")
Back to Journals » OncoTargets and Therapy » Volume 14
Recent Advances in Our Knowledge of mCRC Tumor Biology and Genetics: A Focus on Targeted Therapy Development
Authors Gmeiner WH
Received 12 January 2021
Accepted for publication 11 March 2021
Published 25 March 2021 Volume 2021:14 Pages 2121—2130
DOI https://doi.org/10.2147/OTT.S242224
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Alberto Bongiovanni
William H Gmeiner
Department of Cancer Biology, Wake Forest School of Medicine, Winston-Salem, NC, USA
Correspondence: William H Gmeiner Tel +1 336-716-6216
Fax +1 336-716-0255
Email [email protected]
Abstract: Metastatic colorectal cancer (mCRC) remains a highly lethal malignancy although considerable progress has resulted from characterizing molecular alterations such as RAS mutation status and extent of microsatellite instability (MSI) to guide optimal use of available therapies. The availability of gene expression profiling, next generation sequencing technologies, proteomics analysis and other technologies provides high resolution information on individual tumors, including metastatic lesions to better define intra-tumor and inter-tumor heterogeneity. Recent literature applying this information to further customize personalized therapies is reviewed. Current biomarker-based stratification used to select optimal therapy that is personalized to the mutation profile of individual tumors is described. Recent literature using whole exome sequencing of metastatic lesions and primary CRC tumors and other advanced technologies to more fully elucidate the tumor biology specific to mCRC sub-types and to develop more precise therapies that improve outcomes is also reviewed.
Keywords: colorectal cancer, metastasis, mutation, whole exome sequencing, targeted therapy
Introduction
Colorectal cancer (CRC) is the second leading cause of cancer-related mortality in men and women combined.1 Patients with metastatic CRC (mCRC) have an extremely poor 5-year survival rate, < 14%.2 While overall prognosis remains dismal, progress in more specific targeting of therapy to individual tumor profiles has contributed to significantly improve overall survival for mCRC patients, which now exceeds 30 months.3 Many patients now survive multiple treatments, and treatment with third-line therapy options is increasingly common. Despite these advances, further progress depends on: i) even more detailed delineation of molecular mCRC sub-types that are responsive to specific, targeted therapies; and ii) identifying mutations and other molecular alterations that contraindicate use of established therapies, and developing more effective alternative strategies. Frontline treatment for mCRC continues to rely on fluoropyrimidine (FP)-based chemotherapy that was first shown to provide a survival benefit for post-operative adjuvant chemotherapy in 1988,4 and later shown to improve survival in mCRC.5 While still heavily relied upon, FPs are used to treat >2 million cancer patients world-wide, FP-based doublet (5-fluorouracil (5-FU)/leucovorin (LV)) and triplet (eg, FOLFOX) chemotherapy for CRC is now refined and supplemented by anti-EGFR or anti-VEGF therapy that is based on tumor profiling.6 An exciting recent development has been the use of microsatellite instability (MSI) profiling to identify patients likely to benefit from immunotherapy.7 MSI profiling in CRC was initially implemented to identify patients who were unlikely to benefit from FP-based chemotherapy.8
The availability of comprehensive mutational profile information from whole exome sequencing (WES)9 of tumor samples holds great promise for increasing the precision of therapy for mCRC by identifying mutations associated with metastatic progression. Intriguingly, recent studies employing WES,10 and related technologies,11 have validated the stepwise continuum model of CRC initiation and progression initially put forth by Vogelstein 30 years ago,12 but with nuances that enable refined therapeutic approaches. Importantly, these studies validate that in most instances metastasis is a relatively late event, which largely occurs with preservation of the driver oncogenic profile of the primary tumor indicating that biopsy samples from the primary tumor may be used for therapeutic planning.
CRC evolves according to three main pathways: 1) chromosomal instability (CIN); 2) microsatellite instability (MSI); and 3) CPG island mutator phenotype (CIMP).13 The classic sequential mutational progression described by Vogelstein12 and extended thru detailed multi-omics profiling of CRC tumors10,11 is applicable primarily to the CIN pathway, which constitutes the majority of CRC (>70%), and the overwhelming majority of mCRC (95%).7 Alternatively, CRC can develop due to genetic or epigenetic changes affecting DNA repair capacity. The MSI pathway results from mutations in microsatellites that disable DNA mismatch repair (MMR), resulting in hypermutability.8 In contrast, the CIMP pathway involves aberrant hypermethylation of CpG sequences in promoter regions of genes involved in DNA repair and oncogenesis, and this results in tumor progression following initial mutation in either KRAS or BRAF.14 In general, prognosis and survival for MSI and CIMP CRC is more favorable than for CIN-positive CRC.15
In this review, biomarker-based stratification currently used to select the best available treatment for mCRC patients is first reviewed (Figure 1). Recent developments in tumor biology using WES, proteomics, and other advanced technologies that promise to modify current therapeutic paradigms (Figure 2) and improve outcomes for mCRC patients is then reviewed.
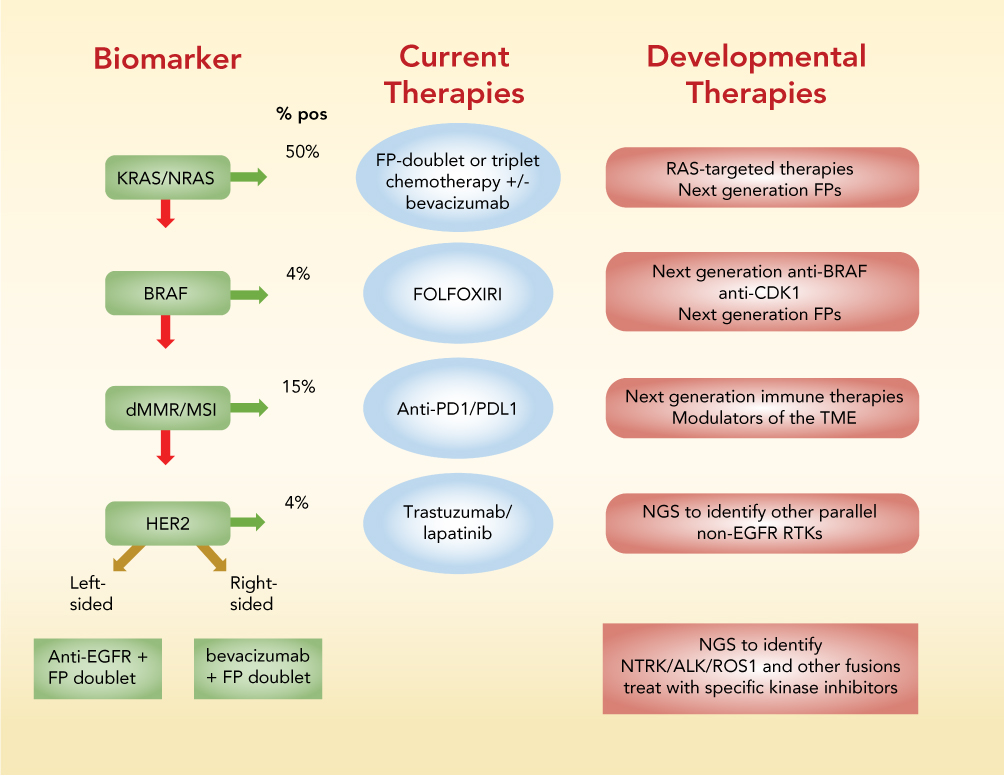 |
Figure 1 Summary of biomarker-based stratification to identify optimal front-line treatment for mCRC patients. Screening for KRAS/NRAS and BRAF activating mutations and for high MSI and deficiencies in MMR is recommended by the NCCN prior to treatment. %-positive indicates percent of patients that test positive for the biomarker indicated. Abbreviations: FP, fluoropyrimidine; FOLFOXFIRI, folinic acid, 5-fluorouracil, oxaliplatin, irinotecan; MMR, mismatch repair; MSI, microsatellite instability; NGS, next generation sequencing; PD1, programmed cell death 1; PDL1, programmed death ligand 1; RTK, receptor tyrosine kinase. |
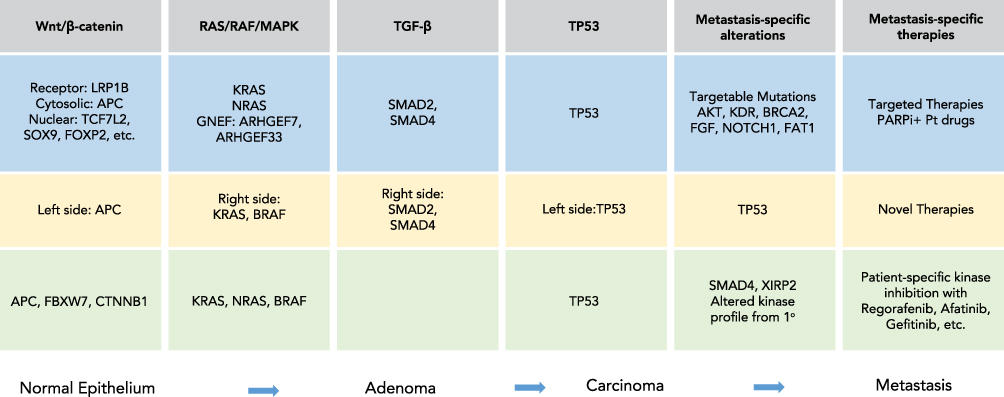 |
Figure 2 Summary of mutations in the Wnt/β-catenin, RAS/RAF/MAPK, TGF-β, and TP53 pathways that occur during the course of CRC tumor progression from normal epithelium to adenoma to carcinoma and to metastasis are listed. Mutations in light blue were reported by Ishaque et al10, light yellow from Yaeger et al11 and light green from Li et al76. Specific metastasis-specific alterations identified in each study are highlighted and potential new therapies for mCRC are indicated. Abbreviations: PARPi, poly ADP ribose polymerase inhibitor; Pt, platinum drugs. |
Current Biomarker-Based Stratification of mCRC
As with other malignancies, such as lung cancer,16 there is increasing emphasis on identifying key oncogenic drivers that define molecular sub-types of metastatic colorectal cancer (mCRC) and in developing effective targeted therapies tailored to the oncogenic drivers.6,17 Epidermal growth factor receptor (EGFR) plays an important role in the normal function of gastrointestinal tissue and a prominent role in the development of neoplasia in colonic epithelium. EGFR regulates proliferation and migration in the intestinal epithelium, which is a highly dynamic tissue.18 EGFR overexpression occurs frequently in CRC, with 60–80% of tumors estimated to overexpress the receptor,19 and overexpression is associated with a poor prognosis. Antibodies targeting EGFR (eg, cetuximab and panitumumab) are a preferred front-line treatment for mCRC,20 provided tumor profiling for biomarkers associated with innate resistance to anti-EGFR therapy does not contraindicate their use.21 However, biomarker screening may yield ambiguous results with tumor heterogeneity a confounding factor.22 Further, acquired resistance to anti-EGFR therapy is also a serious concern.23
EGFR activates multiple signaling pathways24 including RAS-RAF-MAPK and PI3K-Akt to execute its oncogenic function. Activating mutations in these downstream signaling pathways negate the efficacy of anti-EGFR therapy necessitating screening to identify patients who are unlikely to respond to expensive therapy.17 The use of an extended RAS panel that screens not only for the >40% of CRC with activating KRAS mutations in exon 2, but also for mutations in exons 3/4 of KRAS and exons 1/2/3/4 of NRAS (~20% of patients without exon 2 KRAS mutations),25 more broadly detects RAS-related resistance to anti-EGFR therapies,26 and is recommended by the National Comprehensive Cancer Network. In principle, the activating mutations in KRAS that preclude use of anti-EGFR therapy in mCRC could be targeted by RAS-specific agents including AMG510, which targets KRASG12C. While early results with AMG510 and MRTX849 are promising in NSCLC,27 limited response was detected in mCRC.28 Lack of response in mCRC was found due to re-activation of EGFR signaling, and combining AMG510 and anti-EGFR therapy was effective in pre-clinical mCRC models.28 In addition to promoting cell proliferation independent of EGFR activation, the KRASG12D activating mutation promotes an immunosuppressive profile in CRC,29 and is an important modulator of immunotherapy in CRC.29
The BRAFV600E activating mutation also is associated with innate30 and acquired31 resistance to anti-EGFR therapy and poor prognosis overall (OS ~18.2 months vs >38.4 months in RAS/BRAF-WT).32 The detection of BRAF mutations at diagnosis is important because it may stimulate treatment with more aggressive chemotherapy, such as FOLFOXIRI.33 However, targeted anti-BRAFV600E therapy with vemurafenib, dabrafenib, or encorafenib is ineffective in mCRC,34 in part due to increased EGFR signaling thru the MEK/ERK pathway.35 While dual inhibition of BRAF and MEK was effective in melanoma, it was not in mCRC. This resulted in studies showing elevated CDK1 caused apoptosis suppression in BRAF-mutant CRC. CDK1 inhibitors, including R0-3306 and the non-selective CDK inhibitor dinaciclib,36 are being evaluated in pre-clinical studies for treatment of BRAF-mutant CRC. CDK inhibitors show promising activity in CRC with combined targeting of CDK1, 2, and 9 considered the most effective strategy.37
Activation of the PI3K/Akt pathway, which is frequently mutated in CRC with enrichment in mCRC,10 also confers resistance to anti-EGFR therapy and to first-line chemotherapy.38 While multiple clinical trials have evaluated Akt inhibition, Akt-targeted therapies directed in general have not been effective for mCRC treatment.39 PI3K/mTOR dual inhibitors such as BEZ235 and LY3023414, however, display promising activity in pre-clinical models,40 and MTORC1/2 inhibitors such as TAK-228 also show promising activity targeting this pathway.41 Interestingly, MTORC2 activity was not detected in primary human and mouse CRC samples, but was found to be expressed in macrophages where it contributed to anti-tumor activity.42
A number of factors other than KRAS/NRAS and BRAF mutations influence the efficacy of anti-EGFR therapy, including plasma levels of EGFR ligands such as amphiregulin (AREG) and epiregulin (EREG),43 and these may be monitored to provide an indication of resistance to anti-EGFR therapy.24 EGFR gene copy number and acquired mutations in extracellular domains also may contribute anti-EGFR therapy resistance in mCRC. Mutations in Her2, FGFR1, PDGFRA, and MAP2K1 also are associated with acquired resistance to anti-EGFR therapy.44 Resistance to anti-EGFR therapy is associated with gene fusions in actionable targets including ALK (eg, ceritinib) and NTRK1 (eg, larotrectinib), and these may be detected through sequencing of circulating tumor DNA (ctDNA).45 ALK fusions are characteristic of a rare sub-type of mCRC that is highly aggressive, but responsive to ALK inhibitors.46 Other mutations associated with innate resistance to anti-EGFR therapy include HER2 and MET amplification.23 HER2 amplification occurs in 3–5% of mCRC patients47 and promising results have been obtained with anti-HER2 antibody trastuzumab in combination with either the EGFR/HER2 kinase inhibitor lapatinib48 or pertuzumab,49 an antibody that inhibits HER2 dimerization.
MMR Effects on Chemo- and Immunotherapy
Deficiencies in DNA mismatch repair (dMMR) and the occurrence of high microsatellite instability (MSI) occur in about 15% of CRC patients, and the MSI pathway is considered a distinct pathway of oncogenic progression from the more common chromosomal instability (CIN) pathway.50 Approximately 20% of cases with deficiencies in MMR result from germline mutations associated with Lynch syndrome,51 and the remainder are due to sporadic mutations, or frequently epigenetic silencing of MLH1 (CIMP). dMMR/MSI is more common in right-sided tumors and is associated with poor differentiation and increased lymphocyte infiltration.52 Screening for dMMR/MSI has been routinely employed for more than a decade because clinical data indicated dMMR/MSI was associated with lack of response to FP chemotherapy. High MSI is associated with more favorable outcomes for stage II patients, however for patients with mCRC high MSI is associated with worse survival.53 Variable expression of a mutant form of HSP110 (HSP110∆E9) is implicated in differential therapy response in MSI tumors54 and detection of the mutant, while technically challenging, may be beneficial for MSI status determination in ambiguous cases,55 and as a potential therapeutic target.56
A potential explanation for the relationship of MSI with lack of response to 5-FU chemotherapy is that MSI is also associated with elevated thymidylate synthase (TS),57,58 which is the molecular target of FP chemotherapy.59 Elevated TS may also contribute to a pro-metastatic phenotype,60,61 and its increased nuclear localization is associated with poor outcomes in CRC.62 CRC tumors responding to 5-FU chemotherapy display low expression levels of TS (TYMS), thymidine phosphorylase (TYMP), and dihydropyrimidine dehydrogenase (DPYD).63 Germline polymorphisms in DPYD significantly affect 5-FU degradation, and approximately 5% of CRC patients are highly vulnerable to 5-FU-induced systemic toxicities which can be life threatening.64 DPYD testing prior to FP treatment is considered essential in Europe,65 but not yet in the US. Polymeric FPs66,67 are being developed that may overcome limitations of current FPs resulting from elevated TS, altered DPYD, and other factors.
Encouraging results have been obtained using immune checkpoint blockade to treat dMMR/MSI CRC,7 and positive results have largely been restricted to this sub-type.68 Pembrolizumab, which targets PD1, is now used as front-line treatment for mCRC, while Nivolumab (anti-PD1), alone or in combination with ipilimumab (anti-CTLA-4), is used to treat mCRC that has progressed with chemotherapy. The most important indicators of response to immunotherapy with anti-PD1/PDL1 immune checkpoint blockade is dMMR/MSI status, and screening is routinely implemented prior to therapy. The much higher mutation load in MSI vs MSS disease (1782 vs 73 somatic mutations per tumor) results in cancer-specific antigen presentation and increased lymphocyte infiltration into tumors.69 A number of clinical trials are ongoing to extend the benefits of immunotherapy to MSS or MMR-proficient (MMR-p) CRC, which constitutes 95% of mCRC, and these are predominantly in Phase II and being evaluated as second line therapies.7
While dMMR/MSI status is an important indicator of response to immunotherapy, a number of efforts are ongoing to better define factors affecting response to immune checkpoint blockade and to develop therapies directed at alternative targets. Retrospective analysis of immune, fibroblastic, and angiogenic microenvironment of 1388 CRC tumors from transcriptomic analysis resulted in classification into four subgroups that were differentiated with respect to outcomes based on expression of genes associated with immune populations and tumor progression or an anti-tumor immune response.70 The lack of utility for existing CRC immune signatures for extending the benefits of immunotherapy beyond MSI resulted in development of the gastrointestinal immune signature (GAIS-42), that identified four immune-metabolic clusters (IM Cluster 1–4) that differed in expression of cancer-associated fibroblasts (CAFs), myeloid derived suppressor cells (MDSCs), and other immune cell populations together with BRAF and RAS mutations, and metabolic profiling to distinguish tumors and to identify potential therapeutic targets. The authors note the potential for drugs to reprogram tumor metabolism such as pyruvate dehydrogenase kinase 1, or inhibitors of lactate transporters, to increase the capacity of the adaptive immune system to mount an anti-tumor response.71
Defining the Genomic Landscape That Promotes Metastatic Progression
The power of next generation sequencing has been applied by multiple studies in the last few years to characterize oncogenic drivers of metastasis and reveal new therapeutic targets for mCRC and other cancers.72 In general, sequencing studies have demonstrated strong concordance between primary tumors and metastatic lesions with regard to key driver oncogenic mutations.73 Divergence, when it occurs, is generally consistent with it being a late event in cancer progression, which is consistent with the Fearon-Vogelstein model,12 which has recently been updated in a combined approach that also uses mathematical modeling, epidemiological studies, and sequencing data.74 While sequencing studies confirm the validity of the “Vogelstein sequence” of CRC progression with sequential mutations in the Wnt-, Ras-, TGF-β, and TP53 pathways (Figure 2), additional components of these pathways that are vulnerable to activating mutations are still being discovered and as a result classification models are being refined to more accurately predict susceptibility to targeted therapeutic interventions.75 Interestingly, while metastases in general display good concordance with the primary tumor from which they were derived, significant differences are apparent at the proteomic level, indicating multi-omic analysis may be required to more fully understand metastatic progression and optimize treatments.76 Sequencing and multi-omic studies complement differential gene expression analysis that resulted in identification of consensus molecular sub-types (CMS) of CRC based on expression data,77,78 and demonstrated utility in explaining therapeutic response,79 and metastatic progression.80
To identify the genomic determinants of mCRC, Ishaque et al10 performed whole genome sequencing of metastatic lesions vs corresponding primary tumors and normal samples in 12 mCRC patients.10 In general there was concordance between primary and metastases with 65% of all single nucleotide variants (SNVs) shared between primary tumors and metastatic lesions, although their data suggest a higher rate of mutation in metastases relative to matched tumor after truncal separation. Mutations in protein coding regions identified well-established signaling pathways implicated in CRC progression including Wnt/β-catenin, RAS, and TGF-β being important for promoting progression from normal epithelium to adenoma. Interestingly, recurrent mutations were also detected in ARHGEF33, a guanine nucleotide exchange factor, with similar frequency as for KRAS (25% vs 29%), implicating it as an alternative mediator of RAS signaling and a potential modulator of EGFR-targeted therapies. Forty-eight genes were found to be mutated in metastases relative to primary tumors and data supported, in general, a late-stage mutagenic process contributing to metastatic progression. Functional annotation of metastasis-specific genes identified extracellular matrix, PI3K-Akt signaling, and focal adhesion-related pathways being dysregulated during metastatic progression. The authors developed an extended progression model of CRC that includes development of metastasis. In this model, potentially targetable mutations in AKT, BRCA2, and NOTCH1 were enriched in metastatic lesions and, based on DNA repair deficiency mutational signatures in metastases, the authors suggested use of PARP inhibitors and platinum-based therapy for treating mCRC. Hepatic fibrosis/stellate genes were also significantly enriched in metastases consistent with metastasized cells activating pathways that foster organ-specific metastatic colonization.
Yaeger et al11 compared oncogenic genomic alterations in primary tumors from TCGA or from a proprietary cohort sequenced at Memorial Sloan Kettering (MSK) to metastases from patients with mCRC treated at MSK. Sequencing used a hybridization capture-based next generation sequencing assay81 for targeted sequencing of up to 468 genes at all exons and selected introns and was applied to >1100 patient samples. Forty-seven recurrently mutated genes were identified in the primary tumors with APC, TP53, KRAS, PIK3CA and SMAD4 the most frequently mutated genes. A series of potentially novel recurrently mutated genes including PTPRS, PIK3CG, FLT4, MAP2K4, IKZF1, JUN, TBX3, FOXP1, INHBA, and CDKN1B occurred in 1–4% of primary tumors. However, only TP53 mutations were selectively enriched in metastatic disease. Overall, the data indicated strong concordance between primary and metastatic CRC. The study noted significant differences in mutational burden between primary tumors originating in the right and left side of the colon with significant enrichment of KRAS, BRAF, PIK3CA, PTEN, AKT1, RNF43, SMAD2, and SMAD4 in right-sided primary tumors and of APC and TP53 in left-sided primary tumors. >3/4 of right-sided tumors had mutations affecting RAS-MAPK/PI3K pathway activation while the majority of left-sided primary tumors mitogenic pathway activation was limited to EGFR or other receptor tyrosine kinase alterations and survival was longest in such patients. Conversely, RAS mutations which predominated in right-sided primary tumors conferred poor survival and was associated with multiple sites of first metastasis. The authors conclude that the data provide a potential explanation for differential response to EGFR-targeted therapies based on tumor sidedness and suggest inhibitors of other RTKs, such as HER2, may display higher efficacy in left-sided tumors.
Multiple other studies have sought to identify metastasis-related genes affecting survival in colon cancer. For example, whole exome sequencing was performed by Siraj et al82 to identify mutations associated with CRC metastasis. Thirty paired primary CRC and distant metastases revealed high overall genomic concordance between primary CRC and metastasis consistent with use of a single diagnostic biopsy from the primary tumor to characterize genomic variation of metastases. Divergence between primary tumor and metastatic lesions was generally shown to be a late event supporting late dissemination consistent with the Fearon-Vogelstein multistage progression model. Wei et al83 identified differentially expressed genes between primary and metastasis colon cancer tissue. The researchers identified a six gene signature with independent prognostic significance of outcomes in colon cancer. The six genes included aldehyde dehydrogenase 2, neural precursor cell expressed, developmentally down-regulated 9, filamin A, lamin B receptor, twinfilin actin binding protein 1 and arginine-rich splicing factor 1.83 DEG analysis of TCGA data was also undertaken by Zhou et al in a study that identified REG1B, TGM6, NTF4, PNMA5, and HOXC13 as displaying prognostic significance with a potential role in CRC metastasis.84 Other studies investigating genetic alteration in mCRC were recently reviewed.85
While next generation sequencing approaches are providing detailed understanding of mCRC progression and identifying oncogenic driver mutations to guide therapeutic targeting, proteomic analysis has potential to provide an alternative insight into metastasis-specific characteristics that guide therapeutic intervention. Li et al undertook an integrated Omics analysis of mCRC from 146 CRC patients including 70 with metastases.76 Analysis of genomic alterations in mCRC relative to non-mCRC identified SMAD4 and XIRP2 as enriched in primary tumors from mCRC patients. The investigators further applied non-negative matrix factorization to identify single-base substitution (SBS) signatures as classified by Alexandrov et al86 and identified catalogue of somatic mutations in cancer (COSMIC) SBS 1 as having a higher contribution to mCRC than to non-mCRC. Genes enriched in SBS1 include HYDIN, C1QB, and COL22A1 which were previously implicated in CRC metastatic progression.87 Somatic copy number alterations (SCNAs) did not significantly differ between mCRC and non-mCRC.
Omics studies also revealed metastases are similar to the corresponding primary tumor at a genetic level although significant differences emerge upon analysis at a proteome and phosphoproteome levels.76 To determine if differential protein expression could distinguish mCRC from non-mCRC, consensus clustering of 2440 differentially expressed proteins was performed which resulted in identification of 3 consensus clusters. CC2, which included extra-cellular matrix (ECM) and immune-related pathways, and CC3, which included DNA replication and metabolic pathways, showed slight enrichment in mCRC. The investigators then performed phosphoproteomic profiling and identified sub-clusters in each consensus cluster and found that metastatic tumors had more upregulated proteins compared to non-metastatic tumors with proteins important for ECM-receptor interaction, drug metabolism, focal adhesion, and tight junctions upregulated in metastatic tissue. The investigators performed kinase-substrate enrichment analysis and found enrichment for distinct kinases in the different consensus clusters with metastases differing from primary tumors within the same cluster. The investigators went on to test three kinase inhibitors (afatinib, gefitinib, and regorafenib) in 31 mini-PDX models including nine primary-metastatic pairs. Studies revealed that metastatic tissue could respond differently relative matched primary to the same drug and that phosphoproteomic profiling could identify mCRC sub-types that were responsive to particular drugs.
To identify factors affecting the rate of metastasis development, Ottaiano et al88 performed genome profiling and immunohistochemical analysis in primary and matched metastatic tissues. The investigators separately considered patients with oligometastatic disease89 (omCRC) from the more common and more lethal poly-metastatic disease (pmCRC) based on 3 year follow up following detection of primary CRC with synchronous single liver metastasis. There was 91.7% genetic sharing between mutation profile in primary tumors and matched liver metastases. However, there was marked differences in preservation of mutations in key oncogenic drivers in patients with disease that did not progress, including loss of alterations in KRAS and SMAD4 in some patients, while gain of mutations in oncogenic drivers were detected in some patients that developed poly-metastatic disease. The investigators also analyzed differences in immunological microenvironment in matched primary tumor and metastatic disease. CD3+, CD8+, FoxP3+, and GrzB+ were quantified however significant differences related to tumor progression were found only for CD8+ cells at invasive margins of primary tumors and GrzB+ cells in tumor cores of metastatic lesions. The authors concluded that oligo-metastatic disease involves “back” mutations of key driver genes and relatively higher density of GrzB+ T cells while poly-metastatic disease was characterized by “forward” mutations in key oncogenic drivers and low density of GrzB+ T cells.
Discussion
CRC is a heterogeneous disease and considerable progress in extending survival for patients with mCRC has resulted from identification of key molecular driver mutations (eg, KRAS/NRAS/BRAF) and other factors (MSI) that characterize specific CRC molecular sub-types, screening for these in individual patients, and tailoring therapy based on these factors. Recent studies indicate that further progress in developing personalized therapies by implementing screening for alternative genetic aberrations that drive cancer progression are possible. For example detection of mutations in alternative receptor tyrosine kinases beyond EGFR and HER2 and additional fusions beyond ALK and NTRK prior to initiating treatment could further improve outcomes in mCRC. Also, identifying mutations that contraindicate therapy such as ARHGEF33 that similar to KRAS/NRAS activating mutations negates response to anti-EGFR therapy could improve outcomes.
Apart from identifying novel targets thru genomic and proteomic analysis, considerable effort is being dedicated to developing effective therapies targeted at well-established pathways in CRC progression that are currently undruggable. A challenge in developing targeted therapies for improved treatment of mCRC is that many of the oncogenes that are key to driving cancer progression and metastasis are not amenable to targeted therapy. For example, while Wnt90 signaling mutations are ubiquitous in CRC, because of its important role in normal physiology, targeting Wnt is problematic. Another example is Src that is differentially expressed in mCRC and thus of potential utility for mCRC treatment but, because of its important role in the immune system, is not readily targeted for mCRC treatment.91 Detailed analysis of how these pathways are dysregulated in CRC, particularly during metastatic progression, may enable selective targeting that could significantly improve survival.
Acknowledgments
This work was supported by NIH-NCI R21 CA218933, NIH-NCI P30 CA012197 and Wake Forest Baptist Comprehensive Cancer Center.
Disclosure
The author reports no conflicts of interest for this work.
References
1. Siegel RL, Miller KD, Goding Sauer A, et al. Colorectal cancer statistics, 2020. CA Cancer J Clin. 2020;70(3):145–164. doi:10.3322/caac.21601
2. Wang J, Li S, Liu Y, Zhang C, Li H, Lai B. Metastatic patterns and survival outcomes in patients with stage IV colon cancer: a population-based analysis. Cancer Med. 2020;9(1):361–373. doi:10.1002/cam4.2673
3. Bekaii-Saab T, Kim R, Kim TW, et al. Third- or later-line therapy for metastatic colorectal cancer: reviewing best practice. Clin Colorectal Cancer. 2019;18(1):e117–e129. doi:10.1016/j.clcc.2018.11.002
4. Wolmark N, Fisher B, Rockette H, et al. Postoperative adjuvant chemotherapy or BCG for colon cancer: results from NSABP protocol C-01. J Natl Cancer Inst. 1988;80(1):30–36. doi:10.1093/jnci/80.1.30
5. Goodwin RA, Asmis TR. Overview of systemic therapy for colorectal cancer. Clin Colon Rectal Surg. 2009;22(4):251–256. doi:10.1055/s-0029-1242465
6. Punt CJ, Koopman M, Vermeulen L. From tumour heterogeneity to advances in precision treatment of colorectal cancer. Nat Rev Clin Oncol. 2017;14(4):235–246. doi:10.1038/nrclinonc.2016.171
7. Golshani G, Zhang Y. Advances in immunotherapy for colorectal cancer: a review. Therap Adv Gastroenterol. 2020;13:1756284820917527. doi:10.1177/1756284820917527
8. Sinicrope FA, Sargent DJ. Molecular pathways: microsatellite instability in colorectal cancer: prognostic, predictive, and therapeutic implications. Clin Cancer Res. 2012;18(6):1506–1512. doi:10.1158/1078-0432.CCR-11-1469
9. Wei Q, Ye Z, Zhong X, et al. Multiregion whole-exome sequencing of matched primary and metastatic tumors revealed genomic heterogeneity and suggested polyclonal seeding in colorectal cancer metastasis. Ann Oncol. 2017;28(9):2135–2141. doi:10.1093/annonc/mdx278
10. Ishaque N, Abba ML, Hauser C, et al. Whole genome sequencing puts forward hypotheses on metastasis evolution and therapy in colorectal cancer. Nat Commun. 2018;9(1):4782. doi:10.1038/s41467-018-07041-z
11. Yaeger R, Chatila WK, Lipsyc MD, et al. Clinical sequencing defines the genomic landscape of metastatic colorectal cancer. Cancer Cell. 2018;33(1):125–136 e3. doi:10.1016/j.ccell.2017.12.004
12. Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61(5):759–767. doi:10.1016/0092-8674(90)90186-i
13. Ogino S, Goel A. Molecular classification and correlates in colorectal cancer. J Mol Diagn. 2008;10(1):13–27. doi:10.2353/jmoldx.2008.070082
14. Hinoue T, Weisenberger DJ, Pan F, et al. Analysis of the association between CIMP and BRAF in colorectal cancer by DNA methylation profiling. PLoS One. 2009;4(12):e8357. doi:10.1371/journal.pone.0008357
15. Murcia O, Juarez M, Rodriguez-Soler M, et al. Colorectal cancer molecular classification using BRAF, KRAS, microsatellite instability and CIMP status: prognostic implications and response to chemotherapy. PLoS One. 2018;13(9):e0203051. doi:10.1371/journal.pone.0203051
16. Yoda S, Dagogo-Jack I, Hata AN. Targeting oncogenic drivers in lung cancer: recent progress, current challenges and future opportunities. Pharmacol Ther. 2019;193:20–30. doi:10.1016/j.pharmthera.2018.08.007
17. Salem ME, Puccini A, Tie J. Redefining Colorectal Cancer by Tumor Biology. Am Soc Clin Oncol Educ Book. 2020;40:1–13. doi:10.1200/EDBK_279867
18. Miguel JC, Maxwell AA, Hsieh JJ, et al. Epidermal growth factor suppresses intestinal epithelial cell shedding through a MAPK-dependent pathway. J Cell Sci. 2017;130(1):90–96. doi:10.1242/jcs.182584
19. Pabla B, Bissonnette M, Konda VJ. Colon cancer and the epidermal growth factor receptor: current treatment paradigms, the importance of diet, and the role of chemoprevention. World J Clin Oncol. 2015;6(5):133–141. doi:10.5306/wjco.v6.i5.133
20. Battaglin F, Puccini A, Ahcene Djaballah S, Lenz HJ. The impact of panitumumab treatment on survival and quality of life in patients with RAS wild-type metastatic colorectal cancer. Cancer Manag Res. 2019;11:5911–5924. doi:10.2147/CMAR.S186042
21. Wang ZX, Wu HX, He MM, et al. Chemotherapy with or without anti-EGFR agents in left- and right-sided metastatic colorectal cancer: an updated meta-analysis. J Natl Compr Canc Netw. 2019;17(7):805–811. doi:10.6004/jnccn.2018.7279
22. Molinari C, Marisi G, Passardi A, Matteucci L, De Maio G, Ulivi P. Heterogeneity in colorectal cancer: a challenge for personalized medicine? Int J Mol Sci. 2018;19(12):3733. doi:10.3390/ijms19123733
23. Parseghian CM, Napolitano S, Loree JM, Kopetz S. Mechanisms of innate and acquired resistance to anti-EGFR therapy: a review of current knowledge with a focus on rechallenge therapies. Clin Cancer Res. 2019;25(23):6899–6908. doi:10.1158/1078-0432.CCR-19-0823
24. Li QH, Wang YZ, Tu J, et al. Anti-EGFR therapy in metastatic colorectal cancer: mechanisms and potential regimens of drug resistance. Gastroenterol Rep (Oxf). 2020;8(3):179–191. doi:10.1093/gastro/goaa026
25. Taieb J, Jung A, Sartore-Bianchi A, et al. The evolving biomarker landscape for treatment selection in metastatic colorectal cancer. Drugs. 2019;79(13):1375–1394. doi:10.1007/s40265-019-01165-2
26. Al-Shamsi HO, Alhazzani W, Wolff RA. Extended RAS testing in metastatic colorectal cancer-refining the predictive molecular biomarkers. J Gastrointest Oncol. 2015;6(3):314–321. doi:10.3978/j.issn.2078-6891.2015.016
27. Hallin J, Engstrom LD, Hargis L, et al. The KRAS(G12C) inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov. 2020;10(1):54–71. doi:10.1158/2159-8290.CD-19-1167
28. Amodio V, Yaeger R, Arcella P, et al. EGFR blockade reverts resistance to KRAS(G12C) inhibition in colorectal cancer. Cancer Discov. 2020;10(8):1129–1139. doi:10.1158/2159-8290.CD-20-0187
29. Liao W, Overman MJ, Boutin AT, et al. KRAS-IRF2 axis drives immune suppression and immune therapy resistance in colorectal cancer. Cancer Cell. 2019;35(4):559–572e7. doi:10.1016/j.ccell.2019.02.008
30. Mao C, Liao RY, Qiu LX, Wang XW, Ding H, Chen Q. BRAF V600E mutation and resistance to anti-EGFR monoclonal antibodies in patients with metastatic colorectal cancer: a meta-analysis. Mol Biol Rep. 2011;38(4):2219–2223. doi:10.1007/s11033-010-0351-4
31. Ho CC, Liao WY, Lin CA, Shih JY, Yu CJ, Yang JC. Acquired BRAF V600E mutation as resistant mechanism after treatment with osimertinib. J Thorac Oncol. 2017;12(3):567–572. doi:10.1016/j.jtho.2016.11.2231
32. Kayhanian H, Goode E, Sclafani F, et al. Treatment and survival outcome of BRAF-mutated metastatic colorectal cancer: a retrospective matched case-control study. Clin Colorectal Cancer. 2018;17(1):e69–e76. doi:10.1016/j.clcc.2017.10.006
33. Caputo F, Santini C, Bardasi C, et al. BRAF-mutated colorectal cancer: clinical and molecular insights. Int J Mol Sci. 2019;20(21):5369. doi:10.3390/ijms20215369
34. Kopetz S, Desai J, Chan E, et al. Phase II pilot study of vemurafenib in patients with metastatic BRAF-mutated colorectal cancer. J Clin Oncol. 2015;33(34):4032–4038. doi:10.1200/JCO.2015.63.2497
35. Zhang P, Kawakami H, Liu W, et al. Targeting CDK1 and MEK/ERK overcomes apoptotic resistance in BRAF-mutant human colorectal cancer. Mol Cancer Res. 2018;16(3):378–389. doi:10.1158/1541-7786.MCR-17-0404
36. Parry D, Guzi T, Shanahan F, et al. Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol Cancer Ther. 2010;9(8):2344–2353. doi:10.1158/1535-7163.MCT-10-0324
37. Somarelli JA, Roghani RS, Moghaddam AS, et al. A precision medicine drug discovery pipeline identifies combined CDK2 and 9 inhibition as a novel therapeutic strategy in colorectal cancer. Mol Cancer Ther. 2020;19(12):2516–2527. doi:10.1158/1535-7163.MCT-20-0454
38. Wang Q, Shi YL, Zhou K, et al. PIK3CA mutations confer resistance to first-line chemotherapy in colorectal cancer. Cell Death Dis. 2018;9(7):739. doi:10.1038/s41419-018-0776-6
39. Song M, Bode AM, Dong Z, Lee MH. AKT as a therapeutic target for cancer. Cancer Res. 2019;79(6):1019–1031. doi:10.1158/0008-5472.CAN-18-2738
40. Foley TM, Payne SN, Pasch CA, et al. Dual PI3K/mTOR inhibition in colorectal cancers with APC and PIK3CA mutations. Mol Cancer Res. 2017;15(3):317–327. doi:10.1158/1541-7786.MCR-16-0256
41. Fricke SL, Payne SN, Favreau PF, et al. MTORC1/2 inhibition as a therapeutic strategy for PIK3CA mutant cancers. Mol Cancer Ther. 2019;18(2):346–355. doi:10.1158/1535-7163.MCT-18-0510
42. Katholnig K, Schutz B, Fritsch SD, et al. Inactivation of mTORC2 in macrophages is a signature of colorectal cancer that promotes tumorigenesis. JCI Insight. 2019;4(20). doi:10.1172/jci.insight.124164
43. Wakatsuki T, Stintzing S, Zhang W, et al. Single nucleotide polymorphisms in AREG and EREG are prognostic biomarkers in locally advanced gastric cancer patients after surgery with curative intent. Pharmacogenet Genomics. 2014;24(11):539–547. doi:10.1097/FPC.0000000000000087
44. Bertotti A, Papp E, Jones S, et al. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature. 2015;526(7572):263–267. doi:10.1038/nature14969
45. Clifton K, Rich TA, Parseghian C, et al. Identification of actionable fusions as an anti-EGFR resistance mechanism using a circulating tumor DNA assay. JCO Precis Oncol. 2019;3. doi:10.1200/PO.19.00141
46. Yakirevich E, Resnick MB, Mangray S, et al. Oncogenic ALK fusion in rare and aggressive subtype of colorectal adenocarcinoma as a potential therapeutic target. Clin Cancer Res. 2016;22(15):3831–3840. doi:10.1158/1078-0432.CCR-15-3000
47. Sartore-Bianchi A, Marsoni S, Siena S. Human epidermal growth factor receptor 2 as a molecular biomarker for metastatic colorectal cancer. JAMA Oncol. 2018;4(1):19–20. doi:10.1001/jamaoncol.2017.3323
48. Sartore-Bianchi A, Trusolino L, Martino C, et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): a proof-of-concept, multicentre, open-label, Phase 2 trial. Lancet Oncol. 2016;17(6):738–746. doi:10.1016/S1470-2045(16)00150-9
49. Hainsworth JD, Meric-Bernstam F, Swanton C, et al. Targeted therapy for advanced solid tumors on the basis of molecular profiles: results from MyPathway, an open-label, phase IIa multiple basket study. J Clin Oncol. 2018;36(6):536–542. doi:10.1200/JCO.2017.75.3780
50. Pino MS, Chung DC. The chromosomal instability pathway in colon cancer. Gastroenterology. 2010;138(6):2059–2072. doi:10.1053/j.gastro.2009.12.065
51. Lynch HT, Lynch PM, Lanspa SJ, Snyder CL, Lynch JF, Boland CR. Review of the Lynch syndrome: history, molecular genetics, screening, differential diagnosis, and medicolegal ramifications. Clin Genet. 2009;76(1):1–18. doi:10.1111/j.1399-0004.2009.01230.x
52. Kawakami H, Zaanan A, Sinicrope FA. Microsatellite instability testing and its role in the management of colorectal cancer. Curr Treat Options Oncol. 2015;16(7):30. doi:10.1007/s11864-015-0348-2
53. Aasebo KO, Dragomir A, Sundstrom M, et al. Consequences of a high incidence of microsatellite instability and BRAF-mutated tumors: a population-based cohort of metastatic colorectal cancer patients. Cancer Med. 2019;8(7):3623–3635. doi:10.1002/cam4.2205
54. Dorard C, de Thonel A, Collura A, et al. Expression of a mutant HSP110 sensitizes colorectal cancer cells to chemotherapy and improves disease prognosis. Nat Med. 2011;17(10):1283–1289. doi:10.1038/nm.2457
55. Evrard C, Tachon G, Randrian V, Karayan-Tapon L, Tougeron D. Microsatellite instability: diagnosis, heterogeneity, discordance, and clinical impact in colorectal cancer. Cancers (Basel). 2019;11(10):1567. doi:10.3390/cancers11101567
56. Gozzi GJ, Gonzalez D, Boudesco C, et al. Selecting the first chemical molecule inhibitor of HSP110 for colorectal cancer therapy. Cell Death Differ. 2020;27(1):117–129. doi:10.1038/s41418-019-0343-4
57. Ohrling K, Karlberg M, Edler D, Hallstrom M, Ragnhammar P. A combined analysis of mismatch repair status and thymidylate synthase expression in stage II and III colon cancer. Clin Colorectal Cancer. 2013;12(2):128–135. doi:10.1016/j.clcc.2012.11.003
58. Gatalica Z, Vranic S, Xiu J, Swensen J, Reddy S. High microsatellite instability (MSI-H) colorectal carcinoma: a brief review of predictive biomarkers in the era of personalized medicine. Fam Cancer. 2016;15(3):405–412. doi:10.1007/s10689-016-9884-6
59. Gmeiner WH. Chemistry of fluorinated pyrimidines in the era of personalized medicine. Molecules. 2020;25(15):3438. doi:10.3390/molecules25153438
60. Kang M, Zheng W, Chen Q, et al. Thymidylate synthase prompts metastatic progression through the dTMP associated EMT process in pancreatic ductal adenocarcinoma. Cancer Lett. 2018;419:40–52. doi:10.1016/j.canlet.2018.01.026
61. Siddiqui A, Vazakidou ME, Schwab A, et al. Thymidylate synthase is functionally associated with ZEB1 and contributes to the epithelial-to-mesenchymal transition of cancer cells. J Pathol. 2017;242(2):221–233. doi:10.1002/path.4897
62. Gustavson MD, Molinaro AM, Tedeschi G, Camp RL, Rimm DL. AQUA analysis of thymidylate synthase reveals localization to be a key prognostic biomarker in 2 large cohorts of colorectal carcinoma. Arch Pathol Lab Med. 2008;132(11):1746–1752. doi:10.1043/1543-2165-132.11.1746
63. Salonga D, Danenberg KD, Johnson M, et al. Colorectal tumors responding to 5-fluorouracil have low gene expression levels of dihydropyrimidine dehydrogenase, thymidylate synthase, and thymidine phosphorylase. Clin Cancer Res. 2000;6(4):1322–1327.
64. Saif MW, Diasio RB. Benefit of uridine triacetate (Vistogard) in rescuing severe 5-fluorouracil toxicity in patients with dihydropyrimidine dehydrogenase (DPYD) deficiency. Cancer Chemother Pharmacol. 2016;78(1):151–156. doi:10.1007/s00280-016-3063-1
65. Lunenburg C, van der Wouden CH, Nijenhuis M, et al. Dutch Pharmacogenetics Working Group (DPWG) guideline for the gene-drug interaction of DPYD and fluoropyrimidines. Eur J Hum Genet. 2020;28(4):508–517. doi:10.1038/s41431-019-0540-0
66. Gmeiner WH, Dominijanni A, Haber AO, et al. Improved antitumor activity of the fluoropyrimidine polymer CF10 in preclinical colorectal cancer models through distinct mechanistic and pharmacologic properties. Mol Cancer Ther. 2021;20(3):553–563. doi:10.1158/1535-7163.MCT-20-0516
67. Haber AO, Jain A, Mani C, et al. AraC-FdUMP[10] is a next-generation fluoropyrimidine with potent antitumor activity in PDAC and synergy with PARG inhibition. Mol Cancer Res. 2021. doi:10.1158/1541-7786.MCR-20-0985
68. Gmeiner WH. Fluoropyrimidine modulation of the anti-tumor immune response-prospects for improved colorectal cancer treatment. Cancers (Basel). 2020;12(6):1641. doi:10.3390/cancers12061641
69. Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509–2520. doi:10.1056/NEJMoa1500596
70. Becht E, de Reynies A, Giraldo NA, et al. Immune and stromal classification of colorectal cancer is associated with molecular subtypes and relevant for precision immunotherapy. Clin Cancer Res. 2016;22(16):4057–4066. doi:10.1158/1078-0432.CCR-15-2879
71. Pedrosa L, Esposito F, Thomson TM, Maurel J. The tumor microenvironment in colorectal cancer therapy. Cancers (Basel). 2019;11(8):1172. doi:10.3390/cancers11081172
72. Zehir A, Benayed R, Shah RH, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23(6):703–713. doi:10.1038/nm.4333
73. Priestley P, Baber J, Lolkema MP, et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature. 2019;575(7781):210–216. doi:10.1038/s41586-019-1689-y
74. Lahouel K, Younes L, Danilova L, et al. Revisiting the tumorigenesis timeline with a data-driven generative model. Proc Natl Acad Sci U S A. 2020;117(2):857–864. doi:10.1073/pnas.1914589117
75. Allgayer H, Leupold JH, Patil N. Defining the “Metastasome”: perspectives from the genome and molecular landscape in colorectal cancer for metastasis evolution and clinical consequences. Semin Cancer Biol. 2020;60:1–13. doi:10.1016/j.semcancer.2019.07.018
76. Li C, Sun YD, Yu GY, et al. Integrated omics of metastatic colorectal cancer. Cancer Cell. 2020;38(5):734–747 e9. doi:10.1016/j.ccell.2020.08.002
77. Guinney J, Dienstmann R, Wang X, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015;21(11):1350–1356. doi:10.1038/nm.3967
78. Isella C, Brundu F, Bellomo SE, et al. Selective analysis of cancer-cell intrinsic transcriptional traits defines novel clinically relevant subtypes of colorectal cancer. Nat Commun. 2017;8:15107. doi:10.1038/ncomms15107
79. Stintzing S, Wirapati P, Lenz HJ, et al. Consensus molecular subgroups (CMS) of colorectal cancer (CRC) and first-line efficacy of FOLFIRI plus cetuximab or bevacizumab in the FIRE3 (AIO KRK-0306) trial. Ann Oncol. 2019;30(11):1796–1803. doi:10.1093/annonc/mdz387
80. Kamal Y, Schmit SL, Hoehn HJ, Amos CI, Frost HR. Transcriptomic differences between primary colorectal adenocarcinomas and distant metastases reveal metastatic colorectal cancer subtypes. Cancer Res. 2019;79(16):4227–4241. doi:10.1158/0008-5472.CAN-18-3945
81. Cheng DT, Mitchell TN, Zehir A, et al. Memorial sloan kettering-integrated mutation profiling of actionable cancer targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. 2015;17(3):251–264. doi:10.1016/j.jmoldx.2014.12.006
82. Siraj S, Masoodi T, Siraj AK, et al. Clonal evolution and timing of metastatic colorectal cancer. Cancers (Basel). 2020;12(10):2938. doi:10.3390/cancers12102938
83. Wei H, Li J, Xie M, Lei R, Hu B. Comprehensive analysis of metastasis-related genes reveals a gene signature predicting the survival of colon cancer patients. PeerJ. 2018;6:e5433. doi:10.7717/peerj.5433
84. Zhou Y, Zang Y, Yang Y, Xiang J, Chen Z. Candidate genes involved in metastasis of colon cancer identified by integrated analysis. Cancer Med. 2019;8(5):2338–2347. doi:10.1002/cam4.2071
85. Testa U, Castelli G, Pelosi E. Genetic alterations of metastatic colorectal cancer. Biomedicines. 2020;8(10):414. doi:10.3390/biomedicines8100414
86. Alexandrov LB, Kim J, Haradhvala NJ, et al. The repertoire of mutational signatures in human cancer. Nature. 2020;578(7793):94–101. doi:10.1038/s41586-020-1943-3
87. Naba A, Clauser KR, Whittaker CA, Carr SA, Tanabe KK, Hynes RO. Extracellular matrix signatures of human primary metastatic colon cancers and their metastases to liver. BMC Cancer. 2014;14:518. doi:10.1186/1471-2407-14-518
88. Ottaiano A, Caraglia M, Di Mauro A, et al. Evolution of mutational landscape and tumor immune-microenvironment in liver oligo-metastatic colorectal cancer. Cancers (Basel). 2020;12(10):3073. doi:10.3390/cancers12103073
89. Zhao Y, Li J, Li D, et al. Tumor biology and multidisciplinary strategies of oligometastasis in gastrointestinal cancers. Semin Cancer Biol. 2020;60:334–343. doi:10.1016/j.semcancer.2019.08.026
90. Jung YS, Park JI. Wnt signaling in cancer: therapeutic targeting of Wnt signaling beyond beta-catenin and the destruction complex. Exp Mol Med. 2020;52(2):183–191. doi:10.1038/s12276-020-0380-6
91. Chen J, Elfiky A, Han M, Chen C, Saif MW. The role of Src in colon cancer and its therapeutic implications. Clin Colorectal Cancer. 2014;13(1):5–13. doi:10.1016/j.clcc.2013.10.003
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.