")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 13
Recent Advances in Neonatal Diabetes
Received 9 September 2019
Accepted for publication 6 December 2019
Published 12 February 2020 Volume 2020:13 Pages 355—364
DOI https://doi.org/10.2147/DMSO.S198932
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Juei-Tang Cheng
Amanda Dahl, Seema Kumar
Division of Pediatric Endocrinology and Metabolism, Department of Pediatric and Adolescent Medicine, Mayo Clinic, Rochester, MN, USA
Correspondence: Seema Kumar
Division of Pediatric Endocrinology and Metabolism, Department of Pediatric and Adolescent Medicine, Mayo Clinic, 200 First Street SW, Rochester, MN 55590, USA
Tel +1 507-284-3300
Fax +1 507-284-0727
Email [email protected]
Abstract: Neonatal diabetes mellitus (DM) is defined by the onset of persistent hyperglycemia within the first six months of life but may present up to 12 months of life. A gene mutation affecting pancreatic beta cells or synthesis/secretion of insulin is present in more than 80% of the children with neonatal diabetes. Neonatal DM can be transient, permanent, or be a component of a syndrome. Genetic testing is important as a specific genetic mutation can significantly alter the treatment and outcome. Patients with mutations of either KCNJ11 or ABCC8 that encode subunits of the KATP channel gene mutation can be managed with sulfonylurea oral therapy while patients with other genetic mutations require insulin treatment.
Keywords: neonatal diabetes, KCNJ11, sulfonylurea, ABCC8
Introduction
Neonatal diabetes mellitus (DM) is defined by the onset of persistent hyperglycemia within the first six months of life. It is often caused by a mutation in a single gene affecting development and function of pancreatic beta cells leading to diminished insulin secretion/function. While most patients with neonatal DM present within the first 6 months of life, some may present up to 12 months of life.1–5 In contrast, it is unusual for autoimmune type 1 diabetes to present within the first six months of life.5,6
Epidemiology
Incidence of neonatal DM is thought to range from 1:90,000 to 1:160,000 live births.7,8 Hyperglycemia in a neonate is not an uncommon occurrence.9 Therefore, making the diagnosis of neonatal DM can be difficult. Neonatal hyperglycemia is more common to develop in the firs 3–5 days of life and resolve within 2–3 days of onset but can persist up to 10 days.
The difficulty in diagnosis is especially true in the preterm population or low birth weight infants.2,10 The prevalence of hyperglycemia in preterm infants can vary from 25% to 75%.9–11 Common reasons for hyperglycemia in these patients include: sepsis, increased counter-regulatory hormones due to stress, parenteral glucose administration and medications such as steroids and beta-adrenergic agents. Also in critically ill preterm neonates, there is evidence that shows this population has some degree of pancreatic insulin secretion insufficiency and relative insulin resistance.9 However, in a study of 750 patients with diabetes diagnosed before 6 months of age (146 preterm patients born <37 weeks and 604 born ≥37 weeks), a genetic etiology was found in 97/146 (66%) preterm infants compared with 501/604 (83%) born ≥37weeks. A genetic etiology was noted less frequently in early preterm infants (<32 weeks, 31%) than those born between 32–<37 weeks (81%) and ≥37 weeks of gestation (83%). There was no difference in the age at presentation between preterm and term infants (1 week vs 0.7 weeks).10 The diagnosis of neonatal DM, therefore, should be considered in the presence of insulin-dependent hyperglycemia without an alternative causative factor in both preterm infants and in term infants.
Pathogenesis
A genetic mutation is found in approximately 80% of children with neonatal diabetes.1,3 The number of genes that are found in children with neonatal diabetes continues to increase and there are more than 20 known genetic causes for neonatal DM.1,2,12,13 The various genes are associated with specific inheritance pattern, phenotype, and clinical features.2,3,5,13
The genes associated with neonatal diabetes play a vital role in the development of pancreatic beta cells and synthesis and secretion of insulin. In a large series of 1020 patients diagnosed with neonatal DM before 6 months of age, causal mutations were found in 82% of the cases after comprehensive genetic testing including Sanger sequencing, 6q24 methylation analysis, and targeted next-generation sequencing of all known neonatal diabetes genes.1 Mutations in the potassium channel genes (KCNJ11 and ABCC8) were the most common cause (38.2%) of neonatal diabetes but were identified less frequently in consanguineous families (12% in consanguineous families vs 46% in non-consanguineous families).1 Mutations in the INS gene that encodes insulin were found in 10% of patients from non-consanguineous and consanguineous families. A homozygous mutation in EIF2AK3 gene was the most common genetic cause in consanguineous families (Wolcott-Rallison syndrome, 24%).1
The underlying mechanisms for the various gene mutations that lead to development of neonatal diabetes can be classified into three categories:
- Alteration in beta cell function affecting synthesis or secretion of insulin – KCNJ11, ABCC8, GCK, INS, RFX6, SLC2A2, SLC19A2
- Pancreatic hypoplasia or aplasia – PDX1 (IPF1), PTF1A, HNF1B, MNX1, RFX6, GATA4, GATA6, GLIS3, NKX2-2, NEUROG3, NEUROD1, PAX6
- Damage to pancreatic beta cells – INS, EIF2AK3, IER3IPI, FOXP3, WFS1
Presentation
The clinical presentation varies from incidentally detected asymptomatic hyperglycemia to severe dehydration and diabetic ketoacidosis (DKA).5,14 Common clinical manifestations include being small for gestational age due to prenatal intrauterine growth restriction from deficiency of functional insulin which is important for in-utero growth.15 Another common clinical manifestation is poor postnatal growth or failure to thrive and behavioral changes such as irritability and polyuria. Infants with DKA may have few nonspecific symptoms such as tachypnea, lethargy, irritability, and sunken fontanels and eyes. Infants with DKA may develop electrolyte imbalance and dehydration.
The odds of presenting with diabetic ketoacidosis (DKA) increase with age (odds ratio per 1 month increase 1.23 [95% CI 1.04,1.45].5 DKA is also more common in certain monogenic forms of neonatal DM. In one study, DKA was found in 78.8% of patients with mutations of KCNJ11/ABCC8 and in 30% of patients with mutations of INS.5 On the contrary, children with transient DM due to overexpression of 6q24 did not develop DKA.
Malabsorptive diarrhea can occur when the exocrine function of the pancreas is impaired (associated with GATA6, EIF2AK3, PTF1A).2
Extra-pancreatic findings sometimes associated with neonatal DM are usually helpful clues toward presence of underlying genetic mutation.16–25
- Extra-pancreatic findings that can be associated with a specific genetic mutation are:1
- Polycystic kidney disease (associated with HNF-1 beta)
- Neurologic abnormalities and neurodevelopment impairment (associated with KCNJ11, NEUROD1, PTF1A, IER3IP1)
- Immune dysregulation (associated with IPEX)
- Hypothyroidism (associated with GLIS3)
- Deafness (associated with WFS1, SLC19A2)
- Skeletal abnormalities (associated with EIF2A)
- Hepatic dysfunction (associated with EIF2A, SLC2A2)
- Optic abnormalities (associated with WFS1, PAX6)
- Cardiac abnormalities (associated with GATA4, GATA6)
Clinical Phenotypes
Clinically, neonatal DM phenotypic expression can be divided into 3 broad categories: transient neonatal diabetes where diabetes remits and can relapse later in life, permanent neonatal diabetes where it does not remit and is the isolated manifestation, or syndromic neonatal diabetes where neonatal diabetes is only one of the clinical features of the syndrome. In a recent large international cohort study, transient neonatal DM was present in 20% of the subjects with neonatal diabetes.1
Transient Neonatal DM
Children with transient neonatal DM usually have resolution of hyperglycemia in infancy (by 13–18 weeks of age). However, there may be recurrence in adolescence or adulthood.1–3
Overexpression of genes on the 6q24 locus is the most common cause of transient neonatal diabetes. This is a consequence of loss of imprinting at 6q24 by uniparental disomy, by duplication of this region (paternal duplication), or by loss of DNA methylation, and thus activation of the maternal allele.2,26,27 This region includes the genes ZAC and HYMAI. ZAC is a C2H2 zinc-finger transcription factor with multiple functions, including acting as a coactivator with p53 of Apaf1 (apoptotic protease activating factor 1) transcription, regulating the histone acetyltransferase activity of p300, and serving as a coactivator or corepressor of several nuclear hormone receptors.28 HYMAI is an untranslated RNA of undetermined function.26
Patients with 6q24 mutation usually present earlier than those with KCNJ11/ABCC8 mutation.5 Additional features that may present in children with 6q24-related neonatal DM are macroglossia or umbilical hernia. Treatment for patients with 6q24-related neonatal diabetes in the early phase is insulin therapy. However, in older patients, non-insulin therapies such as those used for type 2 diabetes may be effective. As high as 14% of patients in a case series of 6q24-related transient neonatal DM developed hypoglycemia after remission of diabetes.29
The second most common cause of transient neonatal DM is mutations in the two genes encoding the subunits of the voltage-dependent potassium channels.1,2 KCNJ11 encodes for the inner subunit (Kir6.2) of the KATP channel and ABCC8 encodes for the outer subunit (SUR1). Mutations in either of the two genes result in inappropriately open KATP channels despite hyperglycemia leading to the inability of the cell membrane to depolarize and release insulin (Figure 1).
Permanent Neonatal DM
The most common cause of permanent neonatal diabetes is activating heterozygous mutations in KCNJ11 or ABCC8. These mutations account for more than 50% of all cases of neonatal DM.2,30 These two gene mutations are also the second most common cause of transient neonatal diabetes. KATP channels are also present in the brain. Median age at presentation in a recent study was 9.6 weeks (IQR 6.1–18.3 weeks)5 and most patients present before 6 months of age, though presentation after 6 months of age has been reported.
As KATP channels are expressed in the brain, patients with KCNJ11 mutations may have a wide range of neurocognitive disabilities such as decreased ability for reasoning, reading, vocabulary, and auditory working memory compared with sibling controls.31,32 Patients may also demonstrate signs and symptoms of sleep disturbance, attention deficit hyperactivity disorder as well as delays in learning, social-emotional, and behavioral development.33 Severe cases of KCNJ11 mutation may have developmental delay and epilepsy, referred to as the DEND syndrome (Developmental delay, Epilepsy, and Neonatal Diabetes). Effects can vary from mild delays to severe delays with seizures. Patients with mutations in these two genes are sensitive to sulfonylurea (SU) treatment.
The second most common form of permanent neonatal diabetes after KCNJ11/ABCC8 gene mutation is mutationin the insulin gene (INS). Mutations in the INS can be present in up to 20% of infants with permanent neonatal DM.1,34 Mutation in this gene results in misfolding of the insulin protein which results in increased endoplasmic reticulum stress and eventual beta-cell death. The median age for diagnosis is at 10 weeks and 30% of these patients will present with DKA.5 In a study of patients with permanent neonatal diabetes, those with mutations in the INS were diagnosed later than ATP-sensitive K(+) channel mutation carriers (11 vs 8 weeks).34 Patients do not have any other phenotypic features other than permanent neonatal diabetes and require lifelong treatment with insulin.35
Syndromic Forms of Neonatal DM
There are several syndromes that are associated with neonatal DM. The mechanisms leading to presence of neonatal DM in various syndromes include beta-cell destruction, pancreatic hypoplasia or aplasia, impaired beta-cell function or severe insulin resistance.
The most common syndrome is Wolcott-Rallison syndrome which is an autosomal recessive disorder caused by a mutation in EIF2A, a gene encoding the translation initiation factor 2-alpha kinase 3 important in regulation of the endoplasmic reticulum.17 Other features are hepatic dysfunction and skeletal dysplasia. It occurs in about 30% of cases with consanguineous families.
A few of the syndromic forms are more common in consanguineous families, these include EIF23, GCK, GLIS3, RFX6, IER3IP1, and MNX1.2 Other more rare syndromes that present with neonatal DM include IPEX syndrome (X-linked disorder that has immune dysregulation, polyendocrinopathy, and enteropathy – mutation in FOXP3), Fanconi Bickel syndrome (autosomal recessive disorder that has liver dysfunction and hypergalactosemia – mutation of SLC2A2), Rogers syndrome (autosomal recessive disorder that has thiamine-responsive megaloblastic anemia and sensorineural hearing loss – mutation of SLC19A2), Wolfram syndrome (autosomal recessive disorder that has diabetes insipidus, optic atrophy, and deafness – mutation of WFS1), and Rabson-Mendenhall syndrome (post-prandial hyperglycemia, fasting hypoglycemia, poor linear growth, and impaired muscle and adipose development – mutation of INSR).2,6
Differential Diagnosis
Neonatal diabetes needs to be distinguished from autoimmune type 1 diabetes. Most patients diagnosed with diabetes after 6 months of age, and especially after 12 months of age will have autoimmune type 1 diabetes.6 In a study of children with diabetes onset before 13 months of age, the median age for diagnosis for infants with type 1 diabetes was 42.6 weeks (IQR 37.4–50.4) and 87.5% presented in DKA.5 Most of these patients will test positive for at least one of the specific diabetes-related autoantibodies.
Diagnostic Workup
The first approach to hyperglycemia is evaluating if a specific cause could be the contributing factor. This should include assessing the amount of glucose administration. Ideal glucose infusion rates should be 6 to 12 mg/kg/min in a neonate for effective growth and nutrition.2 Blood glucose may normalize with decrease in the glucose infusion rate.9 Medications that can increase blood glucose such as high dose glucocorticoids,epinephrine, norepinephrine or dopamine should be discontinued if possible.
Laboratory Evaluation
The initial assessment for neonates with suspected diabetes should include laboratory assessment of serum glucose, C-peptide, insulin, and urine ketones. At diagnosis, infants typically have high blood glucose and lower HbA1c and C-peptide levels than children aged >2 years suggesting faster beta-cell destruction. A pancreatic ultrasound should also be performed as the presence or absence of pancreas and size of the pancreas will guide diagnosis as pancreatic hypoplasia or agenesis is associated with certain genetic mutations. Neonatal blood contains a high proportion of fetal hemoglobin (HbF), whereas hemoglobin A (HbA) accounts for only 10–20%. During the first 6 months of life, HbF is gradually replaced by HbA. Therefore, HbA1c is less suitable to diagnose diabetes mellitus in infants <6 months of age. Measurement of diabetes-related autoantibodies (glutamic acid decarboxylase, islet cell, insulin, zinc transporter, and tyrosine phosphatases) is helpful in infants between 6 months and 12 months of age. Most children with type 1 diabetes will be positive for at least one autoantibody.
Genetic Testing
Genetic testing should be strongly considered for infants who have persistent hyperglycemia as prognosis and treatment options for monogenic forms of neonatal DM are influenced to a major extent by the specific gene that is mutated. Most patients with mutations in KCNJ11 and ABCC8 are responsive to sulfonylurea therapy and can be transitioned from insulin to sulfonylurea therapy after the genetic basis for their diabetes has been identified. In a large international cohort study with clinically diagnosed diabetes before 6 months of age, 80% had a known genetic diagnosis.1,2 Term infants and premature infants born at more than 32 weeks gestation are more likely to have a monogenic cause. However, Besser et al showed that 31% of all preterm infants with diabetes born at less than 32 weeks gestation were also diagnosed with monogenic cause, suggesting that genetic testing should also be performed in preterm infants.10 Chromosome 6q24 imprinting abnormalities (27% vs 12%) and GATA6 mutations (9% vs 2%) occurred more commonly in preterm than term infants while mutations in KCNJ11 were less common (21% vs 34%). Within their study, they showed that 37% of premature infants have potassium channel mutation and therefore timely genetic testing referral can improve control by replacing insulin with sulfonylurea therapy.10
Targeted gene panels are available in various laboratories for genes associated with neonatal diabetes. Early comprehensive testing has led to a change in the management of patients with neonatal diabetes. The use of exome and genome sequencing has resulted in the identification of 2 novel disease genes (GATA6 and STAT3) and a novel regulatory element of PTF1A, in which mutations cause pancreatic agenesis.12 Genetic testing is recommended for all cases of diabetes diagnosed less than 12 months of age.2 Latest expert guidelines recommend that all patients diagnosed with diabetes in the first 6 months of life should have immediate molecular genetic testing to define their subtype of monogenic neonatal diabetes mellitus (NDM), as type 1 diabetes is extremely rare in this subgroup. In patients diagnosed between 6 and 12 months of age, testing for NDM is recommended in those without islet antibodies as the majority of patients in this age group have type 1 diabetes.13 Median duration of diabetes at the time of genetic testing was noted to decrease from more than 4 years before 2005 to less than 3 months after 2012.1
Treatment
The initial management in a neonate with persistent hyperglycemia is reduction in the glucose infusion rate to physiologic glucose requirements for optimum growth and nutrition (6–12 mg/kg/min). Additionally, underlying conditions such as sepsis need to be treated. An attempt should be made, if medically safe, to decrease the dose of or discontinue medications that can result in hyperglycemia such as epinephrine, norepinephrine, dopamine or glucocorticoids.
If the infant has dehydration, electrolyte imbalance or ketoacidosis, intravenous fluids and electrolytes should be administered with close monitoring of fluid status and electrolytes. All infants with persistent hyperglycemia should initially be started on an intravenous insulin infusion. Some studies showed effective glucose control with insulin rates ranging as low as 0.02 units/kg/hr.36 Other studies in very low birth weight infants often used an initial dose of 0.05 units/kg/hr.9,36 Blood glucose levels need to be monitored closely when on insulin infusion, ideally every hour. Insulin infusion rates should be adjusted in small increments of 0.01 units/kg/hr when glucose levels are less than 100 or greater than 200 mg/dL.
Patients should be transitioned to subcutaneous insulin therapy if hyperglycemia persists after oral feedings have been established. Subcutaneous insulin therapy can be delivered via multiple daily injections or as a continuous subcutaneous insulin infusion (CSII). The initial dose via either of these delivery mechanisms should be conservative to decrease the risk of hypoglycemia. Subcutaneous insulin should be given when blood glucose values are at least above 200–250 mg/dL.
When using the multiple daily injections regimen, rapid-acting insulin analogs such as insulin aspart, insulin lispro or glulisine are recommended 3–4 times per day before a feed.37 The starting dose recommended for these rapid-acting insulin is 0.1 to 0.15 units/kg/dose if the preprandial glucose is above 200–250 mg/dL. It is recommended that all preprandial blood glucose values be checked at least initially, though insulin may be needed with every other feed only (3–4 times per day). Administration of short-acting insulin before each feed may result in stacking of insulin and resultant hypoglycemia. The smallest dose of subcutaneous insulin that can be administered without dilution is 0.5 units. Smaller doses as low as 0.1 units are possible using dilution of the U-100 insulin (100 units of insulin per 1 mL) to up to one-tenth of the original concentration. Insulin should be diluted using insulin specific compatible diluents and the shelf life of the specific diluted insulin should be taken into consideration when decision is made to dilute insulin. Infants can also receive long-acting insulin such as glargine at a dose of 0.2–0.4 unit/kg/day in 1 or 2 injections per day.38,39 Infants with neonatal diabetes are susceptible to hypoglycemia because of the relatively low insulin requirements. Total daily insulin requirements can vary from 0.29 U/kg to 1.4 U/kg/day.14 Intermediate-acting insulins such as regular and NPH should be avoided due to the increased risk of hypoglycemia and erratic control compared with rapid and long-acting insulin. Carbohydrate estimation for breastfed infants is often difficult. Carbohydrate content for breast milk is approximately 2.1 g per ounce of breastmilk.2,14
Continuous subcutaneous insulin infusion (CSII) via an insulin pump offers the advantage of the ability to deliver smaller doses of insulin relative to the multiple daily injections regimen.38,40–43 Some infants may still need dilution of insulin for the insulin pump. The initial dose of basal insulin using the CSII is 0.1 to 0.3 units/kg/day. This can be adjusted to maintain blood glucose values between 100 and 200 mg/dL. Correction doses should be given if preprandial blood glucose values are above 250 mg/dL. The minimum basal rates for insulin pumps range between 0.025 and 0.05 units/hr.2
Sulfonylurea Therapy
Sulfonylurea therapy is effective in treatment of hyperglycemia in patients with neonatal DM who have a mutation in the KCNJ11 and ABCC8 genes. Up to 90–95% of patients with neonatal DM caused by these mutations are able to be taken off of insulin therapy after initiation of SU therapy.2,44 Sulfonylureas (SU) act on the KATP channel to promote closure, allowing for insulin to be released from the beta cells. Glyburide (Glibenclamide) is the SU drug that has been used in a majority of patients with neonatal diabetes.44–50 Other sulphonylureas such as glipizide, gliclazide, tolbutamide, and glimepiride have been used rarely but do not offer an advantage over glyburide.44,45
Since SU therapy increases insulin release, there is a risk for hypoglycemia to occur, especially if an infant or child has decreased oral intake. However, the risk for hypoglycemia with SU therapy is decreased in comparison to insulin therapy. In a 10-year multicenter follow-up study of a large international cohort of patients with KCNJ11 permanent neonatal diabetes, excellent glycemic control was maintained (HbA1c 8.1% before transfer to sulfonylureas, 5.9% at 1 year, and 6.4% at the last follow-up).45 There were no reports of severe hypoglycemia in 809 patient-year follow up for the whole cohort and 93% of the participants remained on sulfonylurea therapy for the 10-year duration. As high as 14% of patients had reported mild, transient side effects which included diarrhea, nausea, weight loss due to reduced appetite, and abdominal pain.44,45 No patients discontinued SU treatment because of the side effects. Additional side effects were hepatitis steatosis and tooth discoloration.45,46,51 As high as 9% patients had microvascular complications but these patients were noted to have been taking insulin longer than those without complications (20.5 years at time of transfer to SU therapy).45 Pearson et al noted among 49 patients with diabetes due to KIR6.2 mutations, 90% of the patients successfully discontinued insulin after receiving sulfonylureas.44 In another study of 14 patients with permanent neonatal diabetes mellitus due to KCNJ11 gene mutation (median age, 12.0 years; range, 5–50 years) who were transferred to SU therapy at least 2 prior, there was a 1.68% (range, 0.3–3.7%) initial reduction of HbA1c after the switch to SU (approximately 3–6 months post-transfer) and good glycemic control was maintained during the follow-up (median 34 months, range, 27–51 months) with average HbA1c level of 6.0% (range, 5.3–6.7%) at the last visit.52 Only 1 patient had rapid progression of retinal changes. This patient was a 34-year-old female with preexisting proliferative diabetic retinopathy.52 Younger age at initiation of SU therapy and shorter duration of diabetes are associated with higher rates of success in transition from insulin therapy to SU.46,53
Importantly, SU therapy can also lead to beneficial effects on neurocognitive development, especially with those who have KCNJ11 mutation.2,54–57 The benefits appear to be most in patients in whom these drugs are started earlier.54,58 SU receptors are widely expressed in the brain and the improvement in neurological status suggests that SU drugs cross the blood-brain barrier in sufficient amount to close the neuronal KATP channels.56 Bowman et al in the 10-year multicenter follow-up study of KCNJ11 permanent neonatal diabetes reported improvement in CNS features in 47% of the patients that had CNS features at the time of transition to SU therapy. Improvements were noted in muscle tone, concentration or ADHD, gross motor skills, epilepsy, muscle weakness, learning difficulties, speech, and tics. However, this effect was usually incomplete and eventually plateaued.45 Bertrand and colleagues also reported improved intelligence scores, hypotonia, visual attention deficits, and gross and fine motor skills after transitioning from insulin to SU therapy.54 CNS features in KCNJ11 permanent neonatal diabetes persist despite long-term treatment with SU which is in contrast with the improved glycemic response.45 The underlying reason for the poor CNS response may be the failure to achieve therapeutic concentration of these drugs in the CSF and brain of patients treated with oral SU to block KATP channels enough to affect neuronal electrical activity.59 The use of SU in pediatric patients is considered an off-label use. It is important to keep in mind that up to 10% of patients with KCNJ11 mutations causing neonatal DM will not respond to sulfonylurea and will require insulin lifelong.53,60 There can be a significant delay between clinical diagnosis of neonatal DM and genetic diagnosis of neonatal diabetes as was noted in the Monogenic Diabetes Registry (mean 10 weeks, range 1.6 to 58.2 weeks).61 Given the potential beneficial effect on neurodevelopmental outcome and glycemic control, an empiric inpatient trial of SU therapy is suggested in the absence of consanguinity, syndromic features, and pancreatic hypoplasia/aplasia.2,61
Several approaches have been utilized for transition from insulin to SU.2,44,50,62–64 Patients can be on multiple daily insulin injections or on CSII prior to transition to SU. Point of care blood glucose should be checked before meals and at bedtime daily during the transition phase.2,44 Continuous glucose monitors can be helpful in monitoring blood glucose values in neonates.65 The transition from insulin to SU can occur in an inpatient setting under the supervision of a physician with expertise in management of infants with diabetes. The transition may also be done as an outpatient provided the family and health providers are comfortable with adjustments in insulin and glyburide dosing based on blood glucose values during the transition phase. Pearson and colleagues recommend a blood glucose values cut off of 126 mg/dL for titrating the dose of glyburide.44 The initial starting dose of glyburide is 0.1 mg/kg/dose twice daily before meals. Glyburide tablets can be crushed and administered as an aqueous suspension. In subsequent days, if the preprandial blood glucose at the time the dose of glyburide is due is greater than 126 mg/dL, the dose of glyburide can be increased by 0.1 mg/kg/dose. If pre-meal point of care glucose values continue to be above 126 mg/dL, dose should be increased to at least 1 mg/kg/day which is usually achieved in 5–7 days. Other experts recommend using a blood glucose cut off of 200 mg/dL to titrate the dose of glyburide due to the concern that the lower blood glucose threshold of 126 mg/dL places the child at a higher risk of hypoglycemia during the transition.2
The dose of the long-acting insulin is decreased the night before giving glyburide or if the patient is using CSII, the basal insulin is decreased by 50% before breakfast on the day of giving the first dose of glyburide. Long-acting and intermediate-acting insulin analogs are discontinued on day 2 of the transition. Once glyburide has been started, the dose of the short-acting insulin dose is adjusted based on the pre-prandial blood glucose. For instance, if blood glucose is >200 mg/dL, the usual dose of short-acting insulin prior to the meal should be given. On the contrary, if blood glucose is <200 mg/dL, the preprandial dose of short-acting insulin should be reduced by at least 50%.2 In most SU-responsive cases, insulin can be discontinued in 5–7 days. The slow outpatient transition begins with a starting glyburide dose of 0.1 mg per kilogram per day and increases by 0.1 mg per kilogram per day once a week.44 The dose of glyburide is increased until insulin independence or until the dose was at least 0.8 mg per kilogram per day.
If the clinical response is unclear, measurement of glucose and C-peptide levels before and 90–120 mins after a meal when glyburide is being used can be helpful in assessing the effect of SU therapy in infants.2
Long-term treatment for neonatal DM requires the involvement of a multidisciplinary team. Blood glucose monitoring is recommended due to the risk of both hyperglycemia and hypoglycemia in children with neonatal diabetes. Hypoglycemia can be difficult to recognize in infants and toddlers and the only sign might be behavioral changes like irritability and inconsolable crying. Hypoglycemia can also be detrimental in regards to cognitive deficits. Infants with neonatal DM should be seen every 3 months. HbA1c should be assessed to measure average 3-month glycemic control but only if the patient is >6 months of age as it does not necessarily reflect average blood glucose values in infants <6 months of age due to higher level of HbF. Screening for microvascular and macrovascular complications of diabetes can be delayed until after the first 5 years after diagnosis since these are uncommon to develop in infants.14
Conclusion
Neonatal diabetes should be diagnosed in infants with persistent hyperglycemia and genetic testing should be considered at the onset in all patients with neonatal diabetes as knowledge of the presence of specific genetic mutations can significantly alter the treatment. Sulfonylurea treatment is the treatment of choice for patients with KATP channel genetic mutation (KCNJ11 and ABCC8) and it has been shown to improve neurocognitive features. Close follow-up is recommended in all patients with neonatal diabetes due to their increased risk for both hyperglycemia and hypoglycemia.
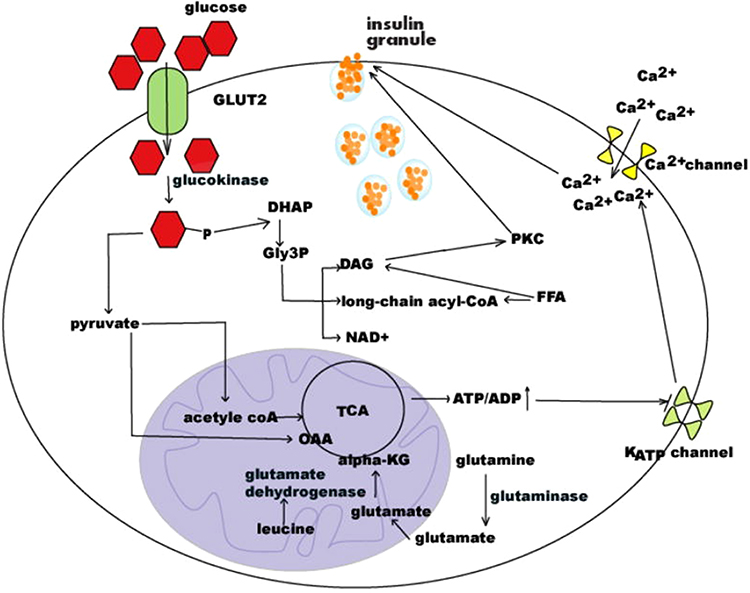 |
Figure 1 Schematic of regulation of insulin secretion. Notes: Republished with permission of EUREKA SCIENCE (FZC), from Regulation of insulin synthesis and secretion and pancreatic Beta-cell dysfunction in diabetes, Fu Z, Gilbert ER, and Liu D, Volume 9(1), 2013; permission conveyed through Copyright Clearance Center, Inc.66 |
Disclosure
Seema Kumar reports personal fees from Rhythm Pharmaceuticals, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. De Franco E, Flanagan SE, Houghton JAL, et al. The effect of early, comprehensive genomic testing on clinical care in neonatal diabetes: an international cohort study. Lancet (London, England). 2015;386(9997):957–963. doi:10.1016/S0140-6736(15)60098-8
2. Lemelman MB, Letourneau L, Greeley SAW. Neonatal diabetes mellitus: an update on diagnosis and management. Clin Perinatol. 2018;45(1):41–59. doi:10.1016/j.clp.2017.10.006
3. Rubio-Cabezas O, Ellard S. Diabetes mellitus in neonates and infants: genetic heterogeneity, clinical approach to diagnosis, and therapeutic options. Horm Res Paediatr. 2013;80(3):137–146. doi:10.1159/000354219
4. Hamilton-Shield JP. Overview of neonatal diabetes. Endocr Dev. 2007;12:12–23.
5. Letourneau LR, Carmody D, Wroblewski K, et al. Diabetes presentation in infancy: high risk of diabetic ketoacidosis. Diabetes Care. 2017;40(10):e147–e148. doi:10.2337/dc17-1145
6. Hattersley A, Bruining J, Shield J, et al. ISPAD clinical practice consensus guidelines 2006–2007. The diagnosis and management of monogenic diabetes in children. Pediatr Diabetes. 2006;7(6):352–360. doi:10.1111/j.1399-5448.2006.00217.x
7. Grulich-Henn J, Wagner V, Thon A, et al. Entities and frequency of neonatal diabetes: data from the diabetes documentation and quality management system (DPV). Diabet Med. 2010;27(6):709–712. doi:10.1111/dme.2010.27.issue-6
8. Nansseu JRN, Ngo-Um SS, Balti EV. Incidence, prevalence and genetic determinants of neonatal diabetes mellitus: a systematic review and meta-analysis protocol. Syst Rev. 2016;5(1):188. doi:10.1186/s13643-016-0369-3
9. Beardsall K, Vanhaesebrouck S, Ogilvy-Stuart AL, et al. Prevalence and determinants of hyperglycemia in very low birth weight infants: cohort analyses of the NIRTURE study. J Pediatr. 2010;157(5):715–719e 711–713. doi:10.1016/j.jpeds.2010.04.032
10. Besser RE, Flanagan SE, Mackay DG, et al. Prematurity and genetic testing for neonatal diabetes. Pediatrics. 2016;138:3. doi:10.1542/peds.2015-3926
11. Sabzehei MK, Afjeh SA, Shakiba M, Alizadeh P, Shamshiri AR, Esmaili F. Hyperglycemia in VLBW infants; incidence, risk factors and outcome. Arch Iran Med. 2014;17(6):429–434.
12. De Franco E, Ellard S. Genome, exome, and targeted next-generation sequencing in neonatal diabetes. Pediatr Clin North Am. 2015;62(4):1037–1053. doi:10.1016/j.pcl.2015.04.012
13. Hattersley AT, Greeley SAW, Polak M, et al. ISPAD clinical practice consensus guidelines 2018: the diagnosis and management of monogenic diabetes in children and adolescents. Pediatr Diabetes. 2018;19(Suppl 27):47–63. doi:10.1111/pedi.12772
14. Karges B, Meissner T, Icks A, Kapellen T, Holl RW. Management of diabetes mellitus in infants. Nat Rev Endocrinol. 2011;8(4):201–211. doi:10.1038/nrendo.2011.204
15. Anderson de la Llana S, Klee P, Santoni F, Stekelenburg C, Blouin J-L, Schwitzgebel VM. Gene variants associated with transient neonatal diabetes mellitus in the very low birth weight infant. Horm Res Paediatr. 2015;84(4):283–288. doi:10.1159/000437378
16. D’Amato E, Giacopelli F, Giannattasio A, et al. Genetic investigation in an Italian child with an unusual association of atrial septal defect, attributable to a new familial GATA4 gene mutation, and neonatal diabetes due to pancreatic agenesis. Diabet Med. 2010;27(10):1195–1200. doi:10.1111/dme.2010.27.issue-10
17. Delepine M, Nicolino M, Barrett T, Golamaully M, Lathrop GM, Julier C. EIF2AK3, encoding translation initiation factor 2-alpha kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nat Genet. 2000;25(4):406–409. doi:10.1038/78085
18. Rigoli L, Lombardo F, Di Bella C. Wolfram syndrome and WFS1 gene. Clin Genet. 2011;79(2):103–117. doi:10.1111/cge.2011.79.issue-2
19. Rubio-Cabezas O, Minton JA, Kantor I, Williams D, Ellard S, Hattersley AT. Homozygous mutations in NEUROD1 are responsible for a novel syndrome of permanent neonatal diabetes and neurological abnormalities. Diabetes. 2010;59(9):2326–2331. doi:10.2337/db10-0011
20. Senee V, Chelala C, Duchatelet S, et al. Mutations in GLIS3 are responsible for a rare syndrome with neonatal diabetes mellitus and congenital hypothyroidism. Nat Genet. 2006;38(6):682–687. doi:10.1038/ng1802
21. Senee V, Vattem KM, Delepine M, et al. Wolcott-Rallison syndrome: clinical, genetic, and functional study of EIF2AK3 mutations and suggestion of genetic heterogeneity. Diabetes. 2004;53(7):1876–1883. doi:10.2337/diabetes.53.7.1876
22. Shalev SA, Tenenbaum-Rakover Y, Horovitz Y, et al. Microcephaly, epilepsy, and neonatal diabetes due to compound heterozygous mutations in IER3IP1: insights into the natural history of a rare disorder. Pediatr Diabetes. 2014;15(3):252–256. doi:10.1111/pedi.2014.15.issue-3
23. Shaw-Smith C, Flanagan SE, Patch AM, et al. Recessive SLC19A2 mutations are a cause of neonatal diabetes mellitus in thiamine-responsive megaloblastic anaemia. Pediatr Diabetes. 2012;13(4):314–321. doi:10.1111/j.1399-5448.2012.00855.x
24. Stanescu DE, Hughes N, Patel P, De Leon DD. A novel mutation in GATA6 causes pancreatic agenesis. Pediatr Diabetes. 2015;16(1):67–70. doi:10.1111/pedi.2015.16.issue-1
25. Yorifuji T, Kurokawa K, Mamada M, et al. Neonatal diabetes mellitus and neonatal polycystic, dysplastic kidneys: phenotypically discordant recurrence of a mutation in the hepatocyte nuclear factor-1 beta gene due to germline mosaicism. J Clin Endocrinol Metab. 2004;89(6):2905–2908. doi:10.1210/jc.2003-031828
26. Temple IK, Shield JP. Transient neonatal diabetes, a disorder of imprinting. J Med Genet. 2002;39(12):872–875. doi:10.1136/jmg.39.12.872
27. Mackay DJ, Callaway JL, Marks SM, et al. Hypomethylation of multiple imprinted loci in individuals with transient neonatal diabetes is associated with mutations in ZFP57. Nat Genet. 2008;40(8):949–951. doi:10.1038/ng.187
28. Kamiya M, Judson H, Okazaki Y, et al. The cell cycle control gene ZAC/PLAGL1 is imprinted–a strong candidate gene for transient neonatal diabetes. Hum Mol Genet. 2000;9(3):453–460. doi:10.1093/hmg/9.3.453
29. Flanagan SE, Mackay DJ, Greeley SA, et al. Hypoglycaemia following diabetes remission in patients with 6q24 methylation defects: expanding the clinical phenotype. Diabetologia. 2013;56(1):218–221. doi:10.1007/s00125-012-2766-z
30. Rafiq M, Flanagan SE, Patch AM, et al. Effective treatment with oral sulfonylureas in patients with diabetes due to sulfonylurea receptor 1 (SUR1) mutations. Diabetes Care. 2008;31(2):204–209. doi:10.2337/dc07-1785
31. Fujimura N, Tanaka E, Yamamoto S, Shigemori M, Higashi H. Contribution of ATP-sensitive potassium channels to hypoxic hyperpolarization in rat hippocampal CA1 neurons in vitro. J Neurophysiol. 1997;77(1):378–385. doi:10.1152/jn.1997.77.1.378
32. Carmody D, Pastore AN, Landmeier KA, et al. Patients with KCNJ11-related diabetes frequently have neuropsychological impairments compared with sibling controls. Diabet Med. 2016;33(10):1380–1386. doi:10.1111/dme.2016.33.issue-10
33. Landmeier KA, Lanning M, Carmody D, Greeley SAW, Msall ME. ADHD, learning difficulties and sleep disturbances associated with KCNJ11-related neonatal diabetes. Pediatr Diabetes. 2017;18(7):518–523. doi:10.1111/pedi.12428
34. Edghill EL, Flanagan SE, Patch AM, et al. Insulin mutation screening in 1044 patients with diabetes: mutations in the INS gene are a common cause of neonatal diabetes but a rare cause of diabetes diagnosed in childhood or adulthood. Diabetes. 2008;57(4):1034–1042. doi:10.2337/db07-1405
35. Dimova R, Tankova T, Gergelcheva I, Tournev I, Konstantinova M. A family with permanent neonatal diabetes due to a novel mutation in INS gene. Diabetes Res Clin Pract. 2015;108(2):e28–30. doi:10.1016/j.diabres.2015.02.021
36. Bottino M, Cowett RM, Sinclair JC. Interventions for treatment of neonatal hyperglycemia in very low birth weight infants. Cochrane Database Syst Rev. 2011;10:CD007453.
37. Park JH, Shin SY, Shim YJ, Choi JH, Kim HS. Multiple daily injection of insulin regimen for a 10-month-old infant with type 1 diabetes mellitus and diabetic ketoacidosis. Ann Pediatr Endocrinol Metab. 2016;21(2):96–98. doi:10.6065/apem.2016.21.2.96
38. Passanisi S, Timpanaro T, Lo Presti D, Mammi C, Caruso-Nicoletti M. Treatment of transient neonatal diabetes mellitus: insulin pump or insulin glargine? Our experience. Diabetes Technol Ther. 2014;16(12):880–884. doi:10.1089/dia.2014.0055
39. Hwang MJ, Newman R, Philla K, Flanigan E. Use of insulin glargine in the management of neonatal hyperglycemia in an ELBW infant. Pediatrics. 2018;141(Suppl 5):S399–S403. doi:10.1542/peds.2016-1638
40. Beardsall K, Pesterfield CL, Acerini CL. Neonatal diabetes and insulin pump therapy. Arch Dis Child Fetal Neonatal Ed. 2011;96(3):F223–224. doi:10.1136/adc.2010.196709
41. Bharucha T, Brown J, McDonnell C, et al. Neonatal diabetes mellitus: insulin pump as an alternative management strategy. J Paediatr Child Health. 2005;41(9–10):522–526. doi:10.1111/jpc.2005.41.issue-9-10
42. Rabbone I, Barbetti F, Marigliano M, et al. Successful treatment of young infants presenting neonatal diabetes mellitus with continuous subcutaneous insulin infusion before genetic diagnosis. Acta Diabetol. 2016;53(4):559–565. doi:10.1007/s00592-015-0828-7
43. Rabbone I, Barbetti F, Gentilella R, et al. Insulin therapy in neonatal diabetes mellitus: a review of the literature. Diabetes Res Clin Pract. 2017;129:126–135. doi:10.1016/j.diabres.2017.04.007
44. Pearson ER, Flechtner I, Njolstad PR, et al. Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. N Engl J Med. 2006;355(5):467–477. doi:10.1056/NEJMoa061759
45. Bowman P, Sulen A, Barbetti F, et al. Effectiveness and safety of long-term treatment with sulfonylureas in patients with neonatal diabetes due to KCNJ11 mutations: an international cohort study. Lancet Diabetes Endocrinol. 2018;6(8):637–646. doi:10.1016/S2213-8587(18)30106-2
46. Thurber BW, Carmody D, Tadie EC, et al. Age at the time of sulfonylurea initiation influences treatment outcomes in KCNJ11-related neonatal diabetes. Diabetologia. 2015;58(7):1430–1435. doi:10.1007/s00125-015-3593-9
47. Oztekin O, Durmaz E, Kalay S, Flanagan SE, Ellard S, Bircan I. Successful sulfonylurea treatment of a neonate with neonatal diabetes mellitus due to a novel missense mutation, p.P1199L, in the ABCC8 gene. J Perinatol. 2012;32(8):645–647. doi:10.1038/jp.2012.46
48. Jesic MM, Jesic MD, Maglajlic S, Sajic S, Necic S. Successful sulfonylurea treatment of a neonate with neonatal diabetes mellitus due to a new KCNJ11 mutation. Diabetes Res Clin Pract. 2011;91(1):e1–3. doi:10.1016/j.diabres.2010.09.027
49. Wambach JA, Marshall BA, Koster JC, White NH, Nichols CG. Successful sulfonylurea treatment of an insulin-naive neonate with diabetes mellitus due to a KCNJ11 mutation. Pediatr Diabetes. 2010;11(4):286–288. doi:10.1111/j.1399-5448.2009.00557.x
50. Zwaveling-Soonawala N, Hagebeuk EE, Slingerland AS, Ris-Stalpers C, Vulsma T, van Trotsenburg AS. Successful transfer to sulfonylurea therapy in an infant with developmental delay, epilepsy and neonatal diabetes (DEND) syndrome and a novel ABCC8 gene mutation. Diabetologia. 2011;54(2):469–471. doi:10.1007/s00125-010-1981-8
51. Kumaraguru J, Flanagan SE, Greeley SA, et al. Tooth discoloration in patients with neonatal diabetes after transfer onto glibenclamide: a previously unreported side effect. Diabetes Care. 2009;32(8):1428–1430. doi:10.2337/dc09-0280
52. Klupa T, Skupien J, Mirkiewicz-Sieradzka B, et al. Efficacy and safety of sulfonylurea use in permanent neonatal diabetes due to KCNJ11 gene mutations: 34-month median follow-up. Diabetes Technol Ther. 2010;12(5):387–391. doi:10.1089/dia.2009.0165
53. Babiker T, Vedovato N, Patel K, et al. Successful transfer to sulfonylureas in KCNJ11 neonatal diabetes is determined by the mutation and duration of diabetes. Diabetologia. 2016;59(6):1162–1166. doi:10.1007/s00125-016-3921-8
54. Beltrand J, Elie C, Busiah K, et al. Sulfonylurea therapy benefits neurological and psychomotor functions in patients with neonatal diabetes owing to potassium channel mutations. Diabetes Care. 2015;38(11):2033–2041. doi:10.2337/dc15-0837
55. Mohamadi A, Clark LM, Lipkin PH, Mahone EM, Wodka EL, Plotnick LP. Medical and developmental impact of transition from subcutaneous insulin to oral glyburide in a 15-yr-old boy with neonatal diabetes mellitus and intermediate DEND syndrome: extending the age of KCNJ11 mutation testing in neonatal DM. Pediatr Diabetes. 2010;11(3):203–207. doi:10.1111/j.1399-5448.2009.00548.x
56. Mlynarski W, Tarasov AI, Gach A, et al. Sulfonylurea improves CNS function in a case of intermediate DEND syndrome caused by a mutation in KCNJ11. Nat Clin Pract Neurol. 2007;3(11):640–645. doi:10.1038/ncpneuro0640
57. Slingerland AS, Nuboer R, Hadders-Algra M, Hattersley AT, Bruining GJ. Improved motor development and good long-term glycaemic control with sulfonylurea treatment in a patient with the syndrome of intermediate developmental delay, early-onset generalised epilepsy and neonatal diabetes associated with the V59M mutation in the KCNJ11 gene. Diabetologia. 2006;49(11):2559–2563. doi:10.1007/s00125-006-0407-0
58. Shah RP, Spruyt K, Kragie BC, Greeley SA, Msall ME. Visuomotor performance in KCNJ11-related neonatal diabetes is impaired in children with DEND-associated mutations and may be improved by early treatment with sulfonylureas. Diabetes Care. 2012;35(10):2086–2088. doi:10.2337/dc11-2225
59. Lahmann C, Kramer HB, Ashcroft FM. Systemic administration of glibenclamide fails to achieve therapeutic levels in the brain and cerebrospinal fluid of rodents. PLoS ONE. 2015;10(7):e0134476. doi:10.1371/journal.pone.0134476
60. Lau E, Correia C, Freitas P, et al. Permanent neonatal diabetes by a new mutation in KCNJ11: unsuccessful switch to sulfonylurea. Arch Endocrinol Metabol. 2015;59(6):559–561. doi:10.1590/2359-3997000000076
61. Carmody D, Bell CD, Hwang JL, et al. Sulfonylurea treatment before genetic testing in neonatal diabetes: pros and cons. J Clin Endocrinol Metab. 2014;99(12):E2709–2714. doi:10.1210/jc.2014-2494
62. Katanic D, Vorgucin I, Hattersley A, et al. A successful transition to sulfonylurea treatment in male infant with neonatal diabetes caused by the novel abcc8 gene mutation and three years follow-up. Diabetes Res Clin Pract. 2017;129:59–61. doi:10.1016/j.diabres.2017.04.021
63. Li X, Xu A, Sheng H, et al. Early transition from insulin to sulfonylureas in neonatal diabetes and follow-up: experience from China. Pediatr Diabetes. 2018;19(2):251–258. doi:10.1111/pedi.2018.19.issue-2
64. Philla KQ, Bauer AJ, Vogt KS, Greeley SA. Successful transition from insulin to sulfonylurea therapy in a patient with monogenic neonatal diabetes owing to a KCNJ11 F333L [Corrected] mutation. Diabetes Care. 2013;36(12):e201. doi:10.2337/dc13-1690
65. Iglesias Platas I, Thio Lluch M, Pociello Alminana N, Morillo Palomo A, Iriondo Sanz M, Krauel Vidal X. Continuous glucose monitoring in infants of very low birth weight. Neonatology. 2009;95(3):217–223. doi:10.1159/000165980
66. Fu Z, Gilbert ER, Liu D. Regulation of insulin synthesis and secretion and pancreatic Beta-cell dysfunction in diabetes. Curr Diabetes Rev. 2013;9(1):25-53 doi:10.2174/157339913804143225
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.