")
Back to Journals » Blood and Lymphatic Cancer: Targets and Therapy » Volume 8
Recent advances in the biology and treatment of B-cell acute lymphoblastic leukemia
Received 6 April 2018
Accepted for publication 28 June 2018
Published 25 September 2018 Volume 2018:8 Pages 47—61
DOI https://doi.org/10.2147/BLCTT.S170351
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor David Dingli
Mehrdad Hefazi, Mark R Litzow
Division of Hematology, Mayo Clinic, Rochester, MN, USA
Abstract: Acute lymphoblastic leukemia (ALL) is a hematologic malignancy arising from precursors of the lymphoid lineage. Conventional cytotoxic chemotherapies have resulted in high cure rates of up to 90% in pediatric ALL, but the outcomes for adult patients remain suboptimal with 5-year survival rates of only 30%–40%. Over the last decade, major advances have been made in our understanding and management of ALL. Identification of new prognostic genomic markers and incorporation of minimal residual diseases’ assessment into therapeutic protocols have improved risk stratification and treatment strategies. The use of pediatric-inspired regimens for adolescent and young adults, and the advent of tyrosine kinase inhibitors and novel targeted therapies, including monoclonal antibodies and chimeric antigen receptor T cells, have redefined the therapeutic paradigm of ALL, and significantly improved the outcomes. In this article, we will provide an overview of the current knowledge regarding the biology and treatment of ALL, and highlight recent diagnostic and therapeutic advances made in this area over the past 5 years.
Keywords: acute lymphoblastic leukemia, minimal residual disease, hematopoietic cell transplantation, Philadelphia chromosome, monoclonal antibodies
Introduction
Acute lymphoblastic leukemia (ALL) is a hematologic malignancy arising from precursors of the lymphoid lineage. It has a bimodal distribution, with the first peak occurring at ~5 years of age (80% of cases) and the second peak occurring around the age of 50 (20% of cases).1 In adults, precursor B-cell ALL (B-ALL) accounts for ~75% of cases and precursor T-cell ALL (T-ALL) comprises the remaining cases. Precursor B-ALL is further classified into early-pre-B (pro-B), common-B, and pre-B ALL (or mature pre-B ALL) according to the B-cell differentiation markers, ranging from the earliest to the latest. All three subtypes express CD19, CD22, and CD79, but the presence of CD10 distinguishes common-B-ALL and the expression of cytoplasmic immunoglobulins (with or without CD10 and CD20) identifies mature pre-B-ALL. Mature B-cell (Burkitt) leukemia, characterized by the expression of surface immunoglobulins, was eliminated from the 2008 WHO classification because it was no longer considered a separate entity from Burkitt lymphoma.2 In the 2016 WHO classification, two provisional entities (BCR-ABL1-like and iAMP21) were added to the list of recurrent genetic abnormalities associated with B-ALL, and the hypodiploid variant was redefined as either low hypodiploid or hypodiploid with TP53 mutations.3 Early T-cell precursor lymphoblastic leukemia was also added as a provisional T-ALL entity.3
In children with B-ALL, current therapies result in long-term survival of 80%–90%.4,5 In adults, while complete remission (CR) rates are similar to those of children, long-term survival is only 40%, with the majority of deaths attributable to disease relapse.6 Adolescents and young adults (AYA), defined as those aged 15–39 years, may benefit from “pediatric-inspired” chemotherapy regimens, but the outcomes for this age group remain substantially inferior to those of children, with 5-year overall survival (OS) rates of 50%–60%.7–9 The need for improved outcomes for adult ALL has led to major advancements in the understanding of the disease biology, the refinement of prognostic markers, and the development of novel therapies. In this review, we will provide an overview of the current knowledge regarding the biology and treatment of ALL and highlight the progress made over the past 5 years in the following five key areas:
- established and emerging prognostic markers for ALL;
- minimal residual disease (MRD) assessment for risk stratification and treatment strategy;
- treatment of AYA with ALL;
- treatment of Philadelphia-positive (Ph+) ALL in the era of tyrosine kinase inhibitors (TKIs);
- salvage therapies with monoclonal antibodies (mAbs);
- role of CAR T-cell therapy in relapsed/refractory (R/R) ALL.
Established and emerging prognostic markers in ALL
Established prognostic markers
Accurate assessment of prognosis is central to the management of ALL. Historically, older age (defined as >35 years), high white blood cell (WBC) count (defined as >30×109 for B-ALL or >100×109 for T-ALL), and Ph chromosome positivity were used as markers of poor outcome.10 Age is at least in part a surrogate for unfavorable intrinsic disease biology. A study of 200 ALL patients aged 15–65 years in the Southwest Oncology Group (SWOG)–9400 study showed that cytogenetic profile was a more important prognostic factor than age or WBC count.11 Ph+ ALL had a 1-year survival of ~10%12 in the pre-TKI era; however, with the advent of TKIs, long-term survival is now achieved in 50%–60% of these patients.13,14
Table 1 details the major genomic abnormalities in B-ALL and their prognostic significance. Recognition of Ph-positive ALL and Burkitt leukemia is essential, because these formerly high-risk subsets require and can greatly benefit from different, highly specific treatments. High hyperdiploidy (51–65 chromosomes) and t(12;21)/ETV6-RUNX1 are well recognized favorable prognostic markers in both pediatric and adult ALL. These two genetic biomarkers account for 60% of pediatric and adolescent ALL but <15% of adult ALL,15 with ETV6-RUNX1 being virtually non-existent in adults aged >30 years.16 Patients with either of these abnormalities have better outcomes compared to their age-matched counterparts, with 5-year survival rates of >90% in pediatric ALL and 55% in adult ALL.17,18
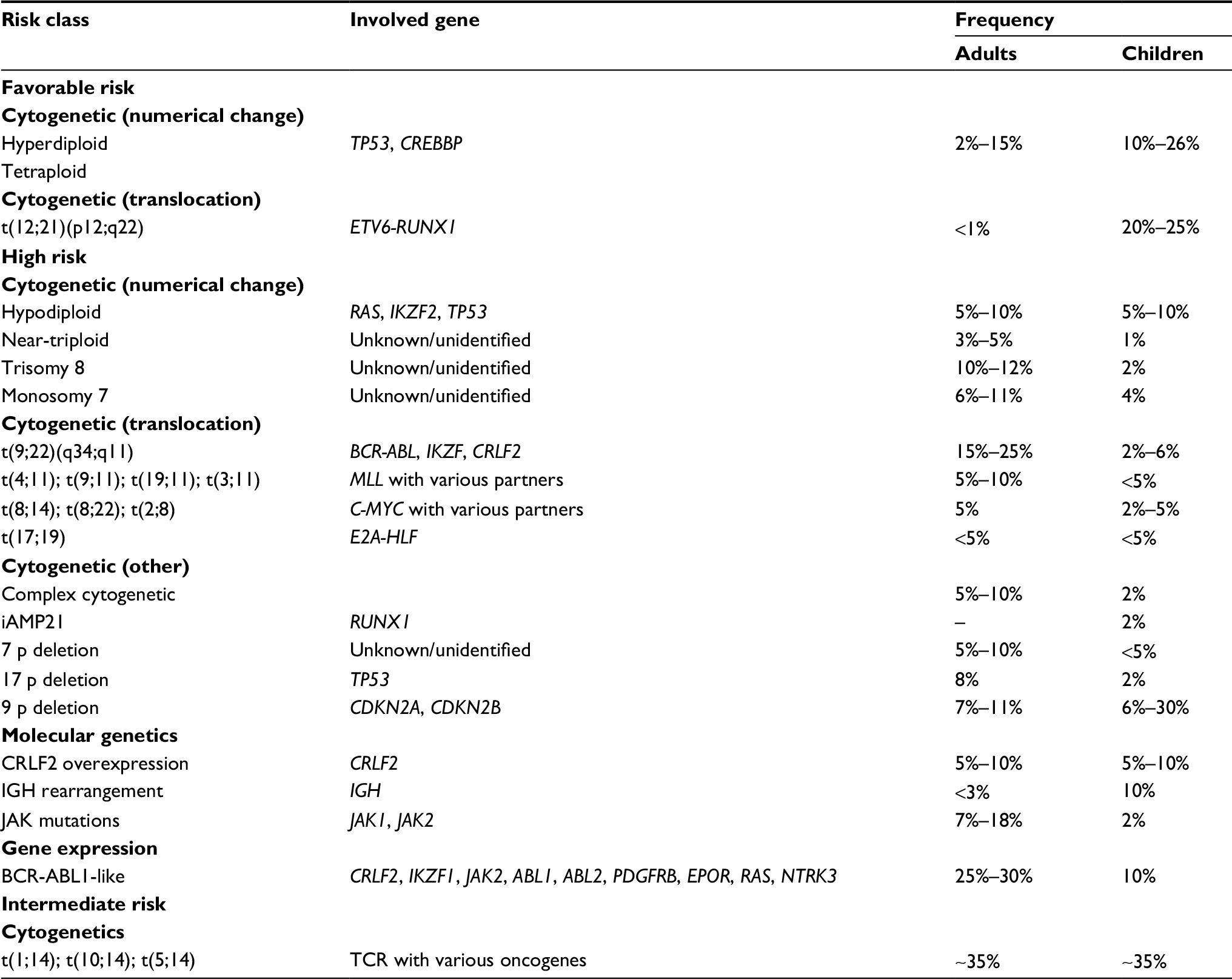 | Table 1 Common genomic abnormalities in B-cell acute lymphoblastic leukemia and their prognostic significance Note: Data from Mrózek et al.118 |
In addition to t(9;22) (based on pre-TKI data), five other cytogenetic abnormalities, including MLL translocations, t(17;19), near-haploidy (24–31 chromosomes), low-hypodiploidy (32–39 chromosomes), near-triploidy (60–78 chromosomes), and complex cytogenetics (≥5 chromosomal abnormalities) are established markers of adverse prognosis. Patients with any of these abnormalities are classified as high risk according to National Comprehensive Cancer Network guidelines and should be considered for treatment with the most intensive regimens.19 Most recently, the presence of CDKN2A/2B deletions in patients with Ph+ ALL were also found to have a negative predictive impact on all endpoints, including OS, disease-free survival (DFS), and duration of remission, despite allogeneic hematopoietic cell transplantation (HCT) in first remission.20
Emerging prognostic markers
Recent discoveries in the genomic landscape of ALL include “Ph-like” ALL, iAMP21, translocations involving immunoglobulin heavy chain (IGH) locus, overexpression of CRLF2, and JAK mutations.
Ph-like ALL
Ph-like ALL is a novel subtype that carries a gene expression signature similar to that of Ph+ ALL without harboring the BCR-ABL1 translocation. This entity represents 10% of ALL cases in children, 15%–20% in AYA, and 25%–30% in adults.21 These patients demonstrate an unfavorable outcome, with a 5-year DFS of only 25% in AYA patients.21,22 Given that Ph-like ALL is defined based on the gene expression profiles, the underlying genetic makeup of this subtype is heterogeneous. Approximately 50% of Ph-like patients harbor CRLF2 rearrangements, with concomitant JAK mutations detected in approximately half of CRLF2 cases.22–24 Other common genetic abnormalities include ABL-class fusions (ABL1, ABL2, PDGFRB) (22%), IKZF1 deletions (28%),22 EPOR and JAK2 rearrangements (18%), RAS pathway (10%), and other mutations that activate JAK-STAT signaling (20%).25 Importantly, in vivo and in vitro studies along with emerging clinical observations indicate that patients with ABL-class fusions may respond to second-generation TKIs such as dasatinib, while patients with a kinase-activating aberration may be amenable to therapy with JAK inhibitors such as ruxolitinib.21 Genomic profiling may therefore expand therapeutic options in this subgroup of patients with poor prognosis, although further studies are needed before these treatments can be incorporated into therapeutic protocols.
iAMP21
Over the last decade, iAMP21 has become an important prognostic marker in pediatric ALL. This structural chromosomal abnormality was discovered during routine screening for the presence of ETV6-RUNX1 fusion by fluorescent in situ hybridization analysis, and is usually defined as ≥3 extra copies of the RUNX1 gene on a single abnormal chromosome (a total of ≥5 RUNX1 signals per cell).26 iAMP21 is found in 1.5%–2% of pediatric ALL patients26,27 and is associated with an inferior outcome when treated with standard therapy and an improved outcome with intensive therapy.28 iAMP21 is thus considered both a prognostic and a predictive biomarker in pediatric ALL. In adult ALL, iAMP21 is extremely rare, and therefore its prognostic significance is unclear in this age group.29
IGH rearrangement, CRLF2 overexpression, and JAK mutations
IGH translocations are well recognized and frequent in lymphoma and mature leukemia. However, recent studies have revealed a variety of IGH rearrangements specific to precursor B-ALL, where the juxtaposition of an oncogene to the IGH enhancer drives its overexpression.30,31 Various partner genes have been identified, with the most common being CRLF2 (~25% of cases) followed by CEBP (~10% of cases). IGH rearrangement frequency is low among children (<3%) but considerably higher (10%) among AYA.31 Patients with IGH translocations have an inferior outcome compared to other patients in the AYA setting.31
The overall frequency of CRLF2 rearrangement in B-ALL is 5%–10%, but the frequency is higher in patients with Down syndrome (>50%).32,33 CRLF2 overexpression can arise from interstitial deletion in the PAR1 region of chromosomes X and Y, as well as in patients who lack clear genetic alterations at this locus.33 Data on the prognostic significance of CRLF2 are conflicting, with some studies suggesting it is a prognostic marker of poor outcome,24 and others concluding it is irrelevant in the context of other risk factors.24 Approximately 50% of patients with CRLF2 overexpression also harbor a JAK mutation.23,24 Although all kinase-activating lesions can theoretically be targeted with appropriate small molecule inhibitors, it remains to be determined which JAK mutations are predictive biomarkers for treatment with such inhibitors. Furthermore, CRLF2 may be a particularly attractive therapeutic target among patients with Down syndrome, as these patients are prone to the toxic side effects of cytotoxic chemotherapy.
MRD assessment for risk stratification and treatment strategy
Several prospective nonrandomized studies have confirmed the strong and independent prognostic impact of MRD after induction and early consolidation in both pediatric and adult ALL.34–40 In the German Multicenter Study Group for Adult ALL (GMALL), molecular MRD analysis was performed in standard risk Ph-negative adult ALL patients after induction (days 11 and 24) and/or consolidation (week 16).39 The researchers identified a small subset of patients (~24%) with a rapid MRD decline to <10-4 by day 11. This group had an excellent prognosis with a 3-year relapse rate of 0%. In contrast, patients with an MRD of ≥10-4 until week 16 had a 3-year relapse rate of 94% and survival of 20%.39 The GMALL group subsequently assessed the prognostic impact of MRD positivity following consolidation therapy, and found that patients who remained in molecular remission after consolidation had a superior 5-year survival compared to those who developed molecular relapse (80% vs 42%).39 Among patients with molecular relapse, those who were still in CR and underwent allogeneic HCT had a higher 5-year CR (66% vs 12%) and a trend toward improved OS (54% vs 33%, P=0.06) compared to those who had molecular relapse but did not undergo HCT. Similarly, MRD was confirmed as the most significant risk factor for relapse in Northern Italy Leukemia Group (NILG), Group for Research on Adult Acute Lymphoblastic Leukemia (GRAALL), and Programa Para El Estudio y Traramiento De Las Hemopatias Malignas (PETHEMA) studies.34,35,40 A PCR-based method with a cutoff value of <10-4 was used in the NILG (at weeks 16 and 22) and GRAALL (at week 6) studies, whereas in the PETHEMA trial, MRD assessment was performed via flow cytometry with a cutoff value of <5×10-4 at week 18. Large-scale AIEOP-BFM-ALL 2000 studies have also shown that MRD-based treatment strategies improve the outcomes in pediatric patients, in both B- and T-ALL.36,37
The previously mentioned studies employed different techniques, cutoff values, and time points for MRD measurement, as there are currently no universally accepted criteria for MRD definition. Thus far, the three most widely used techniques are RT-PCR, multicolor flow cytometry (MFC), and next-generation sequencing (NGS). PCR-based molecular analysis is performed either by tracking the Ig/TCR gene rearrangements present in up to 90% of B- and T-ALL41 or by analyzing the fusion gene transcripts present in 30%–40% of patients with ALL. In addition to these conventional methods, two high-throughput MRD techniques have been introduced in the past 5 years: 1) EuroFlow-based (≥8-color) next-generation flow cytometry42 and 2) high-throughput sequencing (HTS) of Ig/TCR.43 These new approaches are aimed at higher sensitivities, and easy and broad applicability. In a recent study, Wood et al compared HTS of IGH and TRG genes vs MFC for MRD detection at the end of induction chemotherapy in pediatric patients with newly diagnosed B-ALL. HTS and MFC demonstrated similar 5-year event-free survival (EFS) and OS for MRD-positive and MRD-negative patients using a threshold of 0.01% for MRD. However, there was a high discordance rate, with HTS identifying 38% more MRD-positive patients at this threshold. These discordant patients had worse outcomes than MFC-MRD-negative patients. Furthermore, the increased analytic sensitivity of HTS permitted identification of 20% of standard risk patients without MRD at any detectable level who had an excellent 5-year EFS (98%) and OS (100%).44 The advantages and disadvantages of these MRD detection techniques are summarized in Table 2.
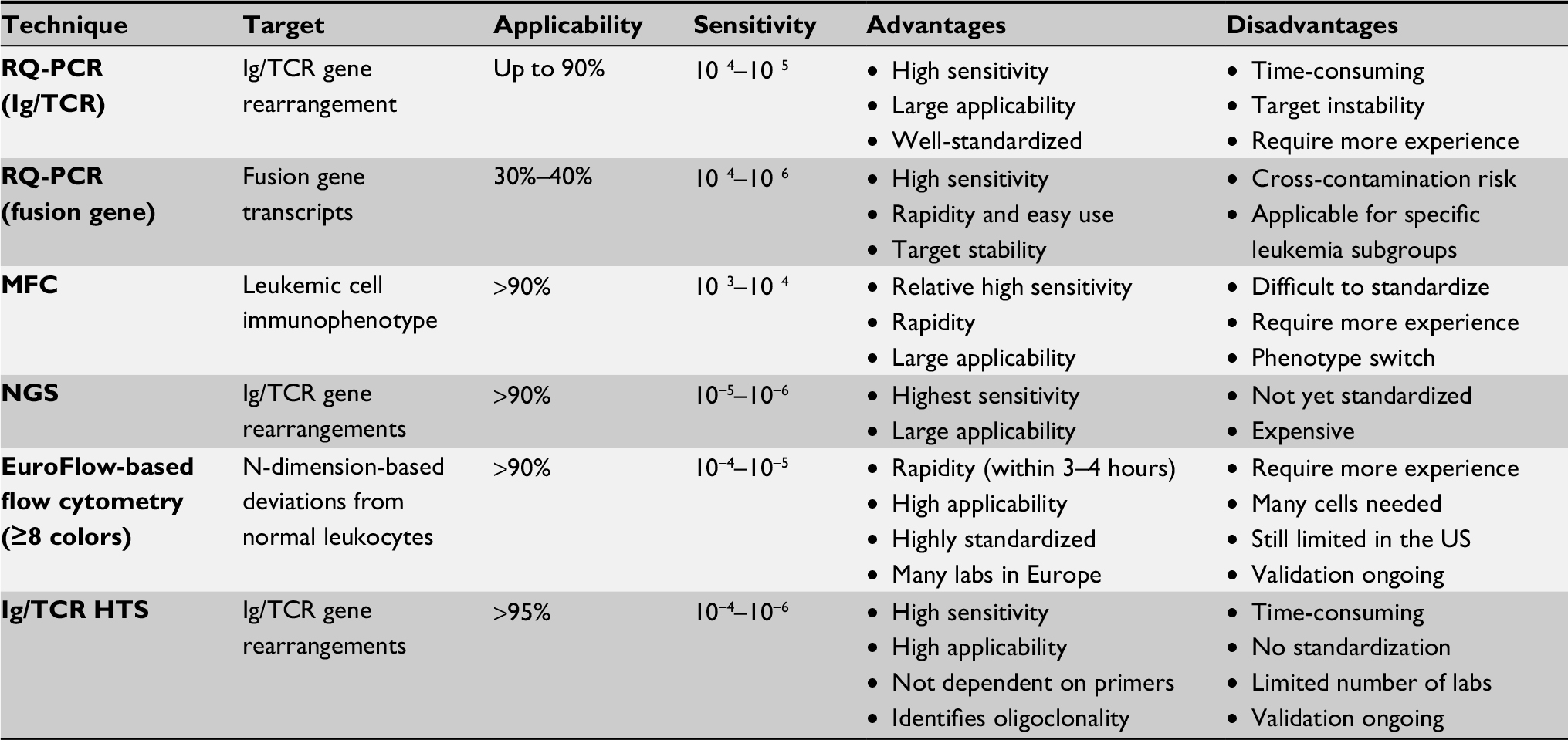 | Table 2 Characteristics of conventional and new high-throughput MRD techniques Abbreviations: HTS, high-throughput sequencing; MFC, multicolor flow cytometry; MRD, minimal residual disease; NGS, next-generation sequencing. |
Various large-scale studies have compared MRD levels in paired blood and bone marrow samples in B- and T-ALL.45–47 These studies have confirmed that for T-ALL, the blood MRD levels are comparable to or up to 1 log lower than the bone marrow MRD level. However, for B-ALL, the blood MRD levels are 1–3 logs lower than the bone marrow MRD levels.45–47 Bone marrow sampling has thus been proposed as the preferred method for MRD assessment in both B-ALL and T-ALL. Although the timing of MRD assessment in adult ALL varies depending on the treatment regimens, it is commonly accepted that the initial measurement should be performed upon completion of induction therapy to facilitate further risk stratification and treatment decisions.48,49 After that, ongoing MRD monitoring during consolidation therapy would serve as a safety net, particularly for patients with MRD-based treatment de-escalation, for whom preemptive salvage therapy in the event of MRD relapse could be a consideration.49 In the GMALL study, MRD-negative patients reconverted to quantifiable MRD positivity a median time of 4.1 months before clinical relapse, supporting the concept that hematologic relapse can be predicted by MRD.50
Treatment of AYA with ALL
The National Cancer Institute (NCI) defines AYA to be those aged 15–39 years. Recent studies have suggested that the AYA population may benefit from treatment with pediatric-inspired regimens and thus are considered separately from adults.9,51 Pediatric-inspired regimens focus on the Berlin-Frankfurt-Munster (BFM) backbone (consisting of corticosteroids, vincristine, and asparaginase), use of prolonged post-remission asparaginase, delayed re-induction, early central nervous system prophylaxis during induction, and reserving allogeneic HCT for very high-risk patients.51,52 In contrast, adult treatment regimens typically consist of intensive use of myelosuppressive agents including daunorubicin, cytarabine, and cyclophosphamide, as well as allogeneic HCT in first remission.53 The GRAALL group tested this concept in 225 patients up to 60 years of age who were treated with a pediatric-inspired regimen and compared the outcomes with those of a historical control group treated with an adult Leucemie aigue lymphoblastique de l’Adult (LALA) regimen.9 The pediatric-inspired regimen resulted in significantly improved survival (66% vs 44%, P<0.001). However, in a subgroup analysis, superior outcomes were seen only in patients younger than 45 years. In patients between the ages of 40 and 60, there was a 23% cumulative incidence of chemotherapy-related deaths, which essentially negated any incremental benefit offered by enhanced anti-leukemic activity. In another study at the MD Anderson Cancer Center, 106 AYA patients were treated with an augmented BFM regimen and compared with the results of those from 102 AYA patients treated with hyper-CVAD (cyclophosphamide, vincristine, adriamycin, dexamethasone) with or without rituximab.8 The results obtained with the augmented BFM regimen were comparable to those achieved with hyper-CVAD ± rituximab (3-year OS rates of 72 % vs 71%, respectively). The pediatric-inspired regimen was more toxic (pancreatitis, liver dysfunction, thrombosis, and osteonecrosis) and worse than the combination of hyper-CVAD with rituximab in patients older than 25 years. Based on the results of this study, one of the adult regimens that is still considered for AYA patients is hyper-CVAD with or without rituximab. The largest prospective study to evaluate the feasibility of a pediatric regimen in AYA ALL patients is the US intergroup trial C10403 (Alliance).54 In this study, 318 AYA ALL patients between 16 and 39 years of age were treated based on the standard arm of the Children’s Oncology Group (COG) regimen (AALL0232).55 The 2-year EFS and OS rates were 66% and 78%, respectively, and toxicities were manageable, with low treatment-related mortality rate of 3%. Based on the results of these studies, the National Cancer Comprehensive Cancer Network recognizes that AYA up to the age of 40 years old may benefit from treatment with pediatric-inspired regimens. Most recently, Huguet et al56 aimed to determine the upper age limit for use of pediatric-inspired regimens and to evaluate the role of hyperfractionated cyclophosphamide vs standard dose of cyclophosphamide during induction and late intensification in adults with newly diagnosed Ph-negative ALL (GRAALL-2005). As a result of worse treatment tolerance, advanced age continuously affected CR rate, EFS, and OS, with 55 years as the best age cutoff. At 5 years, EFS was 55% for patients younger than 55 years of age vs 25% in older patients. Randomization to the hyperfractionated cyclophosphamide arm did not increase the CR rate or prolong EFS or OS.
Treatment of Ph+ ALL
In the pre-TKI era, patients with Ph+ ALL had a poor prognosis with a 5-year OS of 19% for those treated with chemotherapy alone, and 35%–45% for those who underwent allogeneic HCT.57 This resulted in the standard practice of offering allogeneic HCT to all Ph+ patients in first remission. In the current era, however, improved outcomes with TKIs have raised several important questions, including: 1) Is there a best TKI for the treatment of Ph-positive ALL? 2) Could less intensive chemotherapy regimens be used for remission induction in Ph+ ALL? 3) Do all Ph+ ALL patients require allogeneic HCT in first CR? 4) What is the optimum strategy for TKI therapy in the post-HCT settings?
Is there a best TKI for the treatment of Ph+ ALL?
In an important study from the UKALLXII/ECOG2993, Fielding et al demonstrated that the inclusion of imatinib in the standard BFM-type induction chemotherapy significantly improved long-term outcomes.13 The 4-year OS for the imatinib cohort in this study was 38%, which compared favorably to 22% in a historical cohort of patients who were treated with the same regimen, but prior to the advent of imatinib. Similar results were obtained when later-generation TKIs, such as dasatinib, nilotinib, or ponatinib, were added to multi-agent chemotherapy regimens.58–61 However, it remains unclear whether there is a benefit to later-generation TKIs over imatinib, as there are no randomized trials comparing different TKIs head-to-head.
The second- and third-generation TKIs have theoretical advantages over the use of imatinib because of their higher potency, cerebrospinal fluid penetration, activity against non-tyrosine kinases, and altered binding to the BCR-ABL1 kinase domain.62,63 Both nilotinib and dasatinib have demonstrated somewhat better activity than historical controls.60,64 Furthermore, many patients with relapsed Ph+ ALL harbor a T315I clone resistant to imatinib and second-generation TKIs.65 Ponatinib is a more potent third-generation BCR-ABL1 TKI that suppresses the T315I clone.66,67 In a propensity score analysis of two consecutive, prospective, Phase II clinical trials of frontline hyper-CVAD with either ponatinib or dasatinib, patients treated with frontline ponatinib achieved significantly better 3-year OS (83% vs 56%).68 These data suggest that ponatinib may soon have a role in the frontline therapy for Ph+ ALL.
Could less intensive chemotherapy regimens be used for remission induction in Ph+ ALL?
The improved outcomes with TKIs set the stage for the consideration of chemotherapy de-escalation for the treatment of Ph+ ALL to lessen the morbidity and mortality associated with induction chemotherapy. In LAL1201-B and LAL1205 studies from the Italian Gruppo Italiano Malattie EMatologiche dell’Adulto (GIMEMA), patients treated with combinations of corticosteroids and TKIs for induction therapy demonstrated CR rates of 93%–100% and OS of 69%–74% after median follow-ups of 12–25 months. In an ongoing study, the GIMEMA LAL1509 trial, patients received dasatinib combined with corticosteroids as induction therapy, and chemotherapy and/or allogeneic HCT were administered if patients did not reach a sustained complete molecular response (CMR) after induction.69 As previously reported in other GIMEMA trials, no deaths occurred during induction, and 20% of patients achieved CMR. Among patients with CMR, only one experienced a hematologic relapse. Most recently, the GIMEMA investigators reported the initial results of a Phase II study of ponatinib and corticosteroids as frontline therapy for adults with Ph+ ALL aged ≥60 years or deemed unfit for intensive chemotherapy. With this chemotherapy-free regimen, nearly all of the 42 treated patients achieved complete hematologic remission. Further, the CMR rate was 45.8%, which is higher than the 20%–25% rate reported in prior studies with dasatinib plus corticosteroids, suggesting deeper responses with ponatinib.70 Although these studies require longer follow-ups, the results overall suggest that patients who achieve a CMR may not require additional intensive treatments.
Other studies combining dasatinib or nilotinib with intensive or low-intensity chemotherapy have also shown encouraging results.71,72 In the GRAAPH-2005 study, 268 patients aged 18–59 years were randomized to imatinib combined with weekly vincristine and dexamethasone or imatinib combined with hyper-CVAD chemotherapy.73 Patients received similar consolidation, and the ultimate plan was for all patients to undergo allogeneic or autologous HCT. The CR rate after induction was higher in the low-intensity group, mainly because of the higher rate of induction-related mortality in the hyper-CVAD group (7% vs <1%). An equal number of patients in each group proceeded to autologous or allogeneic HCT, and the 3-year OS was similar between the two cohorts (53% for the low-intensity group vs 49% for the hyper-CVAD group). Most recently, Martinelli et al evaluated the efficacy and tolerability of blinatumomab in patients with relapsed or refractory Ph+ ALL. In this Phase II, single-arm study, treatment with single-agent blinatumomab resulted in a 36% CR or CR with partial hematologic recovery (CRh) within the first two cycles. More importantly, 88% of responders also achieved a complete MRD response. These data showed that the proportion of patients who achieved CR/CRh was similar to that of less heavily pretreated patients or those in the Ph– setting, with similar or better response duration than that observed with TKI therapies. Given the high CR rates and minimal toxicity with these low-intensity regimens, we currently favor the enrollment of Ph+ ALL patients in a clinical trial utilizing this approach over induction chemotherapy.
Do all Ph+ ALL patients require an allogeneic HCT in first CR?
Allogeneic HCT has traditionally been considered the standard of care and the only chance for a cure in patients with Ph+ ALL. However, with the introduction of TKIs and improved outcomes, the role of allogeneic HCT in first CR is now being debated. The first group to suggest that allogeneic HCT could be omitted was the COG. In their study, AALL0031, pediatric Ph+ ALL patients treated with imatinib plus chemotherapy had a 3-year EFS that was equal to or better than sibling-related allogeneic HCT (88% vs 57%, respectively).74 A longer follow-up of this study demonstrated similar DFS at 5 years for the chemotherapy plus imatinib group (70%) compared to those who underwent allogeneic HCT (65% for sibling-related and 59% for unrelated donor).75
Several contemporary trials suggest that excellent outcomes with DFS of ~70% can be achieved with or without HCT if patients achieve molecular remission following TKIs.60,69,71 The Korean Society of Hematology Adult ALL Working Party reported the results of a Phase II study of nilotinib combined with multi-agent chemotherapy in the manner of BFM-like induction (KALLA0503).60 Among 76 patients who achieved CMR, the estimated 2-year molecular relapse-free survival was not different between the transplanted and non-transplanted patients (65% vs 53%, respectively), suggesting that allogeneic HCT may not be necessary for patients who achieve deep molecular response with next-generation TKIs. In a different study, Ravandi et al studied the predictive value of MRD in Ph-positive ALL patients treated with chemotherapy plus TKI without allogeneic HCT in first CR.64 Data from this study revealed that the best outcome was observed in patients who achieved CMR within 3 months of therapy; the 4-year OS rates were 66%, 43%, and 32% in patients with 3-month CMR, major molecular remission (MMR), and less than MMR, respectively.64 The Cancer and Leukemia Group B Study 10001 (Alliance) and other publications have also reported encouraging outcomes for autologous HCT as compared to allogeneic HCT, specifically in the setting of deep remissions and low MRD.76,77 Taken together, these studies suggest that the achievement of molecular remission is a goal of treatment in patients with Ph+ ALL and may obviate the need for allogeneic HCT.
What is the optimum strategy for TKI therapy in the post-HCT setting?
TKIs after transplant are commonly considered by most physicians either as prophylaxis or triggered by MRD positivity. However, the benefits of TKIs and the optimal strategy in the post-HCT setting remain unclear. A randomized trial by Pfeifer et al compared prophylactic and MRD-triggered imatinib after allogeneic HCT for Ph+ ALL. With prophylactic imatinib, the incidence of molecular recurrence after HCT was significantly reduced (40%) compared to imatinib given at MRD detection (69%). Five-year OS was high in both groups (80% and 74.5%, respectively), despite premature discontinuation of imatinib in the majority of patients due to poor tolerability.78 In another study, Wassman et al treated 27 Ph+ ALL patients with imatinib upon detection of MRD after allogeneic HCT. All patients who achieved an early molecular response to post-HCT imatinib remained in remission for the duration of imatinib treatment; three patients relapsed after imatinib was discontinued. Failure to achieve MRD negativity shortly after starting imatinib predicted relapse, which occurred in 12 (92%) of 13 patients after a median of 3 months. According to this study, approximately half of patients with Ph+ ALL receiving imatinib for MRD positivity after HCT experienced prolonged DFS, which can be anticipated by the rapid achievement of a molecular CR. Continued MRD positivity after 2–3 months on imatinib identified patients who will ultimately experience relapse and in whom additional or alternative antileukemic treatments should be initiated.79 Conversely, in a retrospective analysis of 102 adults and eleven children with Ph+ ALL who underwent allogeneic HCT in first or later remission, Neither pre- nor post-HCT TKI use were found to significantly impact transplant outcomes after a median follow-up of 5 years.80 According to a consensus statement of the Acute Leukemia Working Party of the European Society for Blood and Marrow Transplantation, patients with undetectable MRD after allogeneic HCT may be treated prophylactically or, alternatively, may be monitored and treated with a TKI only after the detection of MRD in a preemptive strategy. For patients undergoing transplantation during first complete remission (CR1), TKI treatment should be given for 12 months of continuous MRD negativity, whereas patients undergoing HCT at second complete remission (CR2) or a later remission should be treated with TKIs indefinitely unless this is precluded by poor tolerability and toxicities.81
Salvage therapies with mAbs
Despite an exceptionally high rate of initial CR, many adults with ALL will relapse. Cytotoxic chemotherapies can achieve CR rates of 30%–40%, but the OS is dismal with a 5-year survival of 5%–10%.82,83 Targeted therapies using mAbs have shown promising results in this area. mAbs for B-ALL are primarily targeted against CD19, CD20, CD22, and CD52 (Figure 1). The anti-CD20 mAbs have shown to improve outcomes when combined with conventional chemotherapies in the frontline treatment of Burkitt leukemia and CD20+ pre-B-ALL (~30%–50% of pre-B-ALLs).84–87 Other mAbs targeting CD19 and CD22 have shown encouraging results in R/R ALL. Among these, blinatumomab and inotuzumab ozogamicin (InO) are in the most advanced investigational phases (discussed in the following section), while several newer mAbs including ofatumumab, obinutuzumab, epratuzumab, denintuzumab mafodotin, and moxetumomab pasudotox are currently under investigation.
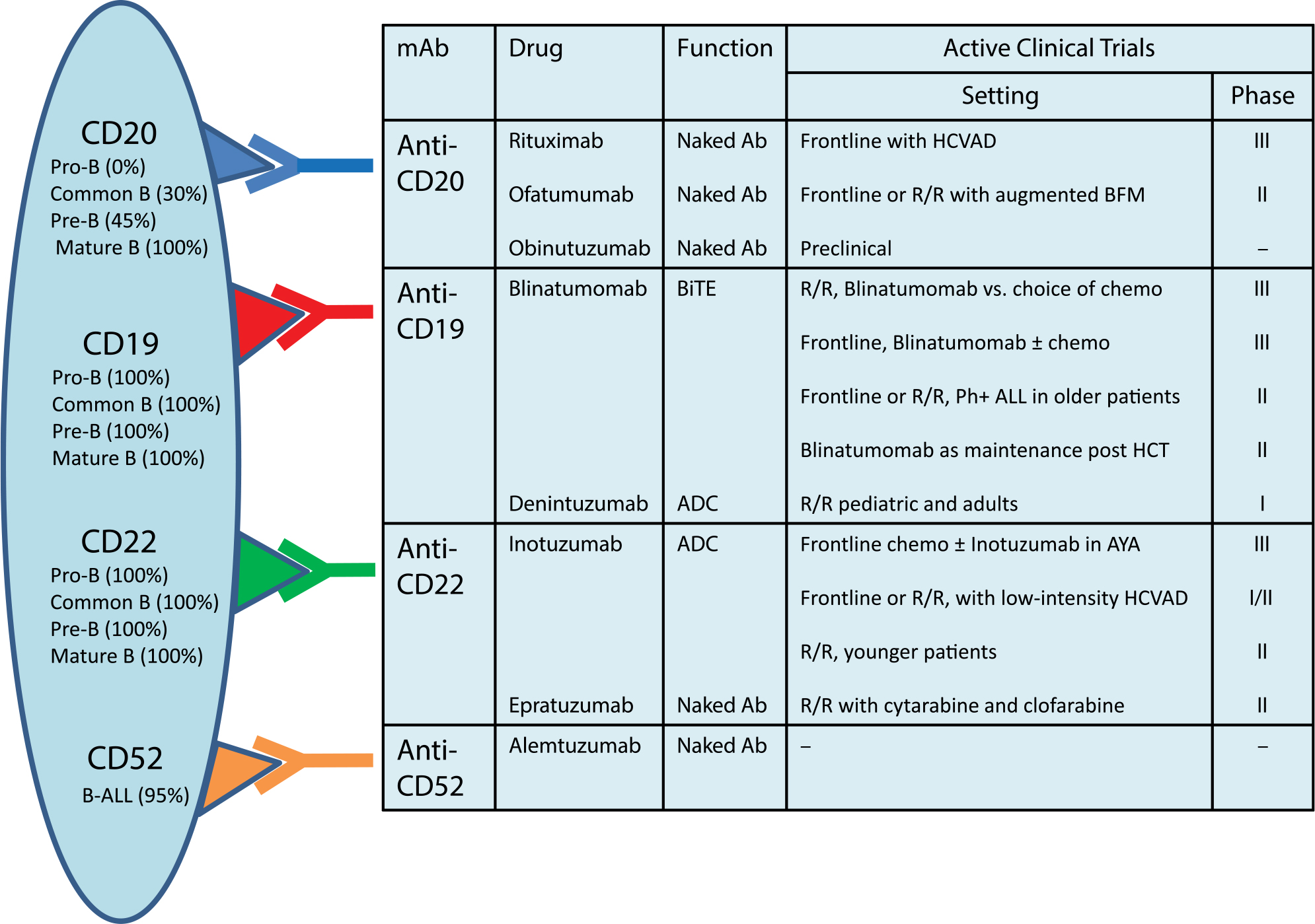 | Figure 1 Monoclonal antibodies under investigation for treatment of B-cell acute lymphoblastic leukemia. Note: List is not comprehensive. Abbreviations: ADC, antibody drug conjugate; AYA, adolescents and young adults; B-ALL, B-cell acute lymphoblastic leukemia; BFM, Berlin-Frankfurt-Munster; BiTE, bispecific T-cell engager; chemo, chemotherapy; HCVAD, hyperfractionated cyclphosphamide, vincristine, adriamycin, dexamethasone; HCT, hematopoietic cell transplantation; mAb, monoclonal antibody; Ph, Philadelphia; R/R, relapsed/refractory. |
Blinatumomab
Blinatumomab is the first-in-class bispecific T-cell engager antibody that has dual specificity for CD19 and CD3 via its two variable antigen-binding domains. On binding to CD19, the cytotoxic T cells become activated and induce cell death via the pore-forming perforin system.88 This drug was initially administered as intermittent infusions 2–3 times per week, but lack of activity and serious adverse events resulted in premature closure of early trials.88 Later in 2004, continuous infusion over several weeks was investigated and showed marked improvement in the drug activity and safety profile. Therefore, subsequent trials were based on continuous infusion rather than short-term infusion.88
The first study of blinatumomab in pre-B-ALL was performed by Topp et al in an attempt to evaluate its efficacy in eradicating MRD.89 In this Phase II, single-arm clinical trial, 80% (16 of 20) MRD-positive patients achieved MRD negativity at the end of the first cycle. Similar results were found in a confirmatory Phase II trial of 116 MRD-positive patients (BLAST study), in which 78% of patients achieved MRD negativity after the first cycle.90,91 Subsequently, a Phase II, multicenter, single-arm study evaluated blinatumomab in 36 patients with R/R B-ALL.92 In this study, 25 of 36 (69%) patients with R/R B-ALL achieved CR with complete or partial CRh at the end of the first cycle. Similarly, in a confirmatory Phase II study of 189 patients with R/R ALL, 81 patients (43%) achieved a CR (33%) or CRh (10%),93 with 82% of the responders also experiencing MRD negativity.
Based on these results, the US Food and Drug Administration (FDA) first approved blinatumomab under accelerated approval in December 2014 for patients with Ph chromosome-negative R/R ALL. Since then, blinatumomab has been studied in a variety of patient populations, including 1) a Phase III, randomized trial comparing blinatumomab to investigator’s choice of chemotherapy in adults with R/R ALL (TOWER study) (Figure 2);94 2) a Phase II, single-arm trial in adults with R/R Ph+ ALL (ALCANTRA study);95 3) a Phase I/II study in pediatric and adolescent patients with R/R ALL (NCT01471782);96 4) a Phase II study of blinatumomab combined with chemotherapy vs dasatinib, prednisone, and blinatumomab in elderly patients with newly diagnosed ALL (NCT02143414);97 and 5) a Phase III study of chemotherapy combined with or without blinatumomab as frontline therapy for adults with ALL aged 30–70 years (NCT02003222).98 Based on these results, blinatumomab was granted full approval by the FDA in July 2017, and the indication was extended to include patients with Ph+ ALL. Most recently, the FDA expanded blinatumomab approval to include MRD-positive ALL. This was based on the results of the MT103 (BLAST) trial that included 86 patients in first to second CR who had detectable MRD ≥10-3 in their bone marrow. MRD-negative status was achieved in 70 patients after one cycle of treatment, and over half of the patients remained alive and in remission for at least 23 months.99
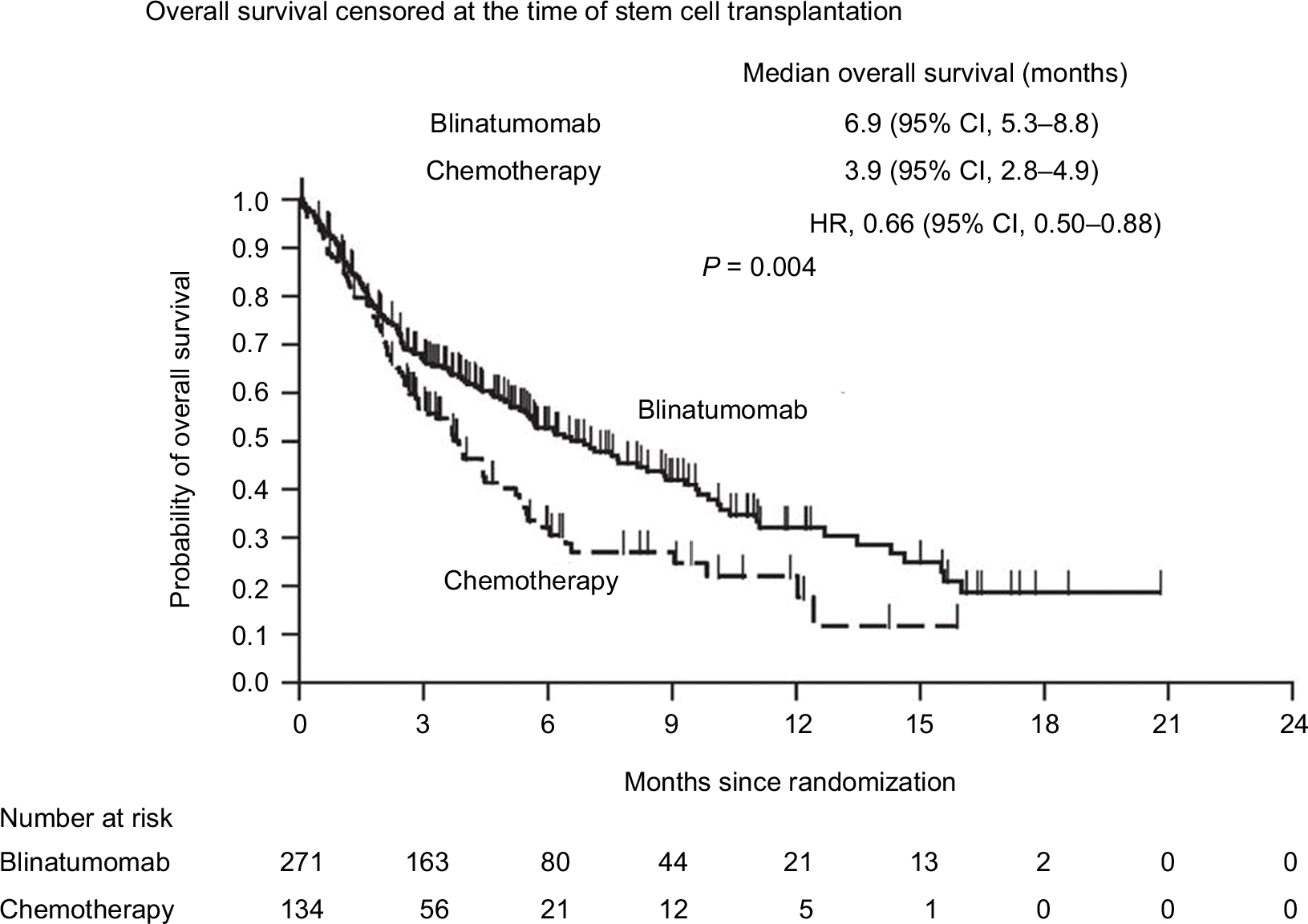 | Figure 2 Overall survival in adults with relapsed or refractory acute lymphoblastic leukemia treated with blinatumomab vs chemotherapy (TOWER Study). Note: From the New England Journal of Medicine, Kantarjian H, Stein A, Gökbuget N, et al, Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia, 376(9):836–847. Copyright © (2017) Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.94 |
Inotuzumab ozogamicin
InO is an mAb against CD22 that is conjugated to calicheamicin, a potent cytotoxic compound.100 Upon internalization of the immunoconjugate, calicheamicin binds with DNA and results in double-stranded DNA breaks, which induce apoptosis.101 Kantarjian et al conducted the first Phase II study of InO as a single agent in 49 patients with R/R ALL aged 6–80 years. In this study, the overall response rate was 57%, and the median OS was 6.7 months.102 A second Phase II study of 90 adult patients with CD22+ ALL in second or later salvage showed similar CR rates (66%) and median OS (7.4 months).103 These promising results in the salvage setting led to a clinical trial incorporating low-dose weekly InO into low-intensity hyper-CVAD in newly diagnosed elderly patients with CD22+ ALL. This study showed encouraging CR rates of 81% and a 1-year OS of 78%.104 Subsequently, the Phase III INO-VATE trial compared single agent InO with standard chemotherapy in 326 adult patients with R/R CD22+ ALL, and found significantly higher CR rate (81% vs 30%), PFS (5 vs 1.8 months), and median OS (7.7 vs 6.7 months) in the InO group (Figure 3).105 The most frequent non-hematologic adverse events with InO were fever, hypotension, and veno-occlusive liver disease (VOD). The majority of VOD in the InO arm was seen after allogeneic HCT. More recently a subset analysis of this trial comparing the safety and efficacy of InO in younger (<55 years) and older (≥55 years) patients demonstrated that InO was tolerable in older patients with R/R ALL. Although OS was longer for younger patients vs older patients (median, 8.6 vs 5.6 months; P=0.003), InO demonstrated high response rates (75% vs 70%; P=0.24) and duration of response (5.4 vs 4.7 months; P=0.09) in both age groups.106 These efforts led to the FDA approval of InO for treatment of R/R B-ALL in August 2017. Following these encouraging results, the US cooperative intergroup has planned a successor AYA trial in which InO is added to the C10403 chemotherapy backbone (NCT03150693), aiming to improve the survival for AYA patients by incorporating targeted agents into frontline chemotherapy.
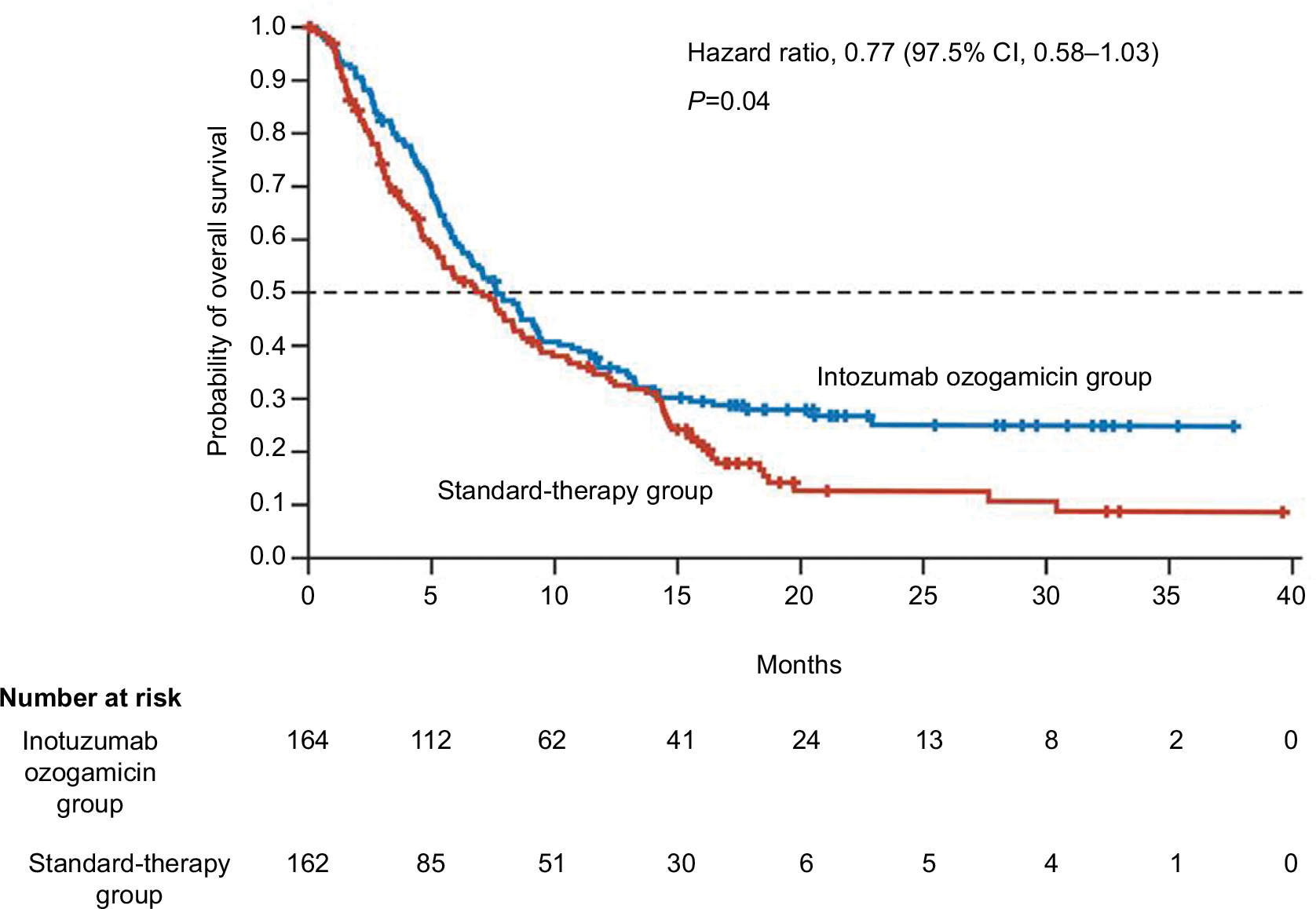 | Figure 3 Overall survival in adults with relapsed or refractory ALL treated with inotuzumab ozogamicin vs standard intensive chemotherapy (INO-VATE Trial). Note: From the New England Journal of Medicine, Kantarjian HM, Deangelo DJ, Stelljes M, et al, Inotuzumab Ozogamicin versus Standard Therapy for Acute Lymphoblastic Leukemia, 375(8):740–753. Copyright © (2016) Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.105 |
Role of CAR T-cell therapy in R/R ALL
CD19-targeted CAR T-cell therapy has shown promising results for R/R ALL in several recent clinical trials.107–110 The majority of these trials are led by three institutions: the University of Pennsylvania (UPenn), the Memorial Sloan Kettering Cancer Center (MSKCC), and the NCI. The first reports came from the MSKCC group and detailed outcomes in adults with R/R B-ALL (16 patients in a first report and 40 in a subsequent larger cohort). This was followed by the reports from the investigators at UPenn and the NCI, who, respectively, treated 25 and 21 children and young adults.108,109 Most recently, the group at the Fred Hutchinson Cancer Research Center reported their outcomes of treating 30 adults.107 Furthermore, two Phase II, single-arm, multicenter trials are currently evaluating the safety and efficacy of CD19-targeted therapy in pediatric patients with R/R ALL (ELIANA111 and ENSIGN112 studies). Although the aforementioned studies differed in CAR designs, T-cell manufacturing, conditioning regimens, patients’ age, and T-cell dosages, each trial was comparably effective in treating R/R ALL, reaching CR rates of 90% even in heavily pretreated patients. In comparison, the expected CR rate for R/R B-ALL treated with salvage chemotherapy is ~30%. Altogether these efforts led to the FDA approval of CAR T-cell therapy for children and young adults up to age 25 with R/R B-ALL in August 2017. Most recently, Park et al have published the long-term outcomes of 53 adults with R/R ALL who were treated with CD19-directed CAR T cells. They reported a strikingly high CR rate of 83% among all patients and a 74% CR rate among the 19 most heavily pretreated patients (≥4 lines of treatment). The key finding in this study was that patients with lower tumor burden experience the highest long-term survival and the lowest toxicities (cytokine release syndrome [CRS] and neurotoxicity). These findings further indicate that CAR T-cell therapy may be most beneficial earlier in the course of treatment for particularly high-risk patients, such as those with MRD positivity after standard induction therapy. Furthermore, it is plausible that CAR T-cell therapy could replace the need for allogeneic HCT for these MRD-positive patients, as the cohort analyzed in this study did not appear to benefit from proceeding to subsequent transplant.113
Toxicities from CAR T cells are mainly related to cell expansion and activation, resulting in CRS and neurotoxicity, which can be severe in ~30% of patients.114 The IL-6 inhibiting agent tocilizumab is effective in this setting.109 The on-tumor off-target toxicity of CD19 CAR T cells can result in B-cell aplasia and agammaglobulinemia with resulting need for long-term immunoglobulin replacement. Most recent advances in the field of CAR T-cell therapy for B-ALL include RNA-based methods (instead of viral vectors) for gene delivery,115 application of CD22-directed (instead of CD19-directed) CAR T cells,116 and use of allogeneic donor-derived (instead of autologous donor) CAR T cells.117 Ongoing research is expected to identify the optimal clinical use and minimize toxicities of this highly innovative therapeutic strategy.
Conclusion
Major advances have been made in our understanding of the pathogenesis and treatment of ALL over the last decade. Genomic discoveries such as the iAMP21 aberration and the Ph-like gene expression profile have become the important prognostic markers in pediatric and adult ALL. MRD assessment following induction and early consolidation has emerged as the strongest prognostic marker for relapse, hence allowing for individualized risk stratification and treatment strategies. The incorporation of TKIs into the treatment of Ph+ ALL has drastically improved outcomes and set the stage for reducing the intensity and minimizing the toxicity of combination chemotherapies in this historically poor prognosis ALL. Blinatumomab and inotuzumab have recently received FDA approval for treatment of R/R B-ALL, while several other promising agents are in different stages of clinical development. CAR T cells have emerged as one of the most promising targeted immunotherapies for R/R ALL, albeit their optimal clinical use and technical challenges are yet to be defined.
Disclosure
The authors report no conflicts of interest in this work.
References
Paul S, Kantarjian H, Jabbour EJ. Adult Acute Lymphoblastic Leukemia. Mayo Clin Proc. 2016;91(11):1645–1666. | ||
Campo E, Swerdlow SH, Harris NL, Pileri S, Stein H, Jaffe ES, The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Blood. 2011;117(19):5019–5032. | ||
Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–2405. | ||
Pui CH, Yang JJ, Hunger SP, et al. Childhood Acute Lymphoblastic Leukemia: Progress Through Collaboration. J Clin Oncol. 2015;33(27):2938–2948. | ||
Pui CH, Campana D, Pei D, et al. Treating childhood acute lymphoblastic leukemia without cranial irradiation. N Engl J Med. 2009;360(26):2730–2741. | ||
Kantarjian HM, O’Brien S, Smith TL, et al. Results of treatment with hyper-CVAD, a dose-intensive regimen, in adult acute lymphocytic leukemia. J Clin Oncol. 2000;18(3):547–561. | ||
Boissel N, Auclerc MF, Lhéritier V, et al. Should adolescents with acute lymphoblastic leukemia be treated as old children or young adults? Comparison of the French FRALLE-93 and LALA-94 trials. J Clin Oncol. 2003;21(5):774–780. | ||
Rytting ME, Jabbour EJ, Jorgensen JL, et al. Final results of a single institution experience with a pediatric-based regimen, the augmented Berlin-Frankfurt-Münster, in adolescents and young adults with acute lymphoblastic leukemia, and comparison to the hyper-CVAD regimen. Am J Hematol. 2016;91(8):819–823. | ||
Huguet F, Leguay T, Raffoux E, et al. Pediatric-inspired therapy in adults with Philadelphia chromosome-negative acute lymphoblastic leukemia: the GRAALL-2003 study. J Clin Oncol. 2009;27(6):911–918. | ||
Rowe JM, Buck G, Burnett AK, et al. Induction therapy for adults with acute lymphoblastic leukemia: results of more than 1500 patients from the international ALL trial: MRC UKALL XII/ECOG E2993. Blood. 2005;106(12):3760–3767. | ||
Pullarkat V, Slovak ML, Kopecky KJ, Forman SJ, Appelbaum FR. Impact of cytogenetics on the outcome of adult acute lymphoblastic leukemia: results of Southwest Oncology Group 9400 study. Blood. 2008;111(5):2563–2572. | ||
Gleissner B, Gökbuget N, Bartram CR, et al. Leading prognostic relevance of the BCR-ABL translocation in adult acute B-lineage lymphoblastic leukemia: a prospective study of the German Multicenter Trial Group and confirmed polymerase chain reaction analysis. Blood. 2002;99(5):1536–1543. | ||
Fielding AK, Rowe JM, Buck G, et al. UKALLXII/ECOG2993: addition of imatinib to a standard treatment regimen enhances long-term outcomes in Philadelphia positive acute lymphoblastic leukemia. Blood. 2014;123(6):843–850. | ||
Ravandi F, O’Brien S, Thomas D, et al. First report of phase 2 study of dasatinib with hyper-CVAD for the frontline treatment of patients with Philadelphia chromosome-positive (Ph+) acute lymphoblastic leukemia. Blood. 2010;116(12):2070–2077. | ||
Moorman AV. New and emerging prognostic and predictive genetic biomarkers in B-cell precursor acute lymphoblastic leukemia. Haematologica. 2016;101(4):407–416. | ||
Jabber Al-Obaidi MS, Martineau M, Bennett CF, et al. ETV6/AML1 fusion by FISH in adult acute lymphoblastic leukemia. Leukemia. 2002;16(4):669–674. | ||
Chilton L, Buck G, Harrison CJ, et al. High hyperdiploidy among adolescents and adults with acute lymphoblastic leukaemia (ALL): cytogenetic features, clinical characteristics and outcome. Leukemia. 2014;28(7):1511–1518. | ||
Moorman AV, Ensor HM, Richards SM, et al. Prognostic effect of chromosomal abnormalities in childhood B-cell precursor acute lymphoblastic leukaemia: results from the UK Medical Research Council ALL97/99 randomised trial. Lancet Oncol. 2010;11(5):429–438. | ||
National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology: Acute Lymphoblastic Leukemia. Version 1 (2016). Available from:https://www.nccn.org/professionals/physician_gls/default.aspx. Accessed September 12, 2018. | ||
Pfeifer H, Raum K, Markovic S, et al. Genomic CDKN2A/2B deletions in adult Ph+ ALL are adverse despite allogeneic stem cell transplantation13. Blood. 2018;131(13):1464–1475. | ||
Roberts KG, Li Y, Payne-Turner D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. 2014;371(11):1005–1015. | ||
Mullighan CG, Su X, Zhang J, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med. 2009;360(5):470–480. | ||
Roberts KG, Morin RD, Zhang J, et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell. 2012;22(2):153–166. | ||
Harvey RC, Mullighan CG, Chen IM, et al. Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric B-progenitor acute lymphoblastic leukemia. Blood. 2010;115(26):5312–5321. | ||
Yokota T, Kanakura Y. Genetic abnormalities associated with acute lymphoblastic leukemia. Cancer Sci. 2016;107(6):721–725. | ||
Harrison CJ, Haas O, Harbott J, et al. Detection of prognostically relevant genetic abnormalities in childhood B-cell precursor acute lymphoblastic leukaemia: recommendations from the Biology and Diagnosis Committee of the International Berlin-Frankfürt-Münster study group. Br J Haematol. 2010;151(2):132–142. | ||
Reichard KK, Kang H, Robinett S. Pediatric B-lymphoblastic leukemia with RUNX1 amplification: clinicopathologic study of eight cases. Mod Pathol. 2011;24(12):1606–1611. | ||
Moorman AV, Richards SM, Robinson HM, et al. Prognosis of children with acute lymphoblastic leukemia (ALL) and intrachromosomal amplification of chromosome 21 (iAMP21. Blood. 2007;109(6):2327–2330. | ||
Leonard JP, Martin P, Roboz GJ. Practical Implications of the 2016 Revision of the World Health Organization Classification of Lymphoid and Myeloid Neoplasms and Acute Leukemia. J Clin Oncol. 2017;35(23):2708–2715. | ||
Dyer MJ, Akasaka T, Capasso M, et al. Immunoglobulin heavy chain locus chromosomal translocations in B-cell precursor acute lymphoblastic leukemia: rare clinical curios or potent genetic drivers? Blood. 2010;115(8):1490–1499. | ||
Russell LJ, Enshaei A, Jones L, et al. IGH@ translocations are prevalent in teenagers and young adults with acute lymphoblastic leukemia and are associated with a poor outcome. J Clin Oncol. 2014;32(14):1453–1462. | ||
Mullighan CG, Collins-Underwood JR, Phillips LA, et al. Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet. 2009;41(11):1243–1246. | ||
Russell LJ, Capasso M, Vater I, et al. Deregulated expression of cytokine receptor gene, CRLF2, is involved in lymphoid transformation in B-cell precursor acute lymphoblastic leukemia. Blood. 2009;114(13):2688–2698. | ||
Bassan R, Spinelli O, Oldani E, et al. Improved risk classification for risk-specific therapy based on the molecular study of minimal residual disease (MRD) in adult acute lymphoblastic leukemia (ALL). Blood. 2009;113(18):4153–4162. | ||
Ribera JM, Oriol A, Morgades M, et al. Treatment of high-risk Philadelphia chromosome-negative acute lymphoblastic leukemia in adolescents and adults according to early cytologic response and minimal residual disease after consolidation assessed by flow cytometry: final results of the PETHEMA ALL-AR-03 trial. J Clin Oncol. 2014;32(15):1595–1604. | ||
Schrappe M, Valsecchi MG, Bartram CR, et al. Late MRD response determines relapse risk overall and in subsets of childhood T-cell ALL: results of the AIEOP-BFM-ALL 2000 study. Blood. 2011;118(8):2077–2084. | ||
Conter V, Bartram CR, Valsecchi MG, et al. Molecular response to treatment redefines all prognostic factors in children and adolescents with B-cell precursor acute lymphoblastic leukemia: results in 3184 patients of the AIEOP-BFM ALL 2000 study. Blood. 2010;115(16):3206–3214. | ||
Gökbuget N, Kneba M, Raff T, et al. Adult patients with acute lymphoblastic leukemia and molecular failure display a poor prognosis and are candidates for stem cell transplantation and targeted therapies. Blood. 2012;120(9):1868–1876. | ||
Brüggemann M, Raff T, Flohr T, et al. Clinical significance of minimal residual disease quantification in adult patients with standard-risk acute lymphoblastic leukemia. Blood. 2006;107(3):1116–1123. | ||
Beldjord K, Chevret S, Asnafi V, et al. Oncogenetics and minimal residual disease are independent outcome predictors in adult patients with acute lymphoblastic leukemia. Blood. 2014;123(24):3739–3749. | ||
Szczepański T, Beishuizen A, Pongers-Willemse MJ, et al. Cross-lineage T cell receptor gene rearrangements occur in more than ninety percent of childhood precursor-B acute lymphoblastic leukemias: alternative PCR targets for detection of minimal residual disease. Leukemia. 1999;13(2):196–205. | ||
van Dongen JJ, Lhermitte L, Böttcher S, et al. EuroFlow antibody panels for standardized n-dimensional flow cytometric immunophenotyping of normal, reactive and malignant leukocytes. Leukemia. 2012;26(9):1908–1975. | ||
Wu D, Sherwood A, Fromm JR, et al. High-throughput sequencing detects minimal residual disease in acute T lymphoblastic leukemia. Sci Transl Med. 2012;4(134):ra63. | ||
Wood B, Wu D, Crossley B, et al. Measurable residual disease detection by high-throughput sequencing improves risk stratification for pediatric B-ALL. Blood. 2018;131(12):1350–1359. | ||
Coustan-Smith E, Sancho J, Hancock ML, et al. Use of peripheral blood instead of bone marrow to monitor residual disease in children with acute lymphoblastic leukemia. Blood. 2002;100(7):2399–2402. | ||
van der Velden VH, Jacobs DC, Wijkhuijs AJ, et al. Minimal residual disease levels in bone marrow and peripheral blood are comparable in children with T cell acute lymphoblastic leukemia (ALL), but not in precursor-B-ALL. Leukemia. 2002;16(8):1432–1436. | ||
Brisco MJ, Sykes PJ, Hughes E, et al. Monitoring minimal residual disease in peripheral blood in B-lineage acute lymphoblastic leukaemia. Br J Haematol. 1997;99(2):314–319. | ||
van Dongen JJ, van der Velden VH, Brüggemann M, Orfao A. Minimal residual disease diagnostics in acute lymphoblastic leukemia: need for sensitive, fast, and standardized technologies. Blood. 2015;125(26):3996–4009. | ||
Al Ustwani O, Gupta N, Bakhribah H, Griffiths E, Wang E, Wetzler M. Clinical updates in adult acute lymphoblastic leukemia. Crit Rev Oncol Hematol. 2016;99:189–199. | ||
Raff T, Gökbuget N, Lüschen S, et al. Molecular relapse in adult standard-risk ALL patients detected by prospective MRD monitoring during and after maintenance treatment: data from the GMALL 06/99 and 07/03 trials. Blood. 2007;109(3):910–915. | ||
Stock W, La M, Sanford B, et al. What determines the outcomes for adolescents and young adults with acute lymphoblastic leukemia treated on cooperative group protocols? A comparison of Children’s Cancer Group and Cancer and Leukemia Group B studies. Blood. 2008;112(5):1646–1654. | ||
Rytting ME, Thomas DA, O’Brien SM, et al. Augmented Berlin-Frankfurt-Münster therapy in adolescents and young adults (AYAs) with acute lymphoblastic leukemia (ALL). Cancer. 2014;120(23):3660–3668. | ||
Stock W, Johnson JL, Stone RM, et al. Dose intensification of daunorubicin and cytarabine during treatment of adult acute lymphoblastic leukemia: results of Cancer and Leukemia Group B Study 19802. Cancer. 2013;119(1):90–98. | ||
Advani A, Sanford B, Luger S, et al. Frontline-Treatment Of Acute Lymphoblastic Leukemia (ALL) In Older Adolescents and Young Adults (AYA) Using a Pediatric Regimen Is Feasible: Toxicity Results of the Prospective US Intergroup Trial C10403 (Alliance). Blood. 2013;122:3903. | ||
Larsen EC, Salzer WL, Devidas M, et al. Comparison of high-dose methotrexate (HD-MTX) with Capizzi methotrexate plus asparaginase (C-MTX/ASNase) in children and young adults with high-risk acute lymphoblastic leukemia (HR-ALL): a report from the Children’s Oncology Group Study AALL0232. Journal of Clinical Oncology. 2011;29(18 Suppl):3. | ||
Huguet F, Chevret S, Leguay T, et al. Intensified Therapy of Acute Lymphoblastic Leukemia in Adults: Report of the Randomized GRAALL-2005 Clinical Trial. J Clin Oncol. 2018:JCO.2017.76.819. | ||
Fielding AK, Rowe JM, Richards SM, et al. Prospective outcome data on 267 unselected adult patients with Philadelphia chromosome-positive acute lymphoblastic leukemia confirms superiority of allogeneic transplantation over chemotherapy in the pre-imatinib era: results from the International ALL Trial MRC UKALLXII/ECOG2993. Blood. 2009;113(19):4489–4496. | ||
Ravandi F, O’Brien SM, Cortes JE, et al. Long-term follow-up of a phase 2 study of chemotherapy plus dasatinib for the initial treatment of patients with Philadelphia chromosome-positive acute lymphoblastic leukemia. Cancer. 2015;121(23):4158–4164. | ||
Ravandi F, Othus M, O’Brien SM, et al. US Intergroup Study of Chemotherapy Plus Dasatinib and Allogeneic Stem Cell Transplant in Philadelphia Chromosome Positive ALL. Blood Adv. 2016;1(3):250–259. | ||
Kim DY, Joo YD, Lim SN, et al. Nilotinib combined with multiagent chemotherapy for newly diagnosed Philadelphia-positive acute lymphoblastic leukemia. Blood. 2015;126(6):746–756. | ||
Jabbour E, Kantarjian H, Ravandi F, et al. Combination of hyper-CVAD with ponatinib as first-line therapy for patients with Philadelphia chromosome-positive acute lymphoblastic leukaemia: a single-centre, phase 2 study. Lancet Oncol. 2015;16(15):1547–1555. | ||
Hu Y, Liu Y, Pelletier S, et al. Requirement of Src kinases Lyn, Hck and Fgr for BCR-ABL1-induced B-lymphoblastic leukemia but not chronic myeloid leukemia. Nat Genet. 2004;36(5):453–461. | ||
Porkka K, Koskenvesa P, Lundán T, et al. Dasatinib crosses the blood-brain barrier and is an efficient therapy for central nervous system Philadelphia chromosome-positive leukemia. Blood. 2008;112(4):1005–1012. | ||
Ravandi F, Jorgensen JL, Thomas DA, et al. Detection of MRD may predict the outcome of patients with Philadelphia chromosome-positive ALL treated with tyrosine kinase inhibitors plus chemotherapy. Blood. 2013;122(7):1214–1221. | ||
Soverini S, de Benedittis C, Papayannidis C, et al. Drug resistance and BCR-ABL kinase domain mutations in Philadelphia chromosome-positive acute lymphoblastic leukemia from the imatinib to the second-generation tyrosine kinase inhibitor era: the main changes are in the type of mutations, but not in the frequency of mutation involvement. Cancer. 2014;120(7):1002–1009. | ||
Cortes JE, Kantarjian H, Shah NP, et al. Ponatinib in refractory Philadelphia chromosome-positive leukemias. N Engl J Med. 2012;367(22):2075–2088. | ||
O’Hare T, Shakespeare WC, Zhu X, et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell. 2009;16(5):401–412. | ||
Sasaki K, Jabbour EJ, Ravandi F, et al. Hyper-CVAD plus ponatinib versus hyper-CVAD plus dasatinib as frontline therapy for patients with Philadelphia chromosome-positive acute lymphoblastic leukemia: A propensity score analysis. Cancer. 2016;122(23):3650–3656. | ||
Chiaretti S, Vitale A, Elia L. First results of the multicenter total therapy gimema LAL 1509 protocol for de novo adult philadelphia chromosome positive (Ph+) acute lymphoblastic leukemia (ALL) patients. Blood. 2014;124:797. | ||
Martinelli G, Piciocchi A, Papayannidis C. First Report of the Gimema LAL1811 Phase II Prospective Study of the Combination of Steroids with Ponatinib As Frontline Therapy of Elderly or Unfit Patients with Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia. ASH. 2017;130:99. | ||
Ottmann OG, Pfeifer H, Cayuela J-M. Nilotinib (Tasigna) and chemotherapy for first-line treatment in elderly patients with de novo Philadelphia chromosome/BCR-ABL1 positive acute lympho- blastic leukemia (ALL): a trial of the European Working Group for Adult ALL (EWALL-PH-02). Blood. 2014;124:798. | ||
Rousselot P, Coudé MM, Gokbuget N, et al. Dasatinib and low-intensity chemotherapy in elderly patients with Philadelphia chromosome-positive ALL. Blood. 2016;128(6):774–782. | ||
Chalandon Y, Thomas X, Hayette S, et al. Randomized study of reduced-intensity chemotherapy combined with imatinib in adults with Ph-positive acute lymphoblastic leukemia. Blood. 2015;125(24):3711–3719. | ||
Schultz KR, Bowman WP, Aledo A, et al. Improved early event-free survival with imatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia: a children’s oncology group study. J Clin Oncol. 2009;27(31):5175–5181. | ||
Schultz KR, Carroll A, Heerema NA, et al. Long-term follow-up of imatinib in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia: Children’s Oncology Group study AALL0031. Leukemia. 2014;28(7):1467–1471. | ||
Wetzler M, Watson D, Stock W, et al. Autologous transplantation for Philadelphia chromosome-positive acute lymphoblastic leukemia achieves outcomes similar to allogeneic transplantation: results of CALGB Study 10001 (Alliance). Haematologica. 2014;99(1):111–115. | ||
Giebel S, Labopin M, Gorin NC, et al. Improving results of autologous stem cell transplantation for Philadelphia-positive acute lymphoblastic leukaemia in the era of tyrosine kinase inhibitors: a report from the Acute Leukaemia Working Party of the European Group for Blood and Marrow Transplantation. Eur J Cancer. 2014;50(2):411–417. | ||
Pfeifer H, Wassmann B, Bethge W, et al. Randomized comparison of prophylactic and minimal residual disease-triggered imatinib after allogeneic stem cell transplantation for BCR-ABL1-positive acute lymphoblastic leukemia. Leukemia. 2013;27(6):1254–1262. | ||
Wassmann B, Pfeifer H, Stadler M, et al. Early molecular response to posttransplantation imatinib determines outcome in MRD+ Philadelphia-positive acute lymphoblastic leukemia (Ph+ ALL). Blood. 2005;106(2):458–463. | ||
Kebriaei P, Saliba R, Rondon G, et al. Long-term follow-up of allogeneic hematopoietic stem cell transplantation for patients with Philadelphia chromosome-positive acute lymphoblastic leukemia: impact of tyrosine kinase inhibitors on treatment outcomes. Biol Blood Marrow Transplant. 2012;18(4):584–592. | ||
Giebel S, Czyz A, Ottmann O, et al. Use of tyrosine kinase inhibitors to prevent relapse after allogeneic hematopoietic stem cell transplantation for patients with Philadelphia chromosome-positive acute lymphoblastic leukemia: A position statement of the Acute Leukemia Working Party of the European Society for Blood and Marrow Transplantation. Cancer. 2016;122(19):2941–2951. | ||
Tavernier E, Boiron JM, Huguet F, et al. Outcome of treatment after first relapse in adults with acute lymphoblastic leukemia initially treated by the LALA-94 trial. Leukemia. 2007;21(9):1907–1914. | ||
Thomas DA, Kantarjian H, Smith TL, et al. Primary refractory and relapsed adult acute lymphoblastic leukemia: characteristics, treatment results, and prognosis with salvage therapy. Cancer. 1999;86(7):1216–1230. | ||
Piccaluga PP, Arpinati M, Candoni A, et al. Surface antigens analysis reveals significant expression of candidate targets for immunotherapy in adult acute lymphoid leukemia. Leuk Lymphoma. 2011;52(2):325–327. | ||
Raponi S, de Propris MS, Intoppa S, et al. Flow cytometric study of potential target antigens (CD19, CD20, CD22, CD33) for antibody-based immunotherapy in acute lymphoblastic leukemia: analysis of 552 cases. Leuk Lymphoma. 2011;52(6):1098–1107. | ||
Thomas DA, Faderl S, O’Brien S, et al. Chemoimmunotherapy with hyper-CVAD plus rituximab for the treatment of adult Burkitt and Burkitt-type lymphoma or acute lymphoblastic leukemia. Cancer. 2006;106(7):1569–1580. | ||
Thomas DA, O’Brien S, Faderl S, et al. Chemoimmunotherapy with a modified hyper-CVAD and rituximab regimen improves outcome in de novo Philadelphia chromosome-negative precursor B-lineage acute lymphoblastic leukemia. J Clin Oncol. 2010;28(24):3880–3889. | ||
Nagorsen D, Kufer P, Baeuerle PA, Bargou R. Blinatumomab: a historical perspective. Pharmacol Ther. 2012;136(3):334–342. | ||
Topp MS, Kufer P, Gökbuget N, et al. Targeted therapy with the T-cell-engaging antibody blinatumomab of chemotherapy-refractory minimal residual disease in B-lineage acute lymphoblastic leukemia patients results in high response rate and prolonged leukemia-free survival. J Clin Oncol. 2011;29(18):2493–2498. | ||
Gökbuget N, Zugmaier G, Klinger M, et al. Long-term relapse-free survival in a phase 2 study of blinatumomab for the treatment of patients with minimal residual disease in B-lineage acute lymphoblastic leukemia. Haematologica. 2017;102(4):e132–e135. | ||
Gökbuget N, Zugmaier G, Klinger M. Long-term relapse-free survival in a phase 2 study of blinatumomab for the treatment of patients with minimal residual disease in B-lineage acute lymphoblastic leukemia. Blood. 2015;126(23):680. | ||
Topp MS, Gökbuget N, Zugmaier G, et al. Phase II trial of the anti-CD19 bispecific T cell-engager blinatumomab shows hematologic and molecular remissions in patients with relapsed or refractory B-precursor acute lymphoblastic leukemia. J Clin Oncol. 2014;32(36):4134–4140. | ||
Topp MS, Gökbuget N, Stein AS, et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. Lancet Oncol. 2015;16(1):57–66. | ||
Kantarjian H, Stein A, Gökbuget N, et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N Engl J Med. 2017;376(9):836–847. | ||
Martinelli G, Boissel N, Chevallier P, et al. Complete Hematologic and Molecular Response in Adult Patients With Relapsed/Refractory Philadelphia Chromosome-Positive B-Precursor Acute Lymphoblastic Leukemia Following Treatment With Blinatumomab: Results From a Phase II, Single-Arm, Multicenter Study. J Clin Oncol. 2017;35(16):1795–1802. | ||
von Stackelberg A, Locatelli F, Zugmaier G, et al. Phase I/Phase II Study of Blinatumomab in Pediatric Patients With Relapsed/Refractory Acute Lymphoblastic Leukemia. J Clin Oncol. 2016;34(36):4381–4389. | ||
National Cancer Institute. Blinatumomab and Combination Chemotherapy or Dasatinib, Prednisone, and Blinatumomab in Treating Older Patients With Acute Lymphoblastic Leukemia. Available from: https://clinicaltrials.gov/ct2/show/NCT02143414. NLM identifier: NCT02143414. Accessed August 16, 2018. | ||
National Cancer Institute. Combination Chemotherapy With or Without Blinatumomab in Treating Patients With Newly Diagnosed BCR-ABL-Negative B Lineage Acute Lymphoblastic Leukemia. Available from: https://clinicaltrials.gov/ct2/show/NCT02003222. NLM identifier: NCT02003222. Accessed August 16, 2018. | ||
Gökbuget N, Dombret H, Bonifacio M, et al. Blinatumomab for minimal residual disease in adults with B-cell precursor acute lymphoblastic leukemia. Blood. 2018;131(14):1522–1531. | ||
Hinman LM, Hamann PR, Wallace R, Menendez AT, Durr FE, Upeslacis J. Preparation and characterization of monoclonal antibody conjugates of the calicheamicins: a novel and potent family of antitumor antibiotics. Cancer Res. 1993;53(14):3336–3342. | ||
Thomas X. Inotuzumab ozogamicin in the treatment of B-cell acute lymphoblastic leukemia. Expert Opin Investig Drugs. 2012;21(6):871–878. | ||
Kantarjian H, Thomas D, Jorgensen J, et al. Inotuzumab ozogamicin, an anti-CD22-calecheamicin conjugate, for refractory and relapsed acute lymphocytic leukaemia: a phase 2 study. Lancet Oncol. 2012;13(4):403–411. | ||
Kantarjian H, Thomas D, Jorgensen J, et al. Results of inotuzumab ozogamicin, a CD22 monoclonal antibody, in refractory and relapsed acute lymphocytic leukemia. Cancer. 2013;119(15):2728–2736. | ||
Kantarjian H, Ravandi F, Short NJ, et al. Inotuzumab ozogamicin in combination with low-intensity chemotherapy for older patients with Philadelphia chromosome-negative acute lymphoblastic leukaemia: a single-arm, phase 2 study. Lancet Oncol. 2018;19(2):240–248. | ||
Kantarjian HM, Deangelo DJ, Stelljes M, et al. Inotuzumab Ozogamicin versus Standard Therapy for Acute Lymphoblastic Leukemia. N Engl J Med. 2016;375(8):740–753. | ||
Jabbour EJ, Deangelo DJ, Stelljes M, et al. Efficacy and safety analysis by age cohort of inotuzumab ozogamicin in patients with relapsed or refractory acute lymphoblastic leukemia enrolled in INO-VATE. Cancer. 2018;124(8):1722–1732. | ||
Turtle CJ, Hanafi LA, Berger C, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. 2016;126(6):2123–2138. | ||
Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385(9967):517–528. | ||
Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–1517. | ||
Davila ML, Riviere I, Wang X, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6(224):224ra25. | ||
Grupp SA, Laetsch TW, Buechner J. Analysis of a Global Registration Trial of the Efficacy and Safety of CTL019 in Pediatric and Young Adults with Relapsed/Refractory Acute Lymphoblastic Leukemia (ALL). Blood. 2016;128:221. | ||
Maude SL, Pulsipher MA, Boyer MW. Efficacy and Safety of CTL019 in the First US Phase II Multicenter Trial in Pediatric Relapsed/Refractory Acute Lymphoblastic Leukemia: Results of an Interim Analysis. Blood. 2016;128:2801. | ||
Park JH, Rivière I, Gonen M, et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N Engl J Med. 2018;378(5):449–459. | ||
Wei G, Wang J, Huang H, Zhao Y. Novel immunotherapies for adult patients with B-lineage acute lymphoblastic leukemia. J Hematol Oncol. 2017;10(1):150. | ||
Riet T, Holzinger A, Dörrie J, Schaft N, Schuler G, Abken H. Nonviral RNA transfection to transiently modify T cells with chimeric antigen receptors for adoptive therapy. Methods Mol Biol. 2013;969:187–201. | ||
Haso W, Lee DW, Shah NN, et al. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood. 2013;121(7):1165–1174. | ||
Cai B, Guo M, Wang Y, et al. Co-infusion of haplo-identical CD19-chimeric antigen receptor T cells and stem cells achieved full donor engraftment in refractory acute lymphoblastic leukemia. J Hematol Oncol. 2016;9(1):131. | ||
Mrózek K, Heerema NA, Bloomfield CD. Cytogenetics in acute leukemia. Blood Rev. 2004;18(2):115–136. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.