")
Back to Journals » Journal of Hepatocellular Carcinoma » Volume 8
Rates of Hepatocellular Carcinoma After Start of Treatment for Chronic Hepatitis C Remain High with Direct Acting Antivirals: Analysis from a Swiss Liver Transplant Center
Authors Karbeyaz F , Kissling S , Jaklin PJ, Bachofner J, Brunner B, Müllhaupt B, Winder T, Mertens JC, Misselwitz B , von Felten S , Siebenhüner AR
Received 4 November 2020
Accepted for publication 13 February 2021
Published 11 June 2021 Volume 2021:8 Pages 565—574
DOI https://doi.org/10.2147/JHC.S289955
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Ahmed Kaseb
Fatih Karbeyaz,1 Seraphina Kissling,2 Paul Julius Jaklin,1 Jaqueline Bachofner,1 Barbara Brunner,1 Beat Müllhaupt,1 Thomas Winder,3 Joachim C Mertens,1 Benjamin Misselwitz,1,4 Stefanie von Felten,5 Alexander R Siebenhüner6,7
1Division of Gastroenterology and Hepatology, University Hospital Zurich and Zurich University, Zurich, Switzerland; 2Master Program in Biostatistics, University of Zurich, Zurich, Switzerland; 3Division of Oncology, Landeskrankenhaus Feldkirch, Feldkirch, Austria; 4Department of Visceral Surgery and Medicine, Inselspital Bern and Bern University, Bern, Switzerland; 5Department of Biostatistics, Epidemiology, Biostatistics and Prevention Institute, University of Zurich, Zurich, Switzerland; 6Department of Medical Oncology and Hematology, University Hospital Zurich and University of Zurich, Zurich, Switzerland; 7Department of Medical Oncology, Cantonal Hospital Schaffhausen, Schaffhausen, Switzerland
Correspondence: Alexander R Siebenhüner
Klinik für Medizinische Onkologie und Hämatologie, Universitätsspital Zürich, Rämistrasse 100, Zürich, CH-8091, Switzerland
Email [email protected]
Background: Direct-acting antivirals (DAA) have revolutionized the therapy of chronic hepatitis C (CHC) and have replaced previous PEG-interferon/ribavirin (PEG-IFN/RBV) treatment. Patients with CHC and advanced liver disease are at increased risk for hepatocellular carcinoma (HCC). However, the effects of DAA-based CHC treatment on subsequent HCC incidence remain poorly understood.
Patients and Methods: This retrospective single-institution cohort study included 243 consecutive patients after PEG-IFN/RBV and 263 patients after DAA treatment. Multivariable cause-specific Cox proportional hazards models were used to compare time to HCC between treatment types, censoring patients who died or had an orthotopic liver transplantation (OLT) at the time of the competing event. Age, gender, BMI, viral load, cirrhosis, fibrosis stage, diabetes, virus genotype and previous PEG-IFN/RBV (before DAA) were used as covariates. In addition, we performed a propensity score-matched analysis.
Results: Nineteen HCC cases were observed after DAA therapy compared to 18 cases after PEG-IFN/RBV treatment. Patients were followed for a median of 4.1 years (IQR: 3.5– 4.7) for DAA and 9.3 years (IQR: 6.6– 12.4) for the PEG-IFN/RBV group. In an unadjusted Cox model, a hazard ratio (HR) of 6.40 (CI: 2.20– 18.61, p=0.006) for HCC following DAA vs PEG-IFN/RBV was estimated. In multivariable Cox proportional hazard models, age and liver cirrhosis were identified as further HCC risk factors but the HR estimates for DAA vs PEG-IFN/RBV still indicate a considerably increased hazard associated with DAA treatment (HR between 7.23 and 11.52, p≤ 0.001, depending on covariates). A HR of 6.62 (CI: 2.01– 21.84, p=0.002) for DAA vs PEG-IFN/RBV was estimated in the propensity score-matched analysis. The secondary outcomes death and OLT did not differ between treatment groups.
Conclusion: In a cohort study from a tertiary care hospital rates of HCC after the start of DAA treatment were higher compared to PEG-IFN/RBV treatment. Our data reinforce the recommendation that surveillance should be continued after successful CHC treatment.
Keywords: hepatocellular carcinoma, chronic hepatitis C, direct-acting antivirals, PEG-interferon and ribavirin, viral load, liver transplantation, sustained virological response
Introduction
Hepatocellular carcinoma is the fifth and ninth most common cancer in men and women, respectively.1 Chronic infection with hepatitis C virus (HCV) is one of the leading risk factors for HCC. The annual risk of HCC is as high as 3% in patients with cirrhosis and untreated HCV.2 In the past decades, the standard of care for chronic HCV was pegylated interferon and Ribavirin (PEG-IFN/RBV). These drug regimens achieved a considerable proportion of sustained virological response (SVR) of about 44–63% and large studies described a reduced risk for HCC following successful HCV treatment with PEG-IFN/RBV.3–5 However, side effects and toxicities forced a substantial percentage of patients to stop treatment early. The recent implementation of highly effective and well-tolerated direct-acting antivirals (DAA) has led to very high SVR of more than 90% in most HCV patient populations. Within the next decade, most patients with known HCV in Western countries are expected to start DAA treatment and achieve SVR.6–8
Recently, DAA have come under scrutiny for a possible increase in HCC incidence after DAA treatment and current data on HCC recurrence or development following DAA-induced SVR are still conflicting. Initial studies reported higher rates of HCC occurrence than expected in patient cohorts treated with DAA,9–11 which were controversially discussed.12 These findings were not confirmed in all studies.13–15 In larger studies, the risk for HCC upon DAA treatment was higher upon DAA treatment compared to controls with PEG-INF/RBV treatment or no treatment; however, this increased risk was eliminated or reversed after correction for confounders.16–20 A recent meta-analysis indicated a reduced or similar risk of HCC upon DAA treatment21 and overall, DAA-induced SVR was protective against the development of HCC.22–24 However, considering the broad use of DAA among HCV patients and the deleterious effect of HCC more data in all possible settings are clearly needed.
With this manuscript, we present a large retrospective single-center cohort study at the University Hospital Zürich, Switzerland examining HCC incidence after DAA treatment in comparison to previous PEG-IFN/RBV data from the same institution.
Patients and Methods
Patients with PEG-IFN/RBV treatment between 16th of January 1998 and 6th of April 2013 and DAA treatment starting between 1st of November 2013 and 31st of May 2016 were included. Pre-existing HCC orthotopic liver transplantation (OLT) before the start of therapy and unknown HCC status before or after treatment were exclusion criteria. PEG-IFN/RBV treatment in a significant fraction of HCV patients is a clinical fact in many hepatology units worldwide and this considerable fraction of patient cannot be ignored in our real-life analysis. Therefore, in a pragmatic approach, patients who received sequential therapies with PEG-IFN/RBV followed by DAA were evaluated in the DAA cohort, but potential confounding by previous PEG-IFN/RBV treatment was accounted for in the statistical analysis (see below).
All time intervals were calculated with the first day of PEG-IFN or DAA treatment as time origin. In one patient who had 2 rounds of DAA, we combined the 2 treatment rounds, with the start of the first round as time origin. Diagnosis of HCC followed established guidelines by the European Association for the Study of the Liver (EASL).25
Our study was approved by the Ethics Committee of Zurich county (KEK Zurich, BASEC 2019-00072). The need for individual patient consent to retrospective data review was waived by the Ethics Committee because patient consent to anonymized data evaluation already existed within the Swiss Hepatitis cohort study. This Swiss Hepatitis cohort study has been approved by KEK Zurich (EK-695). The study was performed in accordance with the declaration of Helsinki. Patient data were anonymized and patient data confidentiality was observed as required by general consent regulations.
Measures
Patients were considered negative for HCC if dedicated imaging (either abdominal ultrasound or contrast-enhanced MRI) at most 6 months prior to treatment was without evidence for HCC. For all other patients, HCC state was considered unknown. Patients within the HCV treatment group received annual ultrasonography of the liver while patients with known cirrhosis received biannual surveillance by ultrasound. Suspicious lesions were followed by MRI.
Sustained virological response (SVR) was defined as negative HCV PCR 12 weeks and 24 weeks after end of treatment for DAA and PEG-IFN/RBV treated patients, respectively. Since PEG-IFN/RBV treatment is frequently accompanied by severe side effects resulting in a difficult decision-making process, whether treatment would need to be stopped, we aimed to understand severity of liver disease and urgency of treatment before start of treatment. For this reason, biopsies were performed close to the start of PEG-IFN/RBV treatment (median 0.47 years, IQR 0.13–2 before start) but at a longer time period before DAA (3.5 years, IQR 1.3–7.2). Liver fibrosis was assessed by liver biopsy following METAVIR criteria (F1-F4)26 or estimated from transient elastography data. For measurement of transient elastography, the Echosens FibroScan 502 Touch machine was used with the following published cut-off values >12.5: F4; >9.5: F3, >7.0: F2, all others F0 or F1.27 The Echosens FibroScan 502 machine was only introduced in 2012, and fibroscan measures were not available before that point in time. For these reasons, fibroscan measures are more prevalent in the DAA group. Transient elastography measurements were acquired at a median of 0.2 years (IQR 0.1–0.4) before DAA and 0.5 years (0.2–1) before PEG-IFN/RBV treatment. Cirrhosis was defined either by biopsy, by a transient elastography (Fibroscan) value >12.5 kPa or according to clinical, endoscopic and/or ultrasound criteria (ie severe liver disease with clear radiographic or endoscopic signs of portal hypertension).
AST to platelet ratio (APRI)28 and FIB-429 were calculated using measurements at the start of HCV therapy. The MELD score was calculated as previously described;30 values <6 and values for individuals without liver cirrhosis were fixed at 6. Since platelet counts are a part of APRI and FIB-4, and bilirubin is contained in the MELD score, no independent correction for platelets and bilirubin as covariates was performed. Further alpha-fetoprotein (AFP) values were missing in 90 patients after DAA and 27 after PEG-IFN/RIB treatment, precluding correction for AFP.
Statistical Analysis
Patient baseline characteristics were summarized per treatment group (PEG-IFN/RBV, DAA) for the eligible patients included in the data analysis. Frequencies and percentages are shown for categorical variables. Medians and interquartile ranges are shown for continuous variables.
To estimate the hazard ratio of DAA vs PEG-IFN/RBV for the primary outcome (time to) HCC we used cause-specific Cox proportional hazards models. Death and OLT were treated as competing risks by right-censoring patients at the competing event. In two cases, discovery of HCC and death or OLT were recorded on the same day. Under the assumption that HCC was present before the other events, we counted both cases as HCC events. Patients without event were right-censored at the last follow-up visit. The minimal model (a) only includes treatment (DAA vs PEG-IFN/RBV) as explanatory variable, our factor of main interest. To address the problem of confounding, other risk factors for HCC were added as covariates to the model. However, due to the limited number of HCC events, we did not include all relevant risk factors in a single model to avoid overfitting. Applying the ten events per variable rule of thumb,31 we limited the number of predictors to four per model. We fitted several alternative adjusted models to assess the robustness of the treatment effect estimate in this observational cohort study and to investigate which risk factors for HCC may be most relevant. Each model includes treatment and age (a well-known risk factor), and either (b) sex and previous treatment with PEG-IFN/RBV, (c) cirrhosis and fibrosis grade (d) BMI and diabetes, (e) viral load (in 1ʹ000ʹ000 international units/mL), or (f) virus genotype as additional covariates. Moreover, we assessed the interaction between age and treatment. The secondary outcomes time to death and time to OLT were analyzed by cause-specific Cox proportional hazards models with age and treatment as explanatory variables.
Times to HCC, death and liver transplantation were visualized using cumulative incidence curves.
Moreover, we performed the following sensitivity analyses to estimate the hazard ratio of DAA vs PEG-IFN/RBV for the primary outcome (time to) HCC, which are presented in the Supplementary Materials: (1) A multivariate Cox model including FIB-4 as continuous variable instead of fibrosis degree, due to the many missing values in fibrosis degree, (2) models a—c described above but excluding DAA patients previously treated with PEG-IFN/RBV and (3) a propensity score matched analysis (detailed description in the Supplementary Materials). All computations were performed using R version 4.0.2.32
Results
We identified 345 patients with DAA treatment and 276 patients with PEG-IFN/RBV treatment. Of the patients with DAA treatment, 105 had received DAA after PEG-IFN/RBV (Figure 1). After exclusion of patients due to preexisting HCC, unknown HCC state after the start of therapy or previous OLT, 263 patients with DAA treatment (thereof 76 who received DAA after PEG-IFN/RBV) and 243 patients with PEG-IFN/RBV treatment were included for further analysis (Figure 1).
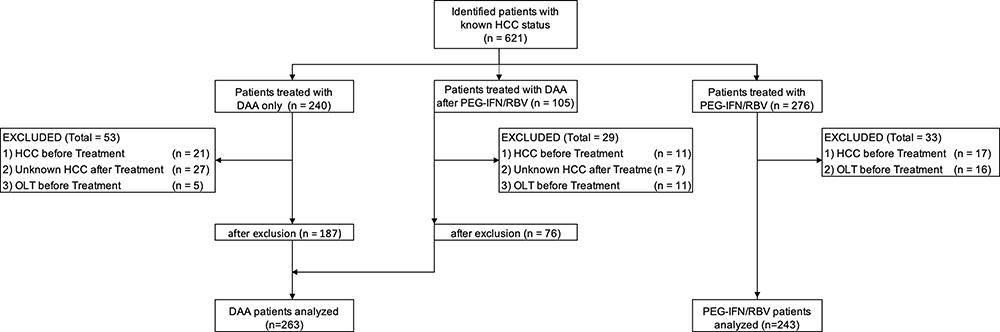 |
Figure 1 Flowchart of patient recruitment. Abbreviations: DAA, directly acting antivirals; HCC, hepatocellular carcinoma; PEG-IFN/RBV, pegylated interferon/ribavirin; OLT, orthotopic liver transplantation. Notes: All patients had known HCC status before treatment. The initial number of patients was 621 (PEG-IFN/RBV + DAA). Following the exclusion criteria, as shown in Figure 1, there were 506 eligible and thus analyzed patients. Patient characteristics are shown in Table 1. |
Patients treated with DAA had a significantly higher body mass index (BMI) than PEG-IFN/RBV patients. Furthermore, genotypes 1 and 4 were more frequent in the DAA cohort (most likely due to higher rates of successful previous PEG-IFN/RBV treatment of patients with genotypes 2 and 3 in our cohort). Liver cirrhosis assessed either by liver biopsy, transient elastography or ultrasound with appropriate clinical context was more prevalent in the DAA cohort (both CHILD class A and B). The APRI and MELD scores were similar in both groups; however, DAA-treated patients had significantly higher FIB-4 scores (Table 1). HCC treatment resulted in SVR in 248/263, 94.3% of DAA treated patients and 133/243, 54.7% of PEG-IFN/RBV patients.
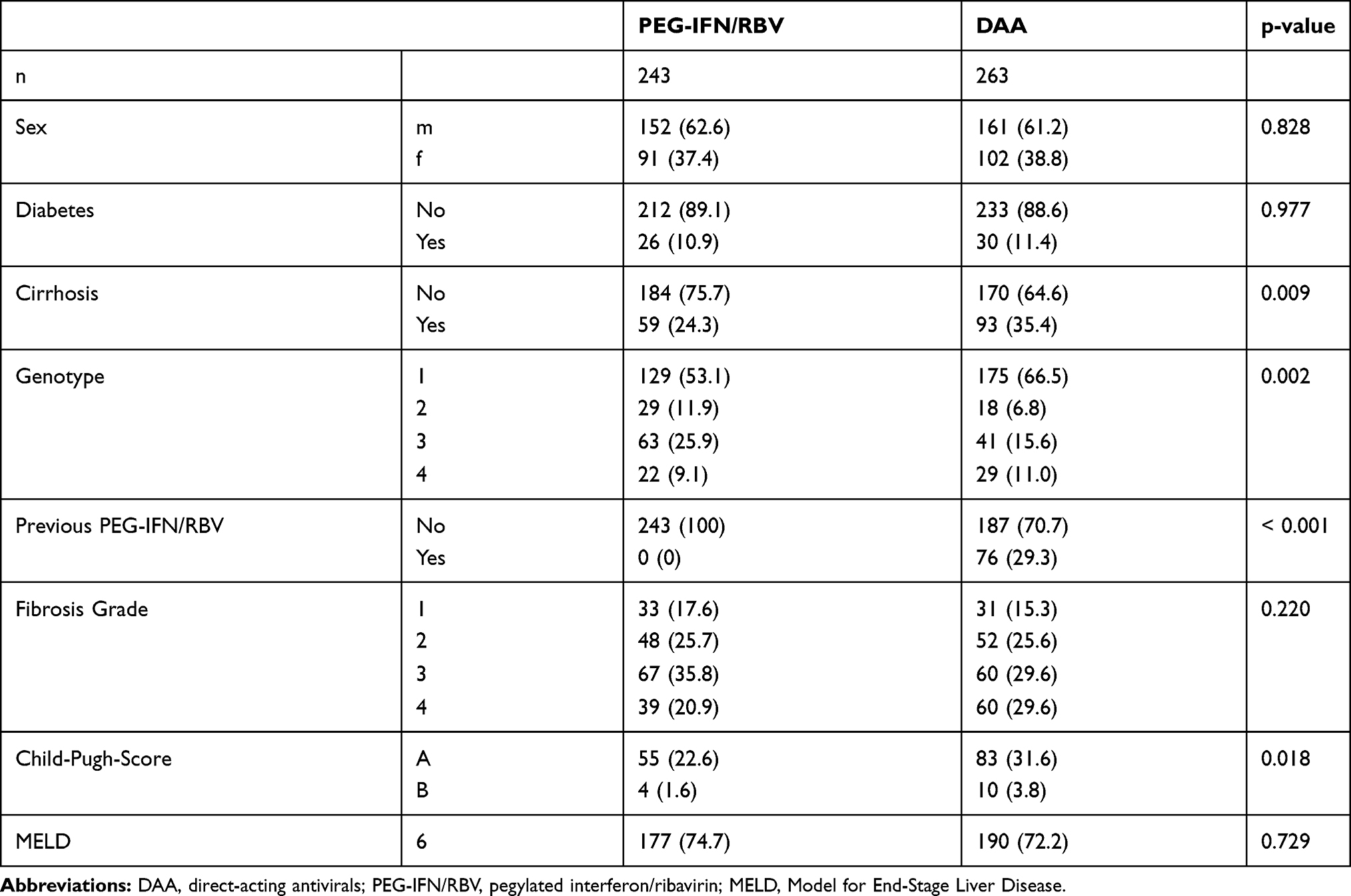 |
Table 1 Overview of the Baseline Characteristics for Both Treatment Groups for Eligible Patients |
After start of treatment, patients were followed for a median of 4.1 years (IQR: 3.5–4.7) for the DAA group and 9.3 years (IQR: 6.6–12.4) for the PEG-IFN/RBV group. Nineteen patients were diagnosed with HCC with DAA compared to 18 patients with PEG-IFN/RBV. However, follow-up upon DAA treatment is significantly shorter in our cohort, and no data beyond about 2000 days (5.5 years) after end of treatment are available. In a simple cause-specific Cox-proportional hazards model with treatment (DAA or PEG-IFN/RBV) as the only explanatory variable, the hazard ratio for DAA vs PEG-IFN/RBV regarding HCC was 6.4 (95% CI: from 2.20 to 18.61, p=0.0006), Table 2A.
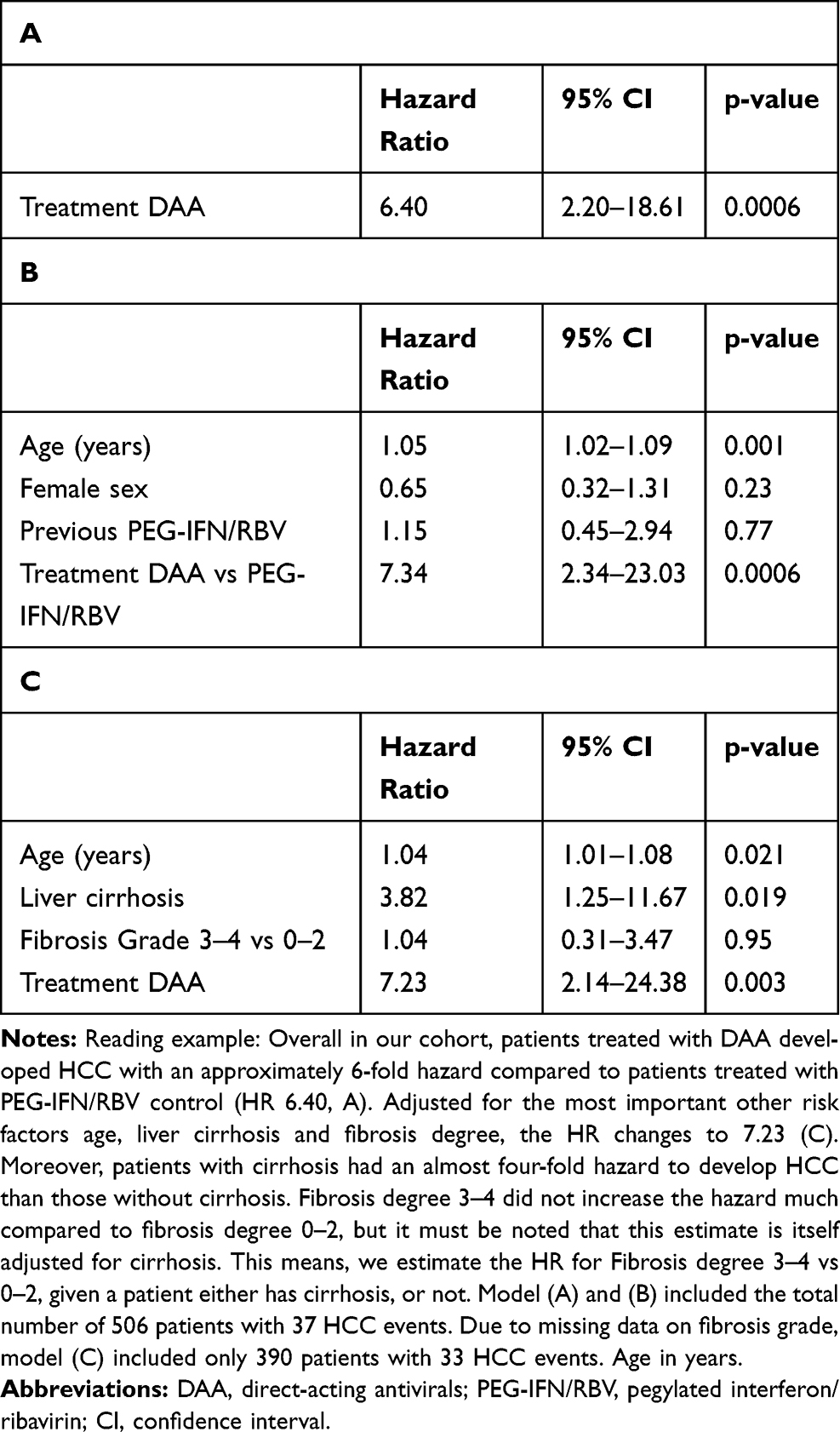 |
Table 2 Hazard Ratio Estimates (with 95% Confidence Intervals) for Treatment (DAA vs PEG-IFN/RBV) and Important Covariates from Cause-Specific Cox Proportional Hazards Models on Time to HCC. The Hazard Ratio for Treatment is Once Estimated without Adjustment for Other Known or Potential Risk Factors for HCC (A), and with Adjustment for Different Sets of Other Risk Factors (B and C) |
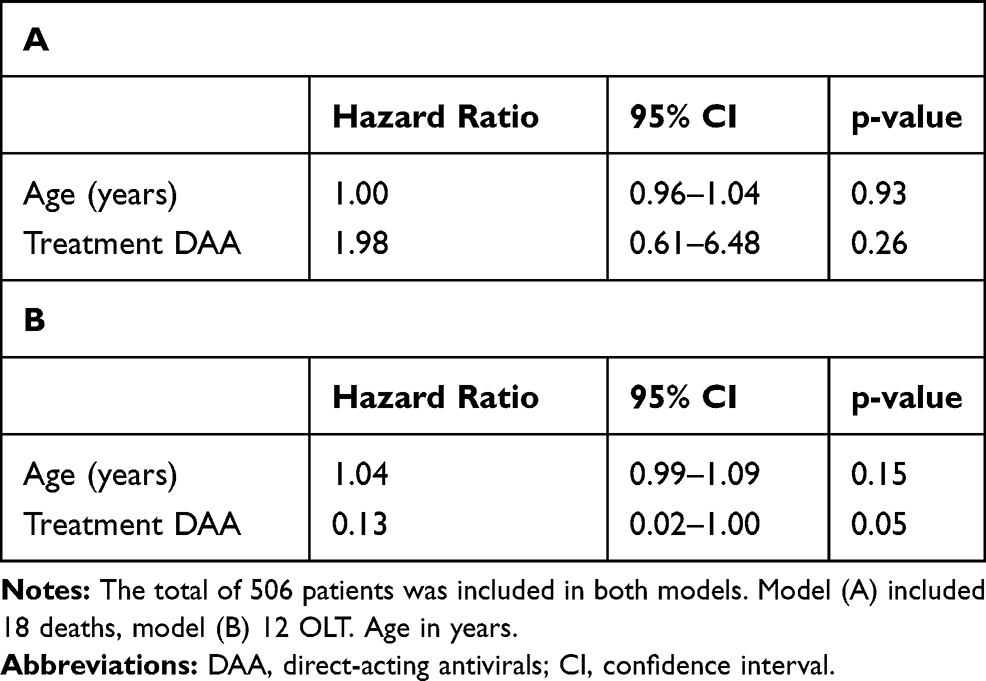 |
Table 3 Hazard Ratio Estimates from a Cause-Specific Cox Proportional Hazards Model on Time to Death (A) and Orthotopic Liver Transplantation (OLT) (B) |
Treatment specific cumulative incidence curves of DAA and PEG-INF/RBV patients for HCC, death and OLT are shown in Figure 2.
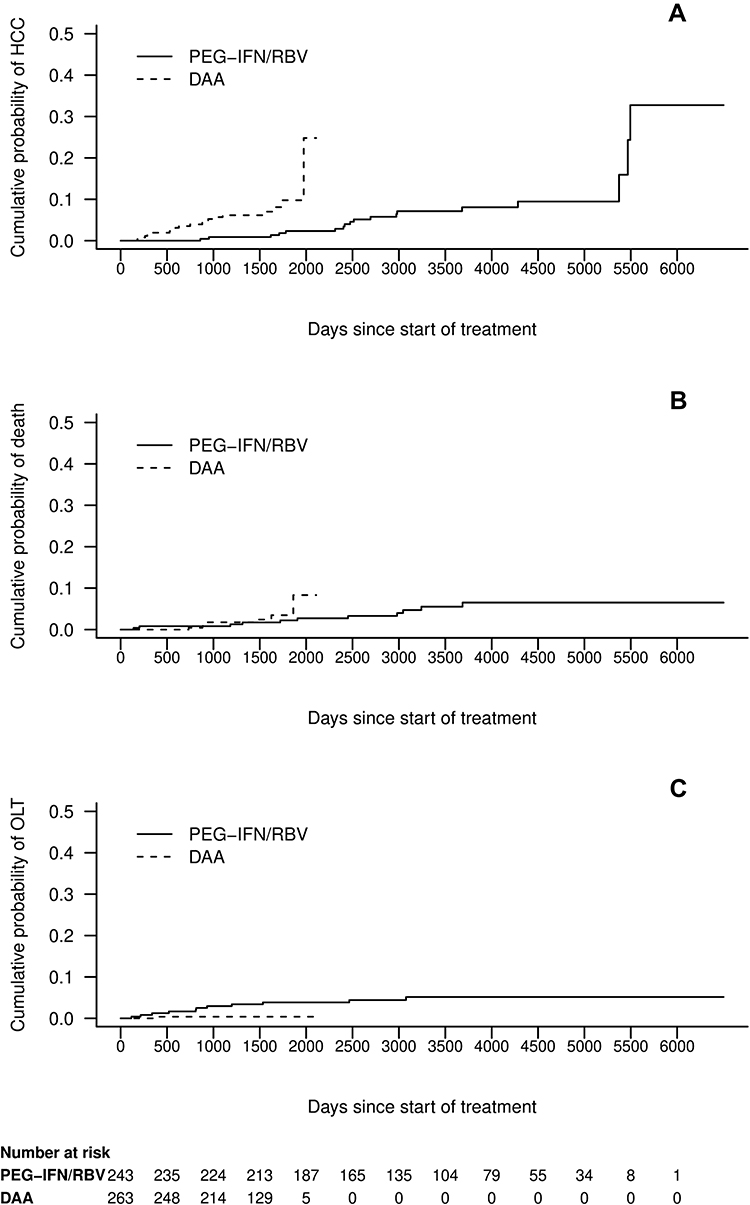 |
Figure 2 Treatment specific cumulative incidence curves for (A) HCC, (B) death and (C) OLT. Numbers at risk are given at the bottom. Abbreviations: DAA, direct-acting antivirals; PEG-IFN/RBV, pegylated interferon/ribavirin; OLT, orthotopic liver transplantation. |
Age is an important risk factor for HCC and was significant in all covariate-adjusted cause-specific Cox-proportional hazards models (HR 1.01–1.06 per additional year of age, Supplementary Table S1A–D). Neither epidemiological parameters such as sex or previous treatment with PEG-IFN/RBV (Table 2B) nor metabolic parameters such as BMI and diabetes mellitus (Table 2C) were significantly associated with time to HCC. However, there is evidence that cirrhosis is associated with shorter time to HCC (HR 3.82, 95% CI: from 1.25 to 11.67). The degree of fibrosis (F3-4 vs F0-2) was not significantly associated with time to HCC (Table 2C).
All our models suggest that patients under DAA treatment developed HCC significantly faster than patients under PEG-IFN/RBV (p≤0.01 for all models). The size of the effect varied slightly (HR 6.4–7.2). The HR of the unadjusted model is in the same range as the adjusted HRs, suggesting a consistent association of DAA treatment with a higher hazard of HCC compared to PEG-IFN/RBV even after adjustment for different potential confounders.
The relatively large HR estimate for DAA vs PEG-IFN/RBV described above was also robust in several sensitivity analyses presented in Supplementary Tables S1, S2 and S3. When FIB-4 was used instead of fibrosis degree, the HR was 3.63 (95% CI: from 1.18 to 11.21) (Supplementary Materials Table S1). Further, adjustment for HCV genotype (1 vs 2–4) or viral load resulted in a HR for DAA vs PEG-IFN/RBV of 7.49 (95% CI: from 2.53 to 22.21) and 11.52 (95% CI: from 2.91 to 45.61), respectively. When DAA patients previously treated with PEG-IFN/RBV were excluded, the HR for the models analogue to Table 2A–C were 6.16–6.97 (Supplementary Table S2A–C). The full and detailed description of the propensity score-matched analysis is sectioned in the Supplementary Materials (detailed description of methods in used as a complementary approach to adjust for confounders led to similar results). A detailed description of the methods used for propensity score matching is given together with Supplementary Figure S1. The unadjusted model on the propensity score matched data set estimated an HR of 6.62 (95% CI 2.01–21.84, p=0.002) and models adjusted for age and genotype (which were not so well balanced after matching, Supplementary Figure S2) or for age, cirrhosis and fibrosis degree event estimated slightly larger HR (Supplementary Table S3A–C).
When looking at an interaction of age and treatment, we found evidence for an increase in the HR for DAA vs PEG-IFN/RBV with patient age, suggesting a stronger DAA-associated HCC risk in older patients (Supplementary Table S1D).
Investigating the associations of age and treatment with the secondary outcomes time to death (Table 3A) and time to OLT (Table 3B) in a cause-specific Cox model, we could not detect any significant effects.
Discussion
We performed a retrospective analysis of HCC incidence in a cohort of HCV-infected patients who received DAA or PEG-IFN/RBV treatment. We would like to emphasize the following key observations: i) In our cohort we observed a high number of HCC cases within the first 2000 days after DAA treatment. Time to HCC was significantly shorter compared to the PEG-IFN/RBV treatment group, even after covariate adjustment. ii) As expected, additional risk factors for HCC after HCV treatment include older age and liver cirrhosis, pointing to subpopulations with highly increased HCC risks. iii) While HCC risk seems highest within the first 2 years following DAA treatment not a single case of HCC was observed within the first 2 years following PEG-IFN/RBV and the incidence rises after well more than 10 years after treatment. iv) The DAA group showed higher rates of liver cirrhosis and higher FIB-4 scores compared to the PEG-IFN/RBV, indicating more severe disease. This might be due to the earlier availability of DAA treatment in younger HCV patients with more severe HCV. However, the association of DAA treatment with time to HCC remained robust even after controlling for these confounders as covariates, or after or in a propensity score matched analysis where these confounders were included in the propensity score model.
Within the last years, the relationship between DAA therapy and new onset of HCC has been intensely examined and discussed. A number of initial studies suggested an increased risk;9–11 nonetheless, a number of large and well-designed studies demonstrated an increased nominal risk upon DAA treatment which was eliminated or reversed after controlling for confounders.16–20 Studies comparing DAA-treated patients with a historical control of PEG-IFN/RBV patients, similar to our study, also showed similar HCC rates with both treatments.15,18,33,34 The largest study cohort consisted of 62,000 patients treated for CHC;17 after controlling for a large set of confounders, eradication of HCV with DAAs was associated with a considerable reduction of HCC risk. Compared to PEG-INF treatment, the authors observed an unadjusted HR for HCC of 2.81 (CI: 2.44–3.22) associated with DAA treatment; however, after adjustment for confounders, the adjusted hazard ratio was reduced to 1.12 (0.95–1.32). In this study, all HCC cases developing within the first 6 months after completion of DAA were excluded to limit study results to truly de novo HCC and the time window immediately following HCV clearance has not been addressed. However, this difference is unlikely to explain the discrepant results completely.
HCC rates after CHC treatment were shown to be independent from the specific DAA used, arguing against direct toxic effects of different DAAs.35 A meta-analysis estimated an incidence rate of new HCC of 2.96/100 person years after DAA and 1.14/100-person years after PEG-IFN/RBV. However, also in this meta-analysis, after controlling for confounders, no significant difference in HCC risk between DAA and PEG-IFN/RBV remained.20 Why controlling for confounders or a propensity score-matched analysis did not eliminate the DAA-associated HCC risk in our study remains unclear.
Age remains a strong risk factor for HCC in our study and in most previous analyses.15,17,18,33,34 In our analyses, we found a statistical interaction of age and treatment, in line with a higher DAA-associated HCC risk in older patients. Somewhat similar, in a previous study liver fibrosis was a strong risk factor for HCC in younger individuals while older patients developed HCC independently from fibrosis state.36 However, even when interactions of treatment and/or fibrosis with age were considered, a DAA-associated risk for HCC remained in our cohort and the age distribution in our cohort is similar as in previous studies.
SVR rates upon DAA treatment were high in our cohort (94.3%), as expected. Previous studies demonstrated a protective effect of SVR regarding new onset of HCC.17,22–24 However, following CHC treatment, SVR should be considered an intermediate variable, a parameter affected by the treatment, which may or may not lie on the causal path to the outcome. To avoid the introduction of bias, correction for intermediate variables is usually discouraged37 and we did not use SVR as an explanatory variable in our analyses.
Recent reviews and guidelines conclude that the risk of de novo HCC after therapy is reduced.25,38 However, it should be noted that a considerable HCC risk remained in all studies even after successful treatment of HCV. In a recent meta-analysis, the incidence rate of new HCC was 3.3% (95% CI 1.2–9%) per year after DAA.39 Some studies noted a much higher HCC risk within the first year after treatment than thereafter.13,23
In our study, a high incidence of HCC after the start of therapy was noted. One possible explanation of these results could be a change in the growth of pre-existing subclinical, undetectable HCC upon DAA treatment. In this scenario, DAA treatment would trigger a boost in HCC within a short time window after HCV clearance despite long-term benefits of DAA. Such an acceleration of HCC growth may be due to changes in immune-surveillance of HCC upon DAA clearance. Our results are in line with a recent report, demonstrating a high HCC risk following DAA treatment, especially in individuals with uncharacterized liver nodules.40 Further, an HCC specific tumor response, which was reduced upon DAA-induced HCV clearance was recently described.41 This T cell-dependent immune response was much weaker in HCV patients who subsequently developed HCC.41 In addition to changes in immune surveillance, other mechanisms like cellular behavior after eradication of HCV for increased HCC growths upon DAA therapy have also been identified in early test models and will be elucidated in future clinical models.38 If confirmed, it would be important to identify patients susceptible to rapid HCC growth. However, it should be noted that our study only provides indirect evidence and neither proves acceleration of HCC growth nor changes in HCC immune surveillance directly.
Our study has several strengths and limitations. We included a relatively large population of HCV-infected individuals and a PEG-IFN/RBV treated control cohort from our center, allowing for direct assessment of confounders. In addition, median follow-up time in our study is 4.1 years for DAA and 9.3 years for PEG-IFN/RBV. The most important limitation of our study is the retrospective study design and for some measures our data are not complete. In addition, since the time of infection with HCV is not known for most of the patients, we cannot correct for duration of disease in our multivariable analysis or for propensity score matching. Finally, our study inevitably compares newer DAA data with older PEG-IFN/RBV data.
For practical purposes, our study serves as a reminder of the remaining HCC risk even after HCV clearance. A patient with long-term CHC is an HCC high-risk patient also after therapy and thus we recommend HCC surveillance in 6-month intervals for patients with cirrhosis and a maximum of 12 months for patients without cirrhosis should be strictly followed.
In summary, we describe a cohort of patients with chronic hepatitis C infection and treatment with DAA or PEG-IFN/RBV. In our cohort, a higher hazard for developing HCC was noted upon DAA treatment compared to PEG-IFN/RBV also after adjustment for confounders. Therefore, a high degree of suspicion for HCC is justified after the start of DAA therapy, especially in high-risk patients.
Abbreviations
AASLD, American Association for the Study of Liver Diseases; APRI, aspartate aminotransferase to platelet ratio index; BMI, body mass index; CHC, chronic hepatitis C; CI, confidence interval; DAA, direct-acting antivirals; EASL, European Association for the Study of the Liver; FIB-4, Fibrosis-4; HCC, hepatocellular carcinoma; HCV, hepatitis C virus; HR, hazard ratio; IFN, interferon; MELD, Model for End-Stage Liver Disease; MRI, magnetic resonance imaging; OLT, orthotopic liver transplantation; PCR, polymerase chain reaction; PEG-IFN, pegylated interferon; RBV, ribavirin; SVR, sustained virological response.
Author Contributions
Fatih Karbeyaz: designed the study, recruited patients, and wrote the manuscript and edited the manuscript for important intellectual content; Seraphina Kissling and Stefanie von Felten: performed statistical analyses and contributed to writing the manuscript and edited the manuscript for important intellectual content, Alexander R. Siebenhüner: designed the study and wrote the manuscript and edited the manuscript for important intellectual content; Paul Jaklin: recruited patients; Jaqueline Bachofner: recruited patients; Barbara Brunner: recruited patients; Beat Müllhaupt: designed the study and edited the manuscript for important intellectual content; Thomas Winder: designed the study and edited the manuscript for important intellectual content; Benjamin Misselwitz: designed the study and wrote the manuscript and edited the manuscript for important intellectual content; Joachim C. Mertens: designed the study and wrote the manuscript and edited the manuscript for important intellectual content.
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
FK and SK are joint first authors; ARS, SvF and BM are joint last authors.
Funding
There is no funding to report.
Disclosure
Alexander R. Siebenhüner has received consulting and speaker honoraria from Amgen, Bayer, BMS, IPSEN, Lilly, Merck, MSD, Pfizer, Sanofi and Servier for work performed outside the current study. Barbara Brunner no disclosures associated with the manuscript. Beat Müllhaupt reports grants and personal fees from Gilead, personal fees from Abbvie, and has received consulting honoraria from Abbvie, BMS, Gilead, Janssen, Merck and MSD for work performed outside the current study. Benjamin Misselwitz has received speaking fees from MSD and Takeda. He has received traveling fees from MSD, Novartis, Vifor, Gilead and Takeda. He has received a research grant from MSD, outside the submitted work. Fatih Karbeyaz, Paul Jaklin, And Jaqueline Bachofner report no disclosures associated with the manuscript. Joachim C. Mertens has received consulting honoraria from Abbvie, Bayer, BMS, Gilead, Janssen, Merck and MSD for work performed outside the current study. Seraphina Kissling, Stephanie von Felten, and Thomas Winder report no disclosures associated with the manuscript. The authors report no other potential conflicts of interest for this work.
References
1. Mittal S, El-Serag HB. Epidemiology of hepatocellular carcinoma: consider the population. J Clin Gastroenterol. 2013;47(Supplement 1):S2–S6. doi:10.1097/MCG.0b013e3182872f29
2. El-Serag HB, Kanwal F. Epidemiology of hepatocellular carcinoma in the United States: where are we? Where do we go? Hepatology. 2014;60(5):1767–1775. doi:10.1002/hep.27222
3. El-Serag HB, Kanwal F, Richardson P, Kramer J. Risk of hepatocellular carcinoma after sustained virological response in Veterans with hepatitis C virus infection. Hepatology. 2016;64(1):130–137. doi:10.1002/hep.28535
4. van der Meer AJ, Veldt BJ, Feld JJ, et al. Association between sustained virological response and all-cause mortality among patients with chronic hepatitis C and advanced hepatic fibrosis. JAMA. 2012;308(24):2584–2593. doi:10.1001/jama.2012.144878
5. Morgan RL, Baack B, Smith BD, Yartel A, Pitasi M, Falck-Ytter Y. Eradication of hepatitis C virus infection and the development of hepatocellular carcinoma: a meta-analysis of observational studies. Ann Intern Med. 2013;158(5 Pt 1):329–337. doi:10.7326/0003-4819-158-5-201303050-00005
6. Maan R, van Tilborg M, Deterding K, et al. Safety and Effectiveness of Direct-Acting Antiviral Agents for Treatment of Patients With Chronic Hepatitis C Virus Infection and Cirrhosis. Clin Gastroenterol Hepatol. 2016;14(12):1821–30 e6. doi:10.1016/j.cgh.2016.07.001
7. Younossi ZM, Stepanova M, Feld J, et al. Sofosbuvir and velpatasvir combination improves patient-reported outcomes for patients with HCV infection, without or with compensated or decompensated cirrhosis. Clin Gastroenterol Hepatol. 2017;15(3):421–30 e6. doi:10.1016/j.cgh.2016.10.037
8. Chhatwal J, Wang X, Ayer T, et al. Hepatitis C disease burden in the United States in the era of oral direct-acting antivirals. Hepatology. 2016;64(5):1442–1450. doi:10.1002/hep.28571
9. Cardoso H, Vale AM, Rodrigues S, et al. High incidence of hepatocellular carcinoma following successful interferon-free antiviral therapy for hepatitis C associated cirrhosis. J Hepatol. 2016;65(5):1070–1071. doi:10.1016/j.jhep.2016.07.027
10. Kozbial K, Moser S, Schwarzer R, et al. Unexpected high incidence of hepatocellular carcinoma in cirrhotic patients with sustained virologic response following interferon-free direct-acting antiviral treatment. J Hepatol. 2016;65(4):856–858. doi:10.1016/j.jhep.2016.06.009
11. Ravi S, Axley P, Jones D, et al. Unusually high rates of hepatocellular carcinoma after treatment with direct-acting antiviral therapy for hepatitis C related cirrhosis. Gastroenterology. 2017;152(4):911–912. doi:10.1053/j.gastro.2016.12.021
12. Reddy KR, Osinusi A, Thompson AJ. The need for appropriate comparisons: a response to Ravi et al. Gastroenterology. 2017;153(1):332–333. doi:10.1053/j.gastro.2017.03.074
13. Cheung MCM, Walker AJ, Hudson BE, et al. Outcomes after successful direct-acting antiviral therapy for patients with chronic hepatitis C and decompensated cirrhosis. J Hepatol. 2016;65(4):741–747. doi:10.1016/j.jhep.2016.06.019
14. Calleja JL, Crespo J, Rincon D, et al. Effectiveness, safety and clinical outcomes of direct-acting antiviral therapy in HCV genotype 1 infection: results from a Spanish real-world cohort. J Hepatol. 2017;66(6):1138–1148. doi:10.1016/j.jhep.2017.01.028
15. Finkelmeier F, Dultz G, Peiffer KH, et al. Risk of de novo hepatocellular carcinoma after HCV treatment with direct-acting antivirals. Liver Cancer. 2018;7(2):190–204. doi:10.1159/000486812
16. Janjua NZ, Wong S, Darvishian M, et al. The impact of SVR from direct-acting antiviral- and interferon-based treatments for HCV on hepatocellular carcinoma risk. J Viral Hepat. 2020;27(8):781–793. doi:10.1111/jvh.13295
17. Ioannou GN, Green PK, Berry K. HCV eradication induced by direct-acting antiviral agents reduces the risk of hepatocellular carcinoma. J Hepatol. 2017.
18. Nagaoki Y, Imamura M, Aikata H, et al. The risks of hepatocellular carcinoma development after HCV eradication are similar between patients treated with peg-interferon plus ribavirin and direct-acting antiviral therapy. PLoS One. 2017;12(8):e0182710. doi:10.1371/journal.pone.0182710
19. Carrat F, Fontaine H, Dorival C, et al. Clinical outcomes in patients with chronic hepatitis C after direct-acting antiviral treatment: a prospective cohort study. Lancet. 2019;393(10179):1453–1464. doi:10.1016/S0140-6736(18)32111-1
20. Waziry R, Hajarizadeh B, Grebely J, et al. Hepatocellular carcinoma risk following direct-acting antiviral HCV therapy: a systematic review, meta-analyses, and meta-regression. J Hepatol. 2017;67(6):1204–1212. doi:10.1016/j.jhep.2017.07.025
21. Huang P, Liu M, Zang F, et al. The development of hepatocellular carcinoma in HCV-infected patients treated with DAA: a comprehensive analysis. Carcinogenesis. 2018;39(12):1497–1505. doi:10.1093/carcin/bgy099
22. Nahon P, Bourcier V, Layese R, et al. Eradication of hepatitis C virus infection in patients with cirrhosis reduces risk of liver and non-liver complications. Gastroenterology. 2017;152(1):142–56 e2. doi:10.1053/j.gastro.2016.09.009
23. Romano A, Angeli P, Piovesan S, et al. Newly diagnosed hepatocellular carcinoma in patients with advanced hepatitis C treated with DAAs: a prospective population study. J Hepatol. 2018;69(2):345–352. doi:10.1016/j.jhep.2018.03.009
24. Shiha G, Mousa N, Soliman R, Nnh Mikhail N, Adel Elbasiony M, Khattab M. Incidence of HCC in chronic hepatitis C patients with advanced hepatic fibrosis who achieved SVR following DAAs: a prospective study. J Viral Hepat. 2020;27(7):671–679. doi:10.1111/jvh.13276
25. European Association for the Study of the Liver. Electronic address eee, European Association for the Study of the L. EASL Clinical Practice Guidelines: management of hepatocellular carcinoma. J Hepatol. 2018;69(1):182–236.
26. Bedossa P, Poynard T. An algorithm for the grading of activity in chronic hepatitis C. The METAVIR Cooperative Study Group. Hepatology. 1996;24(2):289–293. doi:10.1002/hep.510240201
27. Castera L, Forns X, Alberti A. Non-invasive evaluation of liver fibrosis using transient elastography. J Hepatol. 2008;48(5):835–847. doi:10.1016/j.jhep.2008.02.008
28. Lin ZH, Xin YN, Dong QJ, et al. Performance of the aspartate aminotransferase-to-platelet ratio index for the staging of hepatitis C-related fibrosis: an updated meta-analysis. Hepatology. 2011;53(3):726–736. doi:10.1002/hep.24105
29. Sterling RK, Lissen E, Clumeck N, et al. Development of a simple noninvasive index to predict significant fibrosis in patients with HIV/HCV coinfection. Hepatology. 2006;43(6):1317–1325. doi:10.1002/hep.21178
30. Kamath PS, Wiesner RH, Malinchoc M, et al. A model to predict survival in patients with end-stage liver disease. Hepatology. 2001;33(2):464–470. doi:10.1053/jhep.2001.22172
31. Peduzzi P, Concato J, Feinstein AR, Holford TR. Importance of events per independent variable in proportional hazards regression analysis. II. Accuracy and precision of regression estimates. J Clin Epidemiol. 1995;48(12):1503–1510. doi:10.1016/0895-4356(95)00048-8
32. R Core Team. R: a language and environment for statistical computing. R Foundation for Statistical Computing. Vienna, Austria. 2020. Available from: https://www.r-project.org.
33. Lee SH, Jin YJ, Shin JY, Lee JW. Assessment of hepatocellular carcinoma risk based on peg-interferon plus ribavirin treatment experience in this new era of highly effective oral antiviral drugs. Medicine. 2017;96(1):e5321. doi:10.1097/MD.0000000000005321
34. Mashiba T, Joko K, Kurosaki M, et al. Does interferon-free direct-acting antiviral therapy for hepatitis C after curative treatment for hepatocellular carcinoma lead to unexpected recurrences of HCC? A multicenter study by the Japanese Red Cross Hospital Liver Study Group. PLoS One. 2018;13(4):e0194704. doi:10.1371/journal.pone.0194704
35. Mun EJ, Green P, Berry K, Ioannou GN. No difference between direct-acting antivirals for hepatitis C in hepatocellular carcinoma risk. Eur J Gastroenterol Hepatol. 2019;31(1):47–52. doi:10.1097/MEG.0000000000001242
36. Ogawa E, Nomura H, Nakamuta M, et al. Development of hepatocellular carcinoma by patients aged 75–84 with chronic hepatitis C treated with direct-acting antivirals. J Infect Dis. 2020. doi:10.1093/infdis/jiaa359
37. Velentgas P, Dreyer NA, Nourjah P, Smith SR, Torchia MM, editor. Developing a Protocol for Observational Comparative Effectiveness Research: A User’s Guide. Rockville (MD): Agency for Healthcare Research and Quality (US). 2013:Publication No: 12(13)-EHC099.
38. Tampaki M, Savvanis S, Koskinas J. Impact of direct-acting antiviral agents on the development of hepatocellular carcinoma: evidence and pathophysiological issues. Ann Gastroenterol. 2018;31(6):670–679. doi:10.20524/aog.2018.0306
39. Singh S, Nautiyal A, Loke YK. Oral direct-acting antivirals and the incidence or recurrence of hepatocellular carcinoma: a systematic review and meta-analysis. Frontline Gastroenterol. 2018;9(4):262–270. doi:10.1136/flgastro-2018-101017
40. Marino Z, Darnell A, Lens S, et al. Time association between hepatitis C therapy and hepatocellular carcinoma emergence in cirrhosis: relevance of non-characterized nodules. J Hepatol. 2019.
41. Owusu Sekyere S, Schlevogt B, Mettke F, et al. HCC immune surveillance and antiviral therapy of hepatitis C virus infection. Liver Cancer. 2019;8(1):41–65. doi:10.1159/000490360
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.