")
Back to Archived Journals » Research and Reports in Transdermal Drug Delivery » Volume 4
Quality by design approaches for topical dermatological dosage forms
Authors Sivaraman A, Banga A
Received 12 February 2015
Accepted for publication 28 April 2015
Published 10 July 2015 Volume 2015:4 Pages 9—21
DOI https://doi.org/10.2147/RRTD.S82739
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr S. Narasimha Murthy
Arunprasad Sivaraman, Ajay K Banga
Department of Pharmaceutical Sciences, College of Pharmacy, Mercer University, Atlanta, GA, USA
Abstract: The quality of a pharmaceutical dosage form is the foremost criterion during the development of a product. The quality by testing (QbT) technique used by the pharmaceutical industry to ensure the quality of a drug product is a rigid process with tight specifications. The specifications set by QbT are not essentially based upon the critical quality attributes of the materials and critical process parameters involved in development, but based upon recorded observation of manufactured batches. Room for flexibility is narrow as every level change requires submission of a supplement to the US Food Drug and Administration. Unlike QbT, the concept of quality by design (QbD) is a modern approach to ensure the quality of pharmaceutical products. It can identify the critical attributes of the material and the process parameters involved in development of the drug product through substantial scientific understanding with an established design space. QbD tools such as design of experiment, risk assessment, and process analytical technology help to establish a control strategy for every drug product with an option of continual monitoring and improvement for a quality drug product. Implementing the concept of QbD to topical dermatological dosage forms is in the initial stages. For a generic topical dermatological dosage form, establishing the required pharmaceutical and therapeutic equivalence with same components or qualitatively (Q1), same components with same concentration or quantitatively (Q2), and same components in same concentration with same arrangement (Q3) is a cumbersome process. Applying QbD approaches by defining a quality target product profile and identifying critical quality attributes with establishment of a design space and control strategy can guide the design of a quality-based generic topical dermatological product.
Keywords: quality by design, topical dermatological dosage forms, design of experiment, risk assessment, process analytical technology
Introduction
Generally, a pharmaceutical dosage form includes an active pharmaceutical ingredient (API) or APIs in combination with excipient(s), with demonstrated safety and efficacy for ease of administration in patients. A drug product lacking the required quality may not be considered as a therapeutic drug product and may not be either approved or prescribed. As defined by the Center for Drug Evaluation and Research at the US Food and Drug Administration (FDA), a product free of contamination reproducibly delivering the therapeutic benefit promised on the label to the consumer is considered as a high-quality drug product.1 Ensuring and meeting the requirements of quality while developing the drug product is the foremost criterion during product development.2
The current protocol used by the pharmaceutical industry to ensure the quality of a drug product is based upon setting tight specifications for manufactured batches and allows only narrow flexibility for batch-to-batch variations. The number of testing and characterization would be useful to set tight specifications and to identify batch-to-batch differences, but would not be of much help in enhancing the quality of the product. Repeated testing of the finished product without identifying the critical quality attributes (CQAs) of the incorporated materials and process would be of little value in achieving the target quality. Quality should be built into the product by design instead of testing the product, and this is the notion of quality by design (QbD). It is a strategy to design and develop formulations and manufacturing processes to ensure predefined product quality.2
The FDA and the pharmaceutical industry have been emphasizing QbD terminology in recent years. Even so, a vague perception still prevails among scientists about the actual meaning and application of QbD. A clear understanding is essential before applying the concepts of QbD in the development of pharmaceuticals.3 According to the FDA, per the guidelines of International Conference on Harmonisation (ICH) Q8, pharmaceutical QbD is a systematic approach based on sound science and quality risk management that begins with predefined objectives and emphasizes product and process understanding.4
QbD concepts and applications have been recommended for most pharmaceutical dosage forms, and much attention is presently focused on the oral dosage forms. However, this review focuses on QbD approaches to the development of topical dermatological dosage forms (TDDFs), especially the generic forms. Given that developing a generic TDDF is a complex and time-consuming process, employing aspects of QbD could ease the process of development, and this review highlights and discusses basic approaches to QbD. Topical dermatological drug products are medications that are applied directly at the site of action. These are complex drug products designed for topical administration targeted to local tissue and undergo low or limited systemic absorption. For a generic topical dermatological product to secure regulatory approval, a strict similarity with the commercialized reference product is required. Generic TDDFs are required to mimic the reference listed drug (RLD) in terms of pharmaceutical and therapeutic equivalency.5 In addition, critical attributes such as the API, excipients, physicochemical properties of the drug product, container closure system, physical and chemical stability, scalability, and efficacy of the preservatives have to be given adequate weight while developing the product, and play a major role in determining not only the safety and efficacy of the drug product, but also its quality.
QbD emphasizes the development of meaningful TDDF specifications that are based on clinical performance.6 The critical path initiatives for generic drug development identified by the FDA include demonstrating clinical endpoint bioequivalence of locally acting TDDFs as per regulation 21CFR 320.24.7 The non-oral dosage forms, such as TDDFs, will have a significant impact in the future, so it is important to realize the value of QbD approaches during their development.
Quality by testing
Under quality by testing (QbT), components incorporated into the formulation in the manufacturing process are tested for their quality. The raw materials can be used in the manufacturing process if they meet the manufacturer’s proposed and FDA-approved specifications as well as USP standards. The tight specifications set during the manufacturing process ensures the quality of the drug product. Out of specification drug products cannot be taken into consideration and have to be discarded. Any change in operating parameters during manufacturing in a batch record must be notified to the FDA. As a result, the FDA gets a number of Chemistry, Manufacturing, and Controls supplements. Therefore, under QbT, the quality of the drug product is ensured by extensive testing of the finished drug product.
Quality by design
QbD is primarily intended to reduce product variation, improve the efficiency of the process, and reduce costs at different stages. It improves the speed with which the product gets onto the market and enhances the quality process through systematic research and development.8 The FDA guidance, such as pharmaceutical development (ICH Q8), quality risk assessment (ICH Q9), pharmaceutical quality systems (ICH Q10), and the ICH briefly highlight the approach to achieving product quality through QbD.3 QbD has been initiated to design and develop formulations with identification of critical material attributes (CMAs) and to develop robust manufacturing process with identification of critical process parameters (CPPs) to obtain the predefined product quality. CMAs and CPPs are the variables during development of the product. A complete understanding of these variables and knowing how to control these variables within the desired operating range can ensure the quality of the product. Application of QbD provides self-regulatory flexibility for pharmaceutical manufacturers while maintaining quality standards.8 The aim of application of QbD in pharmaceutical development is to create a detailed mechanistic and scientific understanding of the process variability and to provide the capability for a targeted quality product.9 The basic difference between QbT and QbD in pharmaceutical development is shown in Table 1.
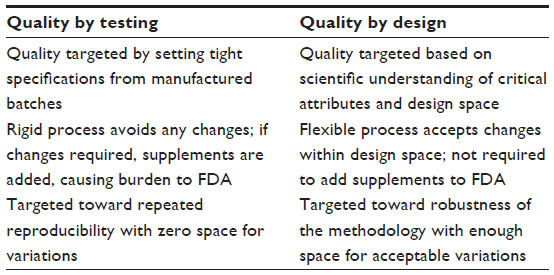 | Table 1 Quality by testing versus quality by design in pharmaceutical development |
The QbD for a generic drug product begins with setting up the target for the desired product profile and defining the quality target product profile (QTPP). The label, packaging insert, patent, literature, and clinical study information of the RLD help in establishing a strategy for QTTP. Taking the safety and efficacy of the drug product into consideration, these characteristics ideally have to be achieved to ensure the targeted quality. A thorough understanding of preformulation studies and the mechanical properties of the drug product and excipients can greatly contribute to design, development, and processing of the drug product.2,3
The physicochemical and biological properties of the raw materials included in the design of a formulation determines the quality of the final product and are known as the CQAs.4 ICH Q8 (R1) defines CQAs as the physicochemical, biological, or microbiological properties or characteristics that should be within the defined limit or distribution to ensure the desired product quality.10 CQA determines the performance of the product.3 Once the QTPP is set, a preliminary formulation and manufacturing process is designed with broad scientific understanding of the components involved in the formulation and process to know the CMAs and CPPs.
The formulation design and process development cannot be separated since a formulation cannot become a product without a defined process. The manufacturing process required to produce a desired quality product usually involves multiple unit operations and operating conditions.2 The outline has to include all the factors that need to be considered for the design of the process. The process parameters which causes direct impact on the quality of the drug product if get varied are considered as the CPPs. Process parameter variability, which has an impact on the critical quality of the material, should be monitored and controlled at all times to ensure the process for the targeted quality.2,4
One way to assure the quality of the product by process design is through identification of CPPs. These can potentially affect the quality of the product and setting up the range to consistently reproduce the same quality product. The process is the one that converts the formulation into a product. An efficient process design with enough understanding about process development and the parameters involved, including the equipment and facility, can help to produce a quality product.2
Understanding through the QbD approach involves detailed experimentation to identify CQAs and CMAs for mitigating the failures associated with different grades of material. Traditional process understanding includes an exhibit batch production followed by a full production batch size with no route to identify the CPPs in the production process. However, the QbD paradigm includes design of experiment (DoE), process analytical technology (PAT), and risk assessment through which the CPPs can be identified. This gives an idea about the design space with the right operating space for scaling up the process from a pilot scale batch to a commercial batch size.
Quality by design tools
Design of experiment
DoE is a structured and organized method for determining the relationship between factors affecting a process and the output of the process. Some examples are Plackett–Burman design, factorial design, and central composite design. As each unit operation has multiple input and output variables, including process parameters, investigating every parameter is practically impossible. Scientists may have to use their prior knowledge and risk management to identify key input and output variables and process parameters while conducting DoE. The results of DoE can help to identify the critical factors associated with CQAs. Details such as the existence of interactions and synergies between the factors can be studied as well. Based on the allowable limit of CQAs, the design space for CPPs can be identified.2,4
Risk management
A combination of the probability of occurrence of harm and severity of the harm is defined as risk assessment according to ICH Q9 quality risk management. Some of the potential risk management tools are: basic risk management facilitation methods (eg, flowcharts, check sheets), failure mode effects analysis, failure mode effects and criticality analysis, fault tree analysis, risk ranking and filtering, supporting statistical tools, Ishikawa diagram, and “what if” analysis.6,11
ICH Q9 quality risk management indicates that the manufacturing and use of a drug product necessarily entail some degree of risk. The risk to quality ratio has to be evaluated and ultimately should be linked to the therapeutic benefit for the patient. A risk assessment before development of a pharmaceutical can help the manufacturer to decide which studies need to be conducted. Risk assessments are often driven by knowledge gaps or uncertainty. Study results determine the critical and non-critical variables that help in establishing a control strategy for in-process, raw material, and final testing.12
Design space
The current ICH Q8 definition of design space is a multidimensional combination and interaction of input variables and process parameters that has been demonstrated to provide assurance of quality. The design space provides regulatory flexibility and can be for a single unit, multiunit, or the entire process. Operating within the design space is not considered as a change, and it confirms a clear process understanding. However, the limitations with the design space are the cost of time and money. In addition, an adverse effect is possible if any critical parameter is left out when constructing the design space. A manufacturing change within the FDA-approved design space is not considered as a change, but movement out of the space is considered as a change. A regulatory post approval change process is required for such changes. Design space varies among the sponsors depending upon their formulation design and on the equipment used for development of the drug product.
Response surface designs
The process variables identified by the screening designs eventually undergo surface optimization. Response surface designs such as Box–Behnken, central composite, and three-level factorial designs can identify the optimum processing conditions. Central composite designs are preferred because they are robust against missing data and corner and center point trials can be included from previously conducted factorial experiments. On the other hand, because of the three levels of each factor, Box–Behnken design is used to simplify the execution of the experiment. The limitation with this design is that for uniform precision, a larger number of center points are required and missing a single datum point will result in an inconclusive result.13,14
The robust operating space is the range that can continuously reproduce a desired quality product within the design space. Deviation from the usual process if it falls within the design space is acceptable. It can have flexibility in regulatory approval only when QbD experimentation is conducted at a manufacturing scale. The variability occurring during the developmental stage at a laboratory scale level provides valuable information in the design space exploration for a scale up and commercial batch size. Evaluation of deviation comparing the laboratory scale equipment versus the commercial scale equipment narrows down the processing variability.3,15 With the CMAs identified from the formulation and CPPs from the process design, a design space for the manufacturing process for the formulation can be identified. The output variables are characterized and compared with the target product profile. If the output is in accordance with the target, a control strategy is created for the entire process. This includes both the product as well as the process understanding followed by continual monitoring to enhance the quality of the product. The QTPP should be redefined and the formulation and manufacturing process should be redesigned with enhanced product and process understanding if the targeted was achieved.
Process analytical technology
PAT is a tool used for continuous inline or online process monitoring of the variable processing parameters to make sure that the process is within the operating space. This is an effective tool to detect failures during processing and the operating parameters that can be controlled or tuned to assure the quality of the product.3,11 It is a system to design, analyze, and control manufacturing through timely measurements with the goal of ensuring the final quality of the product. PAT provides more flexibility, and because of its direct and continuous assessment, it substantiates the robustness of the methodology. An ideal online process monitoring is the one which should detect any out of control situation during manufacturing. PAT can identify, simulate, and control the manufacturing process. Currently, continuous monitoring with an improved and extensive understanding is essentially the application of PAT.2–4 Figure 1 gives an idea of the general QbD approach for pharmaceutical product development.
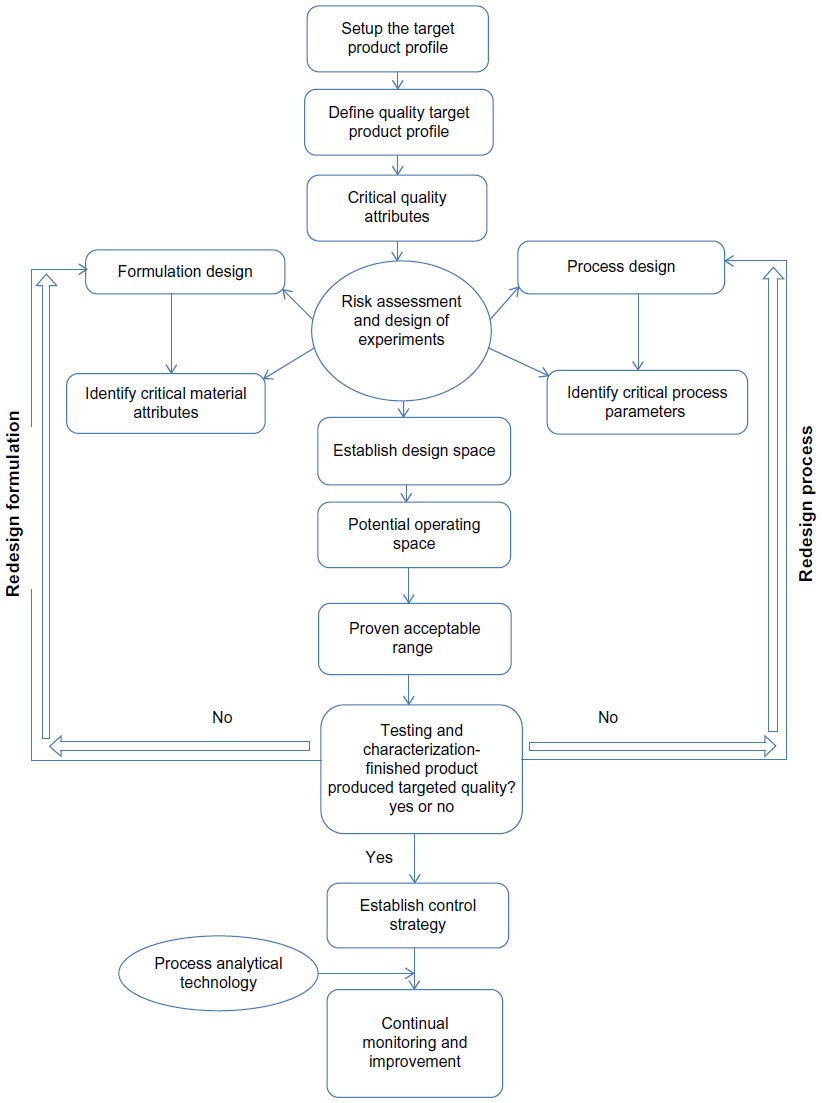 | Figure 1 General quality by design approach for development of a pharmaceutical drug product. |
Topical dermatological drug delivery
The skin is the largest organ in the human body and is composed of three readily distinguishable layers: the stratum corneum, the outermost part of the epidermis; the living epidermis; and the dermis. The stratum corneum, or rate controlling membrane, is the main barrier for diffusion of molecules across the skin. The stratum corneum is a well-recognized heterogeneous two-compartment system composed of keratinized cells embedded in a multilamellar lipid matrix of neutral lipids and ceramides. Corneocytes, ie, keratin-filled dead cells, are entirely surrounded by crystalline lamellar lipid regions. The majority of drugs are delivered through the intracellular and transcellular pathways of the skin, while the skin appendages (sweat glands and hair follicle) play a lesser role.15 Drug accumulation in the dermal layer is critical, and drug transport is a potential approach for topical drug delivery. Most topical dermatological products administered via the skin are intended to be locally active, but some preparations also have systemic effects, and are known as transdermal drug delivery systems. Locally acting preparations exert their action at or on the surface of the skin and alter the function of the epidermis and/or dermis. Drug molecules penetrate the skin primarily through the tortuous and continuous intercellular path.
QbD approaches for development of generic topical dermatological products
Generally, an abbreviated new drug application (ANDA) for a topical dermatological product has to undergo multiple review cycles with the FDA before the product is approved, and the sponsor has to wait for a long time for a decision. Even though the ICH common technical document Q8 describes the generation of a product development report, it is still unclear to most of the ANDA sponsors about its application. One way to overcome this challenge is to have a product development report with verified and justified specifications based on QbD efforts. This can help the Office of Generic Drugs understanding the developed generic product.16,17
Development of a generic topical dermatological product similar to the RLD is a cumbersome process. These products give rise to complex issues in relation to pharmaceutical and therapeutic equivalence. In designing a topical dermatological formulation, various criteria have to be considered, and few of the general critical criteria are given in Table 2. In developing a generic TDDF, it is essential to evaluate the RLD critically based on its physicochemical properties rather than just solely depending on the labeling of the dosage form. A reverse engineering result not only provides information about the number and level of components of the RLD, but also highlights information about impurities, which is valuable when studying the stability of the product. In addition, information from the package insert, patent, and published literature has to be given serious consideration.6
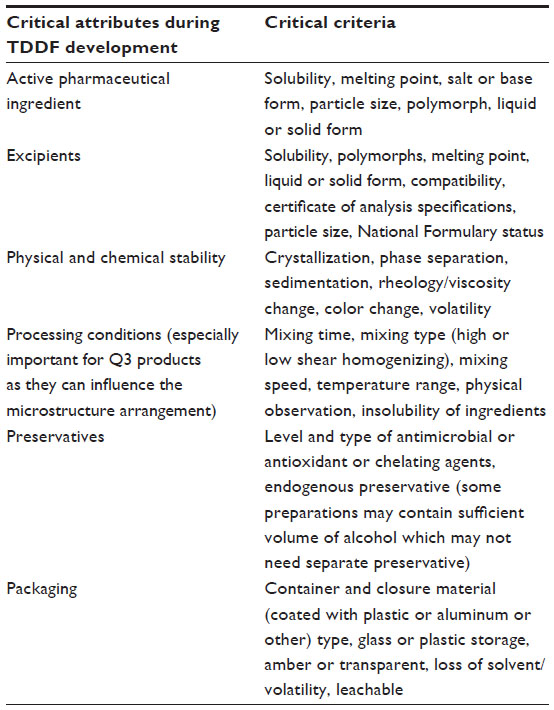 | Table 2 Critical criteria for development of a topical dermatological dosage form |
The foremost criterion in developing a generic topical dermatological product is being able to ensure pharmaceutical and therapeutic equivalency with the RLD. Therapeutic equivalence has the same clinical effect and safety profile as given in the label of the RLD, whereas pharmaceutical equivalence contains the identical amount of the same active ingredient corresponding to the same route of administration in the same dosage form with a similar mechanism of release and a similar rate and extent of absorption of the RLD. The current paradigm to get approval by an ANDA sponsor is testing and demonstrating therapeutic equivalence to the RLD. However, the proposed QbD approach requires the sponsors to meet equivalence on the basis of defined design targets.18
The design of equivalence of a generic dermatological topical product includes meeting the criteria of Q1 and Q2. Depending on the dosage form, the Q3 criterion has to be satisfied in addition to Q1 and Q2. Topical solutions can be stated to be an example for Q3. If the RLD is considerably protected by patents and other mechanisms, the generic manufacturer may be in a position to look for alternate excipients or solvents but not the active ingredient. Still, the generic manufacturer should justify the functionality and amount of excipient used.17
Implementing a QbD approach in developing a TDDF is strongly recommended. Defining the QTPP and identifying the CQAs are essential steps in designing a quality-based generic TDDF.3,17 The general QTPP elements of a TDDF are given in Table 3.19 Meaningful drug product specifications can be obtained through QTPP, CQAs, and clinical performance data. This information can be applied to ensure equivalent safety and efficacy to the RLD when developing a generic TDDF.
 | Table 3 General elements of a QTPP for a TDDF |
The drug manufacturing process is usually associated with some degree of risk. Sound scientific knowledge is the basis for evaluation of quality. The risk assessment is performed to identify the CMAs as well as the CPPs from the formulation design and process design, respectively. Applying the appropriate tool to the manufacturing process used for the dosage form helps to enhance the quality of the product and its attributes. To identify and mitigate the risks associated with the development process is a part of controlling failure modes with the objective of enhancing the quality of the product.17 Failures can occur at any stage during the product development process. Some of the physical and chemical tests where the possibility of failure happens mostly include, but are not limited to, content uniformity, blend uniformity, particle size variation, incompatibility of drug excipients, microbial contamination, lack of homogeneity, uneven rheology, phase separation, and particle size. Identification and applying risk mitigation strategies can ensure that the quality of the product is within acceptable limits. A hypothetical risk assessment using failure mode effects and criticality analysis for lack of viscosity and particle size analysis are provided in Table 4.
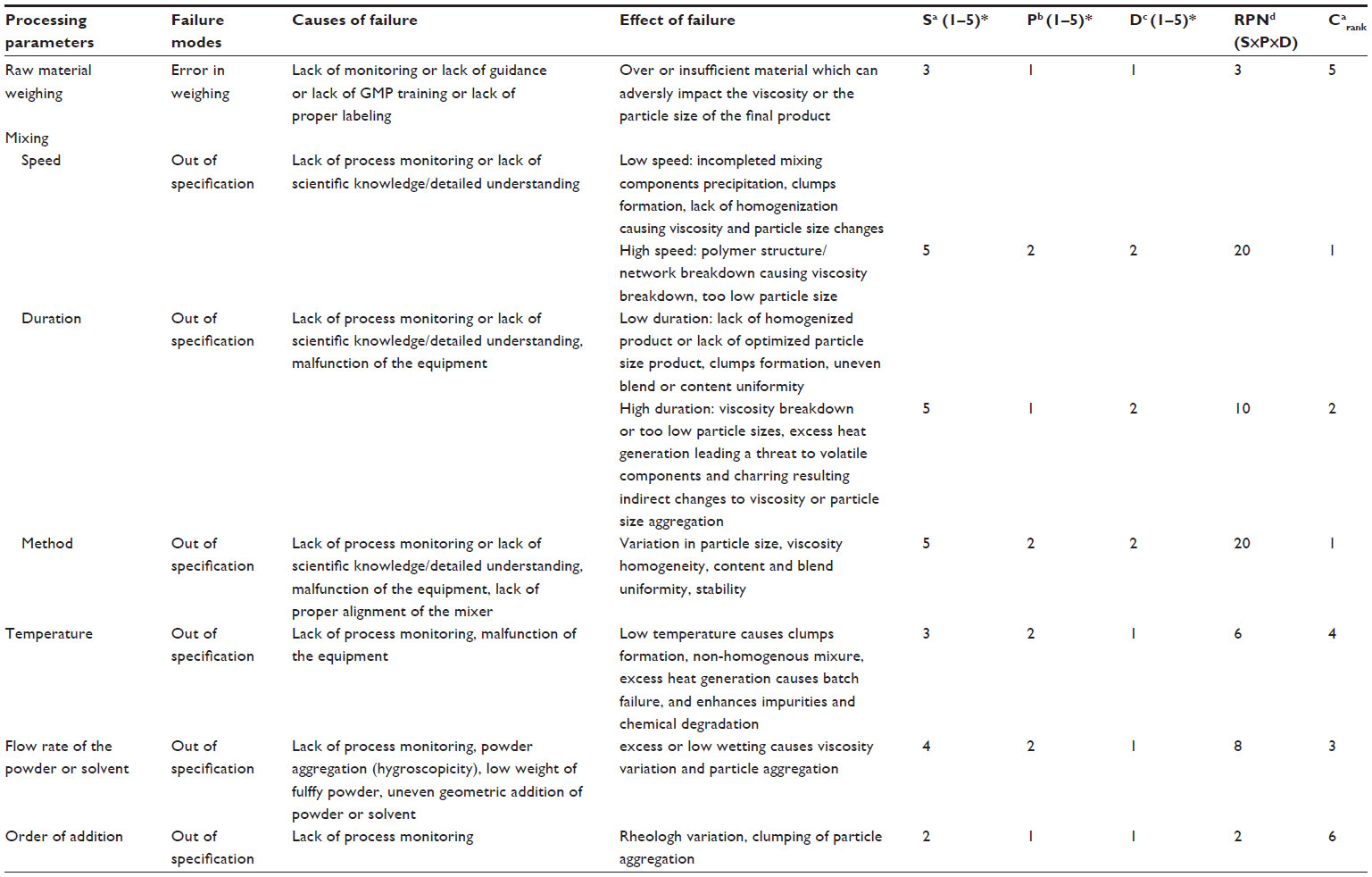 | Table 4 Hypothetical risk assessment using failure mode effects and criticality analysis for lack of viscosity and particle size analysis |
DoE is used to determine the formulation and process spaces for the CMAs and CPPs. The DoE can be used to gain an insight into the influence of processing parameters that can affect the CQAs of the dosage form. In addition, it can be used to implement a Chemistry, Manufacturing, and Controls strategy for evaluating the stability, product performance, and the process control of the dosage form. When DoE is applied to a topical dermatological product development process, the input factors are the raw material attributes and process parameters (eg, mixing time, mixing speed, method of mixing) and the outputs are CQAs, such as the viscosity, pH, uniformity, and microscopic structure of the dosage form. The DoE gives an idea about the optimized manufacturing process within an acceptable range to produce a quality product consistently. For example, variation in the acid value of the excipients may affect the stability of the final product. A hypothetical 22 factorial design was investigated by Chang et al17 to test the difference in the acid value effects for cetyl ester wax and glyceryl monostearate excipients. It was found from these studies that keeping the development process with a low acid value for both these excipients may result in a low level of impurity under accelerated stability storage condition. This type of information is essential to development of a product with acceptable stability.17
A robust process is one that can accept changes within the operating range but still deliver a quality product. The operating space, a CPP, which sets the upper and lower limit has to be well understood by manufacturers to continuously produce a high quality product.20 Importantly, understanding the role and interaction of CQAs and CPPs ensures that a targeted quality product is achieved.2
In general, development of a topical dermatological product includes operating parameters such as weighing of the raw material, order of addition of components, mixer speed, mixing time and duration, flow rate, and heating and cooling rates, and examples of output parameters include the heat and pressure generated during processing. The significance of a parameter is essentially linked to its effect on the quality at the processing and final stage of the product. Depending on its impact, the parameter is considered as a CPP. The potential operating system (POS) sets the maximum and minimum limit for each processing parameter. A parameter would be considered as a non-critical parameter if there is no possibility of failure within the POS. A parameter can be considered significant if there is the potential for failure within the POS. Critical and non-critical parameters can be identified and classified by controlling variations in the process parameters.3 For research and development batches, if the processing parameters are within the design space, validation of the design space is required. In contrast, if the parameters fall outside the design space, it is advisable to create a new design space to evaluate the role of each processing parameter for a commercial batch, which can serve as a valuable tool for regulatory approval.17 With identification of proven acceptable ranges and failure edges, the product outputs are characterized and compared with the target quality product profile. If the output satisfies the target, a control strategy is created followed by continuous monitoring. Using PAT, any deviation in the quality of the product can be monitored throughout the entire manufacturing process. A general QbD approach to development of a TDDF is shown in Figure 2. It is essential to maintain the physical and chemical stability of the product over time, because these dosage forms can have stability issues like phase separation, change in pH, viscosity, appearance, and crystal formation. Processing parameters like weighing of raw materials, method of mixing, duration of mixing, rate of mixing, temperature set or the generated temperature, rate of heating, or cooling temperature are significant observable factors during the manufacture and scalability of a TDDF.
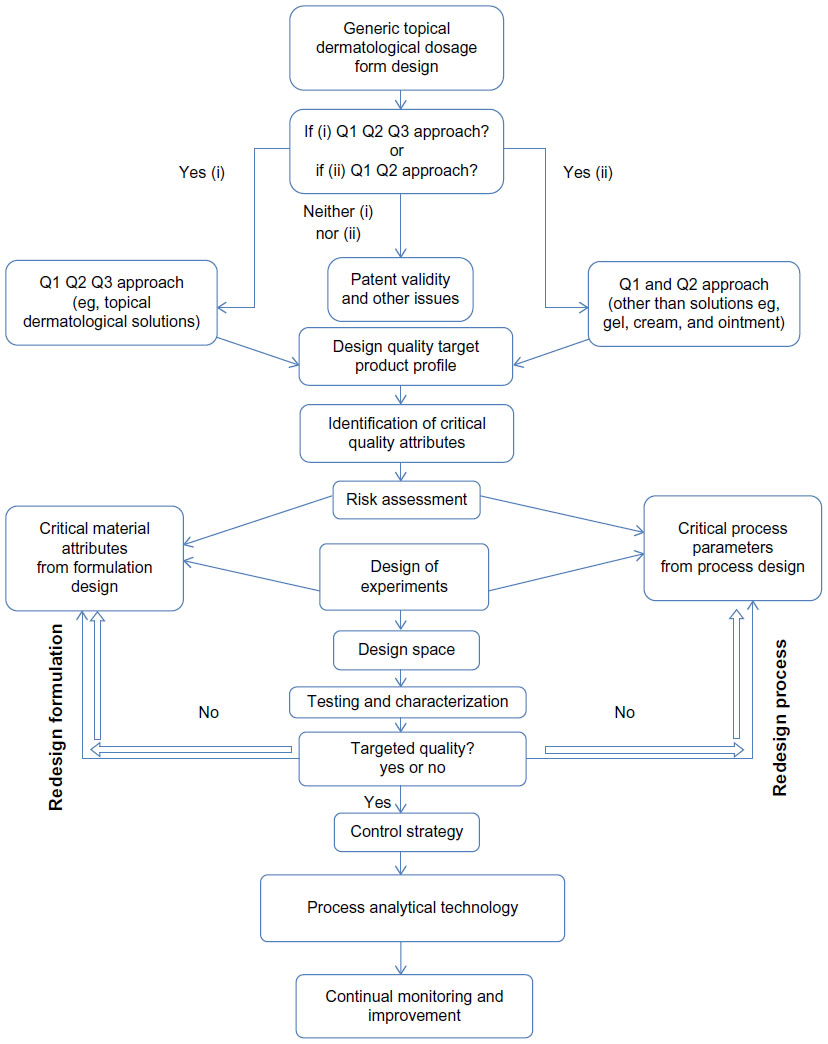 | Figure 2 General quality by design approach for development of a topical dermatological dosage form.25 |
Excipients
Topical formulations include a multitude of excipients. It is important to consider compatibility between the active ingredient and excipients, solvents, and containers. Variation in the grade of the excipients, such as differences in molecular weight, reactive residues can lead to unexpected outcomes. The status of the excipient, ie, whether it is compendial or non-compendial, is an important consideration when selecting an excipient. Additional tests related to pharmacology or toxicology may be required if a non-compendial excipient is chosen other than that specified for the RLD. In most cases, excipients of different grades are compatible with the APIs, but an excipient compatibility test is recommended because there can be variations in the physical and chemical properties of excipients sourced from different vendors. The residual solvent data should meet the USP <467> requirement.6,21
If the manufacturer of a generic product intends to use an excipient not listed in the inactive ingredient guide, it is the responsibility of the sponsor to provide information about the rationale of using this excipient in the formulation including pharmacology and toxicology with clinical data to the FDA unless it is ≤0.1% of the total weight of the drug product.6 Table 5 gives the list of excipients commonly used in topical dosage formulations, with an emphasis on issues faced during processing. The generic drug product is not required to be similar to the RLD in terms of viscosity. However, evaluation of the viscosity of a semi-solid product is encouraged because it is a significant attribute in influencing the drug delivery effect. The ability of a semi-solid product to be retained on the skin is strongly influenced by the viscosity of the product in addition to other physical properties of the dosage form. Factors like the size of the dispersed particles, size of the active ingredient (if dispersed), interfacial tension, partition coefficient of the active ingredient, product rheology, and process temperature influence the physical properties of the dosage form.21 Measurement of viscosity at both the laboratory-scale and scale-up stages is an important criteria for record.
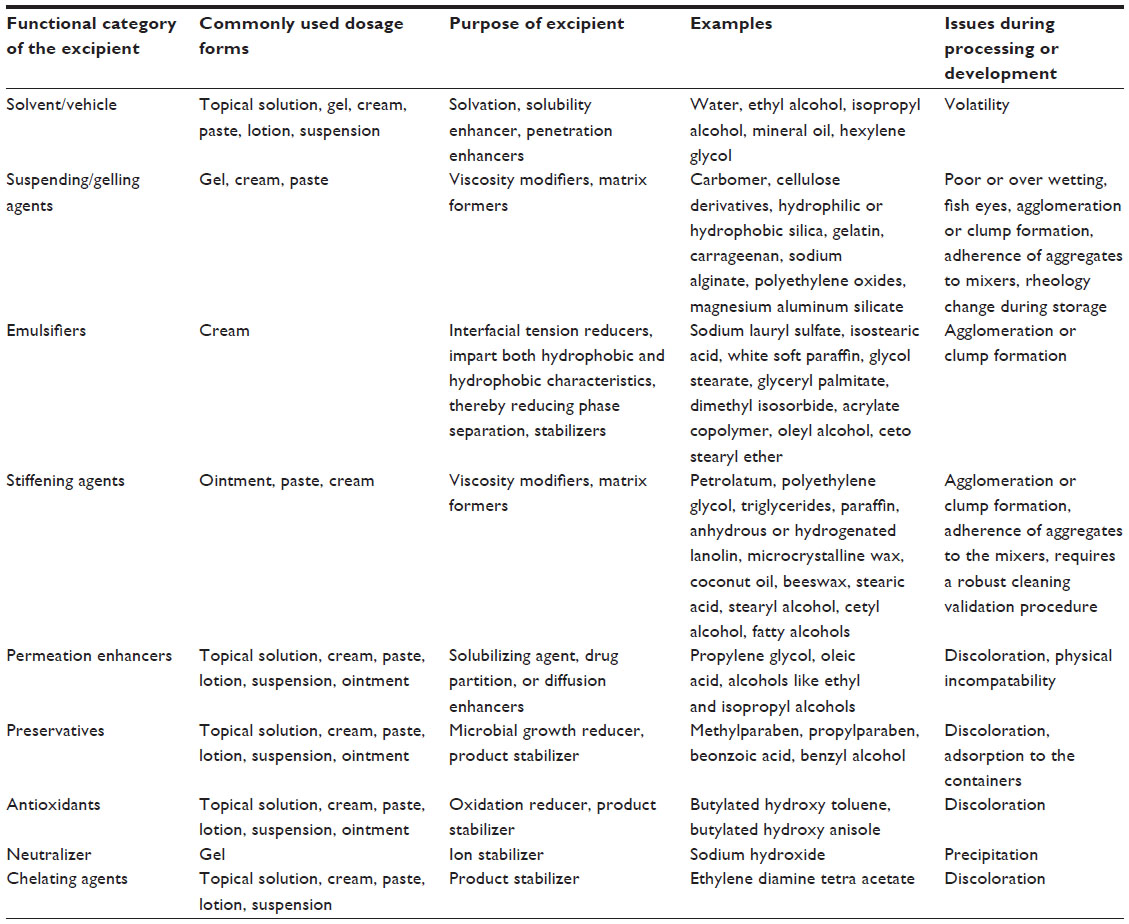 | Table 5 Commonly used excipients and their challenges during development of a topical dermatological dosage form |
Emulsion-based topical semi-solid dosage forms are thermodynamically unstable, requiring cationic or anionic or non-ionic emulsifiers for stabilization. A mixture of emulsifiers of high and low hydrophilic-lipophilic balance values is recommended because such a mixture can produce a complete film around the dispersed droplets or particles so that coalescence can be prevented. Ostwald ripening, ie, merging of smaller dispersed particles into larger particles, is one of the significant stability concerns with most emulsion-based semi-solid products. Crystallization detection in these types of preparations with different temperature cycle is called a freeze-thaw cycle.
Preservatives
For water-based TDDF preparations, an antimicrobial preservative is always a necessary ingredient. It is used to inhibit the growth of bacteria, fungi, and mold. The most commonly used preservative is a combination of methylparaben and propylparaben, which usually ranges from 0.01% to 0.3%. Other additives like antioxidants or antioxidants in combination with a chelating agent may be added for oxidative problems. A generic product manufacturer usually uses the preservative similar to that in the RLD in most cases, but other preservatives can be used if needed. As per the USP <51>, the minimum acceptable limit for preservatives has to be demonstrated. The required concentration and selection of the preservatives must be satisfied.20
Active pharmaceutical ingredient
The source and quality of the API play a significant role in designing a generic topical formulation. It is also important to have a secondary source for the API in the event that the primary source runs into problems. Overage of the API is allowed when there is a manufacturing loss and is allowed for a generic topical product up to the overage of the RLD.
Degradation of the API is a significant stability concern, and knowing the pathway of degradation is a valuable tool during development of a product. As the percentage of the solvent in the topical semi-solid product is relatively higher than in other dosage forms, except for solutions, APIs are susceptible to various instability causing determinants. A detailed understanding of the API degradation pathway via forced degradation studies during development and validation of the manufacturing method is essential to limit the potential degradation routes. It is recommended that the DoE be used if chemical stability is an issue during the developmental stages.21
Packaging
The packaging material, ie, the container and closure system, should be compatible with the components of the formulation. Leaching from the container and closure system can increase the unknown degradable substances. As liquids are more prone to degradation than solids, similarly, semi-solids, which have good rheological properties, may interact with packaging materials and accelerate instability issues. It is wise for the manufacturer of the generic product to use the same material as in the RLD. The ratio of volatile to non-volatile solvents has to be taken into consideration when developing a generic topical product because most of the semi-solid preparations contain an appreciably higher volume of volatile solvent than of other components within the formulation. The rate of evaporation of the solvent is a significant factor because this can cause changes in the performance, stability, and dermal absorption of the product. Added caution is needed if the drug has low solubility in the given solvent. Issues like crystallization, polymorphs, precipitation, and changes to dissolution both in vitro and in vivo may arise due to the variable rate of solvent evaporation.6,21
In vitro release testing
The guidance for sponsors, ie, “Semi-solid dosage forms scale-up and postapproval changes: chemistry, manufacturing and controls; in vitro release testing and in vivo bioequivalence documentation” for nonsterile semi-solid dosage forms given by the FDA represents a significant effort to reduce the regulatory issues involved and their complexity, while still assuring the safety and efficacy of postapproval changes to a pharmaceutical product. This guidance is applicable to pharmaceutical sponsors of new drug applications, ANDAs, and abbreviated antibiotic drug applications. Changes in the composition of a formulation, changes in the category of the components, manufacturing site change, manufacturing process change, equipment change, and changes in the scale of manufacturing require documentation for regulatory approval. These changes are defined at three different levels, as explained in Table 6.22
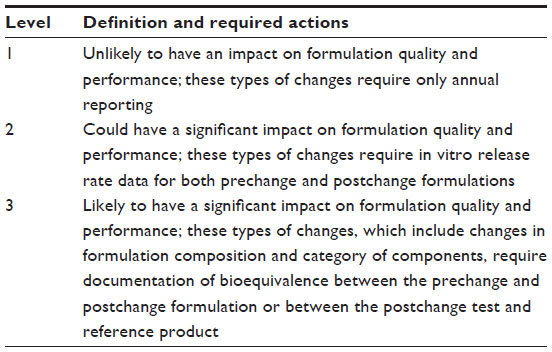 | Table 6 Types of levels and descriptions |
An in vitro diffusion study includes setting up excised human/animal skin or a synthetic membrane between donor and receptor chambers, where the lower or receptor chamber is filled with pH 7.4 phosphate-buffered solution to mimic the physiological milieu and the setup is maintained at 37°C. The required volume or amount of the dosage form is applied on the surface of the membrane or skin, and receptor fluid samples are collected through a side-arm sampling port.23 Diffusion of the drug from the dosage form across the membrane into the receptor medium is quantified by assay. The receptor chamber is replenished with fresh medium after each sampling. To create an adequate drug release profile, at least five sampling points (30, 60, 120, 240, and 360 minutes) are required. Also, based on the Scale-Up and Post-Approval Change Semisolids, at least six cells need to be used for each formulation to determine the drug release profile.24
During development of a formulation, the in vitro diffusion technique is used primarily to choose the best formulation among the various candidate formulations developed. The limitation of this technique is that it does not accept this as a surrogate method to determine in vivo bioavailability or bioequivalence. Another obvious limitation of this technique is the lack of live tissue. Use of full thickness skin can be an issue for poorly water-soluble drugs, but dermatomed skin or isolated epidermis can be an option. Maintaining sink conditions is important for solubilizing agents in the receptor medium. The drawback of using lipophilic solution in the medium is that it may increase percutaneous absorption of lipophilic drugs.
Comparing formulations from different manufacturers using this in vitro technique is not recommended by the FDA. However, semi-synthetic membranes, commercially available synthetic membranes like polysulfone, cellulose acetate/nitrate mixed ester, or polytetrafluoroethylene 70 μm can be used. The test is recommended to determine the sameness of the product under scale-up and when making postapproval changes.24 Differences between formulations with respect to physiological properties such as solubility, rheology, and particle size can be determined with this technique. The in vitro release test is useful for assessing the sameness of the dosage form between prechange and postchange, but is not a surrogate for determining in vivo bioavailability or bioequivalence.23
Conclusion
In recent years, QbD has gained much attention and is being emphasized more than ever before among pharmaceutical manufacturers. However, understanding about its concepts and nomenclature remains vague, which may result in a lack of interest in applying its principles for product development.3 Knowing the demerits of QbT on the one hand and having a detailed understanding of QbD on the other may prompt pharmaceutical manufacturers to apply the principles of QbD in practice. Robust manufacturing of generic TDDFs, with their multiple complex formulation components and the need for strict similarity to the commercial RLD, requires a thorough understanding of CMAs and CPPs. The information gained from development of a topical dermatological product over time at laboratory scale serves as a foundation for pilot or pivotal scale development. QbD helps not only in identifying and understanding CMAs and CPPs in pharmaceutical development, but also in understanding the role and interaction between these in achieving a target quality product.5,8 Therefore, implementing QbD approaches in the development of a TDDF is strongly recommended. From a commercial standpoint, applying QbD reduces costs at all stages of development and accelerates the process of commercializing the product.
Disclosure
The authors report no conflicts of interest in this work.
References
Woodcock J. The concept of pharmaceutical quality. American Pharmaceutical Review. 2004;47(6):1–3. | |
Yu LX. Pharmaceutical quality by design: product and process development, understanding, and control. Pharm Res. 2008;25(4):781–791. | |
Lionberger RA, Lee SL, Lee L, Raw A, Yu LX. Quality by design: concepts for ANDAs. AAPS J. 2008;10(2):268–276. | |
Yu LX, Amidon G, Khan MA, et al. Understanding pharmaceutical quality by design. AAPS J. 2014;16(4):771–783. | |
Boix-Montanes A. Relevance of equivalence assessment of topical products based on the dermatopharmacokinetics approach. Eur J Pharm Sci. 2011;42(3):173–179. | |
Chang R-K, Raw A, Lionberger R, Yu L. Generic development of topical dermatologic products: formulation development, process development, and testing of topical dermatologic products. AAPS J. 2013;15(1):41–52. | |
Lionberger RA. FDA critical path initiatives: opportunities for generic drug development. AAPS J. 2008;10(1):103–109. | |
Weinberg S, Fuqua F. A stochastic model of “quality by design” for the pharmaceutical industry. Clayton State University, 2010. Available from: http://archives.pittcon.org/PittconWeb2010/attendee/pdf/2010/weinberg.pdf. | |
IBM Business Consulting Services. Transforming industrialization: a new paradigm for pharmaceutical development, 2005. Available from: http://www-935.ibm.com/services/us/imc/pdf/ge510-3997-transforming-industrialization.pdf. Accessed May 26, 2015. | |
International Conference on Harmonisation of Technical requirements for registration of pharmaceuticals for human use, Drafts Consensus Guideline, Pharmaceutical Development Annex to Q8, Nov 2007. Available from: http://www.pmda.go.jp/ich/q/step3_q8-a_08_3_17e.pdf. | |
US Food and Drug Administration. Guidance for industry: PAT – a framework for innovative pharmaceutical development, manufacturing, and quality assurance. September 2004. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm070305.pdf. Accessed May 26, 2015. | |
US Food and Drug Administration. Guidance for industry: Q9 quality risk management, ICH, June 2006. Available from: http://www.fda.gov/downloads/Drugs/…/Guidances/ucm073511.pdf. Accessed May 26, 2015. | |
Rosas JG, Blanco M, González JM, Alcalá M. Quality by design approach of a pharmaceutical gel manufacturing process, part 1: determination of the design space. J Pharm Sci. 2011;100(10):4432–4441. | |
Xie L, Wu H, Shen M, et al. Quality-by-design (QbD): effects of testing parameters and formulation variables on the segregation tendency of pharmaceutical powder measured by the ASTM D 6940-04 segregation tester. J Pharm Sci. 2008;97(10):4485–4497. | |
Herkenne C, Alberti I, Naik A, et al. In vivo methods for the assessment of topical drug bioavailability. Pharm Res. 2008;25(1):87–103. | |
US Food and Drug Administration. Guidance for industry: Q8 pharmaceutical development, ICH, May 2006. Available from: http://www.fda.gov/ohrms/dockets/ac/06/briefing/2006-4241B1-02-03-FDA-Q8%20Guidance%206746fnl.pdf. Accessed May 26, 2015. | |
Chang R-K, Raw A, Lionberger R, Yu L. Generic development of topical dermatologic products. Part II: quality by design for topical semisolid products. AAPS J. 2013;15(3):674–683. | |
Lionberger R. Quality by design for topical dosage forms. Office of Generic Drugs, US Food and Drug Administration, 2005. Available from: http://www.fda.gov/ohrms/dockets/ac/05/slides/2005-4137s2_04_lionberger.ppt. | |
Shaw AS. Quality by sesign (QbD) for dermatologic products. Office of Generic Drugs, US Food and Drug Administration, 2013. Available from: http://www.pqri.org/workshops/Topicals2013/Raw.pdf. Accessed May 26, 2015. | |
United States Pharmacopeia (USP) 29-NF 24 Vol 36 (6) [Nov–Dec 2010] page 2499. Available from: http://www.pharmacopeia.cn/v29240/usp29nf24s0_c51.html. Accessed May 26, 2015. | |
Lowenborg M. A troubleshooting guide for topical drug manufacturing. Pharmaceutical Technology. 2012;36:46–49. | |
Shah VP, Flynn GL, Yacobi A, et al. Bioequivalence of topical dermatological dosage forms – methods of evaluation of bioequivalence. Pharm Res. 1998;15(2):167–171. | |
Narkar Y. Bioequivalence for topical products – an update. Pharm Res. 2010;27(12):2590–2601. | |
US Food and Drug Administration. Guidance for industry, nonsterile semisolid dosage forms, scale-up and post approval changes: in vitro release testing and in vivo bioequivalence documentation, May 7, 1997. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070930.pdf. Accessed May 26, 2015. | |
Buhse L, Kolinski R, Westenberger B, et al. Topical drug classification. Int J Pharm. 2005;295(1–2):101–112. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.