")
Back to Journals » Drug Design, Development and Therapy » Volume 13
Qingxin kaiqiao fang ameliorates memory impairment and inhibits apoptosis in APP/PS1 double transgenic mice through the MAPK pathway
Authors Gao SY, Lin J, Wang T, Shen Y , Li Y, Yang W, Zhou K , Hu H
Received 24 September 2018
Accepted for publication 18 December 2018
Published 23 January 2019 Volume 2019:13 Pages 459—475
DOI https://doi.org/10.2147/DDDT.S188505
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jianbo Sun
Shiyu Gao,1,2 Jianwei Lin,1,2 Tianqi Wang,1,2 Yan Shen,1,2 Yan Li,1,2 Wenyu Yang,1,2 Kailiang Zhou,2–4 Haiyan Hu1,2
1Department of Traditional Chinese Medicine, The Second Affiliated Hospital and Yuying Children’s Hospital of Wenzhou Medical University, Wenzhou 325000, Zhejiang, China; 2The Second Clinical Medical College of Wenzhou Medical University, Wenzhou 325003, China; 3Zhejiang Provincial Key Laboratory of Orthopaedics, Wenzhou 325027, China; 4Department of Orthopaedics, The Second Affiliated Hospital and Yuying Children’s Hospital of Wenzhou Medical University, Wenzhou 325000, Zhejiang, China
Background: Qingxin kaiqiao fang (QKF) has been found to treat Alzheimer’s disease (AD) through apoptosis inhibition. The mitogen-activated protein kinase (MAPK) pathway is closely related to apoptosis in the course of AD. This study aimed to investigate whether QKF-induced apoptosis depression is achieved through MAPK pathway.
Materials and methods: C57BL/6 J and APP/PS1 mice were used as control and model groups. APP/PS1 mice were treated with different dosages of QKF (4.75, 9.5, and 19 g·kg-1·d-1·ig, respectively) for 12 weeks as L-QKF, M-QKF, and H-QKF groups. The M-QKF-treated APP/PS1 mice were administrated with 2 µg/kg of U46619 and saline, intra ventricular ventricle injection, as M-QKF+U46619 and M-QKF+saline groups and were injected with PD98059 0.3 mg/kg and the same volume of dimethyl sulfoxide (DMSO), intravenous, as M-QKF+PD98059 and M-QKF+DMSO groups. After 12 weeks treatment, Morris water maze was performed for behavior study. Pathological degeneration was examined by H&E staining, Nissl staining, and transmission electron microscope observation of hippocampus; immunohistochemistry and Western blot (WB) were tested for amyloid β (Aβ) expression. Apoptosis was measured through TUNEL assay; Bax, Bcl-2, and caspase-3 expression through WB; and cleaved caspase-3 expression through ELISA. MAPK pathway was detected via WB for the expressions of ERK1/2, JNK, and p38 MAPK and their phosphorylation patterns.
Results: QKF improved the learning and memory capability, as well as inhibited neuronal apoptosis and then reduced the pathological degeneration of APP/PS1 mice. M-QKF reduced neuron apoptosis by inhibiting p38 MAPK and activating ERK1/2 but had no significant effect on JNK.
Conclusion: QKF, especially at the middle dose, alleviated the learning and memory impairment and played an antiapoptotic role in AD through MAPK pathways.
Keywords: Qingxin kaiqiao, Alzheimer’s disease, apoptosis, APP, PS1 mice, MAPK
Introduction
Alzheimer’s disease (AD) is the most common form of dementia, with a prevalence of 46.8 million affected individuals worldwide, which is expected to reach 131.5 million by 2050.1–4 AD is a neurodegenerative disease closely related to aging; it is characterized by neuritic senile plaque (SP), intraneuronal neurofibrillary tangles (NFTs), and neuronal death.5,6 Apoptosis-induced neuronal death and loss are important factors contributing to the progression of AD.7 Previous studies have shown that neuroprotective agents such as nimodipine and hydergine can inhibit the occurrence of apoptosis and treat AD.8,9 These observations suggest that inhibiting apoptosis in neuronal cells may have a neuroprotective effect against AD.
The apoptosis of neurons in AD is influenced by multiple factors and was shown to be closely related to the MAPK pathway.10,11 MAPKs are serine–threonine kinases that are widely expressed in the central nervous system and mediate intracellular signaling related to a variety of cellular activities, including cell proliferation, differentiation, survival, death, and transformation.12 The mammalian MAPK family consists of p38 MAPK, ERK, and c-Jun NH2-terminal kinase (JNK; also known as stress-activated protein kinase).13 Both p38 and JNK pathways mainly induce apoptosis,14 whereas the ERK pathway is mainly involved in the promotion of cell survival.15 The p38 signaling pathway is involved in inflammatory reactions and apoptosis processes under conditions of oxidative stress.16 Studies on neural and nonneuronal cells have shown that p38 can regulate apoptosis through multiple mechanisms, including phosphorylation of p53,17 activation of c-JUN and c-fos,18 induction of Bax transposition,19 participation in Fas–FasL-mediated apoptosis,20 enhanced expression of c-myc,21 and activation of caspase-3.19 In addition, p38 MAPK can enhance the expression of TNF-α, which in turn activates p38 to induce apoptosis.22 ERK participates in cell activation and migration and plays an important role in synaptic plasticity and memory in vivo.23 It was also reported that amyloid β (Aβ) could induce JNK activation and cell death.24 Therefore, compartment-specific targeting of MAPKs to inhibit apoptosis may provide promising new therapeutic avenues for the treatment of AD.
*Qingxin Kaiqiao Fang (QKF), a well-known classic Chinese herbal prescription for dementia, is based on a formulation named Fumanjian from the medical book Jingyue Quanshu written by Zhang Jing-yue during the Ming Dynasty.25 QKF consists of 10 Chinese herbs: Radix Rehmanniae, Radix Ophiopogonis, Radix Paeoniae, Herba Dendrobii, Cortex Moutan Radicis, Poria Cocos, Pericarpium Citri Reticulatae, Rhizoma Anemarrhenae, Rhizoma Acori Tatarinowii, and Sophorae Flavescentis.26 It has been used clinically for 20 years to alleviate dementia, depression, and anxiety, and its curative effect was shown to be stable, especially for the early symptoms.26 QKF can significantly improve cognitive impairment as well as behavioral and mental symptoms in patients. Our previous study showed that QKF improved learning and memory in a rat model of AD and decreased apoptosis in the hippocampal region by significantly reducing the levels of Bax and cleaved caspase-3, while upregulating the level of Bcl-2 in the hippocampus.25 Therefore, QKF has potential for the treatment of AD. However, the pathological mechanism underlying the apoptosis-inhibiting effect of QKF has yet to be clarified.
Owing to the close relationship of QKF with apoptosis in AD, the present study was performed to investigate the effects of QKF on the MAPK pathway and to further verify the protective effect of QKF against MAPK-mediated apoptosis. Moreover, to accurately reflect the pathological features of AD, an amyloid precursor protein/presenilin 1 (APP/PS1) double transgenic mouse model was used in this study.
Materials and methods
Animals
Specific pathogen-free (SPF) male APP/PS1 transgenic mice, 3 months old, weighing 25±2 g were purchased from Beijing HFK Bioscience Co., Ltd. (Beijing, China; certification number SCXK 2014-0004). Three-month-old male C57BL/6J mice were purchased from Shanghai Slack Laboratory Animal Co., Ltd. (Shanghai, China; certification number SCXK 2012-0002). Mice were reared in the Wenzhou Medical University Laboratory Animal Center, which is a qualified facility meeting clean experimental animal feeding standards. Mice were housed under controlled conditions of 23°C under a 12-hour light/dark cycle and were given free access to food and water. All animal experiments were performed in accordance with the ethical requirements approved by the Chinese Association of Accreditation of Laboratory Animal Care.
Preparation of QKF aqueous extract
QKF is composed of 10 Chinese herbal medicines: Radix Rehmanniae Recens, which consists of unprocessed rehmannia root (Sheng di huang) and dried roots of Radix Rehmanniae Recens; Radix Ophiopogoni, comprising dwarf lilyturf tuber (Maidong) and dried roots of Ophiopogon japonicus; Radix Paeoniae Alba, containing debark peony root (Baishao) and dried roots of Paeonia lactiflora Pall.; Rhizoma Acori Tatarinowii, which consists of grassleaf sweetflag rhizome (Shi chang pu) and dried roots of Acorus Tatarinowii Schott; Herba Dendrobii, which contains Dendrobium (Shihu) and dried roots of Dendrobium officinale Kimura et Migo.; Cortex Moutan Radicis, containing tree peony root bark (Mu Dan Pi) and dried root barks of Paeonia suffruticosa Andr.; (G) Poria; Indian bread (Fuling) and dried sclerotia of Poria cocos (Schw.) Wolf; Pericarpium Citri Reticulatae, containing dried tangerine peel (Chenpi) and dried fruit peel of Citrus reticulata Blanco; Radix Sophorae Flavescentis (Kucen), containing dried roots of Sophora flavescens Ait; and Rhizoma Anemarrhenae, which comprises common anemarrhena rhizome (zhimu) and dried roots of Anemarrhena asphodeloides Bge., in a ratio of 2:2:2:2:2:2:2:1:1.5:1.5 on a dry-weight basis, as recorded in the Chinese Pharmacopeia. All herbal ingredients were provided by the Second Affiliated Hospital of Wenzhou Medical University and verified by the Department of Chinese Materia Medica of Wenzhou Medical University. Before decocting, the herbs were immersed in 10 times volume of distilled water for 30 minutes. After decoction for 2 hours, they were extracted twice, filtered with clean gauze, and concentrated, and the raw herbs were made into a 1 g·mL−1 stock solution, which was stored at 4°C until use.
Reagents and antibodies
Aβ immunohistochemistry kit and DAB staining kit were purchased from Bioss (Beijing, China), and DAB reagent kit was obtained from Zymed (San Diego, CA, USA). Nissl staining solution was purchased from Beyotime (Shanghai, China; C0117). Polyclonal rabbit anti-mouse Bcl-2, Bax, and caspase-3 antibodies were purchased from Wuhan Boster Biological Engineering Co., Ltd. (Wuhan, China; A00040-2, A00183, and PB9188, respectively). Primary antibodies against ERK1/2, p-ERK1/2, p38 MAPK, and p-p38 MAPK were purchased from Cell Signaling Technology (Beverly, MA, USA; #4695, #4370, #8690, and #4511, respectively). Primary antibody against GAPDH was purchased from Biogot Technology (Shanghai, China; AP0063). Primary antibodies against JNK and p-JNK were purchased from Santa Cruz Biotechnology Inc. (Dallas, TX, USA; sc-7345 and sc-6254, respectively). Primary antibody against Aβ1-42 was purchased from Abcam (Cambridge, UK; ab201060;). Horseradish peroxidase (HRP)-conjugated IgG secondary antibody was purchased from Santa Cruz Biotechnology Inc. An inhibitor of ERK1/2, PD98059, was purchased from Santa Cruz Biotechnology Inc. (sc-3532). The p38 MAPK agonist 11α-methanoepoxy prostaglandin F2α (U46619) was purchased from Santa Cruz Biotechnology Inc. (sc-201242). The ECL Plus Reagent kit was purchased from Perkin-Elmer Inc. (Waltham, MA, USA). The Cleaved Caspase-3 ELISA kit was purchased from Boster Biological Technology, Ltd (Wuhan, China).
Animal groups and treatment
Eighty APP/PS1 double transgenic mice were divided into eight groups according to a random number table: model group (n=10), high-dose (19 g·kg−1·d−1) QKF group (H-QKF; n=10), middle-dose (9.5 g·kg−1·d−1) QKF group (M-QKF; n=10), low-dose (4.75 g·kg−1·d−1) QKF group (L-QKF; n=10), M-QKF+saline group (n=10), M-QKF+U46619 group (n=10), M-QKF+dimethyl sulfoxide (DMSO) group (n=10), and M-QKF+PD98059 group (n=10). In addition, 10 C57BL/6J mice were used as controls. Intragastric administration was given in a volume of 20 mL·kg−1·d−1; the high-dose, medium-dose, and low-dose groups received QKF at doses of 19, 9.5, and 4.75 g·kg−1·d−1, respectively. The control group and model group were given the same volume of normal saline. Each group was treated once a day at 10:00 am for a period of 12 weeks. After anesthesia with an intraperitoneal injection of 0.3% pentobarbital, the mice were given an intraventricular injection using a stereotactic apparatus (Kopf Instruments, Tujunga, CA, USA). Taking the former fontanel as the origin, the injection was given 0.5 mm to the rear and 1.0 mm to the right, with an injection depth of 2.0 mm. The injection speed was 2 μL·min−1. U46619 (2 μg·kg−1) and the same concentration of physiological saline were injected in the M-QKF+U46619 group and M-QKF+saline group, respectively, every other week. PD98059 was dissolved in DMSO and prepared into mother liquor at a concentration of 10 mmol·L−1. The mice in the M-QKF+PD98059 group were injected with PD98059 at a dose of 0.3 mg·kg−1 via the tail vein every other week. Mice in the M-QKF+DMSO group received an injection of the same concentration of DMSO at the same time.
Morris water maze (MWM)
Spatial learning memory ability was assessed by the MWM test, which consists of orientation navigation and spatial probe tests. To ensure objective results of the experiment, the test was conducted by two investigators completely blinded to the treatment of the animals. The MWM consists of a circular tank (90 cm in diameter) filled with water at 21°C±2°C with a hidden platform (4.5 cm in diameter) positioned 1–2 cm below the opaque water in the middle of the third quadrant. The orientation navigation tests were performed for 4 days, four times a day, at 30-minute intervals. Each time, the mice were randomly placed in the water at four entry points, facing the wall of the pool. The camera lens above the pool was connected to a computer, and the swimming trajectory of each mouse was recorded and analyzed by a SLY-WMS MWM analysis system (version 2.1; Beijing Sunny Instruments Co. Ltd.; Beijing, China). After finding the hidden platform, the mice were allowed to stay on the platform for 15 seconds. If the mice could not find the platform within 60 seconds, the escape latency (ie, the time needed to find the hidden platform) was recorded as 60 seconds, and the mice were then guided to the platform, where they were allowed to remain for 15 seconds. The spatial probe test was carried out 24 hours after the last orientation navigation test. Investigators removed the platform, placed the mice into the pool, and allowed them to swim freely for 60 seconds. The times for the mice to cross to the original location of the platform were recorded.
Hippocampus collection
At the end of the experiment, the mice were anesthetized with 10% chloral hydrate. In order to conduct different experiments, we divided mice in each group into two parts and each group contained five mice. Random number tables were used in the allocation process. To extract protein from tissues, a part of mice in each group was perfused and quickly decapitated after anesthesia, and the hippocampi were stripped, placed in cryopreservation tubes, and stored at −80°C. To fix tissues and make sections for staining and observation, another part of mice in each group was perfused with 4% paraformaldehyde, and the brains were removed quickly and placed in 4% paraformaldehyde. The tissues were placed in 4% paraformaldehyde fixative for 24 hours and then dehydrated by passage through a graded ethanol series at room temperature. Tissues were then made transparent with xylene, embedded in paraffin, and cut into 5 μm thick sections.
H&E stain
Mouse brains were first fixed in 4% paraformaldehyde for 24 hours and embedded in paraffin wax for transverse sectioning. Sections 4 μm thick were prepared and mounted on poly-L-lysine-coated slides for H&E stain. The number of microvessels per unit area (/mm2) was determined using a light microscope (×400 magnification; Olympus Corporation, Tokyo, Japan) to assess mean microvascular density. Six random fields of three random sections from each tissue specimen were measured.
Nissl staining
The normal paraffin-embedded hippocampi of mice were cut into 4 mm thick sections. After dewaxing and rehydration, the sections were stained for 30 minutes at 60°C with Nissl staining solution. Sections were then subjected to dehydration with anhydrous ethanol, made transparent with xylene, and sealed with neutral gum. The morphology was observed, and images were obtained using a light microscope.
Transmission electron microscopic observation
After the behavior study, the heart was perfused, and the hippocampi were collected. The hippocampi were placed in 3% glutaraldehyde fixative and stored at 4°C. Tissues were cut into pieces of ~1 mm3, placed in 2.5% glutaraldehyde fixative, and fixed for 2 hours at 4°C. The tissues were rinsed in 0.1 mol·L−1 PBS for 30 minutes, immobilized with 1% osmium tetroxide at 4°C for 2 hours, rinsed with 0.1 mol·L−1 PBS for 10 minutes, dehydrated through a graded ethanol series, and stained with 70% ethanol–uranium peroxide acetate block staining for 2 hours. Sections were photographed using the light microscope and then serially cut into 1–2 μm semi-thin sections with a Leica ultramicrotome (EM UC7; Leica Microsystems, Wetzlar, Germany). Selected semi-thin sections were sliced into serial ultrathin sections with a silver–gray interference color, corresponding to a thickness of ~50–70 nm. The ultrathin sections were collected on formvar-coated, single-slot grids and stained with uranyl acetate and lead citrate. Neurons in the hippocampal CA1 region were observed and photographed using a transmission electron microscope (Tecnai Spirit BioTWIN; FEI, Blackwood, NJ, USA).
TUNEL staining
Neuronal apoptosis in the hippocampal CA1 region was assessed by TUNEL staining using an in situ cell death detection kit (Roche China, Shanghai, China) according to the manufacturer’s recommendations. In each animal, six sections were used for quantitative analysis. TUNEL-positive neurons in the hippocampus were observed using a Nikon fluorescence microscope. The number of viable TUNEL-positive neurons in the hippocampus was automatically counted in a predefined area under a high-power magnification (×400).
Immunohistochemistry
Six sections of mouse brain from each group were deparaffinized in xylene and rehydrated through a graded ethanol series. After washing, the sections were blocked with 3% (v/v) H2O2 and treated with 10.2 mM sodium citrate buffer for 20 minutes at 95°C. After blocking with 10% (w/v) BSA in PBS for 10 minutes, the sections were incubated with an antibody against Aβ overnight at 4°C in accordance with the manufacturer’s instructions. Finally, the sections were incubated with HRP-conjugated secondary antibody and counterstained with hematoxylin. The CA1 area of the hippocampus was imaged at ×40 magnification using a DP2-TWAN image acquisition system (Olympus Corporation). Images were analyzed using Image-Pro Plus software (Media Cybernetics, Rockville, MD, USA) for integral absorbance quantification of Aβ; each sample was observed at five random fields in the CA1 area of the hippocampus.
Western blot (WB)
Hippocampal samples of APP/PS1 transgenic mice and C57BL/6 mice were dissected and stored at −80°C before WB analysis. Five samples in each group were processed by extracting proteins with lysis buffer. Protein concentrations were calculated using the bicinchoninic acid assay assay. Proteins were separated by 10% PAGE and subsequently transferred onto polyvinylidene difluoride (PVDF) membranes (Roche Applied Science, Indianapolis, IN, USA). After blocking with 5% (w/v) BSA for 2 hours at room temperature, the membranes were incubated overnight with the following primary antibodies (1:1,000) at 4°C: ERK1/2, p-ERK1/2, JNK, p-JNK, p38 MAPK, p-p38 MAPK, Bcl-2, Bax, caspase-3, Aβ1-42, and GAPDH. The membranes were then incubated with goat anti-rabbit or goat anti-mouse secondary antibody as appropriate for 2 hours at room temperature. Immunoreactive proteins were visualized using the ECL Plus Reagent kit. Finally, the band intensity was quantified using Image Laboratory 3.0 software (Bio-Rad Laboratories Inc., Hercules, CA, USA).
ELISA for cleaved caspase-3
The hippocampal tissues were homogenized in cooled PBS and subjected to repeated freezing and thawing in liquid nitrogen. The homogenate was centrifuged at 10,000g for 10 minutes at 4°C. The supernatant was used for ELISA. The levels of cleaved caspase-3 in the hippocampal tissues and culture medium were determined using commercial ELISA kits according to the manufacture’s protocols (Wuhan Boster Biological Technology, Ltd., Wuhan, China). The quantification of cleaved caspase-3 levels was obtained by a microplate reader at 550 nm with correction wavelength at 450 nm. Data were analyzed by GraphPad Prism software.
Statistical analyses
All data were processed using SPSS 25.0 (IBM Corporation, Armonk, NY, USA), and results are expressed as the mean±standard error of the mean (SEM). The data from the five groups were compared using the independent-samples t-test and one-way ANOVA. Pairwise comparisons were performed using the least significant difference test for homogeneous variance. In all analyses, P<0.05 was taken to indicate statistical significance.
Ethics statement
All surgical procedures, treatments, and postoperative care were performed with the permission of Wenzhou Medical University Ethical Committee and strictly conformed to the International Health and Medical Research Guidelines for Animal Welfare.
Results
QKF ameliorated learning and memory deficits in AD model mice
The MWM, an important method for measuring spatial learning and memory ability in mice, is widely used in the study of AD. Compared with the control group, the double transgenic mice searched for the hidden platform in a random manner. The QKF-treated groups preferentially performed a selective search for the platform and had a shorter search path than did the model group. At a dose of 9.5 g·kg−1·d−1, the M-QKF group performed better than the groups receiving other doses of QKF (Figure 1A). There were no significant differences in escape latency among groups on the first and second days of training. However, significant differences were observed on subsequent days of training. On the third and fourth days, the escape latency of the model group was significantly prolonged compared with that of the control group (P<0.01; Figure 1B). A significant decrease in escape latency was observed in the M-QKF group compared with that in the model group (P<0.05 on the third day and P<0.01 on the fourth day). Moreover, on the third and fourth days, the escape latency of the L-QKF group was not significantly different from that of the control group (P=0.396 and P=0.104, respectively) but that of the H-QKF group was significantly different from the control group on the fourth day (P=0.017; Figure 1C). These data suggested improvement in spatial memory of QKF-treated AD mice from the model groups.
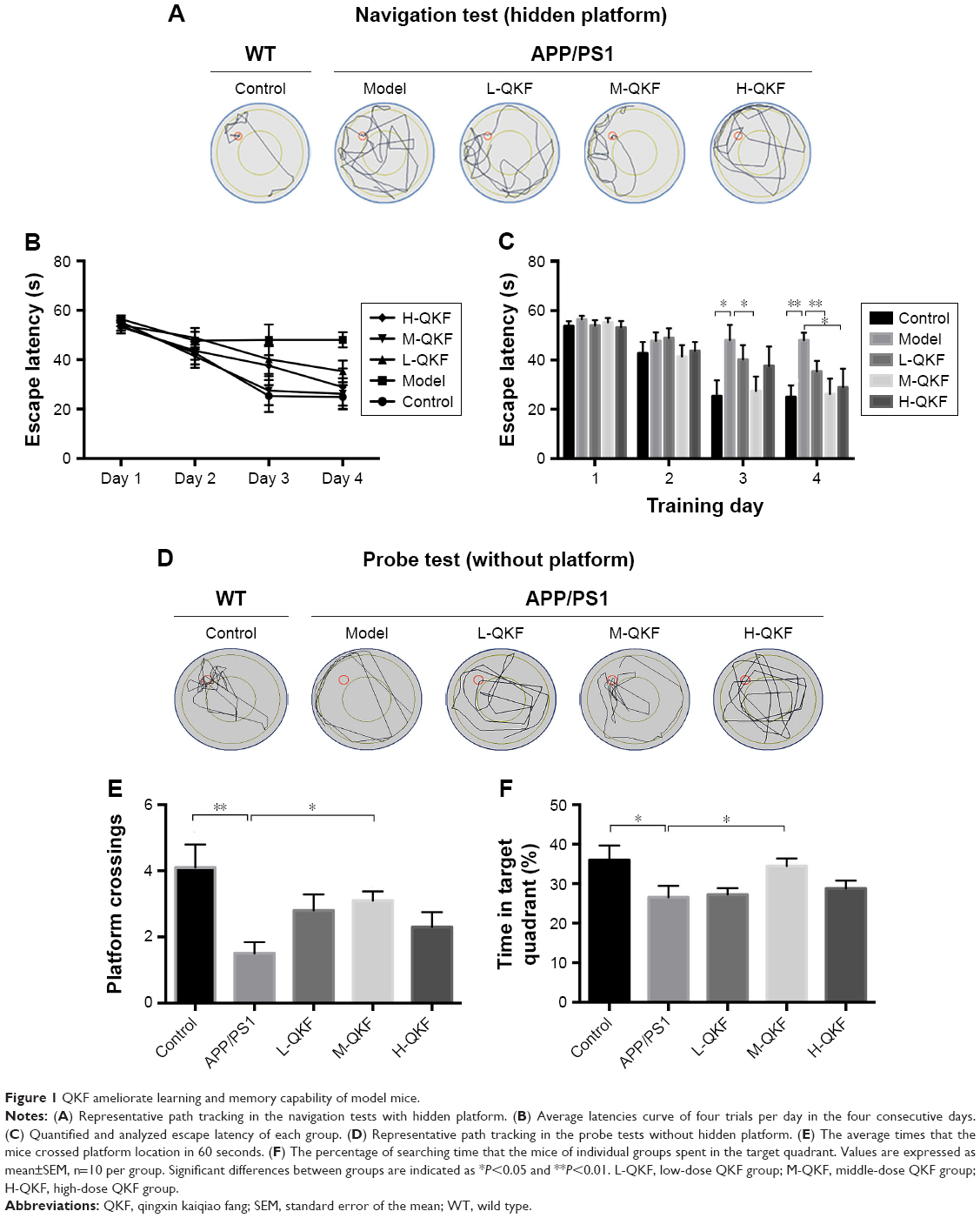 | Figure 1 QKF ameliorate learning and memory capability of model mice. |
In the probe test, although the total path length was similar to that of the vehicle controls, the model group mice swam randomly throughout the tank, indicating poor memory retention of the location of the platform. However, the QKF-treated APP/PS1 double transgenic mice used a spatially biased search strategy to locate the platform, indicating good memory retention (Figure 1D). Furthermore, we calculated the number of times the mice crossed the platform location and the percentage of target quadrant search time, both of which indicated memory retention of the location where the hidden platform had previously been. The control group had a significantly higher number of platform crossings and a greater target quadrant search time compared with the model group (P<0.01 and P<0.05, respectively). Compared with the model group, the M-QKF group showed a higher number of platform crossings (P=0.021). Furthermore, M-QKF mice spent more time in the target quadrant compared with the mice in the model group in the probe test (P=0.034). However, there were no significant differences between the L-QKF or H-QKF groups relative to the model group in platform location crossings or the percentage of target quadrant search time (Figure 1E and F). The results indicated that the middle dose of QKF improved learning and memory capability, thus preventing cognitive deficits in APP/PS1 double transgenic mice.
QKF alleviated pathological degeneration in the brains of APP/PS1 mice
The hippocampal region CA1 in the mice from each group was stained with H&E (Figure 2A). Compared with the model group, the control group showed greater numbers of neurons and a higher degree of order in the hippocampal CA1 area. Neuronal cell density was higher in the QKF-treated groups, especially group M-QKF, compared to the control group. The Nissl body is the main site where neurons synthesize proteins, and it plays an important role in neuron excitation and conduction. Nissl staining was carried out for histological observation in the CA1 region of the hippocampus. As shown in Figure 2B, Nissl bodies disappeared in hippocampal neurons of the model group, some cells were swollen and disrupted, and the arrangement of cells was disordered. After QKF treatment, the degree of Nissl body loss was reduced, and the swelling and disordered arrangement of cells were partially alleviated, especially in the M-QKF group.
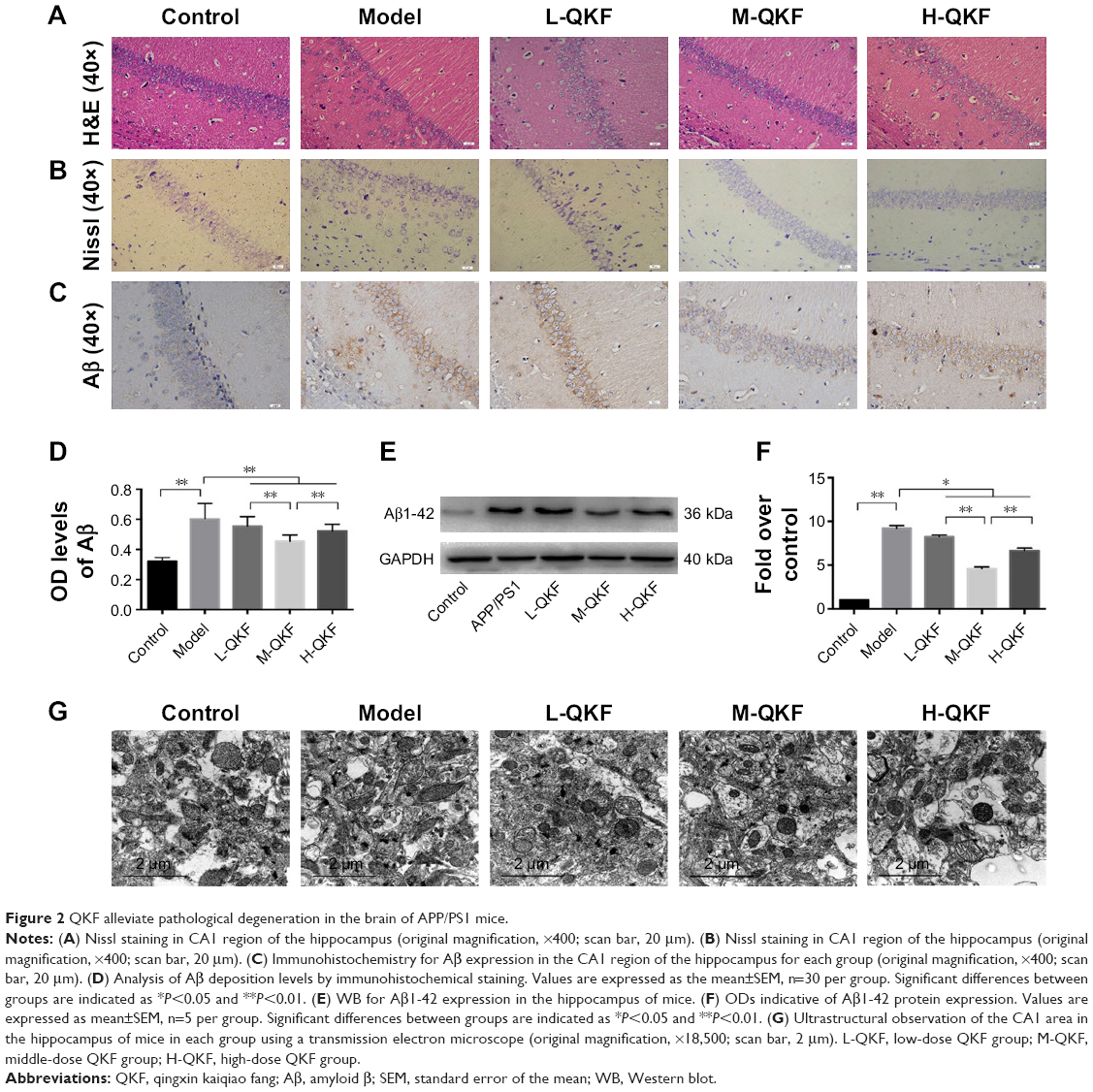 | Figure 2 QKF alleviate pathological degeneration in the brain of APP/PS1 mice. |
Immunohistochemical staining was performed to further determine Aβ-positive granules in the hippocampal CA1 area. As shown in Figure 2C and D, the number of Aβ-positive areas was greater in the model group than that in the control group (P<0.01). The Aβ OD levels of the QKF treatment groups were significantly decreased compared with those of the model group (P<0.01). Among the three doses of QKF, the middle dose resulted in the greatest decrease in Aβ, which was significantly greater than those in the high-dose and low-dose groups (P<0.01). WB indicated significantly higher Aβ1-42 protein levels in the hippocampus of the model group mice compared to those in the hippocampus of the control group (P<0.01). The Aβ1-42 protein expression levels in the QKF groups were significantly decreased compared to those in the model group (P<0.05; Figure 2E and F). Among the three doses of QKF, the M-QKF dose resulted in the greatest decrease, which was significantly greater than those in the H-QKF group and the L-QKF group (P<0.01).
On transmission electron microscopy, the neurons in the CA1 area of the hippocampus in the control group showed complete ultrastructure and well-developed Golgi complex. The neurons showed abundant mitochondria in the cytoplasm, and the mitochondria were round, oval, or rod shaped with complete cristae. In the model group, neurons in the hippocampus showed severe degenerative changes, including decreased rough endoplasmic reticulum, swelling and dilatation of the Golgi complex, reduced number, and blurred or vacuolated cristae of mitochondria. The ultrastructure of neurons in the QKF groups was improved to varying degrees, and the M-QKF group showed the greatest degree of improvement among the dose groups (Figure 2G). These observations indicated that QKF, especially at the middle dose, significantly reduced the pathological degeneration associated with AD and improved the histopathological and morphological changes in APP/PS1 mice.
QKF alleviates neuronal apoptosis in the hippocampus of APP/PS1 mice
The abovementioned results indicated that the medium dose of QKF (M-QKF) had the greatest protective effect against learning and memory degeneration and neuronal apoptosis in the mouse model of AD. Therefore, M-QKF was used in subsequent experiments to examine neuronal apoptosis. Apoptotic cells on TUNEL staining were brown, the nuclei were stained blue, and the background was pale blue (Figure 3A). The number of positive cells in the selected visual field was counted by the positive cell counting method, and the final value was presented as the ratio of the number of positive cells to total cells. The results indicated that the model group showed a significantly increased number of apoptotic cells in the hippocampus compared with the control group (P<0.01). The M-QKF group showed a significant decrease in apoptotic cell number compared with the model group (P<0.01; Figure 3B). As shown in Figure 3C–F, compared with the control group, the Bax and caspase-3 protein levels were markedly increased in the model group (P<0.01), and the levels of Bcl-2 protein were significantly decreased (P<0.01). The protein levels of Bax and caspase-3 were significantly decreased, and the protein levels of Bcl-2 were markedly increased after treatment with QKF at a dose of 9.5 mg·kg−1·day−1 (P<0.01). Levels of cleaved caspase-3 by ELISA showed a significant decrease after M-QKF treatment (P<0.01; Figure 3G). Therefore, QKF treatment at the middle dose reduced neuronal apoptosis by significantly decreasing the levels of Bax and cleaved caspase-3, while upregulating the level of Bcl-2 in the hippocampus of APP/PS1 mice.
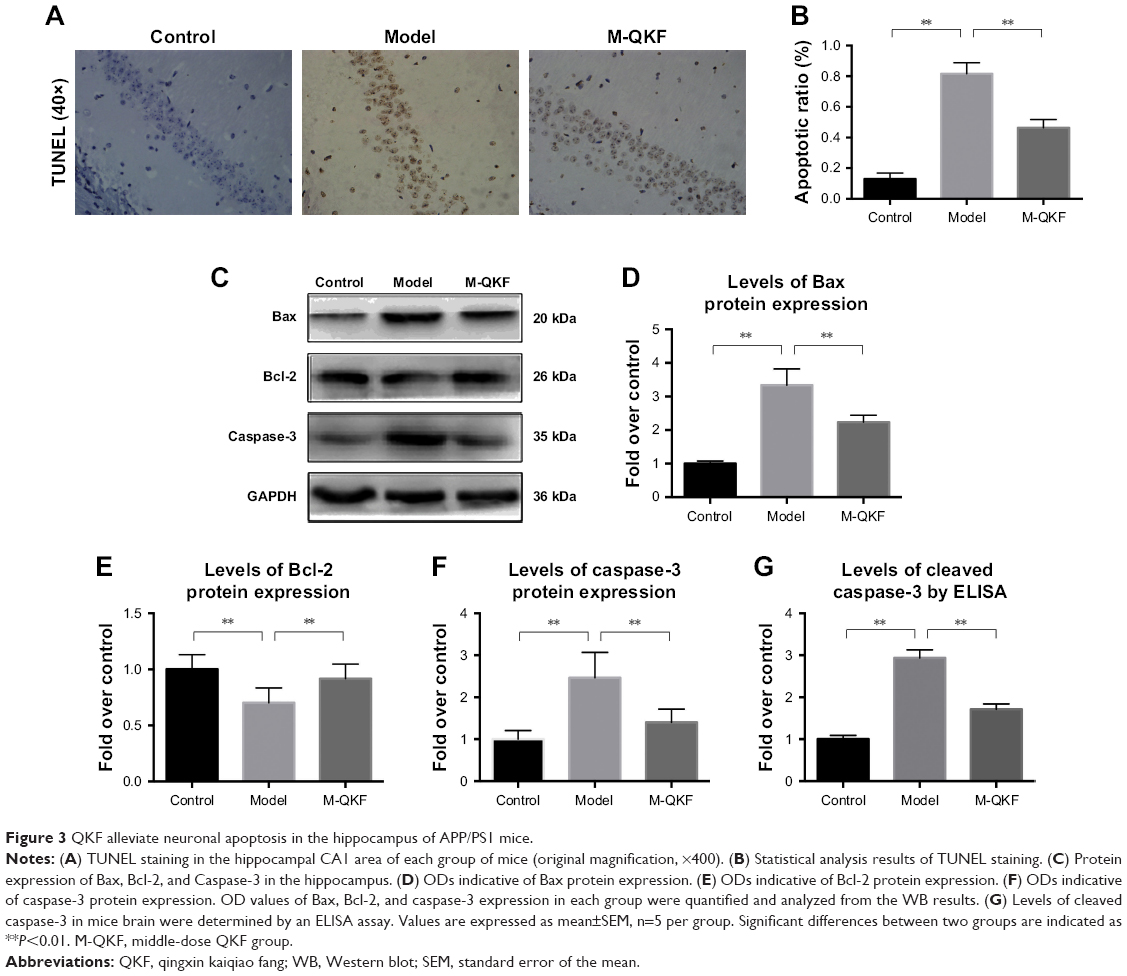 | Figure 3 QKF alleviate neuronal apoptosis in the hippocampus of APP/PS1 mice. |
Effects of QKF on the MAPK pathway
The mammalian MAPK family consists of ERK1/2, JNK, and p38 MAPK. Therefore, we examined the effects of QKF on the MAPK pathway by measuring their protein expression in the mouse hippocampi. WB analysis was performed to detect ERK, JNK, and p38 MAPK expression and their phosphorylation indices. Figure 4A shows ERK1/2 phosphorylation and total ERK1/2 protein in the mouse hippocampi. Compared with the control group, the protein levels of p-ERK1/2 were significantly decreased in the model group (P<0.01). After 3 months of M-QKF treatment, the protein level of p-ERK1/2 was significantly increased (P<0.01; Figure 4C). Total ERK1/2 was not significantly different among the groups (Figure 4B). Figure 4D shows JNK1 and JNK2 phosphorylation and total JNK1 and JNK2 protein levels in the mouse hippocampus (Figure 4D). The protein levels of JNK1 were significantly decreased in the model and M-QKF groups compared with that in the control group (P<0.01; Figure 4E). The same trend was observed in the protein expression of p-JNK1 (P<0.01; Figure 4F). The total JNK2 protein level in the model group was lower than that in the control group (P=0.02; Figure 4G), and there was no significant improvement after M-QKF treatment (P=0.728). The p-JNK2 protein expression level in the model group was lower than that in the control group (P<0.01), but there was no significant improvement in the M-QKF group (P=0.574; Figure 4H). As shown in Figure 4I, although there were no significant differences in total p38 MAPK among groups (Figure 4J), the levels of p-p38 MAPK protein expression in the model group was significantly higher than those in the control group (P<0.01). After M-QKF treatment, the protein levels of p-p38 MAPK were significantly decreased (P<0.01; Figure 4K). These observations indicated that M-QKF acted on the MAPK pathway by inhibiting p38 MAPK and activating ERK1/2 but had no significant effect on JNK.
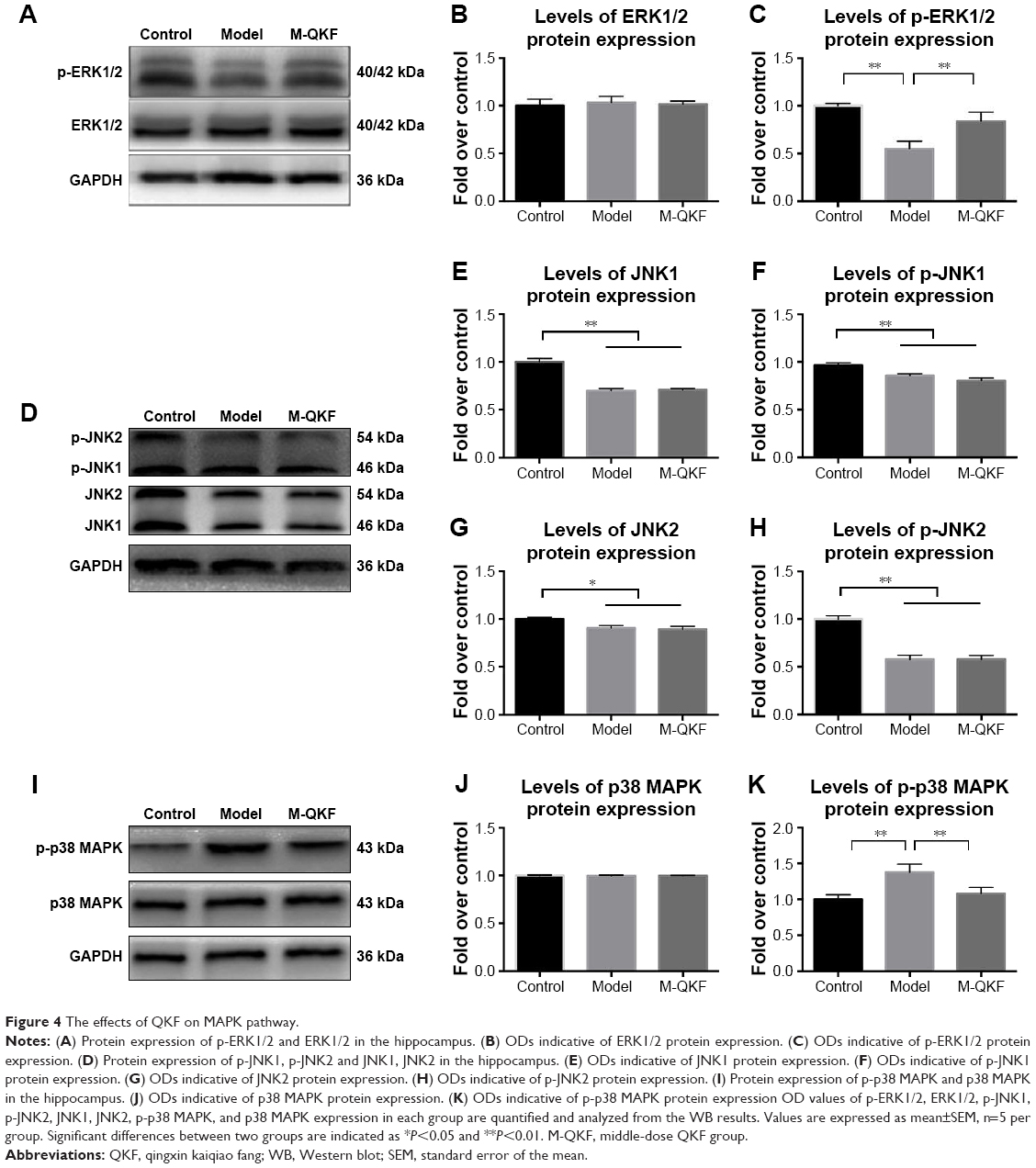 | Figure 4 The effects of QKF on MAPK pathway. |
M-QKF alleviated neuronal apoptosis by acting on ERK1/2
To further determine whether the antiapoptotic effect of QKF on neurons is mediated by activation of ERK1/2, we added the ERK1/2 inhibitor PD98059 to M-QKF treatment. As shown in Figure 5A, the M-QKF+PD98059 group exhibited a more disorganized and longer search path compared with the M-QKF+DMSO group in the navigation tests with the hidden platform. On the third and fourth days of training, the escape latency of the M-QKF+PD98059 group was significantly longer than that of the M-QKF+DMSO group (P<0.05; Figure 5C and D). Figure 5B shows the representative paths in the probe tests without the hidden platform. The locations of mice in the M-QKF+DMSO group were more concentrated, which confirmed that the M-QKF+DMSO group had a better platform exploration strategy and better spatial memory ability than the M-QKF+PD98059 group. These observations suggested that there were fewer platform location crossings and a lower percentage of target quadrant search time in the M-QKF+PD98059 group than in the M-QKF+DMSO group (P<0.05; Figure 5E and F). As shown in Figure 5G, compared to the M-QKF+DMSO group, the expression levels of Bax and caspase-3 protein in the M-QKF+PD98059 group were higher (P<0.01; Figure 5I). The ELISA assay also showed that the M-QKF+PD98059 group had a higher level of cleaved caspase-3 than the M-QKF+DMSO group (P<0.01; Figure 5K). The level of Bcl-2 expression was lower in the M-QKF+PD98059 than in the M-QKF group (P<0.01; Figure 5I). As shown in Figure 5H, M-QKF+PD98059 treatment was associated with a lower level of p-ERK1/2 compared with the M-QKF+DMSO treatment (P<0.01; Figure 5J). These results indicated that M-QKF inhibited neuronal apoptosis by activating ERK1/2.
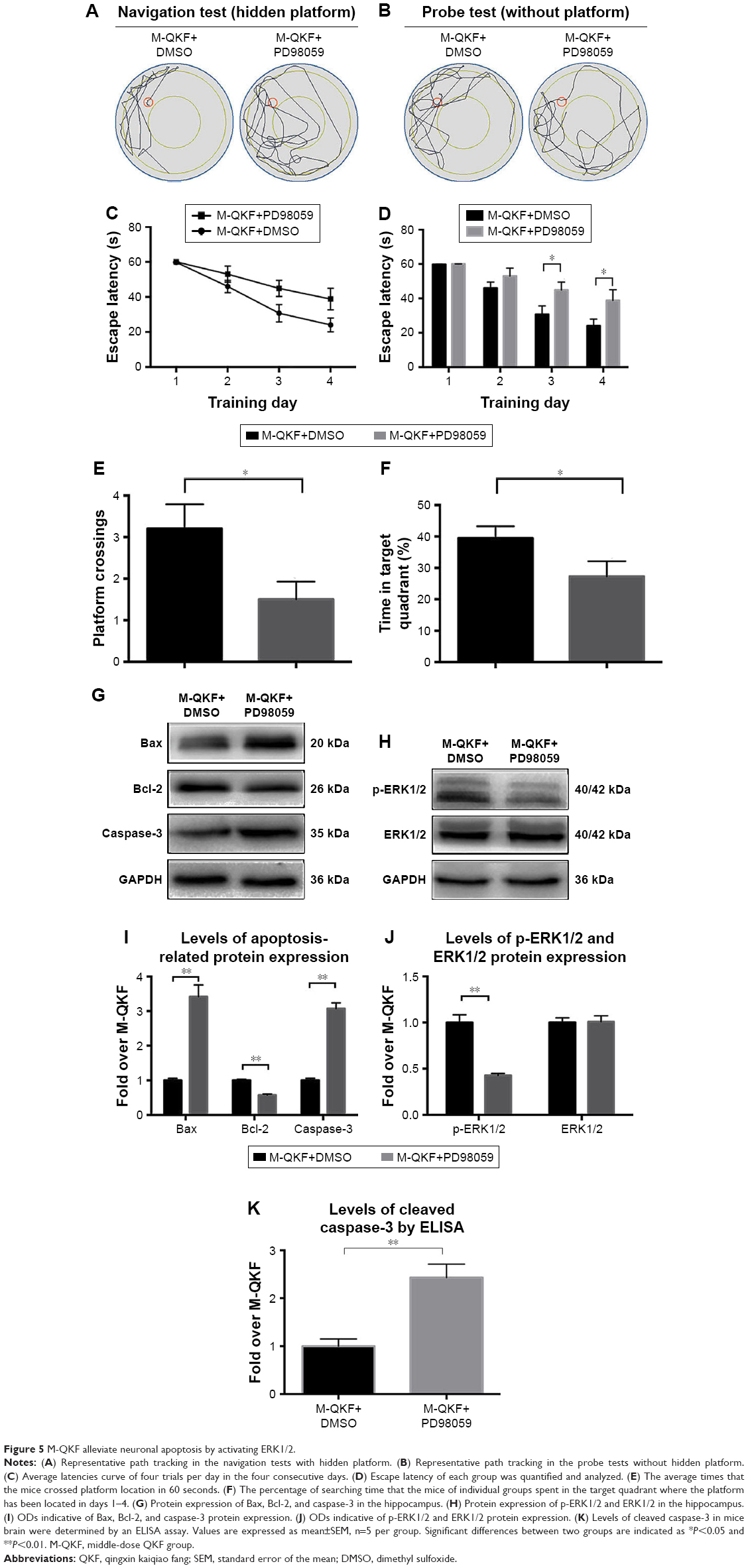 | Figure 5 M-QKF alleviate neuronal apoptosis by activating ERK1/2. |
M-QKF alleviated neuronal apoptosis by inhibiting p38 MAPK
Activation of p38 MAPK activity by U46619 was used to assess whether the antiapoptotic effect of QKF on neurons was mediated by inhibition of p38 MAPK. In the MWM navigation test, the M-QKF+U46619 group exhibited a disordered trajectory and a longer search path than did the M-QKF group (Figure 6A). A significant decrease in escape latency was observed in the M-QKF+saline group compared with the M-QKF+U46619 group on the third and fourth days of training (P<0.05; Figure 6C and D). The locations of mice in the M-QKF+saline group were more concentrated in the probe tests (Figure 6B). The data showed fewer platform location crossings and a lower percentage of target quadrant search time in the M-QKF+U46619 group than in the M-QKF+saline group (P<0.05; Figure 6E and F). Compared with the M-QKF+saline group, the M-QKF+U46619 group showed a higher expression of Bax and caspase-3 protein and a lower expression of Bcl-2 (P<0.01; Figure 6G and H). The level of cleaved caspase-3 by ELISA is higher in the M-QKF+U46619 group. Treatment with M-QKF+U46619 was associated with a higher level of p-p38 MAPK protein expression compared with that in the M-QKF+saline group (P<0.01; Figure 6I and J). These results indicate that M-QKF inhibited neuronal apoptosis by inhibiting p38 MAPK.
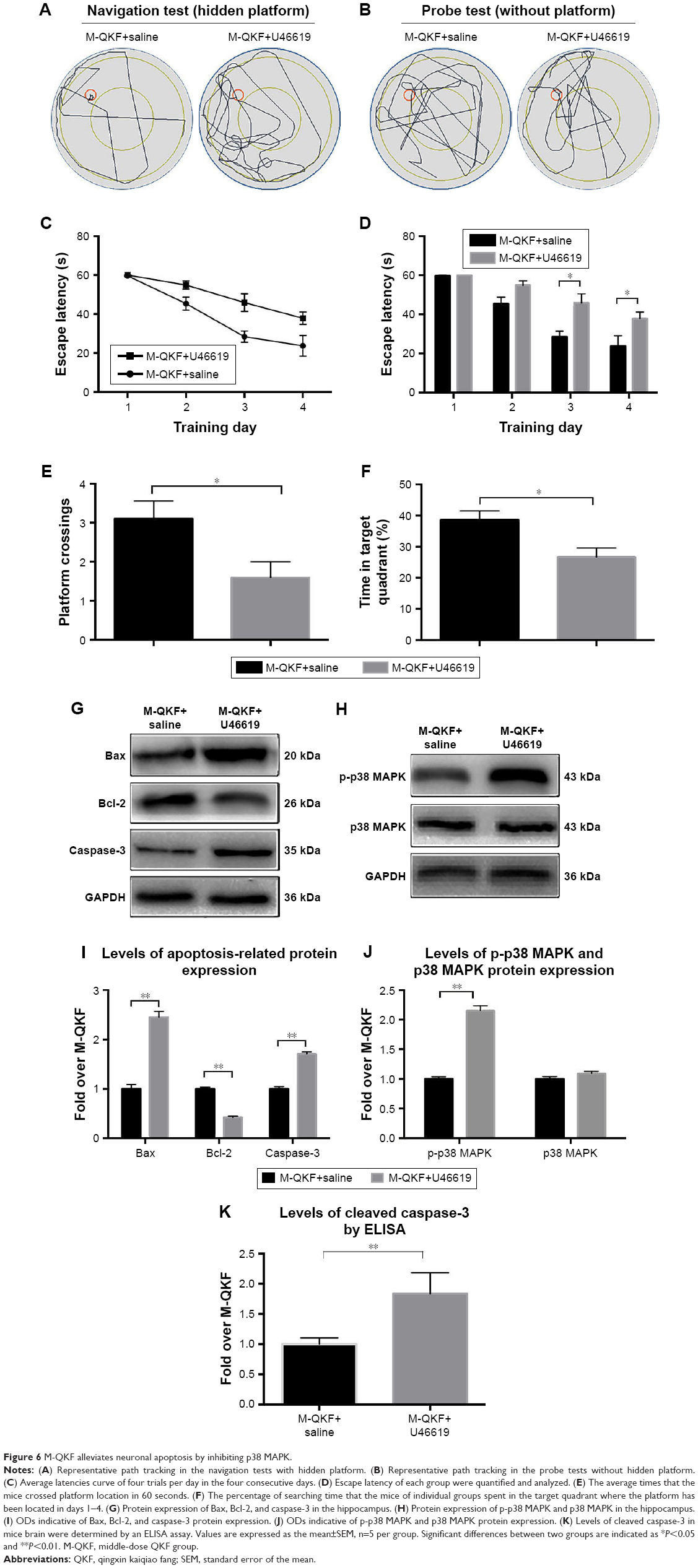 | Figure 6 M-QKF alleviates neuronal apoptosis by inhibiting p38 MAPK. |
Discussion
AD is a devastating age-related neurodegenerative disorder that is the most common cause of dementia worldwide.2–4 According to the amyloid hypothesis, the deposition of aggregated Aβ in brain areas involved in cognitive function is assumed to initiate a pathological cascade resulting in neuronal dysfunction and death in AD.27,28 Neuronal apoptosis, an essential cellular process and a major pathway for neuronal death, plays an important role in neurodegeneration in AD. Our previous study showed that QKF, a classic Chinese herbal formula based on the well-known decoction Fumanjian, significantly improves learning and memory in rats with Aβ1-40 insult and exhibits antiapoptotic effects, suggesting that QKF has therapeutic potential for AD.29 However, the mechanism by which QKF inhibits the apoptosis of AD neurons remains unknown. Therefore, the present study was performed to examine whether QKF-induced inhibition of neuronal apoptosis is mediated by MAPK signaling pathways in APP/PS1 double transgenic mice. Our results indicated that QKF, especially the middle dose of 9.5 g·kg−1·d−1, could alleviate the progression of AD by inhibiting neuronal apoptosis and then inhibiting pathological degeneration in AD mice. Furthermore, the effects of QKF on ERK1/2 and p38 MAPK pathway mediated apoptosis. AD is a specific form of dementia in elderly people that is characterized by cognitive impairment, gradual memory loss, and deterioration of language skills.30 Available data suggest that Aβ, derived from APP, is a crucial initial factor that triggers a complex pathological cascade resulting in AD.31 The accumulation of Aβ can induce neuronal apoptosis, which plays a pivotal role in the pathogenesis of AD.7,32 Inhibition of neuronal apoptosis is often seen as a target for the development of novel drugs for AD, such as donepezil, memantine, and rivastigmine.33 The conditions underlying the induction of apoptosis, including serum starvation, ROS, nitric oxide (NO), UV and γ irradiation, glucocorticoid, and Bax overexpression, are major factors underlying the redistribution of cytochrome c (cyt c) from the intermembrane space of the mitochondria to the cytoplasm. The Bcl-2 family of proteins tightly regulates the release of cyt c. The family is composed of a wide variety of antiapoptotic proteins, including Bcl-2, and proapoptotic proteins, including Bax. The general balance between antiapoptotic and proapoptotic proteins determines the activation of intrinsic apoptotic signals involving mitochondria. Caspase-9 is an important factor in the development of the mammalian nervous system and is the main initiator caspase in the intrinsic apoptotic pathway involving mitochondria. When these upstream caspases are activated, they trigger the cleavage of downstream effector caspases such as caspase-3, which can cleave cytoskeletal and nuclear proteins to induce apoptosis. Caspase-3 is the effector caspase and final executor of apoptosis. In the present study, the WB results showed that QKF enhanced the expression of Bcl-2 and decreased the expression of Bax and caspase-3 in the hippocampus of AD model mice. Moreover, TUNEL staining, which is used to label the 3′-OH terminal produced in the nuclear DNA of apoptotic cells in situ, showed decreased neuronal apoptosis after QKF treatment. Therefore, QKF showed beneficial effects in AD mice by inhibiting neuronal apoptosis. Interestingly, among the three dosages of QKF examined, the middle dose, 9.5 g·kg−1·d−1, showed the greatest treatment effect. A high dosage of QKF did not produce more excellent protective effects probably because it has as-yet-undetermined toxic side effects; therefore, further studies are required. Furthermore, traditional Chinese medicine prescription is a complex system, and different components coordinate with each other. Among the three dosages of QKF, the coordination effect of middle dosage of QKF reaches the best level in ameliorating memory impairment and inhibiting apoptosis, so we considered that middle dosage is the most appropriate dosage. This is also based on the classical theory of Chinese medicine, which suggests that exceeding the limit is as bad as falling short.
The MAPK pathway is closely related to neuron apoptosis in AD.12,34 MAPKs are serine/threonine protein kinases that participate in intracellular signaling during proliferation, differentiation, cellular stress responses, and apoptosis.12 Activation of MAPKs, including ERK1/2, p38 MAPK, and JNK, has been implicated in numerous cellular activities, including cell proliferation, differentiation, death, survival, and transformation. Some previous studies showed that p38 MAPK inhibitors can suppress caspase-3-like activity and partially inhibit cellular apoptosis.35 Activation of the ERK1/2 pathway has been shown to reverse learning and memory impairment in AD due to the engagement of survival mechanisms.36 In addition, some studies showed that phosphorylation of JNK inactivates Bcl-2 and increases the activities of caspase-3 and caspase-8.34,37 Our results demonstrate that enhanced hippocampal caspase activation is associated with phosphorylation of ERK1/2 and p38 MAPK, reinforcing the correlation of the MAPK pathway and neuronal apoptosis and its importance in the pathogenesis of AD. Treatment with QKF enhanced the phosphorylation of ERK1/2, as well as downregulation of p38 MAPK phosphorylation, which suppresses the activation of apoptotic responses. Its neuroprotective effect has a potential therapeutic effect on AD. In the present study, the results of WB analysis indicated that QKF treatment reduced p38 MAPK phosphorylation and increased ERK1/2 phosphorylation. Further studies of the activation of p38 MAPK by U46619 indicated that the antiapoptotic effect of QKF on neurons was accomplished by inhibition of the p38 MAPK pathway. Similarly, using ERK1/2 inhibitors, M-QKF was shown to inhibit neuronal apoptosis by inhibiting ERK1/2. This suggests that QKF inhibits AD apoptosis through the p38 MAPK and ERK1/2 pathways. However, the WB results also showed that M-QKF had a negative effect on the JNK pathway. This suggests that the inhibitory effect of M-QKF on apoptosis is not mediated through the JNK pathway.
Furthermore, measuring the serum and cerebrospinal fluid concentrations of QKF at time points after intragastric administration when steady-state levels are expected to be reached confirms that physiologically relevant levels were administered. To activate ERK1/2 and inhibit p38 MAPK more specifically, adeno-associated virus-induced ERK1/2 and p38 MAPK regulation should be applied in further studies during QKF treatment of AD. In order to improve the quality control of Chinese herbal compound and verify the identity, we will do deeper study to detect QKF in the future.
Conclusion
QKF, especially the middle dose of 9.5 g·kg−1·d−1, may alleviate the progression of AD-like symptoms by inhibiting neuronal apoptosis and reduce pathological degeneration in APP/PS1 double transgenic AD mice, including decreased numbers and disorder of neuronal cells, decreased numbers of mitochondria, and blurred or vacuolated cristae structures, to alleviate the progression of AD. The QKF-induced inhibition of apoptosis is regulated by the ERK1/2 and p38 MAPK pathways.
Acknowledgments
This work was supported by grants from Natural Science Foundation of China (No.81573896 to Haiyan Hu, No. 81601705 to Kailiang Zhou); Zhejiang Provincial Medicine and Health Technology Project (No. LY15H270016 to Haiyan Hu, No. 2017KY472 to Kailiang Zhou). Center of Cryo-Electron Microscopy Zhejiang University provided experimental platform.
Author contributions
SG wrote the manuscript text. SG, JL, TW, and YS prepared figures and collected samples. YL and WY analyzed data. HH and SG designed the experiment. HH and KZ revised the manuscript. All authors contributed toward data analysis, drafting and critically revising the paper, gave final approval of the version to be published and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Courtney C, Farrell D, Gray R, et al. Long-term donepezil treatment in 565 patients with Alzheimer’s disease (AD2000): randomised double-blind trial. Lancet. 2004;363(9427):2105–2115. | ||
Panagaki T, Gengler S, Hölscher C. The novel DA-CH3 dual incretin restores endoplasmic reticulum stress and autophagy impairments to attenuate Alzheimer-like pathology and cognitive decrements in the APPSWE/PS1ΔE9 mouse model. J Alzheimers Dis. 2018;66(1):195–218. | ||
Realdon O, Rossetto F, Nalin M, et al. Technology-enhanced multi-domain at home continuum of care program with respect to usual care for people with cognitive impairment: the Ability-TelerehABILITation study protocol for a randomized controlled trial. BMC Psychiatry. 2016;16(1):425. | ||
Anantha Krishnan D, Gunasekaran K, Iqbal S. Identification of potential PKC inhibitors through pharmacophore designing, 3D-QSAR and molecular dynamics simulations targeting Alzheimer’s disease. J Biomol Struct Dyn. 2017;13:1–16. | ||
Ryu J, Yu HN, Cho H, et al. Neuregulin-1 exerts protective effects against neurotoxicities induced by C-terminal fragments of APP via ErbB4 receptor. J Pharmacol Sci. 2012;119(1):73–81. | ||
Welikovitch LA, do Carmo S, Maglóczky Z, et al. Evidence of intraneuronal Aβ accumulation preceding tau pathology in the entorhinal cortex. Acta Neuropathol. 2018;136(6):901–917. | ||
Lapresa R, Agulla J, Sánchez-Morán I, et al. Amyloid-ß promotes neurotoxicity by Cdk5-induced p53 stabilization. Neuropharmacology. 2018;146:19–27. | ||
Piemontese L, Tomás D, Hiremathad A, et al. Donepezil structure-based hybrids as potential multifunctional anti-Alzheimer’s drug candidates. J Enzyme Inhib Med Chem. 2018;33(1):1212–1224. | ||
Kuskowski MA, Morley G, Malone SM, Dysken MW, Okaya A. Hydergine treatment and psychophysiological measures in primary degenerative dementia. J Geriatr Psychiatry Neurol. 1990;3(1):41–47. | ||
Wang Z, Chen Y, Li X, Sultana P, Yin M, Wang Z. Amyloid-β1-42 dynamically regulates the migration of neural stem/progenitor cells via MAPK-ERK pathway. Chem Biol Interact. 2019;298:96–103. | ||
Lemche E. Early Life stress and epigenetics in late-onset Alzheimer’s dementia: a systematic review. Curr Genomics. 2018;19(7):522–602. | ||
Song J, Park KA, Lee WT, Lee JE. Apoptosis signal regulating kinase 1 (ASK1): potential as a therapeutic target for Alzheimer’s disease. Int J Mol Sci. 2014;15(2):2119–2129. | ||
Yang GH, Jarvis BB, Chung YJ, Pestka JJ. Apoptosis induction by the satratoxins and other trichothecene mycotoxins: relationship to ERK, p38 MAPK, and SAPK/JNK activation. Toxicol Appl Pharmacol. 2000;164(2):149–160. | ||
Sui X, Kong N, Ye L, et al. p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett. 2014;344(2):174–179. | ||
Junttila MR, Li SP, Westermarck J. Phosphatase-mediated crosstalk between MAPK signaling pathways in the regulation of cell survival. FASEB J. 2008;22(4):954–965. | ||
Zhao ZQ. Oxidative stress-elicited myocardial apoptosis during reperfusion. Curr Opin Pharmacol. 2004;4(2):159–165. | ||
Perfettini JL, Castedo M, Nardacci R, et al. Essential role of p53 phosphorylation by p38 MAPK in apoptosis induction by the HIV-1 envelope. J Exp Med. 2005;201(2):279–289. | ||
Schwenger P, Bellosta P, Vietor I, Basilico C, Skolnik EY, Vilček J. Sodium salicylate induces apoptosis via p38 mitogen-activated protein kinase but inhibits tumor necrosis factor-induced c-Jun N-terminal kinase/stress-activated protein kinase activation. Proc Natl Acad Sci U S A. 1997;94(7):2869–2873. | ||
Nakagami H, Morishita R, Yamamoto K, et al. Phosphorylation of p38 mitogen-activated protein kinase downstream of bax-caspase-3 pathway leads to cell death induced by high D-glucose in human endothelial cells. Diabetes. 2001;50(6):1472–1481. | ||
Choi C, Xu X, Oh JW, et al. Fas-induced expression of chemokines in human glioma cells: involvement of extracellular signal-regulated kinase 1/2 and p38 mitogen-activated protein kinase. Cancer Res. 2001;61(7):3084–3091. | ||
Shan ZL, Zhong L, Xiao CL, et al. Shikonin suppresses proliferation and induces apoptosis in human leukemia NB4 cells through modulation of MAPKs and c-Myc. Mol Med Rep. 2017;16(3):3055–3060. | ||
Grethe S, Ares MP, Andersson T, Pörn-Ares MI. p38 MAPK mediates TNF-induced apoptosis in endothelial cells via phosphorylation and downregulation of Bcl-x. Exp Cell Res. 2004;298(2):632–642. | ||
Ko HR, Ahn SY, Chang YS, et al. Human UCB-MSCs treatment upon intraventricular hemorrhage contributes to attenuate hippocampal neuron loss and circuit damage through BDNF-CREB signaling. Stem Cell Res Ther. 2018;9(1):326. | ||
Qin Y, Chen Z, Han X, et al. Progesterone attenuates Aβ(25–35)-induced neuronal toxicity via JNK inactivation and progesterone receptor membrane component 1-dependent inhibition of mitochondrial apoptotic pathway. J Steroid Biochem Mol Biol. 2015;154:302–311. | ||
Mao DD, Yang WY, Li Y, et al. Effect of qingxin kaiqiao fang on hippocampus mRNA expression of the inflammation-related genes IL-1β, GFAP, and Aβ in an Alzheimer’s disease rat model. Evid Based Complement Alternat Med. 2018;2018(7):1–7. | ||
Mao DD, Yang WY, Li Y, et al. Effect of qingxin kaiqiao fang on hippocampus mRNA expression of the inflammation-related genes IL-1, GFAP, and A in an Alzheimer’s disease rat model. Evid Based Complement Alternat Med. 2018. | ||
Rahman SO, Panda BP, Parvez S, et al. Neuroprotective role of astaxanthin in hippocampal insulin resistance induced by Aβ peptides in animal model of Alzheimer’s disease. Biomed Pharmacother. 2018;110:47–58. | ||
Yuan C, Guo X, Zhou Q, et al. OAB-14, a bexarotene derivative, improves Alzheimer’s disease-related pathologies and cognitive impairments by increasing β-amyloid clearance in APP/PS1 mice. Biochim Biophys Acta Mol Basis Dis. 2019;1865(1):161–180. | ||
Hu HY, Cui ZH, Li HQ, Hu H, Cui Z, Li H. Fumanjian, a classic Chinese herbal formula, can ameliorate the impairment of spatial learning and memory through apoptotic signaling pathway in the hippocampus of rats with Aβ1–40 -induced Alzheimer’s disease. Evid Based Complement Alternat Med. 2014;2014(2):1–9. | ||
Rabin LA, Smart CM, Crane PK, et al. Subjective cognitive decline in older adults: an overview of self-report measures used across 19 international research studies. J Alzheimers Dis. 2015;48(s1):S63–S86. | ||
Benny A, Thomas J. Essential oils as treatment strategy for Alzheimer’s disease: current and future perspectives. Planta Med. Epub 2018 Oct 25. | ||
Silva T, Reis J, Teixeira J, Borges F. Alzheimer’s disease, enzyme targets and drug discovery struggles: from natural products to drug prototypes. Ageing Res Rev. 2014;15:116–145. | ||
Kumar A, Singh A, Ekavali. A review on Alzheimer’s disease pathophysiology and its management: an update. Pharmacol Rep. 2015;67(2):195–203. | ||
Jezierski MK, Sturm AK, Scarborough MM, Sohrabji F. NGF stimulation increases JNK2 phosphorylation and reduces caspase-3 activity in the olfactory bulb of estrogen-replaced animals. Endocrinology. 2001;142(6):2401. | ||
Zou W, Zeng J, Zhuo M, et al. Involvement of caspase-3 and p38 mitogen-activated protein kinase in cobalt chloride-induced apoptosis in PC12 cells. J Neurosci Res. 2002;67(6):837–843. | ||
Bartolomé F, de Las Cuevas N, Muñoz U, Bermejo F, Martín-Requero A. Impaired apoptosis in lymphoblasts from Alzheimer’s disease patients: cross-talk of Ca2+/calmodulin and ERK1/2 signaling pathways. Cell Mol Life Sci. 2007;64(11):1437–1448. | ||
Wei Y, Sinha S, Levine B. Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy. 2008;4(7):949–951. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.