")
Back to Journals » Drug Design, Development and Therapy » Volume 8
Quinoxaline-substituted chalcones as new inhibitors of breast cancer resistance protein ABCG2: polyspecificity at B-ring position
Authors Winter E, Gozzi GJ, Chiaradia-Delatorre LD, Daflon-Yunes N, Terreux R, Gauthier C, Mascarello A, Leal PC, Cadena SM, Yunes RA, Nunes RJ, Crecynski-Pasa TB, Di Pietro A
Received 26 October 2013
Accepted for publication 28 February 2014
Published 27 May 2014 Volume 2014:8 Pages 609—619
DOI https://doi.org/10.2147/DDDT.S56625
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Evelyn Winter,1,2,* Gustavo Jabor Gozzi,1,3,* Louise Domeneghini Chiaradia-Delatorre,4 Nathalia Daflon-Yunes,1 Raphael Terreux,5 Charlotte Gauthier,1 Alessandra Mascarello,4 Paulo César Leal,4 Silvia M Cadena,3 Rosendo Augusto Yunes,4 Ricardo José Nunes,4 Tania Beatriz Creczynski-Pasa,2 Attilio Di Pietro1
1Equipe Labellisée Ligue 2013, Université Lyon 1, Institut de Biologie et Chimie des Protéines, Lyon, France; 2Department of Pharmaceutical Sciences, Federal University of Santa Catarina, Florianopolis, Brazil; 3Department of Biochemistry and Molecular Biology, Federal University of Paraná, Curitiba, Brazil; 4Department of Chemistry, Federal University of Santa Catarina, Florianopolis, Brazil; 5Bioinformatique structures et interactions, Université Lyon 1, Institut de Biologie et Chimie des Protéines, Lyon, France
*These authors contributed equally to this work
Abstract: A series of chalcones substituted by a quinoxaline unit at the B-ring were synthesized and tested as inhibitors of breast cancer resistance protein-mediated mitoxantrone efflux. These compounds appeared more efficient than analogs containing other B-ring substituents such as 2-naphthyl or 3,4-methylenedioxyphenyl while an intermediate inhibitory activity was obtained with a 1-naphthyl group. In all cases, two or three methoxy groups had to be present on the phenyl A-ring to produce a maximal inhibition. Molecular modeling indicated both electrostatic and steric positive contributions. A higher potency was observed when the 2-naphthyl or 3,4-methylenedioxyphenyl group was shifted to the A-ring and methoxy substituents were shifted to the phenyl B-ring, indicating preferences among polyspecificity of inhibition.
Keywords: ABC transporters, breast cancer resistance protein (BCRP)/ABCG2, quinoxaline derivatives, structure–activity relationships, drug efflux
Introduction
According to the World Health Organization, cancer is a leading cause of death worldwide.1 Despite the high incidence of cancer, anticancer chemotherapy is limited by cross resistance of tumor cells to these drugs, a phenomenon which is named multidrug resistance (MDR). It is often associated with the overexpression of ATP-binding cassette (ABC) transporters such as P-glycoprotein, also known as ATP-binding cassette, subfamily B, member 1 (ABCB1);2 multidrug resistance protein 1 (MRP1), also known as ATP-binding cassette, subfamily C, member 1 (ABCC1);3 and ATP-binding cassette, subfamily G, member 2 (ABCG2), which is also called breast cancer resistance protein or mitoxantrone-resistance protein.4 These transporters use ATP hydrolysis as a source of energy to transport chemotherapeutics outside the cell, significantly reducing their intracellular accumulation.5
ABCG2 was the most recently discovered among the three main multidrug ABC transporters, as isolated from a MCF7/AdrVp cell line in 1998.4 It is expressed in different physiological membrane barriers protecting brain, placenta, and other sensitive organs. This efflux transporter also plays an important role in MDR of a number of cancer cells such as leukemias and solid tumors.6 For this reason, new and specific ABCG2 inhibitors are being investigated for use in improving the efficiency of chemotherapy of resistant tumors.
Several classes of ABCG2 inhibitors have been characterized: 1) ABCB1 inhibitors such as elacridar and tariquidar were found to be also active toward ABCG2;7 2) selective inhibitors such as fumitremorgin C, from Aspergillus fumigatus, were developed, but their neurotoxicity prevented any clinical assays;8,9 3) new-generation inhibitors included flavonoid derivatives and chemical derivatives of compounds known as ABCG2 inhibitors.10–16
More than 80% of current drugs are obtained from natural sources or are based on natural compounds.17 Chalcones, containing two phenyl A-ring and B-ring moieties, are essential intermediate compounds in flavonoid biosynthesis in plants and have been recently studied for their effects against drug resistance. They were found to inhibit ABCG2 with a marked polyspecificity toward the A-ring moiety where the standard phenyl group could be partly substituted by 2′-OH-naphthyl,18 and quite efficiently by indole19 as well as by 3′,4′-methylenedioxyphenyl or 2′-naphthyl groups.20 Interestingly, indole also appeared to be efficient at the B-ring position when methoxy substituents were present on the phenyl A-ring.19
The aim of this work was to investigate a new series of chalcones containing a quinoxaline unit as B-ring, and to compare the quinoxaline efficiency to other bicyclic B-rings such as a 2-naphthyl or 3,4-methylenedioxy-phenyl unit as inhibitors of ABCG2. The results indicate that quinoxaline potently contributes to inhibition, together with methoxy groups on the other side of the molecule. The intermediate potency of a 1-naphthyl group and functional asymmetry of chalcones further documented the inhibition polyspecificity.
Material and methods
Chemistry
Synthesis of quinoxaline-substituted chalcones
All reagents were purchased from Sigma-Aldrich (St Louis, MO, USA) or Oakwood Products (West Columbia, NC, USA), and solvents from Merck (Whitehouse Station, NJ, USA) or Vetec (Duque de Caxias, RJ, Brazil). The quinoxaline-6-carbaldehyde was synthesized directly from the methyl-quinoxaline as previously described,21 with a yield of 80%. The quinoxaline-substituted chalcones (1–12) were prepared by aldol condensation between quinoxaline-6-carbaldehyde and corresponding acetophenones in methanol and potassium hydroxide 50% w/v, under magnetic stirring at room temperature (Figure 1). Distilled water and 10% hydrochloric acid were added to the reaction for total precipitation of the compounds, which were then obtained by vacuum filtration and recrystallized in dichloromethane/hexane or ethanol. The purity of the synthesized compounds was analyzed by thin-layer chromatography using Merck silica-precoated aluminum plates of 200 μm thickness, with a hexane/ethyl acetate 1:1 system. Compounds were visualized with ultraviolet light (λ=254 nm and 360 nm) and using sulfuric anisaldehyde solution followed by heat application as the developing agent. Compounds 1–4 were previously published by our group,22 and 5–12 were newly synthesized chalcones obtained with yields between 79% and 92%.
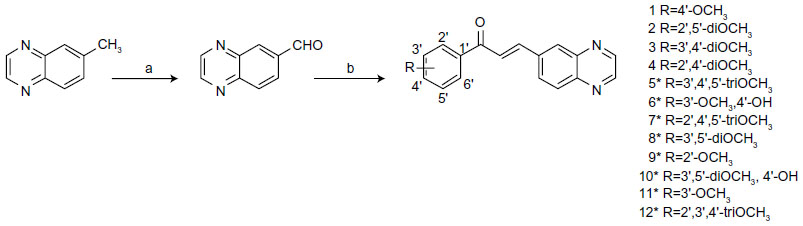 | Figure 1 Synthesis of quinoxaline-substituted chalcones. |
Synthesis of 1-naphthyl-, 2-naphthyl-, and 3,4-methylenedioxyphenyl-substituted chalcones
The chalcones 13–61 were prepared by the same procedure described above and previously published by our group.23,24
Physicochemical data of the compounds
The structures were confirmed by melting point (mp), proton (1H NMR) and carbon (13C NMR) nuclear magnetic resonance spectroscopy. The mps were determined with a Microquimica MGAPF-301 apparatus (Microquímica Equipamentos Ltda., Palhoça, SC, Brazil) and were uncorrected. NMR (1H and 13C) spectra were recorded on a Varian Oxford AS-400 (Agilent Technologies, Santa Clara, CA, USA) (400 MHz) or on a Bruker Ac-200F instrument (Bruker, Billerica, MA, USA) (200 MHz), using tetramethylsilane as internal standard. 1H NMR spectra revealed that all the structures were geometrically pure and E configured (JHα−Hβ =16 Hz). High-resolution mass spectra (HRMS) were recorded on a micrOTOF-QII mass spectrometer (Bruker) equipped with an automatic syringe pump (KD Scientific, Holliston, MA, USA) for sample injection (constant flow of 3 μL/minute) by positive mode of electron spray ionization technique (4.5 kV and 180°C). The instrument was calibrated in the range of 50–3,000 m/z using an internal calibration standard (low concentration tuning mix solution) (Agilent Technologies). An acetonitrile/methanol mixture was used as solvent of the compounds. Data were processed via Bruker Data Analysis Software (version 4.0).
Compound 5: (2E)-1-(3′,4′,5′-trimethoxyphenyl)- 3-(quinoxalin-6-yl)-2-propen-1-one
Light yellow solid, mp 90°C–92°C; 1H NMR (CDCl3) δ 3.96 (s, 3H, p-OCH3), 3.97 (s, 6H, m-OCH3), 7.33 (s, 2H, H2′, H6′), 7.70 (d, 1H, J=16.0 Hz, Hα), 8.01 (d, 1H, J=16.0 Hz, Hβ), 8.07 (dd, 1H, J=8.0/1.0 Hz, H3), 8.16 (d, 1H, J=8.0 Hz, H4), 8.35 (d, 1H, J=1.0 Hz, H1), 8.88 (dd, 2H, J=8.0/1.0 Hz, H6, H7) ppm; 13C NMR (CDCl3) δ 56.41 (m-OCH3), 60.97 (p-OCH3), 106.25 (C2′, C6′), 124.18 (Cα), 128.73 (C3), 130.07 (C1), 130.16 (C4), 133.02 (C1′), 136.64 (C2), 142.74 (Cβ), 143.06 (C4a), 143.87 (C8a), 145.54 (C6), 145.78 (C4′, C7), 153.22 (C3′, C5′), 188.53 (C=O) ppm; HRMS (ESI+) m/z: calculated for C20H18N2O4 [M+]: 351.1339; found, 351.1338. Yield: 89%.
Compound 6: (2E)-1-(3′-methoxy,4′-hydroxy-phenyl)- 3-(quinoxalin-6-yl)-2-propen-1-one
Light yellow solid, mp 179°C–181°C; 1H NMR (CDCl3) δ 4.01 (s, 3H, m-OCH3), 6.26 (sl, 1H, OH), 7.03 (d, 1H, J=8.0 Hz, H5′), 7.67 (d, 1H, J=2.0 Hz, H2′), 7.71 (dd, 1H, J=8.0/2.0 Hz, H6′), 7.76 (d, 1H, J=16.0 Hz, Hα), 7.99 (d, 1H, J=16.0 Hz, Hβ), 8.08 (dd, 1H, J=8.0/1.0 Hz, H3), 8.15 (d, 1H, J=8.0 Hz, H4), 8.33 (d, 1H, J=1.0 Hz, H1), 8.88 (dd, 2H, J=8.0/1.0 Hz, H6, H7) ppm; 13C NMR (CDCl3) δ 55.43 (m-OCH3), 111.06 (C2′), 114.53 (C5′), 123.34 (C6′), 124.07 (Cα), 128.12 (C3), 129.18 (C1′), 129.37 (C4), 129.69 (C1), 136.29 (C2), 1402.80 (Cβ), 142.44 (C4a), 143.08 (C8a), 145.06 (C6), 145.35 (C7), 147.49 (C3′), 151.73 (C4′), 186.83 (C=O) ppm; HRMS (ESI+) m/z: calculated for C18H14N2O3 [M+]: 307.1077; found, 307.1076. Yield: 86%.
Compound 7: (2E)-1-(2′,4′,5′-trimethoxyphenyl)- 3-(quinoxalin-6-yl)-2-propen-1-one
Yellow solid, mp 159°C–161°C; 1H NMR (CDCl3) δ 3.84 (s, 3H, o-OCH3), 3.92 (s, 3H, m-OCH3), 3.98 (s, 3H, p-OCH3), 6.56 (s, 1H, H3′), 7.44 (s, 1H, H6′), 7.86 (d, 1H, J=16.0 Hz, Hα), 7.91 (d, 1H, J=16.0 Hz, Hβ), 8.06 (dd, 1H, J=8.0/1.0 Hz, H3), 8.12 (d, 1H, J=8.0 Hz, H4), 8.29 (d, 1H, J=1.0 Hz, H1), 8.86 (dd, 2H, J=8.0/1.0 Hz, H6, H7) ppm; 13C NMR (CDCl3) δ 56.09 (m-OCH3), 56.18 (p-OCH3), 56.75 (o-OCH3), 96.75 (C3′), 113.10 (C6′), 119.94 (C1′), 127.32 (Cα), 129.79 (C3), 129.99 (C1), 131.01 (C4), 137.49 (C2), 139.74 (Cβ), 142.71 (C5′), 143.18 (C4a), 144.42 (C8a), 145.27 (C6), 145.65 (C7), 153.83 (C2′), 154.67 (C4′), 186.62 (C=O) ppm; HRMS (ESI+) m/z: calculated for C20H18N2O4 [M+]: 351.1339; found, 351.1339. Yield: 89%.
Compound 8: (2E)-1-(3′,5′-dimethoxyphenyl)- 3-(quinoxalin-6-yl)-2-propen-1-one
Cream solid, mp 87°C–88°C; 1H NMR (CDCl3) δ 3.88 (s, 6H, m-OCH3), 6.70 (t, 1H, J=2.0 Hz, H4′), 7.19 (d, 2H, J=2.0 Hz, H2′, H6′), 7.67 (d, 1H, J=16.0 Hz, Hα), 7.99 (d, 1H, J=16.0 Hz, Hβ), 8.06 (dd, 1H, J=8.0/1.0 Hz, H3), 8.15 (d, 1H, J=8.0 Hz, H4), 8.32 (d, 1H, J=1.0 Hz, H1), 8.87 (dd, 2H, J=8.0/1.0 Hz, H6, H7) ppm; 13C NMR (CDCl3) δ 55.63 (m-OCH3), 105.36 (C4′), 106.41 (C2′, C6′), 124.54 (Cα), 128.48 (C3), 130.16 (C4), 130.42 (C1), 136.61 (C2), 139.76 (C1′), 142.96 (Cβ), 143.10 (C4a), 143.91 (C8a), 145.27 (C6), 145.78 (C7), 160.99 (C3′, C5′), 189.53 (C=O) ppm; HRMS (ESI+) m/z: calculated for C19H16N2O3 [M+]: 321.1234; found, 321.1236. Yield: 79%.
Compound 9: (2E)-1-(2′-methoxyphenyl)- 3-(quinoxalin-6-yl)-2-propen-1-one
Cream solid, mp 93°C–95°C; 1H NMR (CDCl3) δ 3.86 (s, 3H, o-OCH3), 7.07 (m, 1H, H3′), 7.38 (m, 1H, H5′), 7.66 (m, 1H, H4′), 7.76 (d, 1H, J=16.0 Hz, Hα), 8.03 (d, 1H, J=16.0 Hz, Hβ), 8.04 (m, 1H, H3), 8.13 (d, 1H, J=8.0 Hz, H4), 8.35 (s, 1H, H1), 8.64 (m, 1H, H6′), 8.77–8.84 (m, 2H, H6, H7) ppm; 13C NMR (CDCl3) δ 55.58 (o-OCH3), 107.45 (C3′), 119.71 (C5′), 124.55 (Cα), 128.02 (C6′), 129.68 (C3), 130.31 (C1), 130.45 (C4), 133.50 (C4′), 136.70 (C2), 138.22 (C1′), 145.62 (C6), 145.85 (C7), 142.87 (Cβ), 143.16 (C4a), 144.02 (C8a), 160.02 (C2′), 189.90 (C=O) ppm; HRMS (ESI+) m/z: calculated for C18H14N2O2 [M+]: 291.1128; found, 291.1130. Yield: 81%.
Compound 10: (2E)-1-(3′,5′-dimethoxy,4′-hydroxy-phenyl)-3-(quinoxalin-6-yl)-2-propen-1-one
Gold yellow solid, mp 83°C–84°C; 1H NMR (CDCl3) δ δ 4.01 (s, 6H, m-OCH3), 6.26 (sl, 1H, OH), 7.39 (d, 2H, J=2.0 Hz, H2′, H6′), 7.72 (d, 1H, J=16.0 Hz, Hα), 8.01 (d, 1H, J=16.0 Hz, Hβ), 8.07 (d, 1H, J=8.0 Hz, H3), 8.17 (d, 1H, J=8.0 Hz, H4), 8.36 (s, 1H, H1), 8.89 (dd, 2H, J=8.0/1.0 Hz, H6, H7) ppm; 13C NMR (CDCl3) δ 56.60 (m-OCH3), 106.16 (C2′, C6′), 133.70 (C1′), 124.20 (Cα), 129.93 (C3), 130.14 (C4), 130.90 (C1), 136.82 (C2), 139.06 (Cβ), 143.32 (C4a), 142.90 (C8a), 145.30 (C6), 145.75 (C4′, C7), 147.03 (C3′, C5′), 194.90 (C=O) ppm; HRMS (ESI+) m/z: calculated for C19H16N2O4 [M+]: 337.1183; found, 337.1180. Yield: 88%.
Compound 11: (2E)-1-(3′-methoxyphenyl)- 3-(quinoxalin-6-yl)-2-propen-1-one
Beige solid, mp 108°C–109°C; 1H NMR (CDCl3) δ 3.90 (s, 3H, m-OCH3), 7.16 (d, 1H, J=8.0 Hz, H6′), 7.44 (t, 1H, J=8.0 Hz, H5′), 7.58 (s, 1H, H2′), 7.65 (d, 1H, J=8.0 Hz, H4′), 7.71 (d, 1H, J=16.0 Hz, Hα), 7.99 (d, 1H, J=16.0 Hz, Hβ), 8.07 (d, 1H, J=8.0 Hz, H3), 8.15 (d, 1H, J=8.0 Hz, H4), 8.32 (s, 1H, H1), 8.87 (d, 2H, J=8.0 Hz, H6, H7) ppm; 13C NMR (CDCl3) δ 55.52 (m-OCH3), 112.83 (C2′), 119.71 (C5′), 121.12 (C4′), 124.49 (Cα), 128.44 (C6′), 129.70 (C3), 130.21 (C1), 130.52 (C4), 136.61 (C2), 139.19 (C1′), 142.91 (Cβ), 143.13 (C4a),143.95 (C8a), 145.58 (C6), 145.81 (C7), 159.98 (C3′), 189.64 (C=O) ppm; HRMS (ESI+) m/z: calculated for C18H14N2O2 [M+]: 291.1128; found, 291.1123. Yield: 80%.
Compound 12: (2E)-1-(2′,3′,4′-trimethoxyphenyl)- 3-(quinoxalin-6-yl)-2-propen-1-one
Beige solid, mp 146°C–148°C; 1H NMR (CDCl3) δ 3.93 (s, 3H, o-OCH3), 3.94 (s, 3H, m-OCH3), 3.95 (s, 3H, p-OCH3), 6.78 (d, 1H, J=8.0 Hz, H5′), 7.56 (d, 1H, J=8.0 Hz, H6′), 7.74 (d, 1H, J=16.0 Hz, Hα), 7.88 (d, 1H, J=16.0 Hz, Hβ), 8.05 (dd, 1H, J=8.0/1.0 Hz, H3), 8.12 (d, 1H, J=8.0 Hz, H4), 8.28 (s, 1H, H1), 8.86 (dd, 2H, J=8.0/1.0 Hz, H6, H7) ppm; 13C NMR (CDCl3) δ 56.16 (m-OCH3), 61.09 (p-OCH3), 62.13 (o-OCH3), 107,46 (C5′), 119.88 (C1′), 126.12 (Cα), 128.60 (C6′), 129.12 (C3), 130.09 (C1), 130.16 (C4), 137.10 (C2), 140.71 (Cβ), 143.16 (C4a), 143.81 (C8a), 145.38 (C6), 145.69 (C7), 154.05 (C3′), 156.51 (C2′), 157.50 (C4′), 190.07 (C=O) ppm; HRMS (ESI+) m/z: calculated for C20H18N2O4 [M+]: 351.1339; found, 351.1340. Yield: 92%.
Biology and biochemistry
Compounds
Mitoxantrone was purchased from Sigma-Aldrich. All commercial reagents were of the highest available purity grade. The chalcones were dissolved in dimethyl sulfoxide and then diluted in Dulbecco’s Modified Eagle’s Medium (DMEM) high glucose medium. The stock solutions were stored at −20°C and warmed to 25°C just before use.
Cell cultures
The human fibroblast HEK293 cell lines transfected with ABCG2 (HEK293-ABCG2) or with the empty vector (HEK293-pcDNA3.1) were obtained as previously described.10 The cells were maintained in DMEM (high glucose) supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, and 0.75 mg/mL geneticin.
ABCG2-mediated mitoxantrone efflux
Cells were seeded at a density of 1.5×105 cells/well into 24-well culture plates. After a 48-hour incubation period, they were exposed to 5 μM mitoxantrone for 30 minutes at 37°C, in the presence or absence of 5 μM of each compound and then washed with phosphate buffer saline and trypsinized. The intracellular fluorescence was monitored with a FACSCalibur cytometer (BD Biosciences, San Jose, CA, USA), using the FL4 channel, and at least 10,000 events were collected. The percentage of inhibition was calculated by using the following equation:
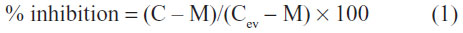
where C is the intracellular fluorescence of resistant cells (HEK293-ABCG2) in the presence of compounds and mitoxantrone, M is the intracellular fluorescence of resistant cells with only mitoxantrone, and Cev corresponds to the intracellular fluorescence of control cells (HEK293-pcDNA3.1) in the presence of compounds and mitoxantrone.
Statistical analysis
Each experiment was performed at least in triplicate. The data are presented as mean ± standard deviation. Statistical significance was assessed by two-tailed Student’s t-test. A P-value lower than 0.05 was considered significant.
Molecular modeling and predictions of absorption, distribution, metabolism, excretion and toxicity
The 61 molecules were modeled using the Sybyl X2.1 suite software (Tripos International, St Louis, MO, USA). Molecules were minimized with the MMFF94 forcefield,25 using a dielectric constant of 80 and an electrostatic cutoff of 16 Å. Minimized molecules were aligned on the central common core and put in a database. Lateral chains were manually checked and aligned on a common position, and the modified conformation was minimized. The differences in internal energy between the two conformations must be lower than 20 kcal · mol−1 to validate the aligned conformations. A three dimensional-quantitative structure–activity relationship using comparative molecular similarity index analysis26 was initiated with the concentration producing 50% inhibition (IC50) values of all 61 molecules. Grids of electrostatic and steric, hydrogen bond acceptor/donor, and hydrophobic potential fields were computed. The grid was filtered with 2.0 kcal · mol−1 as a minimal variation to select probes, and validation by the leave-one-out method was chosen.27 With an optimal number of 12 components, the partial least squares algorithm found coefficients of 0.806 for correlation and 0.912 for calibration. There was no outlier molecule in the calculation. For predicting some ADMET properties (absorption, distribution, metabolism, excretion, and toxicity) of quinoxaline-containing chalcones, ACD/Percepta 14.0.0 software (Advanced Chemistry Development, Inc. [ACD/Labs], Toronto, ON, Canada) was used, including a Passive Absorption Module (five rules of Lipinski, capacity to cross the blood–brain barrier, intraperitoneal tolerance in mice) and an Ames Test Module (genetic toxicity, carcinogenicity, ability to bind to estrogen receptor).
Results
A new series of 12 chalcones containing a quinoxaline unit as B-ring, among a total of 61 studied, showed significant inhibitory effects toward the MDR-conferring protein ABCG2, depending on the number and position of methoxy groups present on the phenyl A-ring (Table 1). The highest potencies of inhibition, given their IC50 values, were obtained with compounds containing two or three methoxy groups at the A-ring. The best derivatives were 4 (2′,4′-diOCH3) and 7 (2′,4′,5′-triOCH3), with an IC50 of 1.4±1.0 μM, by comparison to compounds containing a single methoxy group such as 1 (4′- OCH3), 9 (2′-OCH3), and 11 (3′-OCH3). A very similar potency was observed with 2 (2′,5′-di OCH3), 3 (3′,4′-di OCH3), 5 (3′,4′,5′-tri OCH3), and 8 (3′,5′-di OCH3), the only exception being 12 (2′,3′,4′-tri OCH3), with a lower potency. By contrast, a hydrophilic hydroxyl group at the 4′ position negatively contributed to the inhibition by 6 (3′-OCH3, 4′-OH) versus 3 (8-fold decrease) and by 10 (3′,5′-diOCH3, 4′-OH) versus 5 (5-fold decrease).
 | Table 1 Potent inhibition of ABCG2-mediated mitoxantrone efflux by chalcones containing a quinoxaline group at the B-ring compared to 2-naphthyl and 3,4-methylenedioxyphenyl groups |
The quinoxaline unit replacing the B-ring was more, or much more, active than 3,4-methylenedioxyphenyl or 2-naphthyl, regardless of the number of methoxy groups on the A-ring: in 5 versus 16 and 24 (three methoxy groups); in 2 versus 14 and 22, in 3 versus 15 and 23, and in 8 versus 19 (two methoxy groups); in 1 versus 13 and 21, in 6 versus 25, and in 11 versus 20 (one methoxy group). The only exceptions were 6 versus 17, and 7 versus 18 and 26.
The polyspecificity at the B-ring position was further evaluated with 1-naphthyl, which displayed an intermediate potency between quinoxaline on the one hand, and 2-naphthyl or 3,4-methylenedioxyphenyl on the other hand, regardless of the number of methoxy groups on A-ring (Table 2): the potency of 27 (12.5±2.6 μM) was higher than 30 or 31 (three methoxy groups), the potency of 28 (3.5±1.0 μM) was lower than 2 and higher than 14 (two methoxy groups), and the potency of 29 (13.6±3.1 μM) was lower than 1 and higher than 13 (one methoxy group). An increase in potency was observed for 28 versus 22 and for 29 versus 21, but it was too low to be statistically significant.
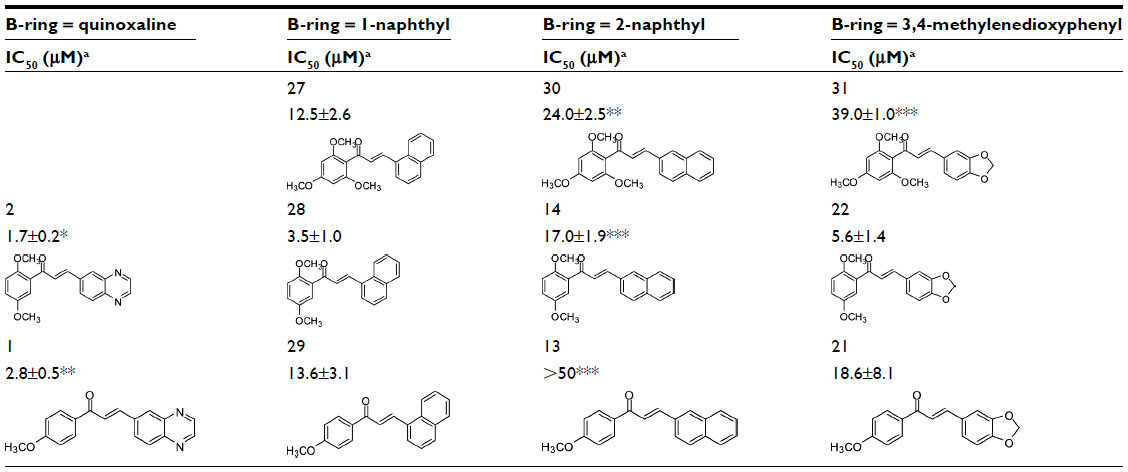 | Table 2 Inhibition efficiency of 1-naphthyl group as chalcone B-ring to inhibit ABCG2-mediated mitoxantrone efflux by comparison to other bicyclic groups |
ADMET predictions performed on a set of nine selected compounds (1–8, 12) indicated mostly optimal values for the five rules of Lipinski, suggesting ability to cross the blood–brain barrier, and a negative Ames tests, related to genotoxicity, carcinogenicity, and binding to estrogen receptor, with a high intraperitoneal tolerance in mice. These quinoxaline-containing compounds therefore display good drug candidate profiles.
It has been experimentally shown that the six best derivatives (2, 3, 4, 5, 7, and 8), displaying IC50 values in the range 1.4–2.2 μM, appeared not to be transported since no evidence of cross-resistance (leading to lower toxicity in ABCG2-transfected cells versus control cells) was observed in cell survival using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, data not shown).
To complete the examination of structure–activity relationships at the B-ring position, two other series of chalcones with a wide panel of substituents on B-ring were investigated (Table 3). The first contained a 3′,4′-methylenedioxy-phenyl unit as A-ring while the second contained a 2′-naphthyl unit.
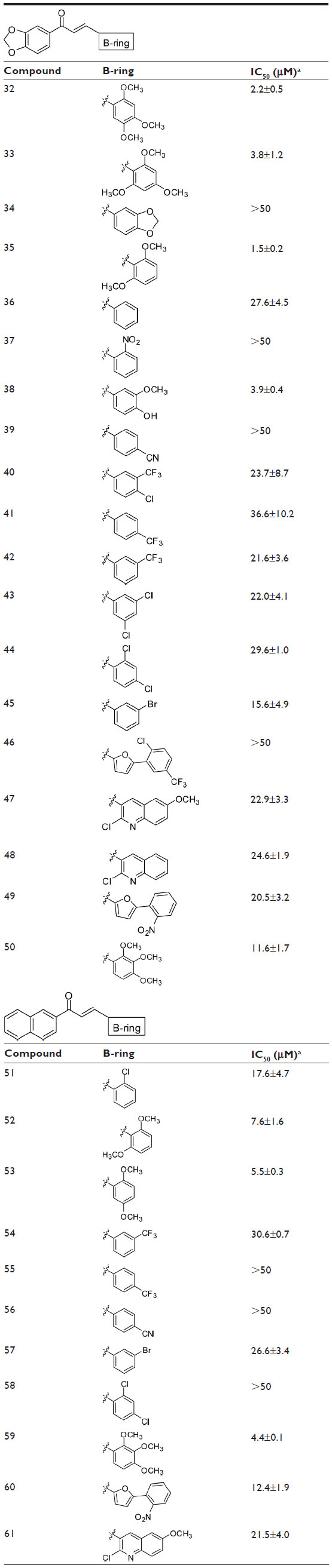 | Table 3 Dependence of the inhibition of ABCG2-mediated mitoxantrone efflux on B-ring substituents in chalcones containing either a 3,4-methylenedioxyphenyl or 2-naphthyl group at the A-ring |
The following substitutions at the B-ring were identified to positively contribute to ABCG2 inhibition:
- At least two methoxy groups were required in all studied chalcones for optimal inhibition when comparing to compounds with only one, or without any, methoxy group. The better inhibitory effects were produced by compounds containing at least two methoxy groups at the B-ring, such as 32, 33, 35, 52, 53, and 59, whereas the contributions were lower in 50.
- 2. By contrast, all other tested substituents on the B-ring were poorly efficient, such as chlorine (Cl) (in 40, 43, 44, 46, 47, 48, 51, 58, and 61), bromine (Br) (in 45 and 57), trifluoromethyl (CF3) (in 40, 41, 42, 46, 54, and 55), nitro group (NO2) (in 37, 49, and 60), cyano group (CN) (in 39 and 56), and hydroxyl (OH) (in 38, 25, 17, 6, and 10).
Although the B-ring could accommodate bicyclic rings (accompanied by OCH3 substituents on the phenyl A-ring) as well as OCH3-substituted phenyl (with bicyclic rings instead of A-ring), the inhibitors were not equivalent in potency. Indeed, the compounds containing a 3′,4′-methylenedioxyphenyl unit as A-ring and methoxy substituents on B-ring were more, or much more, potent than the inverted compounds containing a 3,4-methylenedioxyphenyl unit at B-ring and methoxy substituents on the phenyl A-ring: in 38 versus 25 (12-fold increase), in 33 versus 31 or in D of Figure 2 versus 24 (10-fold increase), and in C of Figure 2 versus 23 or in A of Figure 2 versus 22 (5-to-6-fold increase); however, a slightly opposite effect was observed in 32 versus 26 (2-fold decrease). Moreover, the compounds containing a 2′-naphthyl unit as A-ring and methoxy substituents on phenyl B-ring were more efficient than the inverted compounds containing a 2-naphthyl unit as B-ring and methoxy substituents on phenyl A-ring: in 53 versus 14 (3-fold increase) as well as in G of Figure 2 versus 18 (11-fold increase).
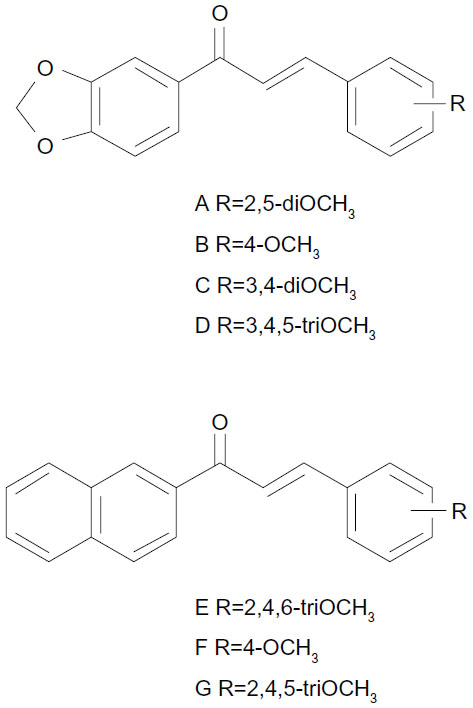 | Figure 2 Structure of chalcones with a 3,4-methylenedioxy-phenyl or a 2-naphthyl group as A-ring, and methoxy substituents on the B-ring. |
A three dimensional quantitative structure–activity relationship molecular model was constructed after structures alignment from the IC50 values of all compounds, as shown on Figure 3. The positive contribution of quinoxaline at B-ring position (as observed in Table 1) appears to be related to electrostatic interactions of the two nitrogen heteroatoms (blue volumes). The better contribution of 1-naphthyl over 2-naphthyl (as observed in Table 2) might correlate both steric negative effects on the right edge of the site (yellow volume) and electrostatic negative effects on the top (red volume). Concerning methoxy contributions (as observed in Tables 1 and 2), the positive effects appear to be related to steric effects (green volume) at position 2′ and to both steric and electrostatic (blue volume) contributions at positions 4′ and 5′, whereas the negative contribution observed in some cases at position 3′ (such as in 12) as well as negative effects of hydroxyl at position 4′ (in 6 and 10) might be due to electrostatic antagonism (red volume). Other electrostatic negative effects related to B-ring positions 2 and 3 (red volume) appear to correlate the low efficiency of compounds containing hydrophilic substituents such as NO2 (in 37), Cl (in 43, 44, 51, and 58), Br (in 45 and 57), or CF3 (in 40, 42, or 54).
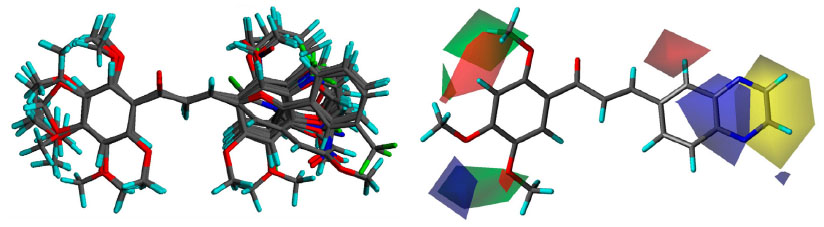 | Figure 3 3D-QSAR analyses. |
Discussion
The present work, performed on a total of 61 chalcones, has allowed the identification of three new characteristic structure–activity relationships of inhibitors toward ABCG2 drug-efflux activity.
Firstly, the polyspecificity at the B-ring position as a consequence of the high efficiency of both newly-synthesized quinoxaline- and 1-naphthyl-derivatives was shown by comparison to that of 3,4-methylenedioxyphenyl- and 2-naphthyl-containing chalcones recently characterized.20 In agreement with previous observations showing that indole was also quite efficient,19 the polyspecific B-ring position appears able to accommodate a number of bicyclic rings. The preference for quinoxaline appears correlated to electrostatic interactions of the two nitrogen heteroatoms. The increase in affinity seems not to be related only to the hydrophobicity, as monitored by logP, since for pairs of inverted compounds (such as 26 versus 32, 31 versus 33, or 14 versus 53), the logP values were the same, whereas the inhibitory activity toward ABCG2-mediated drug-efflux was different. LogP values were also the same among 1-naphthyl and 2-naphthyl derivatives (such as 27 versus 30, 28 versus 14, or 29 versus 13) and among compounds of the same series containing the same number of methoxy groups at different positions (such as 2 versus 3 versus 4 versus 8) (data not shown), which displayed differential inhibition activities.
The quinoxaline-containing chalcones appear not to be transported, as is also observed for other types of chalcones19 as well as for flavones,10 methoxylated trans-stilbenes,14 and chromones.15
Secondly, the requirement for at least two methoxy groups on the phenyl A-ring (through both electrostatic and steric contributions) while a bicyclic unit occupies the B-ring position to provide maximal inhibition, is in agreement with previous observations showing a positive contribution of methoxy groups to the inhibition potency of flavones,10 rotenoids,11 and trans-stilbenes.14 The contribution of methoxy groups at positions 2′ and 4′ was previously observed in other types of chalcones,18–20,28 whereas the effects produced at positions 3′ and 5′ have been characterized here. Many other types of substituents such as Cl, Br, NO2, OH, CN, or CF3 did not contribute to inhibition, likely due to their hydrophilicity, as recently observed with other chalcones.20
Thirdly, a higher potency can be reached by shifting the bicyclic unit to the A-ring and methoxy substituents to the phenyl B-ring, as observed here with either 3′,4′-methylenedioxy or 2-naphthyl, which indicates that the chalcones are functionally asymmetric, and preferences exist among polyspecificity. This is consistent with the high potency previously observed with an indole at A-ring position.19
In conclusion, it would be interesting to synthesize and assay a new-generation of inhibitory chalcones containing either a quinoxaline or 1-naphthyl unit at the A-ring, and a phenyl B-ring substituted by either two or three methoxy groups at various positions, or a p-bromobenzyloxy group, which was recently shown to play a critical role in both chromones15 and chalcones.20 Finally, this chalcone-binding site should be differentiated from that of different inhibitors such as chromones, also known as good drug candidates.
Acknowledgments
EW and GJG were recipients of mobility doctoral fellowships from the Brazilian CAPES (Process number 8792127) and CNPq-CAPES (Science Without Borders Program 245762/2012-4), respectively. NDY is a recipient of a postdoctoral fellowship for the Control of Cancer-CNPq (Science without Borders Program) and CG is the recipient of a doctoral fellowship from the Ligue Nationale Contre le Cancer. Financial support was provided in France by the CNRS and Université Lyon 1 (UMR 5086), the Ligue Nationale Contre le Cancer (Equipe labellisée Ligue 2013), an international grant from French ANR and Hungarian NIH (2010-INT-1101-01), and in Brazil by the CNPq. We acknowledge the Chemistry Department and the Molecular Structural Biology Center from the Federal University of Santa Catarina, Brazil, for the respective NMR and micrOTOF-QII equipment used in the chemical analyses. We also thank PD Neuenfeldt for performing the NMR analyses.
Disclosure
The authors report no conflicts of interest in this work.
References
Global Health Observatory: Cancer mortality and morbidity [webpage on the Internet]. Geneva: World Health Organization; 2011. Available from: http://www.who.int/gho/ncd/mortality_morbidity/cancer/en/index.html. Accessed September 7, 2013. | |
Endicott JA, Ling V. The biochemistry of P-glycoprotein-mediated multidrug resistance. Annu Rev Biochem. 1989;58:137–171. | |
Cole SP, Bhardwaj G, Gerlach JH, et al. Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science. 1992;258(5088):1650–1654. | |
Doyle LA, Yang W, Abruzzo LV, et al. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc Natl Acad Sci U S A. 1998;95(26):15665–15670. | |
Chang G. Multidrug resistance ABC transporters. FEBS Lett. 2003;555(1):102–105. | |
Natarajan K, Xie Y, Baer MR, Ross DD. Role of breast cancer resistance protein (BCRP/ABCG2) in cancer drug resistance. Biochem Pharmacol. 2012;83(8):1084–1103. | |
de Bruin M, Miyake K, Litman T, Robey R, Bates SE. Reversal of resistance by GF120918 in cell lines expressing the ABC half-transporter, MXR. Cancer Lett. 1999;146(2):117–126. | |
Rabindran SK, He H, Singh M, et al. Reversal of a novel multidrug resistance mechanism in human colon carcinoma cells by fumitremorgin C. Cancer Res. 1998;58(24):5850–5858. | |
Allen JD, van Loevezijn A, Lakhai JM, etal. Potent and specific inhibition of the breast cancer resistance protein multidrug transporter in vitro and in mouse intestine by a novel analogue of fumitremorgin C. Mol Cancer Ther. 2002;1(6):417–425. | |
Ahmed-Belkacem A, Pozza A, Muñoz-Martínez F, et al. Flavonoid structure-activity studies identify 6-prenylchrysin and tectochrysin as potent and specific inhibitors of breast cancer resistance protein ABCG2. Cancer Res. 2005;65(11):4852–4860. | |
Ahmed-Belkacem A, Macalou S, Borrelli F, et al. Nonprenylated rotenoids, a new class of potent breast cancer resistance protein inhibitors. J Med Chem. 2007;50(8):1933–1938. | |
Boumendjel A, Macalou S, Valdameri G, et al. Targeting the multidrug ABCG2 transporter with flavonoidic inhibitors: in vitro optimization and in vivo validation. Curr Med Chem. 2011;18(22):3387–3401. | |
Sim HM, Wu CP, Ambudkar SV, Go ML. In vitro and in vivo modulation of ABCG2 by functionalized aurones and structurally related analogs. Biochem Pharmacol. 2011;82(11):1562–1571. | |
Valdameri G, Pereira Rangel L, Spatafora C, et al. Methoxy stilbenes as potent, specific, untransported, and noncytotoxic inhibitors of breast cancer resistance protein. ACS Chem Biol. 2012;7(2):322–330. | |
Valdameri G, Genoux-Bastide E, Peres B, et al. Substituted chromones as highly potent nontoxic inhibitors, specific for the breast cancer resistance protein. J Med Chem. 2012;55(2):966–970. | |
Tan KW, Li Y, Paxton JW, Birch NP, Scheepens A. Identification of novel dietary phytochemicals inhibiting the efflux transporter breast cancer resistance protein (BCRP/ABCG2). Food Chem. 2013;138(4):2267–2274. | |
Harvey AL. Natural products in drug discovery. Drug Discov Today. 2008;13(19–20):894–901. | |
Juvale K, Pape VF, Wiese M. Investigation of chalcones and benzochalcones as inhibitors of breast cancer resistance protein. Bioorg Med Chem. 2012;20(1):346–355. | |
Valdameri G, Gauthier C, Terreux R, et al. Investigation of chalcones as selective inhibitors of the breast cancer resistance protein: critical role of methoxylation in both inhibition potency and cytotoxicity. J Med Chem. 2012;55(7):3193–3200. | |
Rangel LP, Winter E, Gauthier C, et al. New structure-activity relationships of chalcone inhibitors of breast cancer resistance protein: polyspecificity toward inhibition and critical substitutions against cytotoxicity. Drug Des Devel Ther. 2013;7:1043–1052. | |
Zhilina ZI, Vodzinskii SV, Andronati SA. Synthesis and spectral characteristics of porphyrins with hetero and bicyclic meso substituents. Ukrainskii Khimicheskii Zhurnal (Russian Edition). 1990;56:1084–1088. | |
Mielcke TR, Mascarello A, Filippi-Chiela E, et al. Activity of novel quinoxaline-derived chalcones on in vitro glioma cell proliferation. Eur J Med Chem. 2012;48:255–264. | |
Chiaradia LD, Mascarello A, Purificação M, et al. Synthetic chalcones as efficient inhibitors of Mycobacterium tuberculosis protein tyrosine phosphatase PtpA. Bioorg Med Chem Lett. 2008;18(23):6227–6230. | |
Chiaradia LD, Martins PG, Cordeiro MN, et al. Synthesis, biological evaluation, and molecular modeling of chalcone derivatives as potent inhibitors of Mycobacterium tuberculosis protein tyrosine phosphatases (PtpA and PtpB). J Med Chem. 2012;55(1):390–402. | |
Halgren TA. MMFF VII. Characterization of MMFF94, MMFF94s, and other widely available force fields for conformational energies and for intermolecular-interaction energies and geometries. J Comp Chem. 1999;20(7):730–748. | |
Cramer RD III, DePriest SA, Patterson DE, Hecht P. The developing practice of comparative molecular field analysis. In: Kubinyi H, editor. 3D QSAR in Drug Design: Theory, Methods and Applications. Leiden: ESCOM; 1993. | |
Picard RR, Cook RD. Cross-validation of regression models. J Am Stat Assoc. 1984;79(387):575–583. | |
Liu XL, Tee HW, Go ML. Functionalized chalcones as selective inhibitors of P-glycoprotein and breast cancer resistance protein. Bioorg Med Chem. 2008;16(1):171–180. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.