containing taxanes improved efficacy, tolerability and had a favorable pharmacokinetic profile with higher intratumoral taxane exposure compared to the classic drugs. Nevertheless, few of the – in a preclinical setting – promising taxane-containing drug delivery systems have reached clinical practice. Nab-paclitaxel is an example of a taxane-containing drug delivery system which is widely used in the daily clinical care. Several other taxane-containing drug delivery systems failed to improve the efficacy in the clinical setting, while others even failed to improve the tolerability of the treatment. Commonly, side effects described to the drug delivery systems are similar to the toxicity profile of conventional taxanes; however, its severity might be lower. Clinical evidence, proving the hypotheses of nanoparticles leading to an accumulated intratumoral drug exposure, is largely lacking for taxane-containing drug delivery systems, while the development of these systems is based on the assumption that the intratumoral exposure is increased. Furthermore, disappointing results observed in the clinical setting could be caused by the urge of investigators to enhance accumulation of anticancer drug concentrations in the tumor, while in the meantime, the stability of drug delivery systems in the circulation has to be high. However, it should be noted that given the relatively small differences in the physical environment between the tumor and healthy tissue, high stability in the circulation or healthy tissue probably also means a high intratumoral stability. The relatively small difference in the physical environment may also hinder the triggering of stimuli-sensitive drug delivery systems and therefore also their added value over normal drug delivery systems. More recently, several investigations using multifunctional stimulus-responsive drug delivery systems (or nanoreactors) were published.98,99 These systems aim to specifically upregulate stimuli inside the physical environment of the tumor to create an amplified intratumoral stimulus compared to the healthy tissue. This will eventually lead to a release of active drug from the multifunctional stimulus-responsive drug delivery system and therefore intratumoral accumulation of the anticancer drugs.
Another explanation for the disappointing results seen in clinical trials with taxane-containing drug delivery systems might be the focus of most of the currently published manuscripts on total drug exposure, while no data is collected regarding released drug exposure. Nevertheless, only the released drug has antitumor efficacy, and therefore increase of total drug exposure without increasing the released drug could lead to unexpected disappointing results regarding efficacy later on in the drug development.
Further research on these drug delivery systems is necessary and should (also) focus on intratumoral drug exposure of released drugs since this knowledge might explain the reason why improved efficacy could (or could not) be demonstrated in the clinical setting. In conclusion, the development of novel formulations or new drug delivery systems to solve the problem of excessive adverse events and ineffectiveness of the widely used conventional taxanes is still warranted.
Disclosure
This review was performed without external funding. Ron HJ Mathijssen and Stijn LW Koolen received an unrestricted grant of Cristal Therapeutics (producer of CPC634) for a phase I clinical trial with this nanomedicine agent. The authors report no other conflicts of interest in this work.
References
1. Bristol-Myers Squibb Company. Taxol (Paclitaxel). U.S. Food and Durg Administration website. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/020262s049lbl.pdf.
2. Sanofi-aventis U.S. LLC. Taxotere (Docetaxel). U.S. Food and Drug Administration website. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2013/020449s071lbl.pdf.
3. Sanofi-aventis U.S. LLC. Jevtana (Cabazitaxel). U.S. Food and Drug Administration website. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/201023lbl.pdf.
4. Wani MC, Taylor HL, Wall ME, Coggon P, McPhail AT. Plant antitumor agents. VI. The isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J Am Chem Soc. 1971;93(9):2325–2327. doi:10.1021/ja00738a045
5. Ringel I, Horwitz SB. Studies with RP 56976 (taxotere): a semisynthetic analogue of taxol. J Natl Cancer Inst. 1991;83(4):288–291. doi:10.1093/jnci/83.4.288
6. Mita AC, Denis LJ, Rowinsky EK, et al. Phase I and pharmacokinetic study of XRP6258 (RPR 116258A), a novel taxane, administered as a 1-hour infusion every 3 weeks in patients with advanced solid tumors. Clin Cancer Res. 2009;15(2):723–730. doi:10.1158/1078-0432.CCR-08-0596
7. Schiff PB, Horwitz SB. Taxol stabilizes microtubules in mouse fibroblast cells. Proc Natl Acad Sci U S A. 1980;77(3):1561–1565. doi:10.1073/pnas.77.3.1561
8. Cortes JE, Pazdur R. Docetaxel. J Clin Oncol. 1995;13(10):2643–2655. doi:10.1200/JCO.1995.13.10.2643
9. Rowinsky EK, Donehower RC. Paclitaxel (taxol). N Engl J Med. 1995;332(15):1004–1014. doi:10.1056/NEJM199504133321507
10. Weiss RB, Donehower RC, Wiernik PH, et al. Hypersensitivity reactions from taxol. J Clin Oncol. 1990;8(7):1263–1268.
11. Lee M. Greenberger and Deepak Sampath; Chapter: 18 Resistance To Taxanes; Pages: 329-358. Cancer Drug Discovery and Development: Cancer Drug Resistance; 2006; Edited by: B. Teicher - Humana Press Inc., Totowa, NJ.
12. Engels FK, Mathot RA, Verweij J. Alternative drug formulations of docetaxel: a review. Anticancer Drugs. 2007;18(2):95–103. doi:10.1097/CAD.0b013e3280113338
13. Nuijen B, Bouma M, Schellens JH, Beijnen JH. Progress in the development of alternative pharmaceutical formulations of taxanes. Invest New Drugs. 2001;19(2):143–153. doi:10.1023/A:1010682916808
14. Bergh M, Magnusson K, Nilsson JL, Karlberg AT. Contact allergenic activity of Tween 80 before and after air exposure. Contact Dermatitis. 1997;37(1):9–18. doi:10.1111/j.1600-0536.1997.tb00368.x
15. Lorenz W, Schmal A, Schult H, et al. Histamine release and hypotensive reactions in dogs by solubilizing agents and fatty acids: analysis of various components in cremophor El and development of a compound with reduced toxicity. Agents Actions. 1982;12(1–2):64–80. doi:10.1007/BF01965109
16. Ten Tije AJ, Verweij J, Loos WJ, Sparreboom A. Pharmacological effects of formulation vehicles: implications for cancer chemotherapy. Clin Pharmacokinet. 2003;42(7):665–685.
17. Windebank AJ, Blexrud MD, de Groen PC. Potential neurotoxicity of the solvent vehicle for cyclosporine. J Pharmacol Exp Ther. 1994;268(2):1051–1056.
18. Mark M, Walter R, Meredith DO, Reinhart WH. Commercial taxane formulations induce stomatocytosis and increase blood viscosity. Br J Pharmacol. 2001;134(6):1207–1214. doi:10.1038/sj.bjp.0704387
19. Poetker DM, Reh DD. A comprehensive review of the adverse effects of systemic corticosteroids. Otolaryngol Clin North Am. 2010;43(4):753–768. doi:10.1016/j.otc.2010.04.003
20. Sparreboom A, van Zuylen L, Brouwer E, et al. Cremophor EL-mediated alteration of paclitaxel distribution in human blood: clinical pharmacokinetic implications. Cancer Res. 1999;59(7):1454–1457.
21. van Tellingen O, Huizing MT, Panday VR, Schellens JH, Nooijen WJ, Beijnen JH. Cremophor EL causes (pseudo-) non-linear pharmacokinetics of paclitaxel in patients. Br J Cancer. 1999;81(2):330–335. doi:10.1038/sj.bjc.6690696
22. Baker SD, Li J, Ten Tije AJ, et al. Relationship of systemic exposure to unbound docetaxel and neutropenia. Clin Pharmacol Ther. 2005;77(1):43–53. doi:10.1016/j.clpt.2004.09.005
23. Nieuweboer AJ, Hu S, Gui C, et al. Influence of drug formulation on OATP1B-mediated transport of paclitaxel. Cancer Res. 2014;74(11):3137–3145.
24. Atrafi F, van Eerden RAG, van Hylckama Vlieg MAM, et al. Intratumoral comparison of nanoparticle entrapped docetaxel (CPC634) with conventional docetaxel in patients with solid tumors. Clin Cancer Res. 2020;26(14):3537–3545. doi:10.1158/1078-0432.CCR-20-0008
25. Koolen SL, Beijnen JH, Schellens JH. Intravenous-to-oral switch in anticancer chemotherapy: a focus on docetaxel and paclitaxel. Clin Pharmacol Ther. 2010;87(1):126–129. doi:10.1038/clpt.2009.233
26. Veronese ML, Flaherty K, Kramer A, et al. Phase I study of the novel taxane CT-2103 in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2005;55(5):497–501. doi:10.1007/s00280-004-0938-3
27. Silver DA, Pellicer I, Fair WR, Heston WD, Cordon-Cardo C. Prostate-specific membrane antigen expression in normal and malignant human tissues. Clin Cancer Res. 1997;3(1):81–85.
28. Autio KA, Dreicer R, Anderson J, et al. Safety and efficacy of BIND-014, a docetaxel nanoparticle targeting prostate-specific membrane antigen for patients with metastatic castration-resistant prostate cancer: a Phase 2 clinical trial. JAMA Oncol. 2018;4(10):1344–1351.
29. Von Hoff DD, Mita MM, Ramanathan RK, et al. Phase I study of PSMA-targeted docetaxel-containing nanoparticle BIND-014 in patients with advanced solid tumors. Clin Cancer Res. 2016;22(13):3157–3163. doi:10.1158/1078-0432.CCR-15-2548
30. Cardoso MM, Peca IN, Roque AC. Antibody-conjugated nanoparticles for therapeutic applications. Curr Med Chem. 2012;19(19):3103–3127. doi:10.2174/092986712800784667
31. Biffi S, Voltan R, Bortot B, Zauli G, Secchiero P. Actively targeted nanocarriers for drug delivery to cancer cells. Expert Opin Drug Deliv. 2019;16(5):481–496. doi:10.1080/17425247.2019.1604679
32. Hrkach J, Von Hoff D, Mukkaram Ali M, et al. Preclinical development and clinical translation of a PSMA-targeted docetaxel nanoparticle with a differentiated pharmacological profile. Sci Transl Med. 2012;4(128):128ra139. doi:10.1126/scitranslmed.3003651
33. Chang SS, O’Keefe DS, Bacich DJ, Reuter VE, Heston WD, Gaudin PB. Prostate-specific membrane antigen is produced in tumor-associated neovasculature. Clin Cancer Res. 1999;5(10):2674–2681.
34. Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65(1–2):271–284. doi:10.1016/S0168-3659(99)00248-5
35. Sherwood LM, Parris EE, Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285(21):1182–1186. doi:10.1056/NEJM197111182852108
36. Skinner SA, Tutton PJ, O’Brien PE. Microvascular architecture of experimental colon tumors in the rat. Cancer Res. 1990;50(8):2411–2417.
37. Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46(12 Pt 1):6387–6392.
38. Maeda H, Bharate GY, Daruwalla J. Polymeric drugs for efficient tumor-targeted drug delivery based on EPR-effect. Eur J Pharm Biopharm. 2009;71(3):409–419. doi:10.1016/j.ejpb.2008.11.010
39. Hashizume H, Baluk P, Morikawa S, et al. Openings between defective endothelial cells explain tumor vessel leakiness. Am J Pathol. 2000;156(4):1363–1380. doi:10.1016/S0002-9440(10)65006-7
40. Maeda H, Matsumura Y. Tumoritropic and lymphotropic principles of macromolecular drugs. Crit Rev Ther Drug Carrier Syst. 1989;6(3):193–210.
41. Maeda H. Macromolecular therapeutics in cancer treatment: the EPR effect and beyond. J Control Release. 2012;164(2):138–144. doi:10.1016/j.jconrel.2012.04.038
42. Wang J, Sui M, Fan W. Nanoparticles for tumor targeted therapies and their pharmacokinetics. Curr Drug Metab. 2010;11(2):129–141. doi:10.2174/138920010791110827
43. Bawa P, Pillay V, Choonara YE, Du Toit LC. Stimuli-responsive polymers and their applications in drug delivery. Biomed Mater. 2009;4(2):022001.
44. Ruiz ME, Gantner ME, Talevi A. Applications of nanosystems to anticancer drug therapy (Part II. Dendrimers, micelles, lipid-based nanosystems). Recent Pat Anticancer Drug Discov. 2014;9(1):99–128. doi:10.2174/1574891X113089990038
45. ALZA Corporation. Doxil (doxorubicin HCl liposome injection). U.S. Food and Drug Administration website. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2007/050718s029lbl.pdf.
46. Merrimack Pharmaceuticals, Inc. Onivyde (Irinotecan liposome injection). U.S. Food and Drug Administration website. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/207793lbl.pdf.
47. Bhattacharjee H, Balabathula P, Wood GC. Targeted nanoparticulate drug-delivery systems for treatment of solid tumors: a review. Ther Deliv. 2010;1(5):713–734. doi:10.4155/tde.10.47
48. Onoue S, Yamada S, Chan HK. Nanodrugs: pharmacokinetics and safety. Int J Nanomedicine. 2014;9:1025–1037. doi:10.2147/IJN.S38378
49. Singh R, Lillard JW
50. Koudelka S, Turanek J. Liposomal paclitaxel formulations. J Control Release. 2012;163(3):322–334.
51. Juliano RL, Stamp D. Pharmacokinetics of liposome-encapsulated anti-tumor drugs. Studies with vinblastine, actinomycin D, cytosine arabinoside, and daunomycin. Biochem Pharmacol. 1978;27(1):21–27. doi:10.1016/0006-2952(78)90252-6
52. Allen TM. Long-circulating (sterically stabilized) liposomes for targeted drug delivery. Trends Pharmacol Sci. 1994;15(7):215–220.
53. Cattel L, Ceruti M, Dosio F. From conventional to stealth liposomes: a new Frontier in cancer chemotherapy. J Chemother. 2004;16(Suppl 4):94–97. doi:10.1179/joc.2004.16.Supplement-1.94
54. Ishida T, Ichihara M, Wang X, et al. Injection of PEGylated liposomes in rats elicits PEG-specific IgM, which is responsible for rapid elimination of a second dose of PEGylated liposomes. J Control Release. 2006;112(1):15–25. doi:10.1016/j.jconrel.2006.01.005
55. Wang QW, Lu HL, Song CC, Liu H, Xu CG. Radiosensitivity of human colon cancer cell enhanced by immunoliposomal docetaxel. World J Gastroenterol. 2005;11(26):4003–4007. doi:10.3748/wjg.v11.i26.4003
56. Rahman A, Husain SR, Siddiqui J, et al. Liposome-mediated modulation of multidrug resistance in human HL-60 leukemia cells. J Natl Cancer Inst. 1992;84(24):1909–1915. doi:10.1093/jnci/84.24.1909
57. Treat J, Damjanov N, Huang C, Zrada S, Rahman A. Liposomal-encapsulated chemotherapy: preliminary results of a phase I study of a novel liposomal paclitaxel. Oncology (Williston Park). 2001;15(5 Suppl 7):44–48.
58. Soepenberg O, Sparreboom A, de Jonge MJ, et al. Real-time pharmacokinetics guiding clinical decisions; phase I study of a weekly schedule of liposome encapsulated paclitaxel in patients with solid tumours. Eur J Cancer. 2004;40(5):681–688. doi:10.1016/j.ejca.2003.11.027
59. Awada A, Bondarenko IN, Bonneterre J, et al. A randomized controlled phase II trial of a novel composition of paclitaxel embedded into neutral and cationic lipids targeting tumor endothelial cells in advanced triple-negative breast cancer (TNBC). Ann Oncol. 2014;25(4):824–831. doi:10.1093/annonc/mdu025
60. Strieth S, Eichhorn ME, Sauer B, et al. Neovascular targeting chemotherapy: encapsulation of paclitaxel in cationic liposomes impairs functional tumor microvasculature. Int J Cancer. 2004;110(1):117–124. doi:10.1002/ijc.20083
61. Strieth S, Dunau C, Michaelis U, et al. Phase I/II clinical study on safety and antivascular effects of paclitaxel encapsulated in cationic liposomes for targeted therapy in advanced head and neck cancer. Head Neck. 2014;36(7):976–984. doi:10.1002/hed.23397
62. Thurston G, McLean JW, Rizen M, et al. Cationic liposomes target angiogenic endothelial cells in tumors and chronic inflammation in mice. J Clin Invest. 1998;101(7):1401–1413. doi:10.1172/JCI965
63. Schmitt-Sody M, Strieth S, Krasnici S, et al. Neovascular targeting therapy: paclitaxel encapsulated in cationic liposomes improves antitumoral efficacy. Clin Cancer Res. 2003;9(6):2335–2341.
64. Bode C, Trojan L, Weiss C, et al. Paclitaxel encapsulated in cationic liposomes: a new option for neovascular targeting for the treatment of prostate cancer. Oncol Rep. 2009;22(2):321–326.
65. Ignatiadis M, Zardavas D, Lemort M, et al. Feasibility Study of EndoTAG-1, a tumor endothelial targeting agent, in combination with paclitaxel followed by FEC as induction therapy in HER2-negative breast cancer. PLoS One. 2016;11(7):e0154009. doi:10.1371/journal.pone.0154009
66. Fasol U, Frost A, Buchert M, et al. Vascular and pharmacokinetic effects of EndoTAG-1 in patients with advanced cancer and liver metastasis. Ann Oncol. 2012;23(4):1030–1036. doi:10.1093/annonc/mdr300
67. Zhang JA, Anyarambhatla G, Ma L, et al. Development and characterization of a novel Cremophor EL free liposome-based paclitaxel (LEP-ETU) formulation. Eur J Pharm Biopharm. 2005;59(1):177–187. doi:10.1016/j.ejpb.2004.06.009
68. Cabanes A, Briggs KE, Gokhale PC, Treat JA, Rahman A. Comparative in vivo studies with paclitaxel and liposome-encapsulated paclitaxel. Int J Oncol. 1998;12(5):1035–1040.
69. Fetterly GJ, Grasela TH, Sherman JW, et al. Pharmacokinetic/pharmacodynamic modeling and simulation of neutropenia during phase I development of liposome-entrapped paclitaxel. Clin Cancer Res. 2008;14(18):5856–5863. doi:10.1158/1078-0432.CCR-08-1046
70. Slingerland M, Guchelaar HJ, Rosing H, et al. Bioequivalence of Liposome-Entrapped Paclitaxel Easy-To-Use (LEP-ETU) formulation and paclitaxel in polyethoxylated castor oil: a randomized, two-period crossover study in patients with advanced cancer. Clin Ther. 2013;35(12):1946–1954. doi:10.1016/j.clinthera.2013.10.009
71. Mohanty C, Acharya S, Mohanty AK, Dilnawaz F, Sahoo SK. Curcumin-encapsulated MePEG/PCL diblock copolymeric micelles: a novel controlled delivery vehicle for cancer therapy. Nanomedicine (Lond). 2010;5(3):433–449. doi:10.2217/nnm.10.9
72. Cai S, Vijayan K, Cheng D, Lima EM, Discher DE. Micelles of different morphologies–advantages of worm-like filomicelles of PEO-PCL in paclitaxel delivery. Pharm Res. 2007;24(11):2099–2109. doi:10.1007/s11095-007-9335-z
73. Kabanov AV, Alakhov VY. Pluronic block copolymers in drug delivery: from micellar nanocontainers to biological response modifiers. Crit Rev Ther Drug Carrier Syst. 2002;19(1):1–72. doi:10.1615/CritRevTherDrugCarrierSyst.v19.i1.10
74. Li J, Sun L, Liu Y, et al. To reduce premature drug release while ensuring burst intracellular drug release of solid lipid nanoparticle-based drug delivery system with clathrin modification. Nanomedicine. 2019;15(1):108–118. doi:10.1016/j.nano.2018.05.014
75. Liu J, Jiang X, Ashley C, Brinker CJ. Electrostatically mediated liposome fusion and lipid exchange with a nanoparticle-supported bilayer for control of surface charge, drug containment, and delivery. J Am Chem Soc. 2009;131(22):7567–7569. doi:10.1021/ja902039y
76. Hu Q, Rijcken CJ, Bansal R, Hennink WE, Storm G, Prakash J. Complete regression of breast tumour with a single dose of docetaxel-entrapped core-cross-linked polymeric micelles. Biomaterials. 2015;53:370–378. doi:10.1016/j.biomaterials.2015.02.085
77. Atrafi F, Dumez H, Mathijssen RHJ, et al. A phase I dose-escalation and pharmacokinetic study of a micellar nanoparticle with entrapped docetaxel (CPC634) in patients with advanced solid tumours. J Control Release. 2020;325:191–197. doi:10.1016/j.jconrel.2020.06.020
78. Kim SC, Kim DW, Shim YH, et al. In vivo evaluation of polymeric micellar paclitaxel formulation: toxicity and efficacy. J Control Release. 2001;72(1–3):191–202. doi:10.1016/S0168-3659(01)00275-9
79. Park IH, Sohn JH, Kim SB, et al. An open-label, randomized, parallel, Phase III trial evaluating the efficacy and safety of polymeric micelle-formulated paclitaxel compared to conventional cremophor EL-based paclitaxel for recurrent or metastatic HER2-negative breast cancer. Cancer Res Treat. 2017;49(3):569–577. doi:10.4143/crt.2016.289
80. Lee KS, Chung HC, Im SA, et al. Multicenter phase II trial of Genexol-PM, a Cremophor-free, polymeric micelle formulation of paclitaxel, in patients with metastatic breast cancer. Breast Cancer Res Treat. 2008;108(2):241–250.
81. Kim DW, Kim SY, Kim HK, et al. Multicenter phase II trial of Genexol-PM, a novel Cremophor-free, polymeric micelle formulation of paclitaxel, with cisplatin in patients with advanced non-small-cell lung cancer. Ann Oncol. 2007;18(12):2009–2014. doi:10.1093/annonc/mdm374
82. Keam B, Lee KW, Lee SH, et al. A Phase II study of Genexol-PM and cisplatin as induction chemotherapy in locally advanced head and neck squamous cell carcinoma. Oncologist. 2019;24(6):751. doi:10.1634/theoncologist.2019-0070
83. Kim TY, Kim DW, Chung JY, et al. Phase I and pharmacokinetic study of Genexol-PM, a cremophor-free, polymeric micelle-formulated paclitaxel, in patients with advanced malignancies. Clin Cancer Res. 2004;10(11):3708–3716. doi:10.1158/1078-0432.CCR-03-0655
84. Fujiwara Y, Mukai H, Saeki T, et al. A multi-national, randomised, open-label, parallel, phase III non-inferiority study comparing NK105 and paclitaxel in metastatic or recurrent breast cancer patients. Br J Cancer. 2019;120(5):475–480. doi:10.1038/s41416-019-0391-z
85. Hamaguchi T, Matsumura Y, Suzuki M, et al. NK105, a paclitaxel-incorporating micellar nanoparticle formulation, can extend in vivo antitumour activity and reduce the neurotoxicity of paclitaxel. Br J Cancer. 2005;92(7):1240–1246. doi:10.1038/sj.bjc.6602479
86. Kato K, Chin K, Yoshikawa T, et al. Phase II study of NK105, a paclitaxel-incorporating micellar nanoparticle, for previously treated advanced or recurrent gastric cancer. Invest New Drugs. 2012;30(4):1621–1627. doi:10.1007/s10637-011-9709-2
87. Hamaguchi T, Kato K, Yasui H, et al. A phase I and pharmacokinetic study of NK105, a paclitaxel-incorporating micellar nanoparticle formulation. Br J Cancer. 2007;97(2):170–176. doi:10.1038/sj.bjc.6603855
88. Mukai H, Kato K, Esaki T, et al. Phase I study of NK105, a nanomicellar paclitaxel formulation, administered on a weekly schedule in patients with solid tumors. Invest New Drugs. 2016;34(6):750–759. doi:10.1007/s10637-016-0381-4
89. Pekarek KJ, Jacob JS, Mathiowitz E. Double-walled polymer microspheres for controlled drug release. Nature. 1994;367(6460):258–260. doi:10.1038/367258a0
90. Wilczewska AZ, Niemirowicz K, Markiewicz KH, Car H. Nanoparticles as drug delivery systems. Pharmacol Rep. 2012;64(5):1020–1037. doi:10.1016/S1734-1140(12)70901-5
91. Abraxis BioScience, LLC. Abraxane (nanoparticle albumin bound paclitaxel). U.S. Food and Drug Administration website. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/021660s025s026s029lbl.pdf.
92. Desai N, Trieu V, Yao Z, et al. Increased antitumor activity, intratumor paclitaxel concentrations, and endothelial cell transport of cremophor-free, albumin-bound paclitaxel, ABI-007, compared with cremophor-based paclitaxel. Clin Cancer Res. 2006;12(4):1317–1324. doi:10.1158/1078-0432.CCR-05-1634
93. Sparreboom A, Scripture CD, Trieu V, et al. Comparative preclinical and clinical pharmacokinetics of a cremophor-free, nanoparticle albumin-bound paclitaxel (ABI-007) and paclitaxel formulated in Cremophor (Taxol). Clin Cancer Res. 2005;11(11):4136–4143. doi:10.1158/1078-0432.CCR-04-2291
94. Socinski MA, Bondarenko I, Karaseva NA, et al. Weekly nab-paclitaxel in combination with carboplatin versus solvent-based paclitaxel plus carboplatin as first-line therapy in patients with advanced non-small-cell lung cancer: final results of a phase III trial. J Clin Oncol. 2012;30(17):2055–2062. doi:10.1200/JCO.2011.39.5848
95. Lee H, Park S, Kang JE, Lee HM, Kim SA, Rhie SJ. Efficacy and safety of nanoparticle-albumin-bound paclitaxel compared with solvent-based taxanes for metastatic breast cancer: A meta-analysis. Sci Rep. 2020;10(1):530. doi:10.1038/s41598-019-57380-0
96. Gardner ER, Dahut WL, Scripture CD, et al. Randomized crossover pharmacokinetic study of solvent-based paclitaxel and nab-paclitaxel. Clin Cancer Res. 2008;14(13):4200–4205. doi:10.1158/1078-0432.CCR-07-4592
97. Yardley DA. Nab-Paclitaxel mechanisms of action and delivery. J Control Release. 2013;170(3):365–372. doi:10.1016/j.jconrel.2013.05.041
98. Li J, Ke W, Wang L, et al. Self-sufficing H2O2-responsive nanocarriers through tumor-specific H2O2 production for synergistic oxidation-chemotherapy. J Control Release. 2016;225:64–74. doi:10.1016/j.jconrel.2016.01.029
99. Li J, Li Y, Wang Y, et al. Polymer prodrug-based nanoreactors activated by tumor acidity for orchestrated oxidation/chemotherapy. Nano Lett. 2017;17(11):6983–6990. doi:10.1021/acs.nanolett.7b03531
100. Jung KH, Kim K, Yoon DH, et al. A phase I trial to determine the maximum tolerated dose and evaluate the safety and pharmacokinetics (PK) of docetaxel-PNP, polymeric nanoparticle formulation of docetaxel, in subjects with advanced solid malignancies. J Clin Oncol. 2012;30(15_suppl):e13104. doi:10.1200/jco.2012.30.15_suppl.e13104
101. Song SY, Kim KP, Jeong SY, et al. Polymeric nanoparticle-docetaxel for the treatment of advanced solid tumors: phase I clinical trial and preclinical data from an orthotopic pancreatic cancer model. Oncotarget. 2016;7(47):77348–77357.
102. Markman B, De Souza PL, Dees EC, et al. A Phase 1 study of CRLX301, a novel nanoparticle-drug conjugate (NDC) containing docetaxel (DOC), in patients with refractory solid tumors. J Clin Oncol. 2016;34(15_suppl):2526.
103. Deeken JF, Slack R, Weiss GJ, et al. A phase I study of liposomal-encapsulated docetaxel (LE-DT) in patients with advanced solid tumor malignancies. Cancer Chemother Pharmacol. 2013;71(3):627–633. doi:10.1007/s00280-012-2048-y
104. Mahalingam D, Nemunaitis JJ, Malik L, et al. Phase I study of intravenously administered ATI-1123, a liposomal docetaxel formulation in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2014;74(6):1241–1250.
105. Pirollo KF, Nemunaitis J, Leung PK, Nunan R, Adams J, Chang EH. Safety and efficacy in advanced solid tumors of a targeted nanocomplex carrying the p53 gene used in combination with docetaxel: a Phase 1b study. Mol Ther. 2016;24(9):1697–1706. doi:10.1038/mt.2016.135
106. Graziani SR, Vital CG, Morikawa AT, et al. Phase II study of paclitaxel associated with lipid core nanoparticles (LDE) as third-line treatment of patients with epithelial ovarian carcinoma. Med Oncol. 2017;34(9):151.
107. Rochon LS, Devarakonda K, Martinez J, et al. Abstract CT230: A phase I first in human dose escalation trial of MNK-010 in subjects with advanced solid tumors. Cancer Res. 2015;75(15Supplement):CT230.
108. Jain MM, Gupte SU, Patil SG, et al. Paclitaxel injection concentrate for nanodispersion versus nab-paclitaxel in women with metastatic breast cancer: a multicenter, randomized, comparative phase II/III study. Breast Cancer Res Treat. 2016;156(1):125–134. doi:10.1007/s10549-016-3736-9
109. Mita AC, Olszanski AJ, Walovitch RC, et al. Phase I and pharmacokinetic study of AI-850, a novel microparticle hydrophobic drug delivery system for paclitaxel. Clin Cancer Res. 2007;13(11):3293–3301. doi:10.1158/1078-0432.CCR-06-2496
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.