")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 16
Rcn3 Suppression Was Responsible for Partial Relief of Emphysema as Shown by Specific Type II Alveolar Epithelial Cell Rcn3 CKO Mouse Model
Authors Zhang Q, Wang T, Jin J, Shi X, Huang A, Ma Z, Li J, Wang S, Ma RZ , Fang Q
Received 21 July 2020
Accepted for publication 11 December 2020
Published 26 January 2021 Volume 2021:16 Pages 147—158
DOI https://doi.org/10.2147/COPD.S272711
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Russell
Qianyu Zhang,1,* Tong Wang,1,* Jiawei Jin,1,2 Xiaoqian Shi,2 Aiben Huang,3 Zhenru Ma,1 Jiujie Li,4 Shiyu Wang,4 Runlin Z. Ma,4,5 Qiuhong Fang1
1Department of Respiratory and Critical Care Medicine, Beijing Chaoyang Hospital, Capital Medical University, Beijing 100020, People’s Republic of China; 2The Clinical Research Center, Beijing Chaoyang Hospital, Capital Medical University, Beijing 100020, People’s Republic of China; 3Beijing Shijitan Hospital, Capital Medical University, Beijing 100038, People’s Republic of China; 4School of Life Sciences, University of Chinese Academy of Sciences, Beijing 100049, People’s Republic of China; 5State Key Laboratory of Molecular Developmental Biology, Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, Beijing 100101, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Qiuhong Fang
Department of Respiratory and Critical Care Medicine, Beijing Chaoyang Hospital, Capital Medical University, No. 8 Gongren Tiyuchang South Road, Chaoyang District, Beijing 100020, People’s Republic of China
Tel +86 10-85231451
Fax +86 10-85231412
Email [email protected]
Runlin Z. Ma
Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, S2-316 Building #2, West Beichen Road, Chaoyang District, Beijing 100101, People’s Republic of China
Tel +86 10-64806599
Fax +86 10-64806598
Email [email protected]
Background: Chronic obstructive pulmonary disease (COPD), characterized by irreversible airflow limitation, is a highly prevalent lung disease worldwide and imposes increasing disease burdens globally. Emphysema is one of the primary pathological features contributing to the irreversible decline of pulmonary function in COPD patients, but the pathogenetic mechanisms remain unclear. Reticulocalbin 3 (Rcn3) is an endoplasmic reticulum (ER) lumen protein localized in the secretory pathway of living cells. Rcn3 in type II alveolar epithelial cell (AECIIs) has been reported to play a critical role in regulating perinatal lung development and bleomycin-induced lung injury-repair processes. We hypothesized that Rcn3 deficiency is associated with the development of emphysema during COPD, which is associated with the dysfunction of injury-repair modulated by alveolar epithelial cells.
Materials and Methods: We examined Rcn3 expression in lung specimens from COPD patients and non-COPD control patients undergoing lung lobectomy or pneumonectomy. Two mouse models of emphysema were established by cigarette smoke (CS) exposure and intratracheal instillation of porcine pancreatic elastase (PPE). Rcn3 expression was detected in the lung tissues from these mice. Furthermore, conditional knockout (CKO) mice with Rcn3 deletion specific to AECIIs were used to explore the role of Rcn3 in PPE-induced emphysema progression. Rcn3 protein expression in lung tissues was evaluated by Western blot and immunohistochemistry. Rcn3 mRNA expression in lung tissues was detected by qPCR.
Results: Rcn3 expression was significantly increased in the lung specimens from COPD patients versus non-COPD patients and the level of Rcn3 increase was associated with the degree of emphysema. Rcn3 expression were also significantly up-regulated in both CS-induced and PPE-induced emphysematous mouse lungs. Moreover, the selective ablation of Rcn3 in AECIIs significantly alleviated severity of the mouse emphysema in response to intratracheal installation of PPE.
Conclusion: Our data, for the first time, indicated that suppression of Rcn3 expression in AECIIs has a beneficial effect on PPE-induced emphysema.
Keywords: COPD, emphysema, Rcn3, type II alveolar epithelial cell
Introduction
Chronic Obstructive Pulmonary Disease (COPD) is a highly prevalent lung disease worldwide characterized by persistent and progressive airflow limitation.1 The morbidity and mortality of COPD have gone up dramatically in the past decades, and it will become the third leading cause of death by 2020. According to estimates from the Burden of Obstructive Lung Diseases program, more than 384 million people suffer from COPD worldwide.2 In addition, there are around 300 million annual deaths involving COPD worldwide.3 Cigarette smoke (CS) is a major risk factor of the development of COPD.4 Chronic bronchitis and emphysema are the two main pathological features of COPD contributing to airflow limitation.5 Emphysema is characterized by the enlargement of air spaces and destruction of the parenchymal structure, and is believed to be associated with deregulated epithelial injury-repair.1,6 Effective treatment for emphysema is limited, due to the unclear pathogenetic mechanisms, especially the regulation of alveolar structural recovery and reconstruction in emphysema.
Reticulocalbin3 (Rcn3) is a Ca2+-binding ER lumen protein, localized to the secretory pathway.7 It belongs to the Cab45/Rcn/ERC45/Calumenin family,8 the biological function of which is still far from clear. We have previously reported that Rcn3 was expressed in a variety of mouse tissues with the highest expression in the lung.9 Consistently, high expression of Rcn3 in human lung tissue has also been reported by Hou Yu et al.10 We further revealed that Rcn3 knockout mice died from neonatal respiratory distress due to the impaired maturation of type II alveolar epithelial cells (AECIIs), suggesting its potential role in regulating AECIIs function.9 In addition, we demonstrated that selective deletion of Rcn3 in AECIIs exacerbated bleomycin-induced lung fibrosis.11 Since AECIIs play a vital role in the pathogenesis of emphysema, it is therefore our hypothesis that Rcn3 in AECIIs might have a role in the regulation of emphysema development.
In this study, we first investigated the expression of Rcn3 in the lung tissues from COPD patients. Rcn3 expression level was also examined in the emphysema lungs from CS-exposed and PPE-exposed mice. Furthermore, the potential effect of Rcn3 on the development of emphysema was studied by selective Rcn3 knockout in AECIIs. A marked up-regulation of Rcn3 expression was observed in the lung tissues from COPD patients and higher levels of Rcn3 were detected in the lung tissues from both mouse models of emphysema. In addition, the selective deletion of Rcn3 was shown to attenuate the degree of emphysematous changes significantly.
Materials and Methods
The Collection of Surgically Resected Specimens
The human lung tissues were obtained from resected specimens because of lung nodule within 30 minutes after resection from the Department of Thoracic Surgery, Beijing Shijitan Hospital between September 2013 and July 2015. The lung tissues (approximately 0.5–1 cm3) were collected at least 10 cm away from the nodule edge. The results of pulmonary function tests and hematoxylin and eosin (H&E)-stained sections were collected in all specimens. Based on the pathological changes of emphysema and the presence of a post-bronchodilator forced expiratory volume in 1-second to forced vital capacity (FEV1/FVC) < 0.70,12 patients were assigned into the COPD group or the non-COPD group (without other lung diseases except the lung nodule).
Sixteen fresh lung-tissue samples were used for Western blot, qualitative PCR analyses, HE staining and immunohistochemistry and 54 paraffin-embedded samples were used for HE staining and IHC assays. A signed Informed Consent was obtained from each patient. The study was approved by the Ethics Committee for the Clinical Research of the Beijing Shijitan Hospital.
Animals
C57BL/6 mice (6 weeks old) were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. The doxycycline-induced AECII-selective Rcn3 deletion (CKO) mice were established and identified by Runlin Z. Ma Lab in the Institute of Genetics and Developmental Biology, Chinese Academy of Sciences.11 All the animals were housed in a temperature- and humidity-controlled room with free access to water and standard laboratory food. All animal procedures were approved by the Animal Care and Ethics Committee of Beijing Chaoyang Hospital and were performed following the guide for the Care and Use of Laboratory Animals of Beijing Chaoyang Hospital.
Cigarette Smoke-Exposed Emphysema Model
A total of 32 C57 BL/6J (6-week-old) male mice were acclimated for 2 weeks before the experimental procedures (12-h light/dark cycle, 25-26°C, and 50-60% humidity), and then mice were randomly assigned into the following two groups (n=16 per group): (1) Control group; (2) CS (exposed to cigarette smoke) group. The CS groups were exposed to CS generated by burning cigarettes (Baisha, Hunan, China; tar 11 mg, nicotine 0.9 mg, carbon monoxide 12 mg) using the Tobacco Smoke Inhalation Experiment System for small animals (Model SIS-CS, Shibata Scientific Technology Ltd, Tokyo, Japan). A total of 36 cigarettes were used for each exposure procedure and the level of total particulate matter (TSP) was maintained at 400 mg/m3. Mice were exposed to CS 5 days a week for 2 hours a day, for 24 weeks.13 The Control mice were exposed to room air without smoking. Following 24-week CS exposure, the lung tissues were collected for further analyses. This experimental protocol was approved by the Animal Ethics Committee of Beijing Chaoyang Hospital.
Protease-Induced Emphysema Model
C57BL/6 female mice (weight 20–25 g, age 8 weeks) were randomly assigned to the following groups: Control group mice were treated with saline solution (0.9% NaCl, 60 ul) and porcine pancreatic elastase (PPE) groups were treated once intratracheally with PPE (E1250, Sigma-Aldrich, St. Louis, Mo, USA).14 The PPE groups were treated with PPE at three different doses: 0.2 U, 0.3 U, 0.4 U/per mouse in 60 ul of saline solution. All animals were euthanized with cervical dislocation after 4 weeks of PPE administration and lung tissues were collected.
Generation of the Doxycycline-Induced AECII-Selective Rcn3 Deletion (CKO) Mice
The transgenic female mice with selective deletion of Rcn3 in AECIIs were generated using the Cre/loxp method. The tet-on system (triple-transgenic SP-C–rtTAtg/-/(tetO)7CMV-Cretg/-/Rcn3fl/fl, CKO mice) was used to ablate Rcn3 in adulthood by doxycycline (DOX) administration.11 Control mice were the littermates. Two groups of mice both had DOX in the drinking water at the age of 8 weeks old, until they had 0.15 U PPE/per mouse administration at the age of 10 weeks. Four weeks later, the lung tissues were collected.
Lung Histology and Morphometry
The right lower lobe was inflated by 10% phosphate-buffered formalin for at least 48 h before embedding in paraffin and then sectioned at a thickness of 4 μm. For morphometric studies, sections were stained with hematoxylin and eosin (H&E).15 To calculate the average interalveolar distance and the change in alveolar destruction, the mean linear intercept (MLI) was used as a modified method for morphometric assessment. A rectangular grid comprising 20 lines was drawn across 100× magnification H&E staining images (590.8 μm×886.2 μm) and the MLI was determined by dividing the total length of these lines (14,179.2 μm) by the total number of alveolar wall and grid line interceptions (5 fields were assessed at random for each lung).14
Immunohistochemistry (IHC) and Mean Optical Density Analysis
The paraffin-embedded slides were dewaxed, hydrated, and the antigen retrieved with the sodium citrate method for 5 minutes at high pressure.16 The slices were incubated in 3% hydrogen peroxide for 30 minutes and then were blocked for 30 minutes. The Anti-Rcn3 antibody was applied overnight in a wet box at 4°C. Next day, after washing off the primary antibody, the secondary antibody (Zhongshan Golden Bridge, Beijing, China) was applied for 30 minutes. Expression of the Rcn3 was visualized with DAB reagent (Zhongshan Golden Bridge, Beijing, China) after the secondary antibody was washed off. The nucleus was stained with hematoxylin and then the sections were dehydrated and covered.17
The mean optical density of Rcn3 positive cells in IHC assay was calculated by a quantitative dynamic program analysis with Image Pro Plus. Three random views were selected for each IHC image (low or high magnification). The optical density of immunohistochemical staining positive cells was divided by the total area of cells in each view in low magnification, the value representing the mean optical density of Rcn3 positive cells.18
Western Blotting
Protein concentration was quantified by the BCA method (CW Biotech, Beijing, China) and 30 µg of protein extracts from left lung tissue was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (8%). After electrophoresis, proteins were transferred to an NC membrane. The membrane was blocked with 5% skim milk at room temperature for 1 hour. The membranes were probed via overnight incubation at 4°C with each of mouse or rabbit antibodies raised against: (1) Rcn3 (1:500, Santa Cruz Biotechnology, Dallas, TX, USA); (2) GRP78 (1:2000, Santa Cruz Biotechnology, Dallas, TX, USA); (3) Cleaved caspase-3 (1:1000, Cell Signaling Technology, USA); (4) β-actin (1:5000, Santa Cruz Biotechnology, Dallas, TX, USA); (5) β-Tubulin (1:5000, Yeasen, Shanghai, China); (6) GAPDH (1:5000, Santa Cruz Biotechnology, Dallas, TX, USA); at 4°C overnight. After washing with TBST, secondary antibodies (Zhongshan Golden Bridge, Beijing, China) were applied at room temperature for 2 hours. Protein bands were visualized using ECL reagent. Image J software was used for subsequent quantification and statistical analysis.
Quantitative Real-Time PCR
Total RNA was extracted from the right upper lobe and middle lobe, using Trizol (Life Technology-Ambion, Grand Island, NY, USA). Quantitative real-time PCR was performed on an iQ iCycler (BioRad) with SYBR Green qPCR SuperMix (CW Biotech, Beijing, China) in combination with primers specific for mouse Rcn3, GRP78, Collagen I, MMP-9 or with primers specific for human Rcn3, GRP78. GAPDH or RLP19 were used as the internal control for human or mouse specimens. The melting curve was observed to identify the amplification specificity. Three replicates were run using the 2−ΔΔCt method and the cycling threshold (Ct) represented the number of gene amplification cycles when fluorescence threshold is reached.
Statistical Analysis
Statistical analysis was performed by GraphPad Prism 5. Statistical comparisons between two groups were performed using Student’s t-test (unpaired, 2-tailed). Chi-square test was used for comparison of two rates or two constituent ratios in the analysis of characteristics of the patients. Mann–Whitney U-test was also used in the analysis of characteristics of the patients. A p value of <0.05 indicated a significant difference between the groups. Independent experiments were repeated at least in triplicate and quantitative data were presented as mean ± SEM (standard error of the mean).
Results
Rcn3 Expression in the Lung Tissues from COPD Patients
A total of 70 lung specimens (44 from COPD patients and 26 from non-COPD patients) were obtained. The clinical characteristics including average age, gender, smoking history, FEV1 post bronchodilator (% predicted) and FEV1/FVC (%) are shown in Table 1. Thirty-six male COPD patients and 12 male non-COPD patients enrolled in our study. The average age was 68.9 ± 8.3 (years) for COPD patients, and was 61.4 ± 6.9 (years) for controls. Forty-three COPD patients and 3 non-COPD patients had a history of smoking. Compared with the non-COPD group, patients with COPD exhibited marked decreases in FEV1 % predicted (64.07±2.477 compared with 100.2±2.847, p<0.001), and in FEV1/FVC (60.95±1.119 compared with 82.26±0.99, p<0.001).
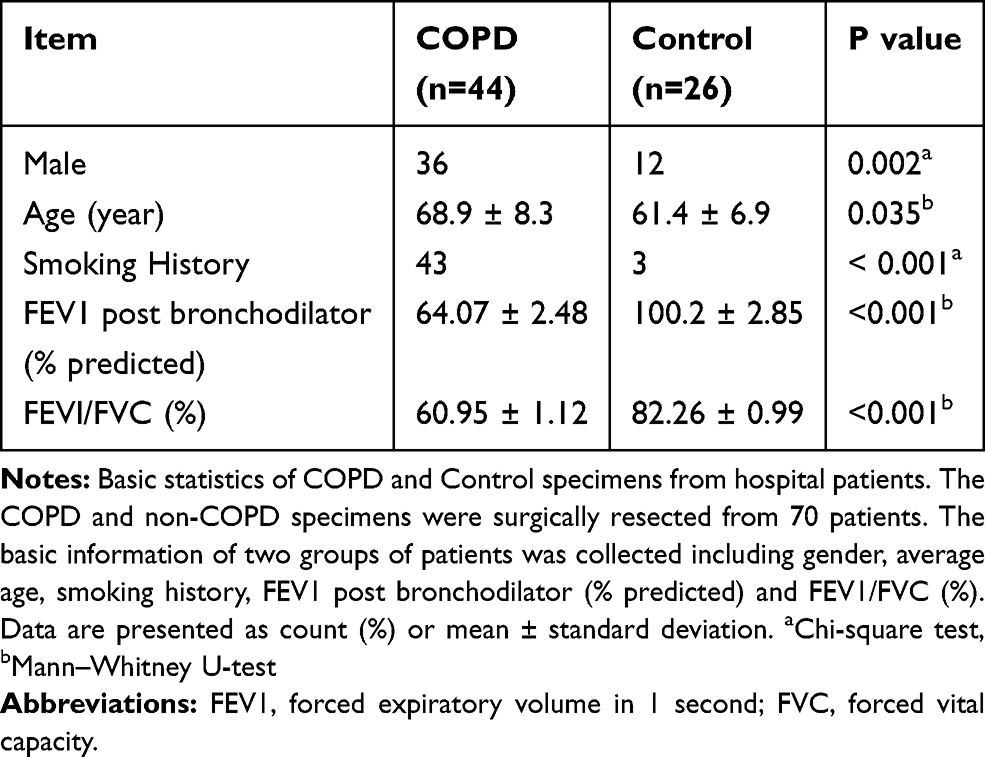 |
Table 1 Basic Information of Patients in COPD Group and Control Group |
H&E analysis of the lung specimens showed destruction of the alveolar walls and enlargement of the alveolar spaces corresponding with the morphological changes of emphysema in COPD patients (Figure 1A). The MLI of COPD patients was 1.3-fold (p<0.05) of that measured in lung sections from control patients (104.8 ± 5.789 μm of COPD patients vs 82.78 ± 9.417 μm of non-COPD patients, p<0.05, Figure 1B). IHC results demonstrated Rcn3 was distributed in airway and alveolar epithelial cells and lung interstitium (Figure 1A). Mean optical density analysis revealed that Rcn3 expression was significantly higher in the COPD patients than in the Control group (p<0.05, Figure 1C). Quantitative PCR and Western blotting further confirmed that both mRNA and protein levels of Rcn3 were significantly higher in the lung specimens from COPD patients than in those from the control patients (both p<0.05, Figure 1D and E). Consistently, both mRNA and protein expression levels of GRP78 were elevated in lungs of these COPD patients (both p<0.05, Figure 1D and E).
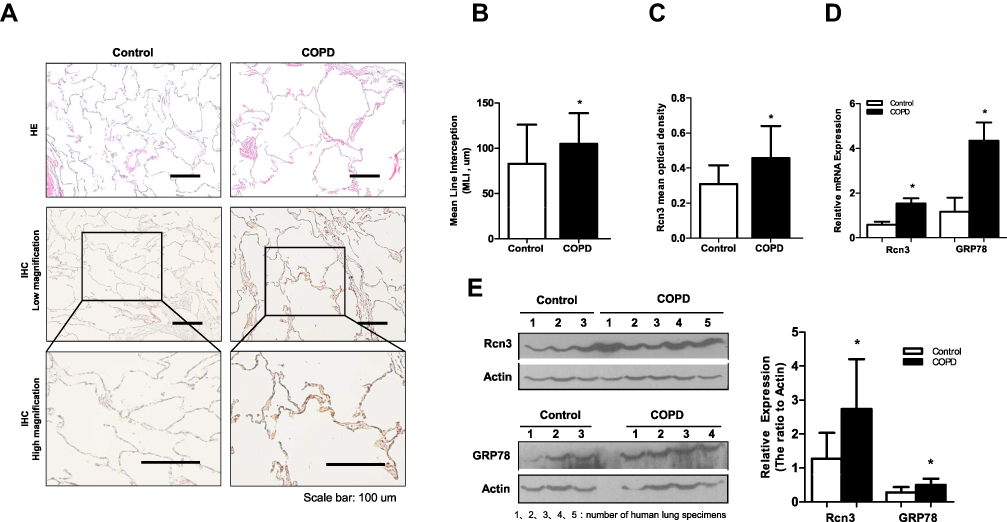 |
Figure 1 Rcn3 expression in lung tissues from COPD patients. A total of 70 specimens were divided into COPD group and Control group according to the pulmonary function test and pathological changes of emphysema. (A) Photomicrography of pulmonary parenchyma stained with H&E (upper panels). Bar size: 100 μm; IHC assays of Rcn3 from COPD group lungs and Control group lungs (low and high magnifications, lower panels). Bar size: 100 μm; (B) The MLI analysis of H&E stained lung sections (82.78 ± 9.417 μm vs.104.8 ± 5.789 μm, n = 21 in Control group and n = 34 in COPD group, p<0.05); (C) Quantification of the mean optical density of Rcn3 expression in the lung tissues according to the Rcn3 IHC assays (3 random views, 100× amplification for one subject, n = 11–12 per group, p<0.05); (D) Qualitative PCR analyses of the mRNA expressions of Rcn3 and GRP78; the data were normalized to the GAPDH content and were analyzed by the 2−ΔΔCt method relative to the Control group; n = 6 in Control group and n = 10 in COPD group; (E) The protein levels of Rcn3 and GRP78 were examined by Western blot analysis and the ratios to Actin presented by bar graph; n = 6 in Control group and n = 10 in COPD group. Data presented as mean ± SEM; *p<0.05 versus the Control group. |
Rcn3 Expression in the Lung Tissues from CS-Exposed Mouse Model of Emphysema
To further characterize the role of Rcn3 in the pathogenesis of emphysema, a well-established experimental model of emphysema in response to chronic CS exposure was used in the current study. C57BL/6 mice displayed mildly damaged alveolar septa and enlarged spaces in response to chronic CS exposure for 6 months (Figure 2A). MLI of CS-exposed mice were obviously larger than those of the air-exposed ones (56.81 ± 3.053 μm of CS group mice vs 44.00 ± 2.528 μm of Control group mice, p<0.01, Figure 2B). Rcn3 expression was significantly up-regulated in CS-exposed emphysema mice compared with that of control animals either by immunohistochemistry in low and high magnification (p<0.01, Figure 2A lower panels and Figure 2C) or by immunoblot (p<0.05, Figure 2D). Consistently, chronic CS exposure caused a significant increase in Rcn3 mRNA expression in the lung tissues (p<0.05, Figure 2E). In addition, both protein and mRNA expressions of GRP78 were significantly increased in the lung tissues from emphysema mice compared with the controls (p<0.05, p<0.01, respectively, Figure 2D, 2E). Protein level of Cleaved caspase-3 was significantly increased in the CS group mice compared with air-exposed mice (p<0.05, Figure 2D).
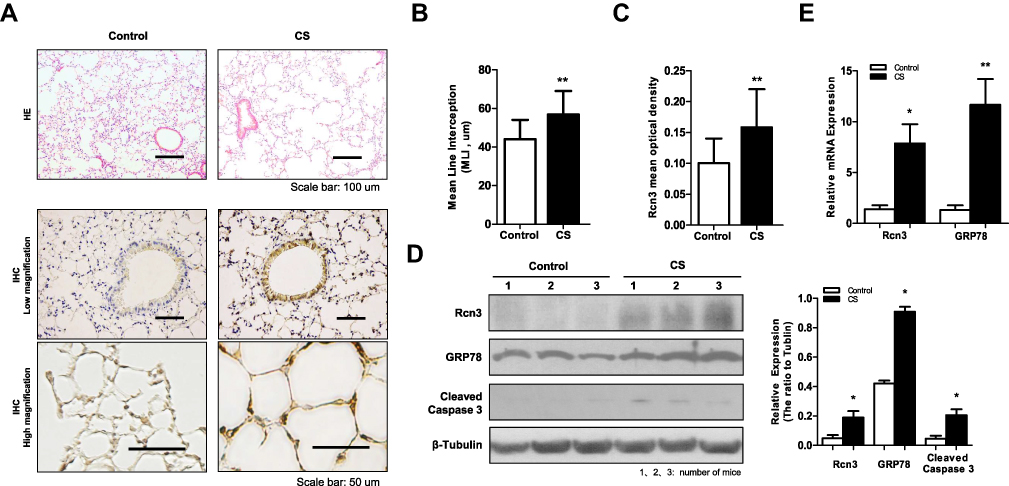 |
Figure 2 Rcn3 expression in lung tissues from CS-induced mouse model of emphysema. A total of 16 C57BL/6 mice (6 weeks old) were exposed to cigarette smoke for 6 months as emphysema mice (CS group) and 16 C57BL/6 mice were exposed to air for 6 months as Control mice (Control group). (A) Photomicrography of pulmonary parenchyma stained with H&E (upper panels). Bar size: 100 μm; IHC assays of Rcn3 (low and high magnifications, lower panels). Bar size: 50 μm; (B) Morphometric analysis of the MLI ((44.00 ± 2.528 μm vs.56.81 ± 3.053 μm, n = 16 per group, p<0.01); (C) The mean optical density of Rcn3 expression was measured in the lung tissues according to the Rcn3 IHC assays (3 random views, 100× amplification for one subject, n = 16 per group, p<0.01); (D) Representative WB for protein levels of Rcn3, GRP78 and Cleaved caspase-3 in lungs and the ratios to β-Tubulin presented by bar graph, n = 8 per group; (E) Expressions of Rcn3 and GRP78 mRNA were determined with Qualitative PCR analysis, n = 9–10 per group. Data presented as mean ± SEM; *p<0.05, **p<0.01 versus the Control group. |
Rcn3 Expression in the Lung Tissues of PPE-Induced Emphysema Model
Rcn3 expression was further examined in the mouse model of emphysema induced by administration of PPE, which was established at day 28 after PPE instillation. Findings of H&E staining showed that all three concentrations (0.2 U, 0.3 U and 0.4 U) of PPE resulted in prominent emphysematous-like changes, while the lungs from the sham mice kept intact alveolar septa with preserved air spaces. Among the three PPE-treatment groups, 0.3U-PPE-group presented the most severe emphysematous alteration (Figure 3A), which was further confirmed by morphometry. In this regard, MLIs (56.10 ± 2.45 μm, 98.23 ± 3.21 μm and 71.22 ± 2.84 μm) measured in the emphysema mice induced by the three concentrations of PPE (0.2 U, 0.3 U and 0.4 U) were significantly larger than that (40.58 ± 0.53 μm) in the saline group mice (p<0.05, p<0.001, and p<0.001, respectively). MLIs of the 0.2U- and 0.4U-PPE groups were lower than that of the 0.3U-PPE group (both p<0.001, Figure 3B). Rcn3 protein expression was significantly increased in PPE-treated groups compared with controls (p<0.01, Figure 3C). The 0.3U-PPE group presented the highest Rcn3 level among the three PPE groups, which was in accordance with the predominant histological changes and MLI measurement in these mice. In addition, enhanced Rcn3 expression occurs at the transcriptional level, evidenced by the fact that the Rcn3 mRNA level was significantly increased by 3.42 times in the 0.3U-PPE-treatment mice versus controls (p<0.001, Figure 3D).
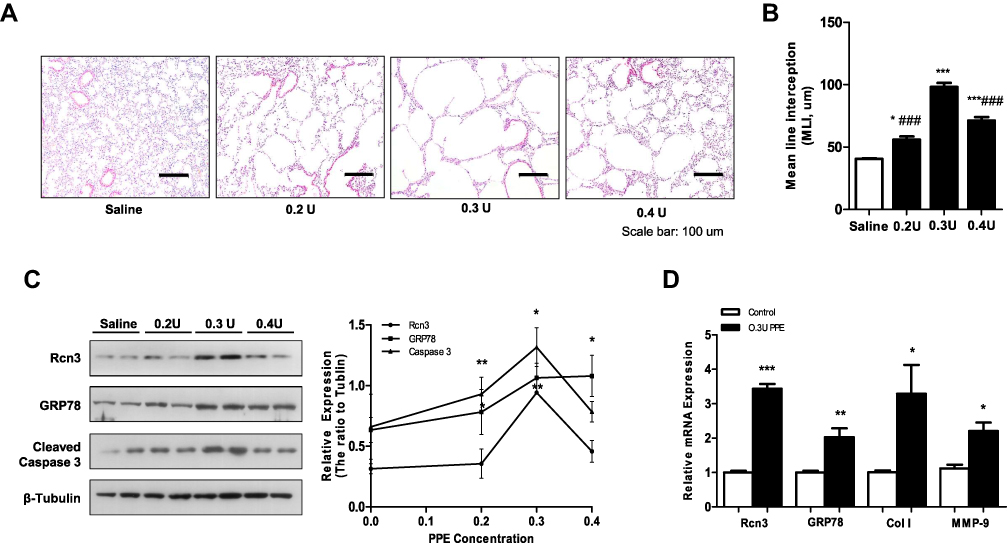 |
Figure 3 Rcn3 expression in lung tissues from the elastase-induced mouse model of emphysema. (A) Lungs after saline or PPE instillation at 0.2 U, 0.3 U and 0.4 U/per mouse were compared in H&E-stained tissue sections. Bar size: 100 μm; (B) The MLI analysis of lung sections after saline or PPE instillation (40.58 ± 0.53 μm, 56.10 ± 2.45 μm, 98.23 ± 3.21 μm and 71.22 ± 2.84 μm, respectively, n = 5 per group); (C) The PPE instillation induced the protein expressions of Rcn3, GRP78, Cleaved caspase-3, and the ratios to β-Tubulin expression are represented by graphs, n = 5 per group; (D) Qualitative PCR analysis of the mRNA expressions of Rcn3, GRP78, Collagen I (Col (I) and MMP-9 in the lungs at 4 weeks after PPE (0.3 U/per mouse) or saline treatment, n = 5 per group. Data presented as mean ± SEM; *p<0.05, **p<0.01, ***p<0.001, versus the Saline group, ###p<0.001, versus the 0.3 U PPE group. |
In addition, protein levels of GRP78 and Cleaved caspase-3 were significantly increased in the three PPE experimental groups compared with that of the saline group, and the 0.3U-PPE group presented the highest level of Cleaved caspase-3 (p<0.05, Figure 3C). Interestingly, compared with the groups of lower PPE concentrations, 0.4U-PPE-treatment augmented protein level of GRP78 (p<0.05, Figure 3C). In addition, mRNA expressions of GRP78, Collagen I (Col I) and MMP-9 were up-regulated in the 0.3U-PPE-treatment mice (p<0.01, p<0.05, and p<0.05, respectively, Figure 3D) compared with the Saline group mice.
Selective Deletion of Rcn3 in the Mouse AECIIs Alleviated Severity of PPE-Induced Emphysema
To further investigate the role of Rcn3 in the development of emphysema, AECII-selective Rcn3 knockout (CKO) mice were successfully established in our laboratory and used to induce a mouse model of emphysema by PPE instillation. In the current study, the immunohistochemical staining in CKO mice displayed a virtually complete loss of Rcn3 staining primarily observed in the corner of alveoli, in a distribution consistent with AECIIs (Figure 4A). Findings of H&E staining showed that the lungs from saline-treated Control mice and CKO mice kept intact alveolar septa with preserved air spaces (Figure 4B and C). In addition, no statistical difference of Rcn3, GRP78, Cleaved caspase-3, MMP-9 and Collagen I expression at protein or mRNA level was observed between saline-treated two groups (Figure 4D and E).
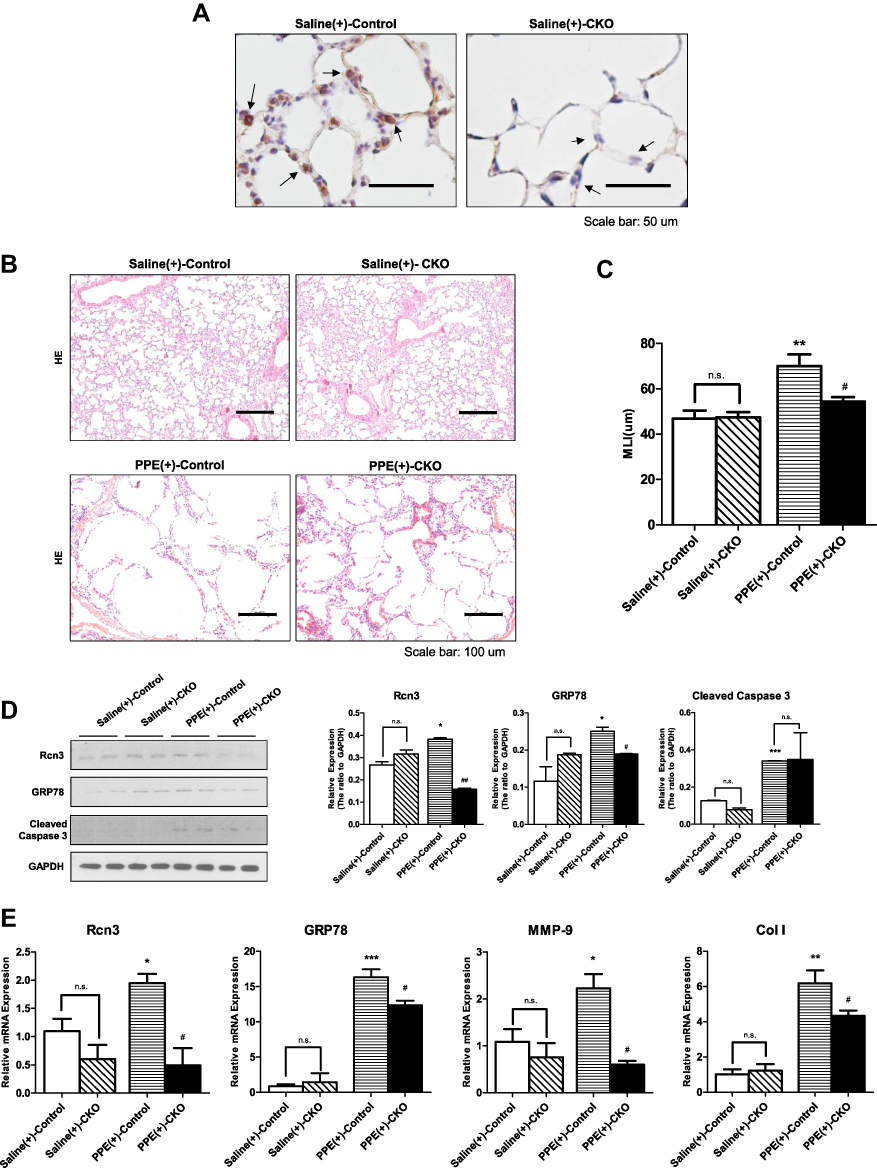 |
Figure 4 The selective deletion of Rcn3 in AECIIs alleviated elastase-induced emphysema. (A) IHC assays of Rcn3 from lungs in saline-treated WT mice or saline-treated CKO mice (arrows indicate the corners of alveoli, suggestive of AECIIs). Bar size: 50 μm; (B) H&E staining of lung tissues from mice in Saline(+)-Control, Saline(+)-CKO, PPE(+)-Control and PPE(+)-CKO groups. Bar size: 100 μm; (C) The MLI analysis of lung sections from the four groups of mice (46.85 ± 3.56 μm, 47.45 ± 2.268 μm, 70.07 ± 5.067 μm, and 54.34 ± 1.977 μm, respectively, n = 9–10 per group); (D) The protein levels of Rcn3, GRP78 and Cleaved caspase-3 in Saline(+)-Control, Saline(+)-CKO, PPE(+)-Control, and PPE(+)-CKO mice were examined by Western blot analysis and the ratios to GAPDH presented by bar graph, n = 5 per group; (E) Qualitative PCR analysis of the mRNA expressions of Rcn3, GRP78, MMP-9, and Collagen I in Saline(+)-Control, Saline(+)-CKO, PPE(+)-Control, and PPE(+)-CKO mice, n = 5 per group. Data presented as mean ± SEM; *p<0.05, **p<0.01, ***p<0.001, versus Saline(+)-Control group; #p<0.05, ##p<0.01, versus PPE(+)-Control group. Abbreviations: n.s., not significant; DOX, doxycycline. |
The H&E staining of sections after PPE instillation showed more severe emphysematous alteration in both CKO and Control mice compared with the saline-treated mice (Figure 4B). MLI measured in PPE(+)-Control (70.07 ± 5.067 μm) were significantly larger than that in the Saline(+)-Control mice (46.85 ± 3.56 μm, p<0.01). Meanwhile, PPE(+)-CKO mice showed fewer lung mechanical changes than PPE-induced Control mice, as evidenced by the MLI values (52.91±1.48 μm of CKO mice vs 70.07±5.07 μm of wild type mice, p<0.05, Figure 4C). The expressions of mRNA as well as protein of Rcn3 were significantly increased in PPE-treated Control mice compared with saline-treated mice (both p<0.05, Figure 4D and E). In PPE-treated CKO mice, Rcn3 expression was reduced in whole lung homogenates compared with that of PPE-treated Control mice (p<0.05 in mRNA and <0.01 in protein, Figure 4D and E). Figure 4D and E show that PPE instillation (0.15 U) significantly enhanced expressions of GRP78, Cleaved caspase-3, MMP-9 and Collagen I in Control mice compared with saline-treated mice at mRNA or protein level (p<0.05, p<0.001, p<0.05, and p<0.01, respectively), which are consistent with our previous findings, shown in Figure 3. Protein level of GRP78 was significantly decreased in PPE-treated CKO mice compared with Control mice (p<0.05), while no significant alteration of Cleaved caspase-3 protein was found in the two PPE groups (Figure 4D). Accordingly, mRNA expressions of GRP78, MMP-9 and Col I were significantly down-regulated in PPE-treated CKO mice lungs (all p<0.05, Figure 4E).
Discussion
In the present study, for the first time, we demonstrated that Rcn3 expression was significantly increased in the lungs from COPD patients. Consistently, Rcn3 expression was up-regulated in the lung tissues from both CS-exposed and elastase-induced mouse models of emphysema. Furthermore, the selective ablation of Rcn3 in AECIIs alleviated the degree of PPE-induced emphysema. These results indicate that increased Rcn3 expression in the lungs might facilitate the progression of emphysema, and thus participate in the development of COPD.
In the current study, lung specimens were obtained from patients who had experienced lobectomy or pneumonectomy, and were either COPD or non-COPD control subjects without any other airway and lung diseases except the lung nodule. The detailed clinical characteristics and demographic information about the control and COPD subjects are provided in Table 1, including the average age, gender, smoking history, FEV1 post bronchodilator (% predicted) and FEV1/FVC (%). Patients enrolled in the COPD group had histological emphysema changes in the lung sections in comparison with those of the non-COPD control patients, which was also confirmed by MLI measurement (Figure 1B). Consistent with our previous report,10 Rcn3 expression in human lung tissue was demonstrated, predominantly in the alveolar epithelial cells. Furthermore, Rcn3 expression was significantly higher in the lungs from COPD patients compared with those from the Control group. Although there were statistical differences in demographics and patient characteristics of the two groups of patients, Rcn3 required further study because it might be involved in the progression of COPD.
To further investigate the role of Rcn3 in emphysema, we established a mouse model of emphysema by chronic cigarette smoke exposure. Cigarette smoke has been known to be the main cause of COPD19 and Rabe et al. have demonstrated that a CS-induced mouse model of COPD usually promotes morphological changes characteristic of mild emphysema.20 Figure 2A and B show the emphysema pattern and increased MLI of CS group mice indicated the well-established mouse model of emphysema. Consistent with the mentioned findings in the patients, significant up-regulation of Rcn3 protein and mRNA expressions were confirmed in the lung tissues from CS-exposed mice compared with those from the Control group, suggesting Rcn3 may play a role in the pathogenesis of emphysema in response to cigarette smoke exposure.
In addition to emphysema, chronic bronchial and small airway inflammation are also responsible for the pathological changes in both COPD patients and chronic CS-exposed mice. To focus on the role of Rcn3 in emphysema, we then selected another mouse model of emphysema by intra-tracheal administration of PPE, which is now widely used in the study of emphysema. The emphysema model induced by PPE is mainly based on the currently accepted hypothesis of protease-antiprotease imbalance.21 PPE instillation induced more severe morphological alterations, corresponding to the histological features of emphysema and bullae in lungs, than those in chronic CS-exposure mice.22 Our study demonstrated that 0.4 U PPE instillation produced a less severe degree of emphysema than that of 0.3 U PPE instillation, which is similar to the results of Ip MP et al.,23 probably because 0.3 U PPE was the optimum concentration of enzyme to substrate (Figure 3C). Nevertheless, Rcn3 expression was also significantly increased in the emphysematous lungs induced by all three concentrations of PPE (0.2, 0.3, and 0.4 U) compared with the sham one, and the level of Rcn3 seemed to be correlated with the degree of emphysema, with the highest Rcn3 expression corresponding to the most severe emphysematous alterations (Figure 3C).
In our study, we detected that Rcn3 expression in the lung tissues was up-regulated in both the CS-exposed model and PPE-induced model of emphysema. To further confirm the role of Rcn3 in emphysema progression, gene targeting technology was needed in the study. Our previous study found that mice with whole body Rcn3 knockout died after birth because of respiratory failure.9 Therefore, selective Rcn3 knockout in AECIIs (CKO) mice were successfully established in our laboratory, and have been employed to study the function of Rcn3 in the development of lung fibrosis.11 However, the stability of AECII-selective Rcn3 knockout at more than 28 days was not clear in the previous study. In addition, elastase-induced emphysematous lesions in the lung tissues can be more prominent and induced in a shorter period of time by alternating the dose of elastase than in CS-exposed lungs. We hereby established the PPE-induced emphysema animal model in the AECII-selective Rcn3 knockout mice to further explore if the alteration of Rcn3 expression in lung tissue affected the histological changes of emphysema. Similar to our previous report, immunohistochemical staining in CKO mice (Figure 4A) displayed a virtually complete loss of Rcn3 in AECIIs with lung architecture kept normal, which confirmed the AECII-selective Rcn3 knockout mouse model was established successfully. Figure 4D and E showed that whole lung Rcn3 expression was similar between saline-treated CKO and WT mice at baseline.
Figure 4 shows that Rcn3 expression was significantly reduced in whole lung homogenates from PPE-treated CKO mice compared with those of PPE-treated WT mice. Moreover, selective deletion of Rcn3 in the AECIIs resulted in significant remission in severity of emphysematous changes in mice response to PPE instillation as evidenced by a 26.4% reduction in MLI. The data indicated that Rcn3 knockout specific to AECIIs was able to inhibit the whole lung Rcn3 expression challenged by PPE instillation, and further alleviate the degree of emphysema. These findings strongly illustrated that the increased expression of Rcn3 in lung tissue may promote the progression of emphysema and thus participate in the development of COPD.
The mechanisms behind emphysema formation are multifactorial. In this regard, deficient tissue repair was known to be involved in addition to the central role of inflammatory processes.1 Similar to the results of previous reports,24–26 elevated MMP-9 level was found in the PPE-induced emphysematous lungs along with augmented expression of Collagen I in the current animal models, which was considered to result from over-synthesis of ineffective collagen, implying the pathogenesis of over-injury or relative deficiency of repair.27 Therefore, MMP-9 and Collagen I expression were then detected in the lungs from emphysema mice with AECII-selective Rcn3 ablation. We found that MMP-9 and Collagen I were down-regulated in the lungs of PPE-treated CKO mice compared to those in the wild-type mice. Our previous study has shown that deletion of Rcn3 in ACEIIs facilitates bleomycin-induced lung fibrosis.11 In another in vitro study,3 Rcn3 was identified as a novel negative mediator of collagen production in human cardiac fibroblasts. These preliminary data indicated that Rcn3 might be involved in the injury-repair process by inhibiting repairing function. The present data suggested that up-regulation of Rcn3 in the lungs from COPD patients as well as from animal models of CS-exposed and PPE-induced emphysematous lungs might exacerbate emphysema through restraining the repair process.
An accumulation of misfolded and unfolded proteins in ER lumen under physiological or pathological stress results in ER stress,28 of which GRP78 is a marker protein.29,30 A previous study showed that ER stress was involved in the lung injury-repair process. Recently, it has been shown that ER stress was induced in mouse lungs in response to CS and the lungs of chronic smokers.31 In our study, we observed the up-regulation of GRP78 in the lungs not only from COPD patients but also from two mouse models of emphysema. In the PPE-induced emphysema mice, the lung expressed less GRP78 when Rcn3 was deleted selectively in the AECIIs, which was in the same tendency with the changes of Rcn3 expression. The data indicated that ER stress was possibly involved in the Rcn3 signal transduction pathway. However, this result seemed to conflict with the previous findings, in which lung specimens from the mouse model of lung fibrosis induced by bleomycin administration expressed higher levels of GRP78 when Rcn3 was deleted selectively in AECIIs.11 This is probably due to the difference of duration needed to establish each typical animal model, only 7 days for lung fibrosis formation after bleomycin challenge, while the elastase-induced emphysema model experienced a more chronic process of 28 days after PPE instillation.
Our present study revealed that Rcn3 deficiency in AECIIs may play a role in restoring the repair process of lung, at least partially by regulating MMP-9 and type I Collagen production as aforementioned. We speculated that interfering with ER stress probably was involved in the lung injury-repair behind the process of emphysema regulated by Rcn3, although the downstream signal transduction pathway of ER stress affected by Rcn3 is not clear.
In addition, while Cleaved caspase-3 protein level was enhanced in PPE group compared with the Control group, there was no significant difference in protein level of Cleaved caspase-3 in emphysema mice lungs of AECII-selective Rcn3 ablation versus wild-type mice (Figure 4D), suggesting Rcn3 might not participate in apoptosis-related pathways of emphysema progression.
In conclusion, the findings of the current study, for the first time, indicated that Rcn3 is involved in the development of emphysema. Suppression of Rcn3 expression in AECIIs has a protective effect on emphysema progression. Rcn3 deficiency in AECIIs may stimulate repair processes in the lung, but the downstream signal transduction pathway of ER stress affected by Rcn3 needs further study. Additional biological functions of Rcn3 on alveolar epithelial cells and its mechanisms remain to be further investigated.
Abbreviations
COPD, chronic obstructive pulmonary disease; Rcn3, Reticulocalbin 3; ER, endoplasmic reticulum; AECIIs, type II alveolar epithelial cells; CS, cigarette smoke; PPE, porcine pancreatic elastase; CKO mice, conditional knockout (CKO) mice with Rcn3 deletion specific to AECIIs; BALF, bronchial alveolar lavage fluid; H&E, hematoxylin and eosin; MLI, mean linear intercept; IHC, immunohistochemistry.
Data Sharing Statement
Material samples collected from both clinical and the CKO model animals are available to the Rcn3 research communities upon request.
Ethics Approval
Human data in the study has been performed in accordance with the Declaration of Helsinki. Ethical approval for the clinical study was obtained from the Research Ethics Committee for the Clinical Research of the Beijing Shijitan Hospital.
All the animal usage was approved by both of the Animal Care and Ethics Committees in Beijing Chaoyang Hospital and in the Institute of Genetics and Developmental Biology, Chinese Academy of Sciences. Procedures were performed following the guide for the Care and Use of Laboratory Animals of Beijing Chaoyang Hospital.
Consent for Publication
All authors gave the consent for open publication.
Acknowledgments
We thank Prof. Yu Kuang for the assistance in part of the experimental design using the animal models, and also for assistance in performing parts of the experiments.
Author Details
QZ, TW, JL, SW: Graduate students; JJ: Associate Professor; XS: Assistant Professor; AH, ZM: clinical doctor; FQ and MR: Professor and Principal Investigator.
Department of Respiratory and Critical Care Medicine, Beijing Chaoyang Hospital, Capital Medical University, Beijing, 100,020, China. The Clinical Research Center, Beijing Chaoyang Hospital, Capital Medical University, Beijing, 100,020, China. Beijing Shijitan Hospital, Capital Medical University, Beijing, 100,038, China. College of Life Sciences, University of Chinese Academy of Sciences, Beijing, 100,049, China. Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, Beijing, 100,101, China.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
This work was supported by National Natural Science Foundation of China (No. 81641004 to Qiuhong Fang), and was supported in part by National Natural Science Foundation of China (No. 81770062 to Jiawei Jin).
Disclosure
The authors declare that they have no competing or conflicts of interest.
References
1. Singh D, Agusti A, Anzueto A, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease. GOLD Science Committee Report 2019. 2019;53.
2. Adeloye D, Chua S, Lee C, et al. Global and regional estimates of COPD prevalence. Systematic Review Meta-Analysis. 2015;5:020415.
3. GBD 2013 Mortality and Causes of Death Collaborators. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990–2013: a systematic analysis for the global burden of disease study 2013. J Lancet:. 2015;385:117–171. doi:10.1016/S0140-6736(14)61682-2
4. Miniño A, Xu J. National vital statistics reports: from the centers for disease, prevention national center for health statistics, national vital statistics system: deaths: preliminary data for 2008. Control Kochanek Kd. 2010;59:1–52.
5. Vogelmeier C, Criner G, Martínez F, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease 2017 report. GOLD Executive Summary. 2017;53:128–149.
6. Stolz D, Barandun J, Borer H, et al. Diagnosis, prevention and treatment of stable COPD and acute exacerbations of COPD. Swiss Recommendations 2018. 2018;96:382–398.
7. Honoré B. The rapidly expanding CREC protein family: members, localization, function, and role in disease. Bioessays. 2009;31:262–277.
8. Tsuji A, Kikuchi Y, Sato Y, et al. A proteomic approach reveals transient association of reticulocalbin-3, a novel member of the CREC family, with the precursor of subtilisin-like proprotein convertase, PACE4. Biochem J. 2006;396:51–59. doi:10.1042/BJ20051524
9. Jin J, Li Y, Ren J, et al. Neonatal respiratory failure with retarded perinatal lung maturation in mice caused by reticulocalbin 3 disruption. J American J Respiratory, Biology Molecular. 2016;54:410–423. doi:10.1165/rcmb.2015-0036OC
10. Hou Y, Li Y, Gong F, et al. A preliminary study on RCN3 protein expression in non-small cell lung cancer. Clin Lab. 2016;62:293–300. doi:10.7754/Clin.Lab.2015.150411
11. Jin J, Shi X, Li Y, et al. Reticulocalbin 3 deficiency in alveolar epithelium exacerbated bleomycin-induced pulmonary fibrosis. J American J Respiratory, Biology Molecular. 2018;59:320–333. doi:10.1165/rcmb.2017-0347OC
12. López-Campos J, Tan W. Global burden of COPD. Respirology Soriano Jb. 2016;21::14–23. doi:10.1111/resp.12660
13. Rashid K. Lung cellular senescence is independent of aging in a mouse model of COPD/emphysema. J Scientific. 2018;8:9023.
14. Anciães A, Olivo C, Prado C, et al. Respiratory mechanics do not always mirror pulmonary histological changes in emphysema. Clinics Lopes Fd. 2011;66:1797–1803.
15. Taguchi L, Pinheiro N, Olivo C, et al. A flavanone from Baccharis retusa (Asteraceae) prevents elastase-induced emphysema in mice by regulating NF-κB, oxidative stress and metalloproteinases. Respir Res. 2015;16:79.
16. Zou Y, Li S, Zou W, et al. Upregulation of gelatinases and epithelial-mesenchymal transition in small airway remodeling associated with chronic exposure to wood smoke. J Plos. 2014;9:e96708. doi:10.1371/journal.pone.0096708
17. Yang C, Li L, Guo J, Zhang W, Zhu W, Rao X. Up-regulation of Pim-3 in chronic obstructive pulmonary disease (COPD) patients and its potential therapeutic role in COPD rat modeling. J Pathology Research, Practice. 2017;213:322–326. doi:10.1016/j.prp.2017.01.018
18. Cai X, Xc N, Yi Y, et al. Overexpression of CD39 in hepatocellular carcinoma is an independent indicator of poor outcome after radical resection. Medicine (Baltimore). 2016;95:e4989.
19. Murray C. Measuring the global burden of disease. J New England. 2013;369:448–457. doi:10.1056/NEJMra1201534
20. Rabe K, Hurd S, Anzueto A, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2007;176:532–555.
21. Boyer L, Plantier L, Dagouassat M, et al. Role of nitric oxide synthases in elastase-induced emphysema. Lab Invest2011;91:353–362.
22. Rodrigues R. A murine model of elastase- and cigarette smoke-induced emphysema. J Jornal Brasileiro De Pneumologia. 2017;43:95–100. doi:10.1590/s1806-37562016000000179
23. Mp I, Kleinerman J, Ranga V, Sorensen J. The effects of small doses of oligopeptide elastase inhibitors on elastase-induced emphysema in hamsters: a dose-response study. J American Review Respiratory. 1981;124:714–717.
24. Lopes FD, Toledo AC, Olivo CR, et al. A comparative study of extracellular matrix remodeling in two murine models of emphysema. Histol Histopathol. 2013;28:269–276. doi:10.14670/HH-28.269
25. Lanone S, Zheng T, Zhu Z, Liu W. Overlapping and enzyme-specific contributions of matrix metalloproteinases-9 and −12 in IL-13-induced inflammation and remodeling. J Clin Investigation. 2002;110:463–474. doi:10.1172/JCI14136
26. Zheng T, Zhu Z, Wang Z. Inducible targeting of IL-13 to the adult lung causes matrix metalloproteinase- and cathepsin-dependent emphysema. J Clinical. 2000;106:1081–1093.
27. Shifren A. The stumbling block in lung repair of emphysema: elastic fiber assembly. J Proceedings American Thoracic. 2006;3:428–433. doi:10.1513/pats.200601-009AW
28. Barreiro E, Salazar-Degracia A, Sancho-Munoz A, Gea J. Endoplasmic reticulum stress and unfolded protein response profile in quadriceps of sarcopenic patients with respiratory diseases. J Cell Physiol. 2019;234:11315–11329. doi:10.1002/jcp.27789
29. Wang Y. Inhibition of endoplasmic reticulum stress alleviates cigarette smoke-induced airway inflammation and emphysema. Oncotarget. 2017;8:77685–77695. doi:10.18632/oncotarget.20768
30. Jorgensen E, Stinson A, Shan L, Yang J, Gietl D. Cigarette smoke induces endoplasmic reticulum stress and the unfolded protein response in normal and malignant human lung cells. J Bmc. 2008;8:229.
31. Naiel S, Tat V, Padwal M, et al. Protein misfolding and er stress in chronic lung disease: will cell-specific targeting be the key to the cure? Chest. 2019. doi:10.1152/ajpheart.00523.2018
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.