")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 14
Rare complications of neurofibromatosis 1 diagnosed incidentally in two children
Authors Lazea C , Asavoaie C , Al-Khzouz C , Popa L
Received 15 May 2018
Accepted for publication 13 June 2018
Published 31 August 2018 Volume 2018:14 Pages 1547—1552
DOI https://doi.org/10.2147/TCRM.S173237
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Cecilia Lazea,1 Carmen Asavoaie,2 Camelia Al-Khzouz,3 Lenuta Popa1
1Department of Pediatrics I, Emergency Clinic Hospital for Children, “Iuliu Hatieganu” University of Medicine and Pharmacy, 2Department of Imaging and Radiology, Emergency Clinic Hospital for Children, 3Medical Genetics Department, Emergency Clinic Hospital for Children, University of Medicine and Pharmacy, Cluj-Napoca, Romania
Abstract: Neurofibromatosis 1 (NF1) is an autosomal-dominant disorder with various clinical expressivity and complications. Arterial hypertension may be present in patients with NF1 and is secondary to vasculopathy or pheochromocytoma. We herein describe two children (17 and 4 years old) diagnosed late with NF1 after severe arterial hypertension had been discovered due to pheochromocytoma and middle aortic syndrome. Routine measurement of arterial pressure is mandatory in children with NF1, in order to diagnose the complications of this disease.
Keywords: neurofibromatosis, pheochromocytoma, middle aortic syndrome, arterial hypertension, children
Introduction
Neurofibromatosis 1 (NF1) is a common neurocutaneous condition with an autosomal-dominant pattern of inheritance, affecting about 1 in 3,000 individuals.1 It is caused by mutations in the NF1 gene that lead to the production of nonfunctional neurofibromin that cannot regulate cell growth or division, which generates increased risk for various benign and malignant tumors.2,3 This disease has varied expression, even within families. Diagnosis criteria are: six or more café au lait macules (>0.5 cm at largest diameter in a prepubertal child or >1.5 cm in postpubertal individuals), axillary or groin freckling, two or more neurofibromas or one or more plexiform neurofibromas, two or more Lisch nodules, bony dysplasia (sphenoid wing dysplasia, bowing of the long bone ± pseudarthrosis), optic pathway glioma, and a first-degree relative with neurofibromatosis type 1. Two or more features are required for diagnosis.4 Hypertension is common among NF1 patients, and may be secondary to vasculopathy (renovascular disease or coarctation of the aorta) or pheochromocytoma, but recent research has suggested that in most cases of NF1, hypertension has no association with pheochromocytoma or renal artery stenosis.5 Here, we describe two children diagnosed with NF1 after severe secondary hypertension induced by pheochromocytoma and obstructive lesions of the middle aorta had been discovered. Written informed consent was obtained from the patients’ parents for publication of this case series and accompanying images.
Case presentation
Patient 1
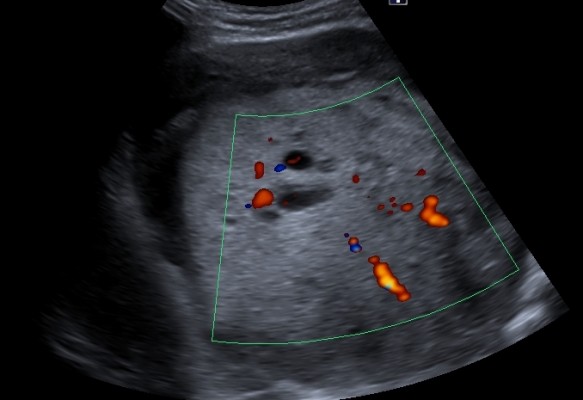
After a car accident without traumatic consequences, elevated arterial pressure of 190/130 mmHg (95th percentile for his age, sex, and height = 136/87 mmHg) were discovered in a 17-year-old male. Initially, arterial hypertension was attributed to his emotional state after the car accident, but during hospitalization the patient continued to be anxious and tachycardic, and arterial pressure remained high. Physical examination revealed normal somatic and pubertal development, numerous café au lait spots (13 spots with diameter >1.5 cm), axillary freckling and one plexiform neurofibroma. He had a 2-year history of headache, and brain magnetic resonance imaging (MRI) revealed an unprogressive arachnoid cyst. The adolescent’s mother presented numerous café au lait spots and neurofibromas, and these modifications, which were also present in the patient’s grandfather and great-grandfather, were considered a family trait. Abdominal ultrasound showed a parenchymal, well-defined, inhomogeneous mass lesion of 126×110×118 mm3 at the area superior to the left upper kidney pole, and adrenal gland origin could not be ruled out (Figure 1). The left kidney had pelvicalyceal dilatation, and the splenic vein was compressed by the tumor mass. The right adrenal gland and right kidney were normal. Contrast abdominal MRI confirmed the adrenal origin of the tumor (Figure 2). Hormonal assays revealed elevated plasma-free metanephrines and urinary catecholamines (Table 1). Pheochromocytoma was diagnosed, and 123I-metaiodobenzylguanidine (123I-MIBG) scintigraphy ruled out extra-adrenal uptake and metastatic localization (Figure 3). Echocardiography revealed left-ventricle hypertrophy. Ophthalmological findings were unremarkable. Endocrinological consultation was consistent with NF1 and pheochromocytoma, and surgical treatment was recommended. Prior to surgery, therapy with daily prazosin 2 mg was started, and after 7 days daily propranolol 60 mg was added. After 6 weeks of treatment, arterial pressure was 120/60 mmHg and heart rate had decreased to 60 bpm. Surgical removal of the tumor mass of the adrenal gland was performed, and microscopy of the adrenal mass showed histological features of Pheochromocytoma of the Adrenal gland Scaled Score of 2. After surgery, blood pressure was within normal range, and 1 week later antihypertensive therapy was stopped. Postoperative plasma free metanephrines had normalized.
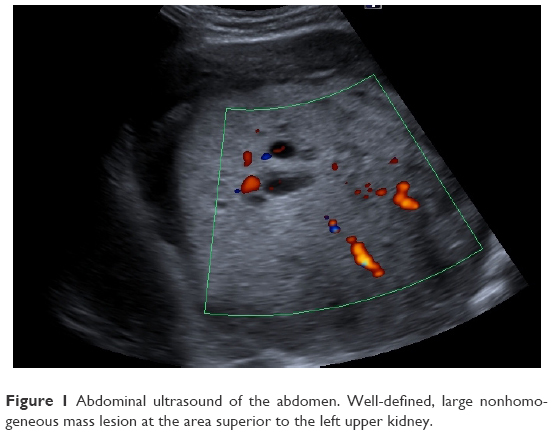 | Figure 1 Abdominal ultrasound of the abdomen. Well-defined, large nonhomogeneous mass lesion at the area superior to the left upper kidney. |
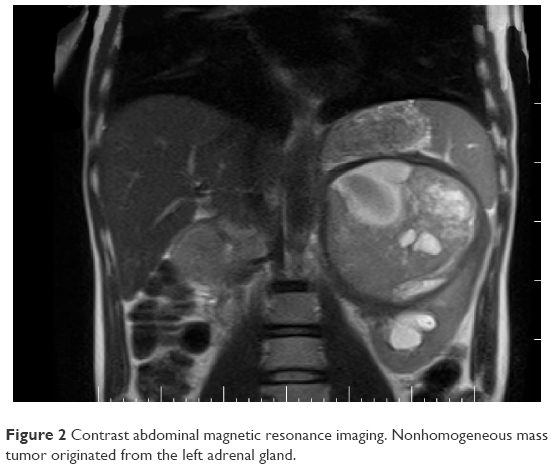 | Figure 2 Contrast abdominal magnetic resonance imaging. Nonhomogeneous mass tumor originated from the left adrenal gland. |
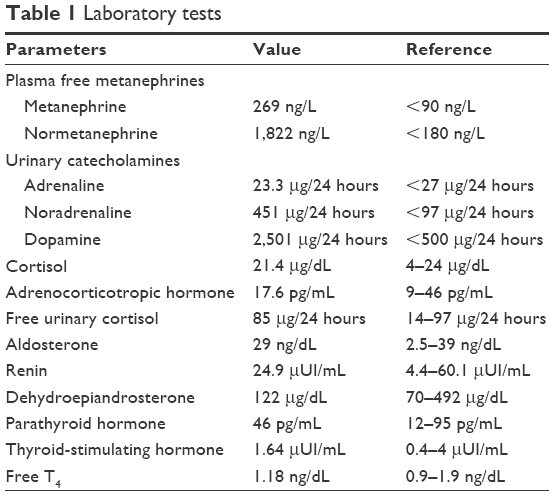 | Table 1 Laboratory tests |
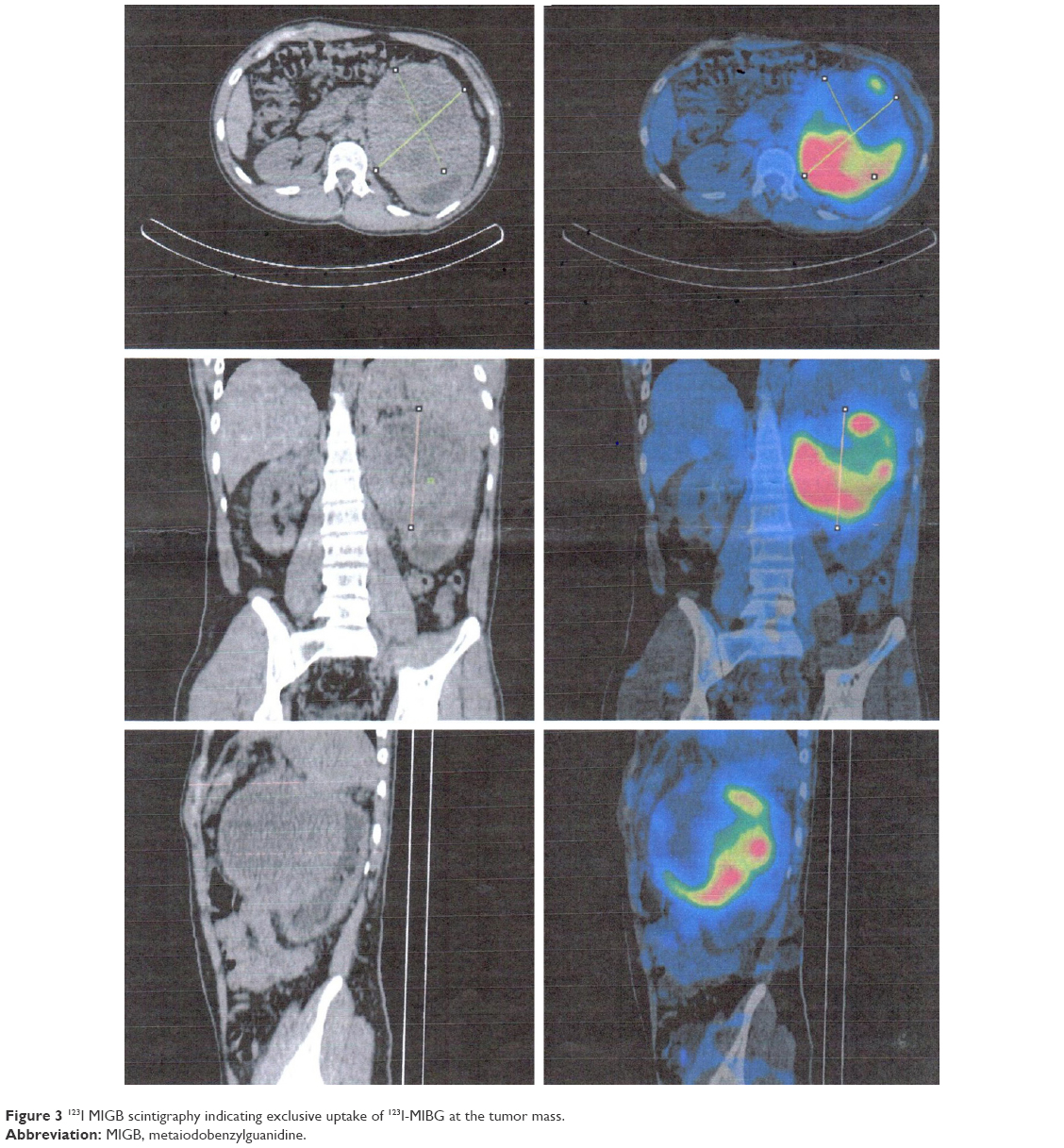 | Figure 3 123I MIGB scintigraphy indicating exclusive uptake of 123I-MIBG at the tumor mass. |
Patient 2
A 4-year-old boy diagnosed at birth with dextrocardia presented with an incidental finding of severe hypertension 170/90 mmHg (95th percentile for his age, sex, and height = 107/67 mmHg) after a minor cranial trauma. He had a 2-year history of idiopathic abdominal pain. Physical examination revealed normal weight, height in the 10th percentile, numerous café au lait spots (eight spots with diameter >0.5 cm), axillary and groin freckling, and one plexiform neurofibroma. All peripheral pulses were palpable, there was no detectable abdominal murmur, and blood pressure was recorded in all four limbs (right arm 145/95 mmHg, left arm 150/100 mmHg, right leg 160/100 mmHg, left leg 155/95 mmHg). Laboratory investigations revealed that renal- and liver-function tests, blood-cell counts, urinalysis, serum electrolytes, aldosterone, free plasma metanephrines, and 24-hour urine levels of catecholamines were all within normal ranges. Plasma-renin level was elevated, suggesting a renovascular cause of hypertension (Table 2). Renal ultrasound examination of the abdomen was normal, with both kidneys normal size. Doppler-derived renal resistive index revealed elevated values (0.78 on the left and 0.8 on the right). Thoracic and abdominal MR angiogram revealed reduced caliber of the abdominal aorta and celiac trunk, stenosis of the upper mesenteric artery, irregular right renal artery with reduced caliber, and narrowed left renal artery at its emergence (Figure 4). Echocardiography revealed mild left-ventricle hypertrophy and dextrocardia. Ophthalmological findings were unremarkable. The final diagnosis of NF1 with middle aortic syndrome (MAS) and secondary hypertension was then made. For 1 month, the patient took a calcium-channel blocker, an α-adrenergic agonist, and a diuretic agent. This regimen failed to control blood pressure, so angioplasty was scheduled.
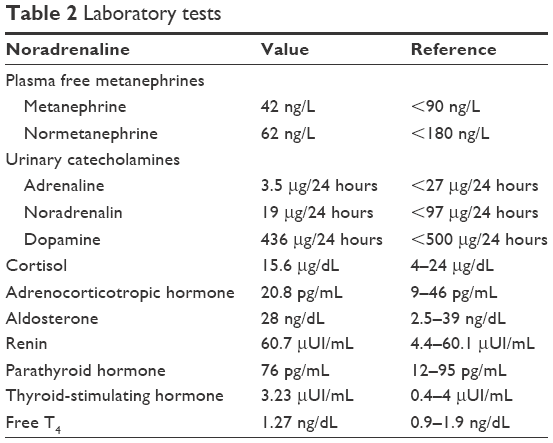 | Table 2 Laboratory tests |
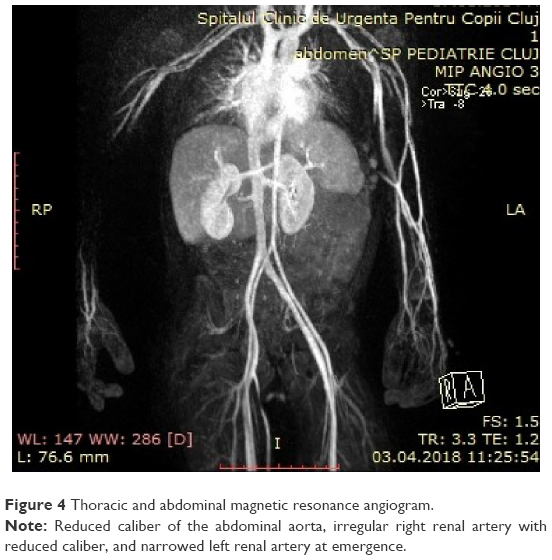 | Figure 4 Thoracic and abdominal magnetic resonance angiogram. |
Discussion
We present two patients who were diagnosed with NF1 only after elevated blood pressure values had been detected, although the clinical markers of this disease (café au lait spots) were present at birth. In the first case, cutaneous markers were present in other generations in the family but considered a family trait, so the diagnosis was delayed. In the second case, the café au lait spots had been overlooked by both the family and medical staff who examined the child. Usually, these spots are multiple and present at birth or in early infancy and observed in children with NF1 by age 3 years, so it is important to increase awareness of NF1 diagnosis among medical staff that come into contact with such patients to prevent the various complications of this disease.
Studies have shown that hypertension is much more frequent in children with NF1 (14%–16%) compared to the general pediatric population (3.5%).5,6 According to some studies, hypertension may be secondary to renal artery stenosis or pheochromocytoma, which have a prevalence of 2%, but these findings cannot explain the high prevalence of increased blood pressure in children with NF1, suggesting that hypertension could be a common primary feature of this disease.4,5 Detection of elevated blood pressure values in children with NF1 requires complete evaluation for secondary causes (pheochromocytoma and vasculopathy).
The first patient reported was diagnosed with NF1 and pheochromocytoma after detection of severe hypertension at the scene of a car accident in which he was involved. Pheochromocytoma is a tumor arising from adrenomedullary chromaffin cells that commonly produces one or more catecholamines (epinephrine, norepinephrine, and dopamine), and prevalence in children with hypertension is approximately 1.7%.7 This type of tumor is variable in NF1 patients (0.1%–7.7%) and the first tumor of the adrenal gland in a patient with NF1 was mentioned in 1910.8,9 The mean age of presentation in the general population is in the fourth decade, and the majority of tumors are unilateral and benign.10 Pheochromocytoma detected in NF1 patients can have no clinical manifestations, but different outcomes are possible: from well-controlled hypertension to multiorgan failure.11
Plasma free metanephrines are the ideal test for diagnosis, because these metabolites of catecholamines are released continuously from the tumor. In our patient, norepinephrine production was higher than epinephrine. This finding is in line with studies that in patients with negligible epinephrine-producing tumors, age at diagnosis was earlier than in epinephrine-producing tumors.12,13 Computed tomography can localize the adrenal tumors, and MRI is superior for the detection of extra-adrenal tumors. 123I-MIBG scintigraphy and 18F-dopamine or 18F-dopa positron-emission tomography are used to detect or exclude multifocal or metastatic disease.7,13 In our patient, ultrasonography and MRI revealed the tumor mass, and 123I-MIBG scintigraphy ruled out multifocal disease and malignancy. The size of secreting pheochromocytoma is correlated with the amount of catecholamines released, and this finding was demonstrated in our case, who presented plasma concentration of metanephrines ten times that of the upper reference limit.9,14
The treatment of pheochromocytoma involves a multidisciplinary approach. An α-blocker (phenoxybenzamine or prazosin) is the initial drug of choice. β-Blockers (propranolol, metoprolol, atenolol) should never be used in the absence of an α-blocking agent, as that could precipitate a hypertensive crisis. Tumor removal consists in laparoscopic adrenalectomy (retroperitoneal approach is preferred) or open resection for large or invasive pheochromocytoma.13 Careful intraoperative and postoperative monitoring of the patient’s blood pressure, heart rate, and volume replacement are crucial. Postoperatively, plasma or urinary metanephrine-concentration measurements are required.3,13 In our patient, treatment was started with prazosin and propranolol added after 7 days, with considerable reduction in blood pressure and heart rate. Open surgery was required because of the large dimensions of the tumor. Blood pressure normalized after 1 week, and at follow-ups over the next 12 months, he had not experienced any relapses. Microscopy of the tumor revealed histological features of benign pheochromocytoma. The Pheochromocytoma of the Adrenal gland Scaled Score was developed to distinguish benign from malignant, but the use of this score has not been validated in all studies, and a new scoring system was proposed.3,15
The second patient reported was a 4-year-old boy diagnosed with NF1 in whom severe blood pressure values recorded during hospital admission for minor trauma to the head led to detection of MAS as an underlying vascular condition. Vascular abnormalities that can occur in NF1 patients are stenosis, aneurysms, and arteriovenous malformations due to cellular proliferation, degeneration, healing, and smooth-muscle loss and fibrosis caused by deficiency in neurofibromin with proliferation of the vessel wall. The renal artery is most often affected and arterial hypertension the most common presentation.16 Patients can be asymptomatic despite multiple-vessel involvement. MAS consists in obstructive lesions of the middle aorta, involves renal and splanchnic branches of the aorta, and is more common in children. Besides NF1, there are other causes, such as genetic (Alagille syndrome or Williams syndrome), inflammatory (Takayasu’s arteritis), and intrauterine infection (rubella), or it can be idiopathic.17
In our case, Doppler ultrasonography of the renal arteries suggested a diagnosis of renal artery stenosis, and MR angiography, considered an alternative to conventional angiography, revealed reduced caliber of the abdominal aorta and celiac trunk, stenosis of the upper mesenteric artery, and bilateral renal artery stenosis. The right renal artery had several narrow areas, and the left renal artery had segmental narrowing at emergence. Our 4-year-old patient was diagnosed early compared to the common mean age at presentation (6.7–9.1 years).17,18 Extra-aortic vessel involvement in our case was in line with other studies, in which 60%–69% of cases had bilateral renal artery stenosis, 30%–60% superior mesenteric artery stenosis, and 22%–60% narrowing of the celiac trunk.17,18
Current management of MAS can be pharmacological with multiple antihypertensive drugs, endovascular, or surgical. Pharmacological management is not satisfactory in many cases, and surgical procedures, such as aorto-aortic bypass grafting, graft vascular replacement, patch angioplasty, renal autotransplantation, or endovascular procedures are required. Percutaneous transluminal angioplasty with or without stenting are endovascular procedures used in this condition with various outcomes, but are associated with complications more frequently in children with NF1. Restenosis and impaired flow may develop due to fibrosis or lack of growth expected from grafts or stents. Residual hypertension is also common after surgical or endovascular procedures and requires antihypertensive medication or secondary intervention.17–19 In our case, a regimen with three antihypertensive drugs failed in blood pressure control, and an endovascular procedure was considered.
Conclusion
In many cases, NF1 diagnosis is often delayed until other symptoms and clinical problems arise. NF1 can have severe complications due to severe hypertension, and regular measurement of blood pressure is mandatory in all children with NF1 to diagnose as early as possible the manifestations of this disease. Screening with plasma or urinary metanephrines and Doppler examinations of the renal vessels and suprarenal aorta may be justified in children with NF1.
Author contributions
CL directed this study. CL, CA, and CAK collected patient data. CL wrote the main manuscript. LP and CA gave suggestions on this study. All authors contributed toward data analysis, drafting and critically revising the paper, reviewed the manuscript, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Reynolds RM, Browning GG, Nawroz I, Campbell IW. von Recklinghausen’s neurofibromatosis: neurofibromatosis type 1. Lancet. 2003;361(9368):1552–1554. | ||
Hamilton SJ, Friedman JM. Insights into the pathogenesis of neurofibromatosis 1 vasculopathy. Clin Genet. 2000;58(5):341–344. | ||
Shah U, Giubellino A, Pacak K. Pheochromocytoma: implications in tumorigenesis and the actual management. Minerva Endocrinol. 2012;37(2):141–156. | ||
Ferner RE, Huson SM, Thomas N, et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet. 2007;44(2):8–18. | ||
Dubov T, Toledano-Alhadef H, Chernin G, Constantini S, Cleper R, Ben-Shachar S. High prevalence of elevated blood pressure among children with neurofibromatosis type 1. Pediatr Nephrol. 2016;31(1):131–136. | ||
Flynn JT, Kaelber DC, Baker-Smith CM, et al. Clinical practice guideline for screening and management of high blood pressure in children and adolescents. Pediatrics. 2017;140(3):e20171904. | ||
Lenders JW, Duh QY, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2014;99(6):1915–1942. | ||
Walther MM, Herring J, Enquist E, Keiser HR, Linehan WM. von Recklinghausen’s disease and pheochromocytomas. J Urol. 1999;162(5):1582–1586. | ||
Képénékian L, Mognetti T, Lifante JC, et al. Interest of systematic screening of pheochromocytoma in patients with neurofibromatosis type 1. Eur J Endocrinol. 2016;175(4):335–344. | ||
Costa MH, Ortiga-Carvalho T, Volante AD, Valsman M. Pheochromocytomas and paragangliomas: clinical and genetic approaches. Front Endocrinol. 2015;6:126. | ||
Ahmed R, Darrat Y, Hamoudeh E, Elhamdani MO, Yaqub A. Acute cardiomyopathy and multiorgan failure in a patient with pheochromocytoma and neurofibromatosis type 1. J Pak Med Assoc. 2014;64(2):214–216. | ||
Eisenhofer G, Timmers HJ, Lenders JW, et al. Age at diagnosis of pheochromocytoma differs according to catecholamine phenotype and tumor location. J Clin Endocrinol Metab. 2011;96(2):375–384. | ||
Cecchin D, Lumachi F, Marzola MC, et al. A meta-iodobenzylguanidine scintigraphic scoring system increases accuracy in the diagnostic management of pheochromocytoma. Endocr Relat Cancer. 2006;13(2):525–533. | ||
Eisenhofer G, Lenders JW, Goldstein DS, et al. Pheochromocytoma catecholamine phenotypes and prediction of tumor size and location by use of plasma free metanephrines. Clin Chem. 2005;51(4):735–744. | ||
Lloyd RV. Adrenal cortical tumors, pheochromocytomas and paragangliomas. Mod Pathol. 2011;24 Suppl 2:S58–S65. | ||
Oderich GS, Sullivan TM, Bower TC, et al. Vascular abnormalities in patients with neurofibromatosis syndrome type I: clinical spectrum, management, and results. J Vasc Surg. 2007;46(3):475–484. | ||
Rumman RK, Nickel C, Matsuda-Abedini M, et al. Disease beyond the arch: a systematic review of middle aortic syndrome in childhood. Am J Hypertens. 2015;28(7):833–846. | ||
Porras D, Stein DR, Ferguson MA, et al. Midaortic syndrome: 30 years of experience with medical, endovascular and surgical management. Pediatr Nephrol. 2013;28(10):2023–2033. | ||
Barral X, de Latour B, Vola M, Lavocat MP, Fichtner C, Favre JP. Surgery of the abdominal aorta and its branches in children: late follow-up. J Vasc Surg. 2006;43(6):1138–1144. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.