")
Back to Journals » OncoTargets and Therapy » Volume 7
Recent advances in the ARID family: focusing on roles in human cancer
Authors Lin C, Song W, Bi X, Zhao J, Huang Z, Li Z, Zhou J, Cai J, Zhao H
Received 4 November 2013
Accepted for publication 3 January 2014
Published 18 February 2014 Volume 2014:7 Pages 315—324
DOI https://doi.org/10.2147/OTT.S57023
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Chen Lin,1 Wei Song,2 Xinyu Bi,1 Jianjun Zhao,1 Zhen Huang,1 Zhiyu Li,1 Jianguo Zhou,1 Jianqiang Cai,1 Hong Zhao1
1Department of Abdominal Surgical Oncology, Cancer Hospital, 2National Laboratory of Medical Molecular Biology, Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, People's Republic of China
Abstract: The human AT-rich interaction domain (ARID) family contains seven subfamilies and 15 members characterized by having an ARID. Members of the ARID family have the ability to regulate transcription and are involved in cell differentiation and proliferation. Accumulating evidence suggests that ARID family members are involved in cancer-related signaling pathways, highly mutated or differentially expressed in tumor tissues, and act as predictive factors for cancer prognosis or therapeutic outcome. Here we review the molecular biology and clinical studies concerned with the role played by the ARID family in cancer. This may contribute to our understanding of the initiation and progression of cancer from a novel point of view, as well as providing potential targets for cancer therapy.
Keywords: AT-rich interaction domain, human cancer, cancer-related signaling pathway, therapy, potential targets
Introduction
The human AT-rich interaction domain (ARID) family is a superfamily of 15 members. The family is divided into seven subfamilies based on the degree of sequence identity between individual members (Figure 1). Members of the ARID family all contain a DNA-binding domain that was initially found to interact with AT-rich DNA elements.1 Further studies demonstrated that AT-rich binding was not an intrinsic property of ARID and that members of the ARID family might be involved in a wider range of DNA interactions.2 As transcription regulators, the ARID family has been found to affect cell growth, differentiation, and development.3 Recently, an increasing number of studies have suggested that the ARID family may play a role in human cancer. Our aim was to review the literature to date regarding the relationship between the ARID family and cancer (Table 1).
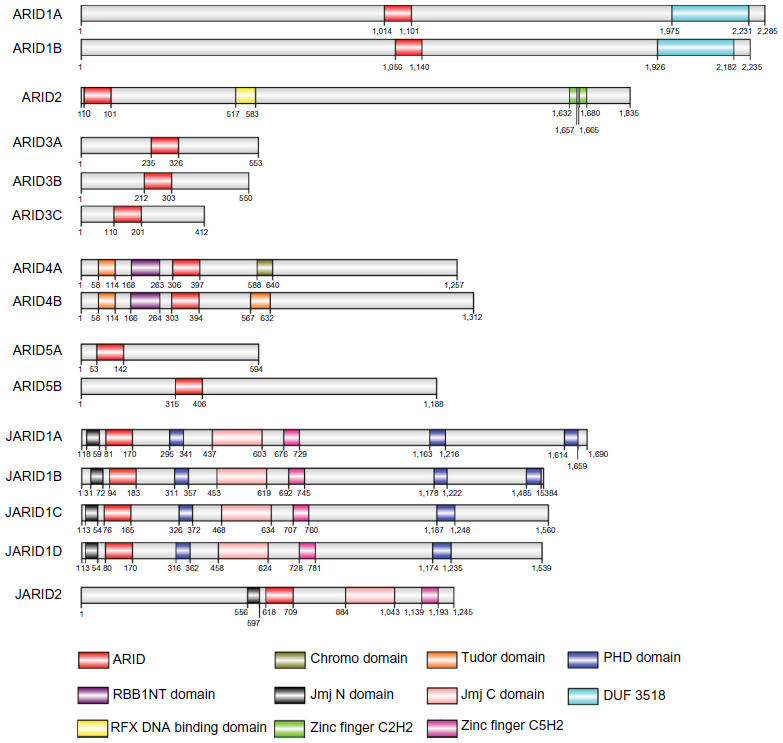 | Figure 1 Brief overview of the ARID family proteins and the domains in each member. ARID family proteins share structure similarity within the subfamilies. The domains were identified through the SMART databases. |
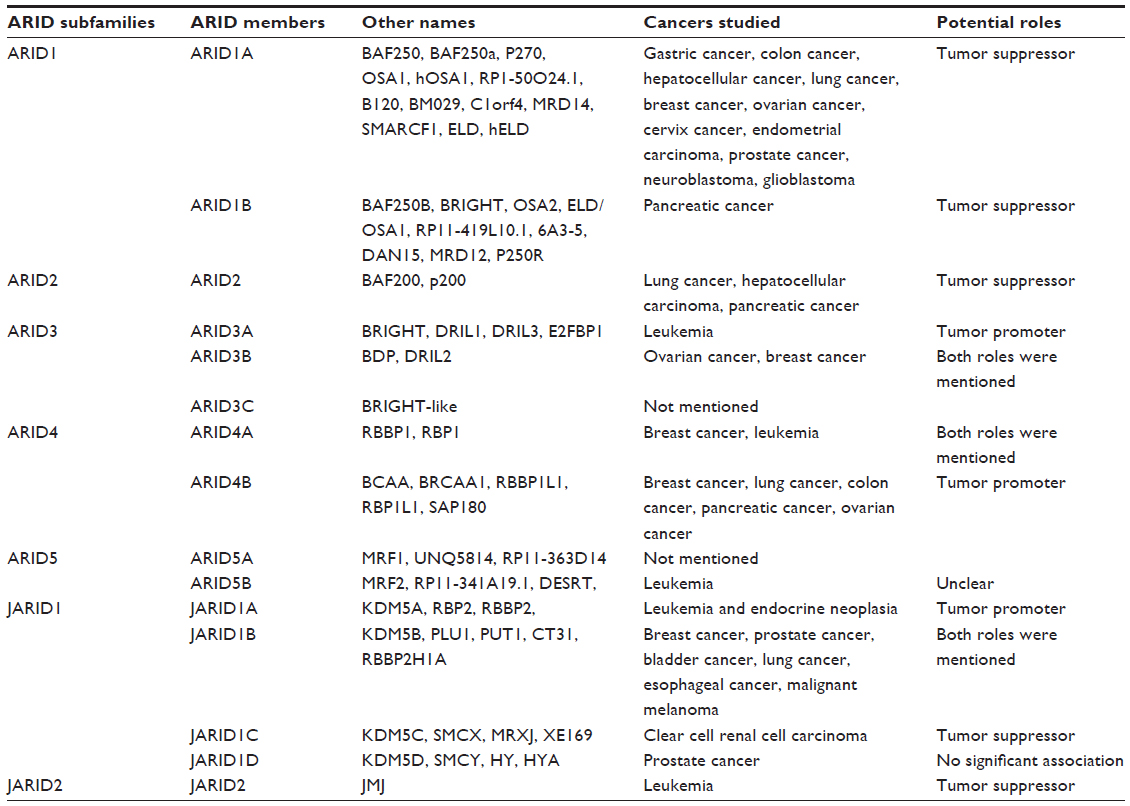 | Table 1 ARID family and related cancers |
Members of the ARID family
ARID1
The ARID1 subfamily has two members, ie, ARID1A and ARID1B.1 ARID1A and ARID1B are located on 1p35.3 and 6q25.1, respectively. Their encoding proteins share 66% overall similarity and are two alternative, mutually exclusive subunits of BRG1-associated factor (BAF), a subclass of the human SWI/SNF complex involved in ligand-dependent transcriptional activation by nuclear receptors.4,5 In addition to ARID, ARID1 proteins each bear a domain of unknown function 3518. Even though they are similar in structure, ARID1A and ARID1B containing complexes are functionally distinct from each other.6
ARID1A
ARID1A is found in both the cytoplasm and the nucleus. Nuclear ARID1A is less stable than cytoplasmic ARID1A because it is rapidly degraded by the ubiquitin-proteasome system in the nucleus. ARID1A mutation has been shown to affect the consensus nuclear export signal and reduce steady-state protein levels of ARID1A.7 ARID1A is widely expressed in many normal human tissues, but is markedly reduced in human tumor tissues and cell lines.8 Recent studies indicate that ARID1A is a tumor suppressor. ARID1A interacts with hypermethylated in cancer-1, a tumor suppressor that is epigenetically silenced in many human cancers, and represses transcription of E2F transcription factor-1 and E2F transcription factor-1-dependent cell survival and stress responses.9 ARID1A is essential for cell cycle arrest, with ARID1A-depleted cells continuing synthesis of DNA and failing to show other characteristics typical of arrested cells, including upregulation of p21 and downregulation of cyclins.6 ARID1A collaborates with p53 to regulate p21 and Smad family member 3 transcription and alters tumor growth in gynecologic cancers.10 Further, colony formation in breast and gastric cancer cell lines has been shown to be significantly inhibited after re-expression of ARID1A.11,12
Accumulating evidence shows that ARID1A is highly mutated in a number of different cancers (Table 2). Several studies have shown that decreased ARID1A expression is related to a variety of types of cancer. Loss of ARID1A expression has been found to be significantly more frequent in certain types of cancers, including Epstein-Barr virus-positive gastric carcinoma, informative ovarian clear-cell carcinoma, and aggressive phenotypes of breast cancer.11,13,14 ARID1A has also been found to be related to the prognosis of some cancers. ARID1A and ARID1B mutations have been associated with early treatment failure and decreased survival in children with neuroblastoma.15 Reduced ARID1A expression has been shown to be related to poor prognosis in ovarian clear-cell carcinoma and gastric cancer,12,13,16 although other studies have suggested the opposite, eg, the level of ARID1A expression was not significantly associated with survival in endometrial carcinoma17,18 and was not related to the prognosis of uterine cervix cancer.19
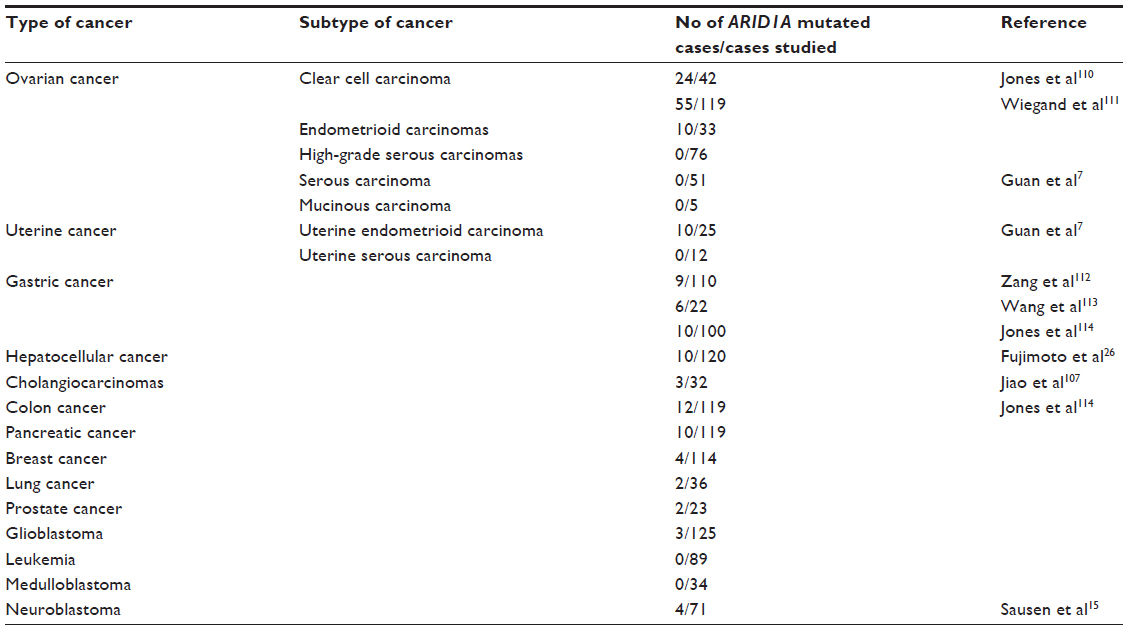 | Table 2 Gene sequencing of ARID1A in different cancers |
ARID1B
Like ARID1A, ARID1B is widely distributed in tissues. Although structurally similar, ARID1B appears to play an opposite role to that of ARID1A. ARID1B-containing BAF complexes have been shown to activate the cell cycle.20 In contrast with its role as a cell cycle activator, ARID1B seems to be a tumor suppressor in pancreatic cancer. It is also less expressed in advanced-stage pancreatic cancer tissues and was found to inhibit colony formation in a pancreatic cancer cell line.21 The role of ARID1B protein remains unclear and needs further study.
Within the ARID1 subfamily, ARID1A and ARID1B play opposite roles in the cell cycle and are two mutually exclusive subunits of BAF complex. The roles played by the BAF complex in human cancer may be decided by the dominant subunit. Studies focused on both members are needed in the future to clarify how the same ARID1A status can have different clinicopathological characteristics.
ARID2
The ARID2 subfamily has only one member. ARID2 is located on 12q12. As with the other members of the ARID1 subfamily, its encoding proteins also comprise a subunit of the human SWI/SNF complex, but in an alternative subclass containing complex polybromo-associated BAF (PBAF).22 ARID2 protein contains an ARID, another DNA-binding domain known as RFX DNA, and two conservative C-terminal C2H2 Zn fingers. Among these domains, mutations have been found to disrupt the Zn finger motifs only, suggesting the importance of these motifs in the biological activity of ARID2.23
ARID2 is widely expressed. Gene sequencing results show that ARID2 is highly mutated in certain types of cancers, including non-small-cell lung cancer, hepatocellular carcinoma (HCC), and pancreatic cancer.24–27 Interestingly, ARID2 has been shown to be highly mutated in cases of hepatitis C virus (HCV)-associated HCC, while some studies indicate that ARID2 might play a role in infection with the virus. ARID2 is essential for regulation of interferon-induced gene expression, especially IFITM1, a tight junction protein that inhibits entry of HCV.22,28 Viral hepatitis is important in the tumorigenesis of HCC. We can hypothesize that ARID2 prevents HCV infection, leading to its tumor suppressor role in HCV-associated HCC. Further studies should focus in more detail on ARID2 in the pathways of viral carcinogenesis.
ARID3
The ARID3 subfamily contains three members, ie, ARID3A, ARID3B, and ARID3C.1 Their genes are located on 19p13.3, 15q24, and 9p13.3, respectively. Their encoding proteins are similar in their amino acid sequences and characterized by extended ARID (with additional alpha-helices at the N- and C-termini of the core ARID).1,29 The members of this subfamily play a role in embryonic patterning, cell lineage gene regulation, and cell cycle control.
ARID3A
ARID3A protein is mainly expressed in B-lymphocytes and the testes.30 Studies have related ARID3A to the p53 tumor suppressor to some degree. A potential p53 binding site was found in the second intron of the ARID3A gene.31 A further study by the same group of investigators found a cooperative role for ARID3A and p53 in the transcriptional activation of p21, a p53 target gene important in cell cycle arrest.32 Another study found ARID3A to disrupt cellular protection against RASV12-induced proliferation downstream of the p19/p53 pathway.33
There is some evidence that ARID3A can block cell differentiation and promote proliferation, indicating a tumor promoter role for ARID3A. One study found that ARID3A was required to maintain cell survival potential by downregulation of promyelocytic leukemia nuclear bodies.34 Further research revealed ARID3A to act as a critical antagonist of the p16 retinoblastoma protein (RB) tumor suppressor machinery by regulating the stability of promyelocytic leukemia.35 However, direct knockdown of human ARID3A resulted in colonies expressing multiple lineage markers, suggesting that repression of ARID3A promoted developmental plasticity.36 Repression of ARID3A by miR-125b blocked cell differentiation and apoptosis, and increased cell proliferation in B-cell acute lymphoblastic leukemia.37
ARID3B
The testes and leukocytes show the highest levels of ARID3B.38 The role of ARID3B in cancer remains controversial. Some studies suggest that ARID3B tends to act as an oncogene. ARID3B is a target of miR-125a, promoting invasion of ovarian cancer and migration of breast cancer.39,40 Coexpression of ARID3B and Mycn has been observed in a proportion of germ cell tumors. ARID3B and Mycn have been shown to cooperate in proliferation of mouse stem cells, with ARID3B avoiding cell death and Mycn driving progression of the cell cycle.41 Another study reported the opposite, ie, overexpression of full-length ARID3B increased tumor necrosis factor alpha (TNF-α) levels and TNF-related apoptosis inducing ligand (TRAIL)-induced cell death in ovarian cancer cells.42
There might be some factors contributing to this discrepancy. First, ARID3B Sh, a splice form of ARID3B has been demonstrated, overexpression of which did not induce cell apoptosis.42 The expression levels of full-length ARID3B and ARID3B Sh may together contribute to the effect of ARID3B in human cancer. Second, there has been a study suggesting that cellular localization of ARID3B is required for its function.43 Different cellular localization of endogenous and exogenous ARID3B may result in different roles in human cancer. Further study is needed to understand the underlying mechanisms of ARID3B in carcinogenesis. Increasing the expression of full-length ARID3B and altering its cellular location may become a novel treatment strategy for cancer.
ARID3C
ARID3C is a newly discovered member of the ARID3 subfamily. It is expressed preferentially in B lineage lymphocytes. ARID3C encodes two isoforms, ie, Brightlike and Brightlike delta 6, that include and exclude the REKLES-encoding exon 6, respectively.44 No study has focused on the relationship between ARID3C and cancer as yet. Further research is needed to understand ARID3C better.
ARID4
The ARID4 subfamily has two members, ie, ARID4A and ARID4B.1 ARID4A and ARID4B are located on 14q23.1 and 1q42.1-q43, respectively. Their encoding proteins are 40%–50% identical in sequence. They contain ARIDs, Tudor domains, and N-terminal domain in retinoblastoma-binding protein-1 family domains. ARID4A contains an additional LXCXE motif, which is an RB binding motif.45
ARID4A
ARID4A has been found to play a role in cell growth and migration via interaction with pRB and the mSIN3-histone deacetylase (mSIN3-HDAC) complex. ARID4A represses E2F-dependent transcription by recruiting the mSIN3-HDAC complex to pRB family members and plays a central role in arresting cell growth.46 Breast cancer metastasis suppressor 1, which suppresses the metastasis of multiple types of human cancer cells, complexes with ARID4A and mSIN3-HDAC, thereby repressing transcription.47
However, the role of ARID4A in cancer remains controversial. KASIFLK, a peptide epitope identical to ARID4A, was found to be expressed preferentially in breast cancer cell lines and tissues, indicating that ARID4A may be an oncogene.48 However, another study found ARID4A to be a leukemia suppressor gene, with ARID4A-deficient mice showing ineffective hematopoiesis followed by transition to leukemia.49 In this study, ten of twelve ARID4A−/− ARID4B+/− mice progressed to acute myeloid leukemia, while only five of 42 ARID4A−/− mice did so, indicating that ARID4B might influence the role of ARID4A in carcinogenesis. However, this study was based on an animal model of leukemia, in which the mechanisms may be different from those in solid tumors in humans.
ARID4B
Like ARID4A, ARID4B has been found to be an mSIN3-HDAC complex-associated protein. The ARID4B gene is also known as breast cancer-associated antigen 1 (BRCAA1) and is highly expressed in human breast, lung, colon, pancreatic, and ovarian cancers, and in normal testes, but has limited expression in other normal tissues.50 There is some evidence to suggest its involvement in breast cancer. A causative role of ARID4B in progression of metastatic breast cancer has been observed in cell lines and animal models, as well as in patients.51 SSKKQKRSHK, a peptide epitope identical to ARID4B, was shown to be associated with prognosis in patients with breast cancer, in whom its serum titer was significantly elevated.52
ARID5
The ARID5 subfamily contains two members, ie, ARID5A and ARID5B. Their genes are located on 2q11.2 and 10q21.2, respectively. ARID5A and ARID5B share more than 70% identity within the ARID domain, but outside this domain their amino acid sequences are not similar.3
ARID5A
ARID5A is widely distributed in human tissue. Its DNA binding activity has been found to be significantly reduced in differentiated cells and it acts as a trans-acting factor in repressing enhancer activity.53 Studies had found ARID5A to be involved in the inflammatory process.54,55 An association between ARID5A and cancer has not been reported as yet.
ARID5B
ARID5B is also widely expressed in human tissue. Histone deacetylase-1, which plays a role in cell proliferation, differentiation, and apoptosis, was found to be associated with ARID5B.56 Of all the malignant diseases, leukemia is the one most closely linked with ARID5B. Mounting evidence shows that mutations and single nucleotide polymorphisms of ARID5B are associated with the development of acute lymphoblastic leukemia and the treatment outcome, and may be associated with acute myeloblastic leukemia in childhood.57–62
JARID1
Jumonji ARID1 (JARID1) is the biggest subfamily, with four members previously named JARID1A through D and now known as lysine (K)-specific demethylase (KDM) 5A through D.63 Their genes are located on 4q42, 1q32.1, Xp11.22–p11.21, and Yq11, respectively. The name Jumonji (meaning “cross” in Japanese) comes from the morphology produced by the normal neural groove and abnormal grooves on the neural plates of Jumonji mutant mice.64 The subfamily members contain ARID, Jumonji N, and Jumonji C domains, as well as plant homeodomain and Zf-C5HC2 Zn fingers. Plant homeodomains can bind methylated lysine residues,65 and all members have been found to be histone demethylases.66,67 An increasing number of studies have found the JARID1 subfamily to be associated with cancer, especially JARID1A and JARID1B.
JARID1A
Like ARID4A, JARID1A is also a pRB-binding protein, and binding of JARID1A to pRB promotes cell differentiation.68 JARID1A interacts physically with RBP-J, which activates transcription of Notch target genes and is crucial for Notch-induced growth and tumorigenic responses.69 JARID1A protein was found to regulate demethylation of histone H3 lysine 4,70 and since then, a number of studies have indicated that the demethylase activity of JARID1A is associated with cancer-related signaling pathways. JARID1A protein-mediated demethylation of H3 lysine 4 has been associated with silencing of target genes for retinoblastoma and the Myc/Max/Mad network.71,72 The histone demethylase activity of JARID1A was also found to be related to tumorigenesis in hematopoietic malignancies and endocrine neoplasia.73,74 JARID1A was identified as the fusion partner of NUP98 in samples from leukemia patients, and this gene fusion induced acute myeloid leukemia in animal models requiring its demethylase activity.73,75 Other studies have found that chronic drug exposure causing an alteration in the chromatin state is associated with JARID1A in drug-tolerant cancer cells.76,77
JARID1B
Sharing 56% overall similarity to JARID1A, JARID1B is a histone demethylase that interacts with several proteins.78 Unlike other JARID1 subfamily members, JARID1B has limited expression in normal tissue. It has been found to be upregulated in primary breast, prostate, bladder, lung, and esophageal cancers, but downregulated in malignant melanoma.79–83 JARID1B may promote proliferation of breast cancer cell lines by demethylation of histone H3 lysine 4.84 JARID1B downregulates breast cancer-1 (BRCA1), which is a tumor suppressor gene.85 JARID1B downregulates chemokine (C-C motif) ligand 14 (CCL14) which suppresses the angiogenic and metastatic potential of breast cancer cells.86 It also downregulates genes involved in regulating the M phase of the mitotic cell cycle which induces cells to fail to arrest in G2/M in breast cancer.85 A dramatic decrease in E2-stimulated tumor growth was observed in nude mice with downregulated JARID1B.87 In bladder cancer, knockdown of JARID1B suppresses the proliferation of cancer cells and increases the number of cells in sub-G1 phase via the E2F/RB pathway.81,88 In colorectal cancer, JARID1B-positive cells showed greater tumorigenicity and depletion of JARID1B-induced cellular senescence.89 However, in melanoma, JARID1B tends to be a tumor suppressor. It has been shown to inhibit proliferation, increase caspase activity, and induce partial cell cycle arrest in G1/0 phase by modulation of pRB.90 Expression of a set of melanoma-relevant genes was altered after increasing the levels of JARID1B.91 Interestingly, expression of JARID1B was found to be dynamically regulated in melanoma cells without following a hierarchical cancer stem cell model.92
JARID1B seems to play different roles in different malignant tumors. As a histone demethylase, JARID1B is a transcription regulator, but its exact role may depend on the different genes it regulates.
JARID1C
JARID1C mutation has been widely studied in X-linked mental retardation.93–95 In addition to mental disorders, JARID1C has also been associated with cancer. JARID1C might contribute to tumorigenesis via the transforming growth factor-beta/Smad family member 3 signaling pathway.96 Inactivating mutations of JARID1C have been found in clear-cell renal cell carcinoma specimens;97 von Hippel–Lindau−/− clear-cell renal cell carcinoma cells with suppressed JARID1C were found to form much larger tumors in a xenograft model, suggesting a tumor suppressor role for JARID1C in clear-cell renal cell carcinoma.98
JARID1D
The evidence presently available suggests that JARID1D contributes to rejection of male tissues by female transplant recipients.99 There is limited research on the association between JARID1D and cancer. JARID1D is expressed to a similar extent in prostate cancer and benign prostatic hypertrophy tissues.100 Even though JARID1D is similar to JARID1C in its amino acid sequence, no significant relationship between JARID1D and cancer has been identified as yet.
JARID2
The JARID2 subfamily contains only one member, which is located on 6p24p23. Its encoding protein has an ARID domain, JmjN and JmjC domains, and a Zf-C5HC2 Zn finger domain.101 Unlike members of the JARID1 subfamily, JARID2 has histone methyltransferase activity.102
Recent studies suggest that JARID2 has a tumor suppressor role. JARID2 protein was found to regulate cell proliferation in a negative manner103 and to suppress transcription of cyclin D1 by methylation of histone H3-K9.102 Deletions of JARID2 have been detected in leukemic transformation of chronic myeloid malignancies.104 Reticuloendotheliosis virus strain T, an oncogenic retrovirus, promotes cell survival by downregulation of targets that include JARID2.105
Summary
Members of the ARID family have DNA binding ability, are likely to be involved in modification of chromatin structure, and regulate targeted gene transcription positively or negatively.106 A number of the members of this family have been found to be altered in cancer tissues, at both the DNA and protein levels. Accumulating evidence suggests an important role for the ARID family in the development, progression, and treatment of cancer. Members of this family may play either a tumor promoter or a tumor suppressor role. Interestingly, ARID3B, ARID4A, and JARID1B seem to have conflicting roles in different types of cancer. Their transcription-regulating role may be specific to particular types of cancer and depend on the target gene they regulate. Their signaling pathways remain unclear, and clarifying their biological function may lead to an answer.
The ARID family is divided into subfamilies according to the protein structure of its members. Similar in structure, their roles in human cancer may be influenced by other members of the same subfamily. Members of the ARID1 and ARID2 subfamily are all components of a chromatin regulation complex. ARID1A and ARID2 were both found to be mutated in a study of cholangiocarcinoma.107 Another study of a series of genes in HCC, including ARID1A, ARID1B, and ARID2, showed that loss-of-function mutations were more enriched in these chromatin-regulating genes than in genes from other areas of the HCC genome.26 Members of the JARID1 subfamily are all histone demethylases, while JARID2 is a histone methyltransferase, and may have an opposite role to that of members of the JARID1 subfamily. The evidence indicates that these members may be involved in the same cancer-related pathways within ARID1, ARID2, JARID1 and JARID2 subfamilies.
There is little evidence to suggest that members of the ARID3 and ARID4 subfamilies interact with each other in human cancer. However, current research suggests that members of these two subfamilies may interact in pathways other than those for carcinogenesis. ARID3C significantly co-activates ARID3A-mediated immunoglobulin gene transcription.44 ARID4A and ARID4B both function as transcriptional coactivators for the androgen receptor and RB involved in male fertility.108 Currently, most of the published studies focus on individual members of the ARID family, and have sometimes yielded conflicting results. Studying subfamily members together may give us a more complete picture of the ARID family.
Recently, several small molecules were found to be JARID1B inhibitors and to inhibit proliferation of cells overexpressing JARID1B.109 This suggests that the ARID family could be a potential therapeutic target in the treatment of cancer. More studies focusing on the relationship between the ARID family and cancer are needed in the future, and may lead to a new understanding of the prevention, diagnosis, and treatment of cancer.
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (81201967), the Beijing Natural Science Foundation (7132193) and Beijing Nova Program (No. 2009A69).
Disclosure
The authors report no conflicts of interest in this work.
References
Wilsker D, Probst L, Wain HM, Maltais L, Tucker PW, Moran E. Nomenclature of the ARID family of DNA-binding proteins. Genomics. 2005;86(2):242–251. | |
Dallas PB, Pacchione S, Wilsker D, Bowrin V, Kobayashi R, Moran E. The human SWI-SNF complex protein p270 is an ARID family member with non-sequence-specific DNA binding activity. Mol Cell Biol. 2000;20(9):3137–3146. | |
Wilsker D, Patsialou A, Dallas PB, Moran E. ARID proteins: A diverse family of DNA binding proteins implicated in the control of cell growth, differentiation, and development. Cell Growth Differ. 2002;13(3):95–106. | |
Wang XM, Nagl NG, Wilsker D, et al. Two related ARID family proteins are alternative subunits of human SWI/SNF complexes. Biochemical. 2004;383:319–325. | |
Hurlstone AF, Olave IA, Barker N, van Noort M, Clevers H. Cloning and characterization of hELD/OSA1, a novel BRG1 interacting protein. Biochem J. 2002;364(Pt 1):255–264. | |
Nagl NG, Patsialou A, Haines DS, Dallas PB, Beck GR, Moran E. The p270 (ARID1A/SMARCF1) subunit of mammalian SWI/SNF-related complexes is essential for normal cell cycle arrest. Cancer Res. 2005;65(20):9236–9244. | |
Guan B, Gao M, Wu C-H, Wang T-L, Shih I-M. Functional analysis of in-frame indel ARID1A mutations reveals new regulatory mechanisms of its tumor suppressor functions. Neoplasia. 2012;14(10):986–993. | |
Wang X, Nagl NG Jr, Flowers S, Zweitzig D, Dallas PB, Moran E. Expression of p270 (ARID1A), a component of human SWI/SNF complexes, in human tumors. Int J Cancer. 2004;112(4):636. | |
Van Rechem C, Boulay G, Leprince D. HIC1 interacts with a specific subunit of SWI/SNF complexes, ARID1A/BAF250A. Biochem Biophys Res Commun. 2009;385(4):586–590. | |
Guan B, Wang TL, Shih IM. ARID1A, a factor that promotes formation of SWI/SNF-mediated chromatin remodeling, is a tumor suppressor in gynecologic cancers. Cancer Res. 2011;71(21):6718–6727. | |
Mamo A, Cavallone L, Tuzmen S, et al. An integrated genomic approach identifies ARID1A as a candidate tumor-suppressor gene in breast cancer. Oncogene. 2012;31(16):2090–2100. | |
Wang D-d, Chen Y-b, Pan K, et al. Decreased expression of the ARID1A gene is associated with poor prognosis in primary gastric cancer. PLoS One. 2012;7(7). | |
Abe H, Maeda D, Hino R, et al. ARID1A expression loss in gastric cancer: pathway-dependent roles with and without Epstein-Barr virus infection and microsatellite instability. Virchows Archiv. 2012; 461(4):367–377. | |
Ayhan A, Mao TL, Seckin T, et al. Loss of ARID1A expression is an early molecular event in tumor progression from ovarian endometriotic cyst to clear cell and endometrioid carcinoma. Int J Gynecol Cancer. 2012;22(8):1310–1315. | |
Sausen M, Leary RJ, Jones S, et al. Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat Genet. 2013;45(1):12–17. | |
Katagiri A, Nakayama K, Rahman MT, et al. Loss of ARID1A expression is related to shorter progression-free survival and chemoresistance in ovarian clear cell carcinoma. Mod Pathol. 2012;25(2):282–288. | |
Werner HMJ, Berg A, Wik E, et al. ARID1A loss is prevalent in endometrial hyperplasia with atypia and low-grade endometrioid carcinomas. Mod Pathol. 2013;26(3):428–434. | |
Samartzis EP, Samartzis N, Noske A, et al. Loss of ARID1A/BAF250a-expression in endometriosis: a biomarker for risk of carcinogenic transformation? Mod Pathol. 2012;25(6):885–892. | |
Katagiri A, Nakayama K, Rahman MT, et al. Frequent loss of tumor suppressor ARID1A protein expression in adenocarcinomas/adenosquamous carcinomas of the uterine cervix. Int J Gynecol Cancer. 2012;22(2):208–212. | |
Nagl NG, Wang XM, Patsialou A, Van Scoy M, Moran E. Distinct mammalian SWI/SNF chromatin remodeling complexes with opposing roles in cell-cycle control. Embo J. 2007;26(3):752–763. | |
Khursheed M, Kolla JN, Kotapalli V, et al. ARID1B, a member of the human SWI/SNF chromatin remodeling complex, exhibits tumour-suppressor activities in pancreatic cancer cell lines. Brit J Cancer. 2013;108(10):2056–2062. | |
Yan ZJ, Cui KR, Murray DM, et al. PBAF chromatin-remodeling complex requires a novel specificity subunit, BAF200, to regulate expression of selective interferon-responsive genes. Genes Dev. 2005;19(14):1662–1667. | |
Zhao H, Wang J, Han Y, et al. ARID2: a new tumor suppressor gene in hepatocellular carcinoma. Oncotarget. 2011;2(11):886–891. | |
Manceau G, Letouze E, Guichard C, et al. Recurrent inactivating mutations of ARID2 in non-small cell lung carcinoma. Int J Cancer. 2013;132(9):2217–2221. | |
Li M, Zhao H, Zhang X, et al. Inactivating mutations of the chromatin remodeling gene ARID2 in hepatocellular carcinoma. Nat Genet. 2011;43(9):828–829. | |
Fujimoto A, Totoki Y, Abe T, et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet. 2012;44(7):760–764. | |
Biankin AV, Waddell N, Kassahn KS, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491(7424):399–405. | |
Wilkins C, Woodward J, Lau DTY, et al. IFITM1 is a tight junction protein that inhibits hepatitis C virus entry. Hepatology. 2013; 57(2):461–469. | |
Iwahara J, Clubb RT. Solution structure of the DNA binding domain from Dead ringer, a sequence-specific AT-rich interaction domain (ARID). Embo J. 1999;18(21):6084–6094. | |
Webb CF, Smith EA, Medina KL, Buchanan KL, Smithson G, Dou SS. Expression of bright at two distinct stages of B lymphocyte development. J Immunol. 1998;160(10):4747–4754. | |
Ma KW, Araki K, Ichwan SJA, Suganuma T, Tamamori-Adachi M, Ikeda MA. E2FBP1/DRIL1, an AT-rich interaction domain-family transcription factor, is regulated by p53. Mol Cancer Res. 2003; 1(6):438–444. | |
Lestari W, Ichwan SJA, Otsu M, et al. Cooperation between ARID3A and p53 in the transcriptional activation of p21WAF1 in response to DNA damage. Biochem Biophys Res Commun. 2012;417(2):710–716. | |
Peeper DS, Shvarts A, Brummelkamp T, et al. A functional screen identifies hDRIL1 as an oncogene that rescues RAS-induced senescence. Nat Cell Biol. 2002;4(2):148–153. | |
Fukuyo Y, Mogi K, Tsunematsu Y, Nakajima T. E2FBP1/hDril1 modulates cell growth through downregulation of promyelocytic leukemia bodies. Cell Death Differ. 2004;11(7):747–759. | |
Fukuyo Y, Takahashi A, Hara E, Horikoshi N, Pandita TK, Nakajima T. E2FBP1 antagonizes the p16(INK4A)-Rb tumor suppressor machinery for growth suppression and cellular senescence by regulating promyelocytic leukemia protein stability. Int J Oral Sci. 2011;3(4):200–208. | |
An GY, Miner CA, Nixon JC, et al. Loss of bright/ARID3a function promotes developmental plasticity. Stem Cells. 2010;28(9):1560–1567. | |
Puissegur MP, Eichner R, Quelen C, et al. B-cell regulator of immunoglobulin heavy-chain transcription (Bright)/ARID3a is a direct target of the oncomir microRNA-125b in progenitor B-cells. Leukemia. 2012;26(10):2224–2232. | |
Numata S, Claudio PP, Dean C, Giordano A, Croce CM. Bdp, a new member of a family of DNA-binding proteins, associates with the retinoblastoma gene product. Cancer Res. 1999;59(15):3741–3747. | |
Dahl KDC, Dahl R, Kruichak JN, Hudson LG. The epidermal growth factor receptor responsive miR-125a represses mesenchymal morphology in ovarian cancer cells. Neoplasia. 2009;11(11):U1208–U1124. | |
Akhavantabasi S, Sapmaz A, Tuna S, Erson-Bensan AE. miR-125b targets ARID3B in breast cancer cells. Cell Struct Funct. 2012;37(1):27–38. | |
Kobayashi K, Jakt LM, Nishikawa SI. Epigenetic regulation of the neuroblastoma genes, Arid3b and Mycn. Oncogene. 2013;32(21):2640–2648. | |
Joseph S, Deneke VE, Dahl KDC. ARID3B induces tumor necrosis factor alpha mediated apoptosis while a novel ARID3B splice form does not induce cell death. PLoS One. 2012;7(7). | |
Kim D, Tucker PW. A regulated nucleocytoplasmic shuttle contributes to Bright’s function as a transcriptional activator of immunoglobulin genes. Mol Cell Biol. 2006;26(6):2187–2201. | |
Tidwell JA, Schmidt C, Heaton P, Wilson V, Tucker PW. Characterization of a new ARID family transcription factor (Brightlike/ARID3C) that co-activates Bright/ARID3A-mediated immunoglobulin gene transcription. Mol Immunol. 2011;49(1–2):260–272. | |
Gong WB, Zhou T, Mo JJ, Perrett S, Wang JF, Feng YG. Structural insight into recognition of methylated histone tails by retinoblastoma-binding protein 1. J Biol Chem. 2012;287(11):8531–8540. | |
Lai A, Kennedy BK, Barbie DA, et al. RBP1 recruits the mSIN3-histone deacetylase complex to the pocket of retinoblastoma tumor suppressor family proteins found in limited discrete regions of the nucleus at growth arrest. Mol Cell Biol. 2001;21(8):2918–2932. | |
Meehan WJ, Samant RS, Hopper JE, et al. Breast cancer metastasis suppressor 1 (BRMS1) forms complexes with retinoblastoma-binding protein 1 (RBP1) and the mSin3 histone deacetylase complex and represses transcription. J Biol Chem. 2004;279(2):1562–1569. | |
Cao J, Gao T, Giuliano AE, Irie RF. Recognition of an epitope of a breast cancer antigen by human antibody. Breast Cancer Res Treat. 1999;53(3):279–290. | |
Wu MY, Eldin KW, Beaudet AL. Identification of chromatin remodeling genes Arid4a and Arid4b as leukemia suppressor genes. J Natl Cancer Inst. 2008;100(17):1247–1259. | |
Cao JN, Gao TW, Stanbridge EJ, Irie R. RBP1L1, a retinoblastoma-binding protein-related gene encoding an antigenic epitope abundantly expressed in human carcinomas and normal testis. J Natl Cancer Inst. 2001;93(15):1159–1165. | |
Winter SF, Lukes L, Walker RC, Welch DR, Hunter KW. Allelic variation and differential expression of the mSIN3A histone deacetylase complex gene Arid4b promote mammary tumor growth and metastasis. PLoS Genetics. 2012;8(5). | |
Cui DX, Jin GQ, Gao TW, et al. Characterization of BRCAA1 and its novel antigen epitope identification. Cancer Epidem Biomar. 2004;13(7):1136–1145. | |
Huang TH, Oka T, Asai T, et al. Repression by a differentiation-specific factor of the human cytomegalovirus enhancer. Nucleic Acids Res. 1996;24(9):1695–1701. | |
Masuda K, Ripley B, Nishimura R, et al. Arid5a controls IL-6 mRNA stability, which contributes to elevation of IL-6 level in vivo. Proc Natl Acad Sci U S A. 2013;110(23):9409–9414. | |
Masuda K, Ripley B, Kishimoto T. ARID5a is an IL-6 mRNA stability protein. Chlorpromazine mediates its inhibitory effect on IL-6 production in macrophages through inhibition of ARID5A expression. Cytokine. 2012;59(3):544–544. | |
Joshi P, Greco TM, Guise AJ, et al. The functional interactome landscape of the human histone deacetylase family. Mol Syst Biol. 2013;9:672. | |
Xu H, Cheng C, Devidas M, et al. ARID5B genetic polymorphisms contribute to racial disparities in the incidence and treatment outcome of childhood acute lymphoblastic leukemia. J Clin Oncol. 2012;30(7):751–757. | |
Rudant J, Orsi L, Bonaventure A, et al. Are ARID5B and IKZF1 polymorphisms also associated with childhood acute myeloblastic leukemia: the ESCALE study (SFCE)? Leukemia. 2013;27(3):746–748. | |
Linabery AM, Blommer CN, Spector LG, Davies SM, Robison LL, Ross JA. ARID5B and IKZF1 variants, selected demographic factors, and childhood acute lymphoblastic leukemia: a report from the Children’s Oncology Group. Leuk Res. 2013;37(8):936–942. | |
Peyrouze P, Guihard S, Grardel N, et al. Genetic polymorphisms in ARID5B, CEBPE, IKZF1 and CDKN2A in relation with risk of acute lymphoblastic leukaemia in adults: a Group for Research on Adult Acute Lymphoblastic Leukaemia (GRAALL) study. Br J Haematol. 159(5):599–602. | |
Wang Y, Chen J, Li J, et al. Association of three polymorphisms in ARID5B, IKZF1 and CEBPE with the risk of childhood acute lymphoblastic leukemia in a Chinese population. Gene. 2013;524(2):203–207. | |
Yang W, Trevino LR, Yang JJ, et al. ARID5B SNP rs10821936 is associated with risk of childhood acute lymphoblastic leukemia in blacks and contributes to racial differences in leukemia incidence. Leukemia. 2010;24(4):894–896. | |
Allis CD, Berger SL, Cote J, et al. New nomenclature for chromatin-modifying enzymes. Cell. 2007;131(4):633–636. | |
Takeuchi T, Watanabe Y, Takano-Shimizu T, Kondo S. Roles of jumonji and jumonji family genes in chromatin regulation and development. Dev Dyn. 2006;235(9):2449–2459. | |
Shi XB, Hong T, Walter KL, et al. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature. 2006; 442(7098):96–99. | |
Iwase S, Lan F, Bayliss P, et al. The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell. 2007;128(6):1077–1088. | |
Huang F, Chandrasekharan MB, Chen YC, Bhaskara S, Hiebert SW, Sun ZW. The JmjN domain of Jhd2 is important for its protein stability, and the plant homeodomain (PHD) finger mediates its chromatin association independent of H3K4 methylation. J Biol Chem. 2010;285(32):24548–24561. | |
Benevolenskaya EV, Murray HL, Branton P, Young RA, Kaelin WG Jr. Binding of pRB to the PHD protein RBP2 promotes cellular differentiation. Mol Cell. 2005;18(6):623–635. | |
Liefke R, Oswald F, Alvarado C, et al. Histone demethylase KDM5A is an integral part of the core Notch-RBP-J repressor complex. Genes Dev. 2010;24(6):590–601. | |
Christensen J, Agger K, Cloos PAC, et al. RBP2 belongs to a family of demethylases, specific for tri- and dimethylated lysine 4 on histone 3. Cell. 2007;128(6):1063–1076. | |
Chicas A, Kapoor A, Wang X, et al. H3K4 demethylation by Jarid1a and Jarid1b contributes to retinoblastoma-mediated gene silencing during cellular senescence. Proc Natl Acad Sci U S A. 2012;109(23):8971–8976. | |
Ge Z, Li WJ, Wang N, et al. Chromatin remodeling: recruitment of histone demethylase RBP2 by Mad1 for transcriptional repression of a Myc target gene, telomerase reverse transcriptase. Faseb J. 2010;24(2):579–586. | |
Wang GG, Song JK, Wang ZX, et al. Haematopoietic malignancies caused by dysregulation of a chromatin-binding PHD finger. Nature. 2009;459(7248):U847–U846. | |
Lin W, Cao J, Liu J, et al. Loss of the retinoblastoma binding protein 2 (RBP2) histone demethylase suppresses tumorigenesis in mice lacking Rb1 or Men1. Proc Natl Acad Sci U S A. 2011;108(33):13379–13386. | |
van Zutven LJCM, Onen E, Velthuizen SCJM, et al. Identification of NUP98 abnormalities in acute leukemia: JARID1A (12p13) as a new partner gene. Genes Chromosomes Cancer. 2006;45(5):437–446. | |
Sharma SV, Lee DY, Li BH, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141(1):69–80. | |
Yan H, Chen X, Zhang QP, et al. Drug-tolerant cancer cells show reduced tumor-initiating capacity: depletion of CD44(+) cells and evidence for epigenetic mechanisms. PLoS One. 2011;6(9):e24397. | |
Roesch A, Becker B, Meyer S, et al. Retinoblastoma-binding protein 2-homolog 1: a retinoblastoma-binding protein downregulated in malignant melanomas. Mod Pathol. 2005;18(9):1249–1257. | |
Lu PJ, Sundquist K, Baeckstrom D, et al. A novel gene (PLU-1) containing highly conserved putative DNA chromatin binding motifs is specifically up-regulated in breast cancer. J Biol Chem. 1999;274(22):15633–15645. | |
Roesch A, Becker B, Meyer S, et al. Retinoblastoma-binding protein 2-homolog 1: a retinoblastoma-binding protein downregulated in malignant melanomas. Mod Pathol. 2005;18(9):1249–1257. | |
Hayami S, Yoshimatsu M, Veerakumarasivam A, et al. Overexpression of the JmjC histone demethylase KDM5B in human carcinogenesis: involvement in the proliferation of cancer cells through the E2F/RB pathway. Mol Cancer. 2010;9:5. | |
Xiang Y, Zhu Z, Han G, et al. JARID1B is a histone H3 lysine 4 demethylase up-regulated in prostate cancer. Proc Natl Acad Sci U S A. 2007;104(49):19226–19231. | |
Sun L-L, Sun X-X, Xu X-E, et al. Overexpression of Jumonji AT-rich interactive domain 1B and PHD finger protein 2 is involved in the progression of esophageal squamous cell carcinoma. Acta Histochemica. 2013;115(1):56–62. | |
Yamane K, Tateishi K, Klose RJ, et al. PLU-1 is an H3K4 dernethylase involved in transcriptional repression and breast cancer cell proliferation. Mol Cell. 2007;25(6):801–812. | |
Scibetta AG, Santangelo S, Coleman J, et al. Functional analysis of the transcription repressor PLU-1/JARID1B. Mol Cell Biol 2007; 27(20):7220–7235. | |
Li Q, Shi L, Gui B, et al. Binding of the JmjC demethylase JARID1B to LSD1/NuRD suppresses angiogenesis and metastasis in breast cancer cells by repressing chemokine CCL14. Cancer Res. 2011;71(21):6899–6908. | |
Catchpole S, Spencer-Dene B, Hall D, et al. PLU-1/JARID1B/KDM5B is required for embryonic survival and contributes to cell proliferation in the mammary gland and in ER+ breast cancer cells. Int J Oncol. 2011;38(5):1267–1277. | |
Nijwening JH, Geutjes EJ, Bernards R, Beijersbergen RL. The histone demethylase jarid1b (Kdm5b) is a novel component of the Rb pathway and associates with E2f-Target genes in MEFs during senescence. PLoS One. 2011;6(9). | |
Ohta K, Haraguchi N, Kano Y, et al. Depletion of JARID1B induces cellular senescence in human colorectal cancer. Int J Oncol. 2013;42(4):1212–1218. | |
Roesch A, Becker B, Schneider-Brachert W, Hagen I, Landthaler M, Vogt T. Re-expression of the retinoblastoma-binding protein 2-homolog 1 reveals tumor-suppressive functions in highly metastatic melanoma cells. J Invest Dermatol. 2006;126(8):1850–1859. | |
Roesch A, Mueller AM, Sterapfl T, Moehle C, Landthaler M, Vogt T. RBP2-H1/JARID1B is a transcriptional regulator with a tumor suppressive potential in melanoma cells. Int J Cancer. 2008;122(5):1047–1057. | |
Roesch A, Fukunaga-Kalabis M, Schmidt EC, et al. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell. 2010;141(4):583–594. | |
Jensen LR, Amende M, Gurok U, et al. Mutations in the JARID1C gene, which is involved in transcriptional regulation and chromatin remodeling, cause X-linked mental retardation. Am J Hum Genet. 2005;76(2):227–236. | |
Abidi FE, Holloway L, Moore CA, et al. Mutations in JARID1C are associated with X-linked mental retardation, short stature and hyperreflexia. J Med Genet. 2008;45(12):787–793. | |
Santos C, Laia RR, Madrigal I, Badenas C, Pineda M, Mila M. A novel mutation in JARID1C gene associated with mental retardation. Eur J Hum Genet. 2006;14(5):583–586. | |
Kim TD, Shin S, Janknecht R. Repression of Smad3 activity by histone demethylase SMCX/JARID1C. Biochem Biophys Res Commun. 2008;366(2):563–567. | |
Dalgliesh GL, Furge K, Greenman C, et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010;463(7279):360–363. | |
Niu X, Zhang T, Liao L, et al. The von Hippel-Lindau tumor suppressor protein regulates gene expression and tumor growth through histone demethylase JARID1C. Oncogene. 2012;31(6):776–786. | |
Wang W, Meadows LR, Denhaan JMM, et al. Human H-Y – a male-specific histocompatibility antigen derived from the smcy protein. Science. 1995;269(5230):1588–1590. | |
Lau YFC, Zhang JQ. Expression analysis of thirty one Y chromosome genes in human prostate cancer. Mol Carcinogen. 2000;27(4):308–321. | |
Patsialou A, Wilsker D, Moran E. DNA-binding properties of ARID family proteins. Nucleic Acids Res. 2005;33(1):66–80. | |
Shirato H, Ogawa S, Nakajima K, et al. A jumonji (Jarid2) protein complex represses cyclin D1 expression by methylation of histone H3-K9. J Biol Chem. 2009;284(2):733–739. | |
Toyoda M, Kojima M, Takeuchi T. Jumonji is a nuclear protein that participates in the negative regulation of cell growth. Biochem Biophys Res Commun. 2000;274(2):332–336. | |
Puda A, Milosevic JD, Berg T, et al. Frequent deletions of JARID2 in leukemic transformation of chronic myeloid malignancies. Am J Hematol. 2012;87(3):245–250. | |
Bolisetty MT, Dy G, Tam W, Beemon KL. Reticuloendotheliosis virus strain T induces miR-155, which targets JARID2 and promotes cell survival. J Virol. 2009;83(23):12009–12017. | |
Kortschak RD, Tucker PW, Saint R. ARID proteins come in from the desert. Trends Biochem Sci. 2000;25(6):294–299. | |
Jiao Y, Pawlik TM, Anders RA, et al. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat Genet. 2013;45(12):1470–1473. | |
Wu RC, Jiang M, Beaudet AL, Wu MY. ARID4A and ARID4B regulate male fertility, a functional link to the AR and RB pathways. Proc Natl Acad Sci U S A. 2013;110(12):4616–4621. | |
Sayegh J, Cao J, Zou MR, et al. Identification of small molecule inhibitors of jumonji AT-rich interactive domain 1B (JARID1B) histone demethylase by a sensitive high throughput screen. J Biol Chem. 2013;288(13):9408–9417. | |
Jones S, Wang T-L, Shih I-M, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010;330(6001):228–231. | |
Wiegand KC, Shah SP, Al-Agha OM, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med. 2010;363(16):1532–1543. | |
Zang ZJ, Cutcutache I, Poon SL, et al. Exome sequencing of gastric adenocarcinoma identifies recurrent somatic mutations in cell adhesion and chromatin remodeling genes. Nat Genet. May 2012; 44(5):570–574. | |
Wang K, Kan J, Yuen ST, et al. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat Genet. 2011;43(12):1219–1223. | |
Jones S, Li M, Parsons DW, et al. Somatic mutations in the chromatin remodeling gene ARID1A occur in several tumor types. Hum Mutat. 2012;33(1):100–103. | |
Letunic I, Doerks T, Bork P. SMART 7: recent updates to the protein domain annotation resource. Nucleic Acids Res. 2012;40(Database issue):D302–D305. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.