")
Back to Journals » Cancer Management and Research » Volume 12
Pyrrolidine Dithiocarbamate Facilitates Arsenic Trioxide Against Pancreatic Cancer via Perturbing Ubiquitin-Proteasome Pathway
Authors Yu S, Wu N, Zhu J, Liu Y, Han J
Received 26 August 2020
Accepted for publication 21 November 2020
Published 22 December 2020 Volume 2020:12 Pages 13149—13159
DOI https://doi.org/10.2147/CMAR.S278674
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Eileen O'Reilly
Simin Yu,1,* Ning Wu,2,* Jianmin Zhu,3 Ying Liu,4 Jinbin Han1
1Department of Traditional Chinese Medicine, Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, People’s Republic of China; 2Department of Oncology, Shanghai Pudong New Area Gongli Hospital, Shanghai, People’s Republic of China; 3Shanghai Clinical Center, Chinese Academy of Sciences/Xuhui Central Hospital, Shanghai, People’s Republic of China; 4Department of Oncology, Yunnan Provincial Hospital of Chinese Medicine, Kunming, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jinbin Han Tel +862123271699
Fax +862163136856
Email [email protected]
Purpose: To investigate whether pyrrolidine dithiocarbamate (PDTC) could facilitate arsenic trioxide (ATO) to induce apoptosis in pancreatic cancer cells via perturbing ubiquitin-proteasome pathway.
Methods: Mass spectrometry was performed to examine the interaction between PDTC and ATO, and the data showed they could form a complex termed PDTC-ATO. Inhibiting effects on cell viability were examined by CCK-8 test, and apoptosis was examined by flow cytometry. Four treatment arms (n = 6), including the control, PDTC, ATO, and PDTC-ATO, were evaluated using BALB/c nude mouse models bearing a xenograft tumor of SW1990 human pancreatic cancer line. Western blot, immunohistochemistry assays were to detect the mechanism.
Results: The results showed that PDTC-ATO had higher inhibiting effects on proliferation of pancreatic cancer cells than ATO in vitro. In bearing-tumor mice, PDTC-ATO inhibited tumor growth by 79%, being more potent than ATO (by 46%) or PDTC (by 35%) compared to the control. Results of Western blot and immunohistochemistry showed proteasome inhibition and apoptotic cell death, together with obvious suppression of associating E3 ubiquitin ligase activity, occurred more frequently in tumors treated with PDTC-ATO than those with ATO.
Conclusion: PDTC demonstrated the function to facilitate ATO against pancreatic cancer due to forming a stable complex to perturb ubiquitin-proteasome pathway.
Keywords: arsenite, ubiquitin ligase, pancreatic cancer cell, dithiocarbamate, complex
Introduction
Pancreatic cancer accounts for 1% to 12% of all malignant tumors with 90% of pancreatic tumors arising from the glandular duct epithelium. As a very lethal form of gastrointestinal tumors, it is hardly possible to detect before an advanced stage and is especially resistant to current therapies.1,2 Less than 10% of pancreatic cancer patients survive for 5 years after diagnosis.
The ubiquitin-proteasome pathway is involved in a range of mammalian cellular processes including differentiation, proliferation, angiogenesis, and apoptosis, contributing to the pathology of multiple malignancies such as pancreatic cancer, in which deregulated cell proliferation requires more protein degradation and production than that of non-transformed tissues.3–6 As the essential role in ubiquitin-proteasome pathway, ubiquitination involves ubiquitin-activating (E1), ubiquitin-conjugating (E2), and ubiquitin-ligating (E3) enzymes.7 The RING family of E3 ligases charactered by a conserved arrangement of cysteine and histidine residues coordinating two zinc ions is the active sequences of E3 in ubiquitin transfer.8,9 Recent evidences suggest that arsenites could induce apoptosis in pancreatic cancer cells by perturbing ubiquitin-proteasome pathway led by their action on E3 ligases.10–12
Arsenic trioxide (ATO), a traditional drug of the eastern cultures and the western, was uncovered to have significant curative effects in patients with acute promyelocytic leukemia (APL).13 Previous studies including our own showed ATO could induce apoptosis in pancreatic cancer stem cells (PCSCs) via blocking sonic hedgehog (SHH) pathway; however, the evidences from laboratories or clinics could hardly support ATO for clinical translation in pancreatic cancer therapy so far, and this is particularly true when it is used as monotherapy.14–16
It is well known that there are sulfhydryl compounds such as glutathione (GSH) to aid detoxification biologically, and the effects of ATO could be reduced because arsenic ions are captured by these compounds within the body.17,18 To improve anticancer effects of ATO, we hypothesized that a proper sulfhydryl compound might be capable of facilitating ATO via forming a complex to keep arsenic ions away from being captured by detoxicating compounds biologically, and then the ions could be released owing to the interaction between the target proteins and the complex in pancreatic cancer cells.
Dithiocarbamates are organic compounds containing sulfhydryl groups that could combine metals to form complexes.19 Some dithiocarbamates have been approved for clinical treatment of bacterial, fungal, and viral infections, including the human immunodeficiency virus (HIV).20–23 Pyrrolidine dithiocarbamate (PDTC) is a member of dithiocarbamates, functioning as an antioxidant and NF-κB inhibitor biologically.24 Several complexes constituted by PDTC and metals demonstrate capacity to induce apoptosis in human cancer cell lines.25,26
We had investigated the combination of PDTC and ATO, and the results demonstrated that they could form a stable complex which was termed PDTC-ATO in this study. PDTC-ATO showed obvious inhibition to pancreatic cancer cell viability in cultures and in xenografts. Further cellular, animal, and molecular biological analysis indicated that PDTC-ATO could function as a potential anticancer agent due to perturbing the ubiquitin-proteasome pathway.
Materials and Methods
Cell Lines and Reagents
Pancreatic cancer cell lines SW 1990, PANC-1, MIA-PaCa 2, and BxPC-3 were obtained from the cell bank of the Chinese Academy of Science (Shanghai, China). All cell lines were tested negative for mycoplasma contamination and authenticated to match their genic characters. RPMI medium and fetal bovine serum (FBS) were obtained from Gibco Invitrogen (Carlsbad, CA, USA). Cells were cultured in RPMI with 10% FBS. Clinical injections of 1 mg/mL ATO and 0.9% sodium chloride were from Harbin Yida Pharmaceutical Co, Ltd (lot no. 20,171,103, 20,190,102). Ammonium 1-pyrrolidinecarbodithioate was from MilliporeSigma (catalog no. P8765-5G). The proteasome 20S assay kit was from Enzo Life Sciences, Inc. (catalog no. BML-AK740-0001).
Preparation of the Complex
PDTC-ATO was prepared just before use. The concentration of PDTC-ATO was determined by ATO content. Because each ATO molecule contained two arsenic ions, ATO and PDTC in water were mixed in tubes at an ATO:PDTC molar ratio of 1:4 to prepare the complex, and stored at 4°C. Supercritical fluid chromatography-mass spectrometry (SFC-MS) analysis was performed with a triple quadrupole instrument (Xevo G2-XS QTOFMS; Waters, Milford, MA, USA) for detection of the possible product structures.
Cell Viability Assay
Cells were grown in 96-well plates in triplicate at a starting density of 5000 cells per well. Fresh standard culture medium containing PDTC, ATO, PDTC-ATO or control normal saline was added at designed concentrations on the morning after cell plating when cells were attached to well bottoms. CCK-8 reagent (catalog no.CK04, Dojindo Laboratories) was carried out after 48 hours to quantify viable cells. Fresh standard culture medium containing 10% CCK-8 reagent was added to wells for 2-hour incubation at 37°C, protected from light. A multi-plate reader (ELX-800; BioTek, Winooski, VT, USA) was used to determine the absorbance at 450 nm. Cell viability was expressed as a percentage of viable cells compared to the control. The IC50 values were calculated with probit analysis using software SPSS 19.0.
Cellular and Nuclear Morphologic Analysis
Microscopic imaging was by an Eclipse Ni-U Model microscope (Nikon, Tokyo, Japan). Phase-contrast cellular morphology and Hoechst 33,258 fluorescence nuclear morphology were performed as previously described.27 Nuclei that were brightly stained, granular, and punctate were considered apoptotic.
Cell Apoptosis Analysis
Pancreatic cancer cells were treated for 48 hours as described above for the cell viability assay and harvested by trypsinization, then followed by being washed at 4°C. An annexin V-FITC/PI apoptosis kit (catalog no. 556,547, BD) was used to stain cells for flow cytometry. Detached cells were resuspended in binding buffer and propidium iodide (PI) as well as annexin V reagents were added for 15-minute incubation followed by the addition of more binding buffer. Flow cytometry was carried out using CytoFLEX (Beckman Coulter Inc., Brea, CA, USA) and the data analysis of the cytometric files was performed by CytExpert Software (v1.2, Beckman Coulter, Brea, CA, USA).
Proteasome Activity Assay
Per the protocol supplied with the assay kit, human pancreatic cell pellets were lysed on ice and assay aliquots prepared by centrifugation at 4°C. Supernatant aliquots were added to microplate wells containing Suc-LLVY-AMC. After 1-hour incubation at 37°C, free AMC caused by proteasome activity was examined with a microplate reader (Synergy H4 Hybrid Multi-Mode Microplate Reader; Bio-TEK Instruments Inc. Winooski, VT, USA) with an excitation filter of 360 nm and an emission filter of 460 nm. Control activity was defined as 100% and sample activity was calculated by comparison with the control.28
Pancreatic Cancer Xenograft Experiments
Protocols for animal experiments of this study were approved by the Animal Experimental Ethics Committee of the Ninth People’s Hospital, School of Medicine, Shanghai Jiao Tong University (HKDL2018328), in compliance with the National Institutes of Health (NIH) guidelines for the care and use of laboratory animals. Female BALB/c nude mice, 5 weeks of age, were obtained from Shanghai SLAC Laboratory Animal Co., Ltd (Shanghai, China) and maintained with specific pathogen-free conditions. Mice were raised in laminar flow rooms with stable humidity and steady temperature. SW 1990 cells (5 × 106) were injected subcutaneously (s.c.) into the flank of 5 mice. After 3 weeks, bearing-tumor mice were sacrificed and tumors were removed. Two large tumors were cut into 1 mm × 1 mm × 1 mm pieces and transplanted into 24 mice.29 After 2 weeks, the 24 bearing-tumor mice were randomly divided into 4 groups (n = 6): Control, PDTC, ATO, and PDTC-ATO. Models were treated with intraperitoneal injection as follows: Control group, normal saline at the same volume as the PDTC-ATO group; PDTC group, 200 mg/kg PDTC dissolved in normal saline; ATO group, 8 mg/kg ATO; and PDTC-ATO group, 8 mg/kg ATO added to PDTC at 1:4 molar ratio. Injections were performed once every other day and tumor sizes were measured before injection. Tumor volumes (V) were calculated as V = (L × W2) × 0.5, where L was tumor length and W was width. Mice received 4-week treatment and were euthanized humanely by inhaling CO2 on day 28. Tumors were removed and photographed before Western blot analysis, hematoxylin and eosin (H&E) staining, immunohistochemistry (IHC), as well as terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labeling (TUNEL) assay.
Western Blot Analysis
Proteins were extracted from tumor samples by grinding, lysis, and centrifugation. Protein quantification was by the bicinchoninic acid procedure (Cell Signaling Technology, Danvers, MA, USA). Protein samples (30 μg) were separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes (EMD Millipore, Hayward, CA, USA). Membranes were blocked for 1 hour with 5% nonfat milk at room temperature, followed by incubation in buffers. Primary antibodies were incubated overnight at 4°C and included: anti-human cleaved caspase-3 (catalog no. 9664S, CST), anti-ubiquitin (catalog no. ab7780, Abcam), anti-ZFAND2A (catalog no. HPA019469, Sigma-Aldrich), anti-Pirh2 (catalog no. ab189907, Abcam) anti-p40-ΔNp63 (catalog no. ab203826, Abcam) and anti-α1a-tubulin (catalog no. sc-134,237, Santa Cruz Biotechnology). Membranes were incubated with corresponding peroxidase-labeled anti-mouse (catalog no. P0217, DAKO) or anti-rabbit (catalog no. P0260, DAKO) IgG secondary antibody for 1 hour. The Western blot gel images were obtained with an Minichemi 610 chemiluminescent imager (Sagecreation, Beijing, China), and analyzed by ImageJ 15.1 software (National Institutes of Health, USA).
H&E Staining, IHC, and TUNEL Assays
Paraffin-embedded tumor samples prepared on slides were dewaxed and rehydrated for pretreatment with hydrogen peroxide and then being washed in PBS. Samples were stained with H&E, followed by rinsing and coverslip mounting with Permount (Fisher Scientific, San Francisco, CA, USA). For IHC, endogenous peroxidase was blocked with 3% hydrogen peroxide. Samples were incubated with a primary antibody, anti-human cleaved caspase-3 (catalog no. 9664S, CST). Then, color visualization was done with a SPlink Detection Kit (catalog no. SP-9000, ZSGB-BIO, Beijing, China) in accordance with the manufacturer's instruction. An in situ cell death detection kit (catalog no. 11,684,817,910, Roche Applied Science) was used for TUNEL assay per the protocol provided by the manufacturer. Briefly, after dewaxing and hydration, tissue slides were incubated with Working Strength TdT Enzyme and Working Strength Stop/Wash Buffer, conjugated with anti-digoxigenin and stained with diaminobenzidine peroxidase substrate. Slides were mounted under glass coverslips in Permount and analyzed as well as photographed with a microscope.
Statistics
GraphPad Prism software (GraphPad Software Inc., La Jolla, CA, USA) was applied for statistical analysis. All data were statistically analyzed using one-way ANOVA and unpaired t-test for independent analysis was carried out to judge differences between groups for comparison. Significant differences between groups were set at P < 0.05.
Results
PDTC and ATO Formed a Stable Complex
An ATO molecule could deliver double arsenic ions. When ATO and PDTC were mixed at the molar ratio ATO:PDTC = 1:4 in water, the solution was clear without obvious change either in color or in temperature. The peak value detected by mass spectrometry which indicated the main chemical member in solution was 366.94 m/z, consistent with the mass value of the supposed structural of PDTC-ATO complex (Figure 1A and B), and its molecular formula was determined as C10H16AsN2S4. As a new chemical structure, the dosage of PTDC-ATO was defined by the quantity of ATO contained owing to the stable construction and the certain number of arsenic ions chelated by the ligand.
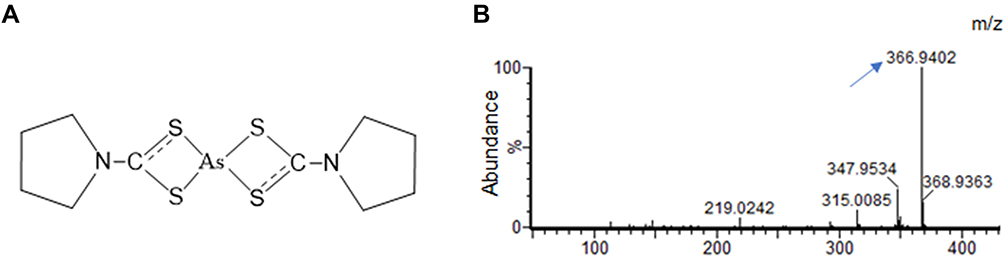 |
Figure 1 PDTC and arsenic ions (As3+) released from ATO constituted a stable complex termed PDTC-ATO. (A) Chemical structure of PDTC-ATO. (B) QTOF-MS spectrum resulted from PDTC-ATO examination. The arrow points the molecular ion peak generated by the complex. Abbreviations: PDTC, pyrrolidine dithiocarbamate; ATO, arsenic trioxide. |
PDTC-ATO Inhibited Proliferation of Pancreatic Cancer Cells
Effects of PDTC-ATO on the proliferation of human pancreatic cell lines, including SW 1990, PANC-1, MIA PaCa-2, and BxPC-3, were examined by CCK-8 test. Concentrations were PDTC 20 μM, ATO 5 μM, and PDTC-ATO 5 μM (defined by ATO). PDTC, ATO and PDTC-ATO showed capacity to inhibit proliferation of pancreatic cancer cell lines when the cells were cultured with them for 48 hours. Among the three drugs, PDTC inhibited cell viability from 10% to 23%, ATO from 13% to 58%, and PDTC-ATO from 79% to 90% for the cell lines studied (Figure 2A). To detect the effects of PDTC-ATO furtherly, the pancreatic cancer cell lines with either vehicle or dosages ranged from 0.1 μM to 10 μM for 24 or 48 hours were applied to determined IC50 values, and the data is shown in Table 1, indicating a credible function of PDTC-ATO to suppress pancreatic cancer cell viability.
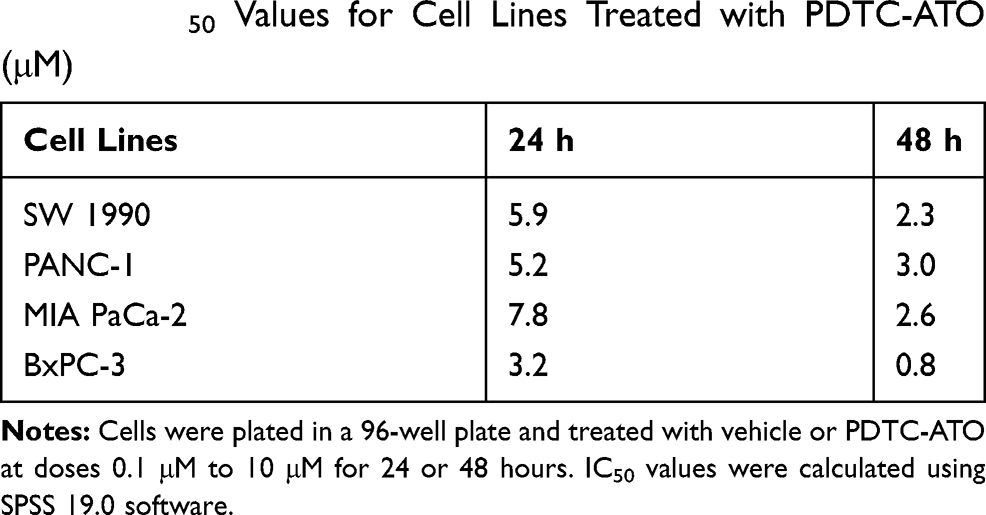 |
Table 1 IC50 Values for Cell Lines Treated with PDTC-ATO (μM) |
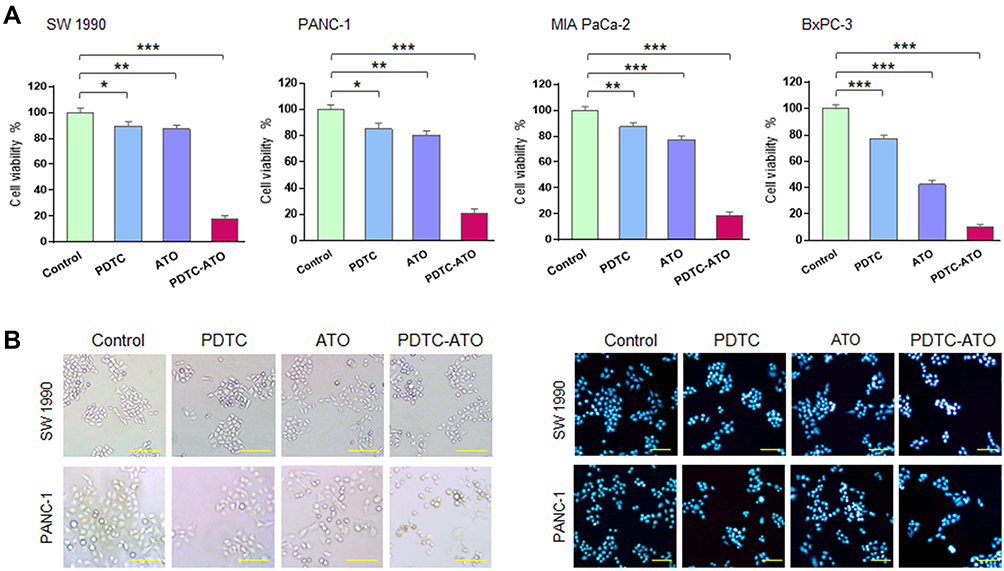 |
Figure 2 PDTC-ATO inhibited pancreatic cancer cell viability in cultures. (A) Decreased viability of pancreatic cancer cells induced by PDTC-ATO in cultures. Cell lines were cultured with PDTC (20 µmol/L PDTC), ATO (5 µmol/L ATO), or PDTC-ATO (5 µmol/L ATO + 20 µmol/L PDTC) for 48 hours, followed by CCK-8 test. Columns, mean of measured viability; bars, SD. (B) PDTC-ATO led to morphologic changes in pancreatic cancer cells. SW 1990 and PANC-1 cell lines were treated as in (A). The left, cells treated with PDTC-ATO were spherical and detached; the right, apoptotic nuclei by Hoechst 33,258 staining (brighter, granular, or punctate) of the treated cells. *P < 0.05, **P < 0.01, ***P < 0.001. Scale bars, 100 µm. |
The effects of PDTC-ATO on cellular and nuclear morphology of pancreatic cancer cell lines, SW 1990 and PANC-1, were evaluated. Cells treated with PDTC-ATO were detached and had apoptotic nuclear characteristics when compared to those treated with either ATO or PDTC (Figure 2B), indicating that PDTC-ATO potentially induced apoptosis of pancreatic cancer cells.
Based on the data of CCK-8 test, the effect of a concentration of 5 μM PDTC-ATO on SW 1990 and PANC-1 cell lines for 48 hours was examined by flow cytometry. Apoptosis was assessed by annexin V and PI staining. Treatment with 5 μM PDTC-ATO significantly increased the percentage of late apoptotic cells by 43% for SW 1990 and 50% for PANC-1 compared to the control, and 17% for SW 1990 and 29% for PANC-1 compared to ATO (Figure 3A and B).
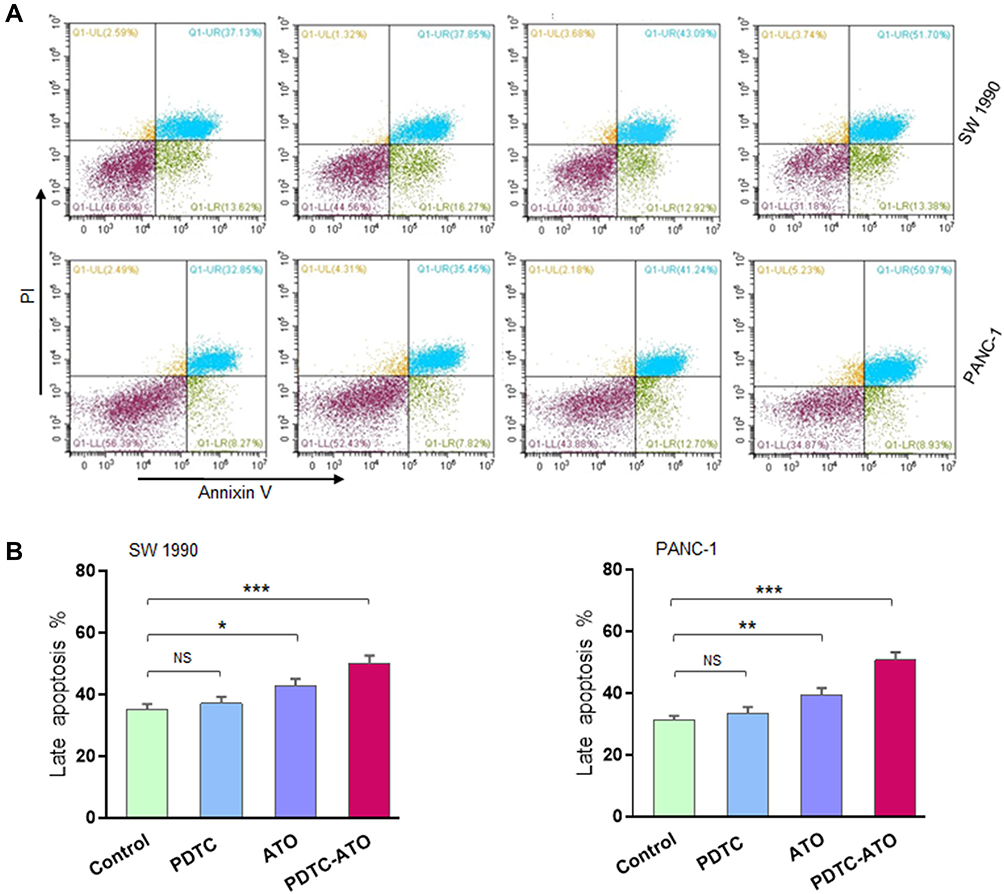 |
Figure 3 PDTC-ATO induced apoptotic death in pancreatic cancer cell cultures. SW 1990 and PANC-1 were treated in vitro for 48 hours (Figure 2 legend), harvested, annexin V-FITC/PI double-stained, and analyzed by flow cytometry. (A) Representative measurement of stained cells by flow cytometry. For all panels, cells in Q1-LL were alive, Q1-LR early apoptosis, Q1-UL death, and Q1-UR late apoptosis. (B) Late apoptotic cell numbers identified in Q1-UR. Values are mean fluorescence intensities ± standard errors, determined for three independent samples. *P < 0.05, **P < 0.01, ***P < 0.001. Abbreviations: PI, propidium iodide; NS, no significance. |
PDTC-ATO Retarded Pancreatic Cancer Cell Tumorigenesis in vivo
To verify the anticancer effect, the in vivo experiment was performed with SW1990 xenograft mouse models. The results showed that in vivo tumor growth was inhibited by 79% for PDTC-ATO, 46% for ATO, and 35% for PDTC compared to the control, respectively (Figure 4A and C).
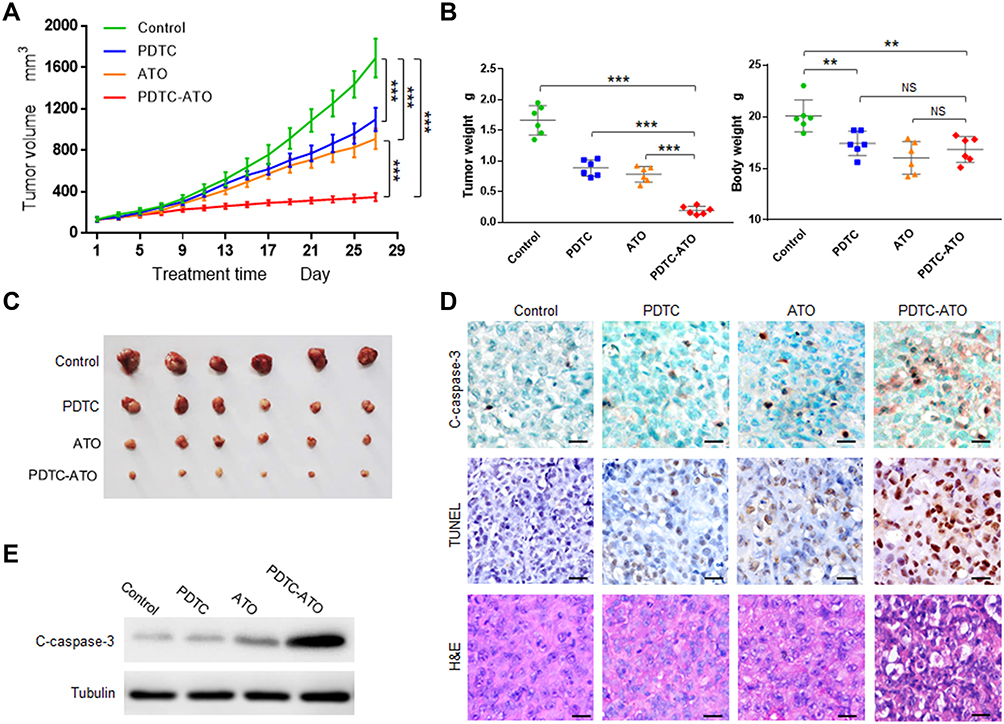 |
Figure 4 PDTC-ATO retarded tumorigenesis of pancreatic cancer cells in vivo. Mouse models with SW 1990 xenografts were divided randomly into 4 groups and given normal saline, PDTC, ATO, or PDTC-ATO treatment respectively. Mice were euthanized and tumors were harvested on day 28. (A) Tumor growth chart. Tumor growth was inhibited by about 79% after 4-week treatment with PDTC-ATO compared to the control. Points, mean of tumor volumes, bars, SD. (B) The left, tumor weight comparisons were consistent with size differences among groups; the right, body weights of mouse models at the end of experiments indicated PDTC-ATO had similar systemic toxicity to ATO. Bars, SD. (C) Photos of harvested tumors for size comparison. Tumors from PDTC-ATO-treated mice were significantly smaller than those from the other groups. (D) IHC, TUNEL, and H&E assays on tumor samples. Tumor tissue sections were processed for immunostaining with cleaved caspase-3 antibody, and for TUNEL as well as H&E assay. Stronger or more positive stained cells associated with cleavage of caspase-3 (C-caspase-3) were detected in tumor tissues from PDTC-ATO group as judgement by IHC. Positive and/or apoptotic condensed nuclei were observed in TUNEL assay. Apoptotic cell characteristics were observed by H&E staining. Scale bars, 20 μm. (E) Western blot of tumor tissue extracts using antibody against cleaved caspase-3. **P < 0.01, ***P < 0.001. Abbreviation: NS, no significance. |
ATO was toxic for animals and the system toxicity could be reflected by changes in body weight.30 Before treatment, the body weights of mice in each group were about 19.6 ± 0.9 g, no significant difference among them (P > 0.1). On the treatment day 28, mice treated with ATO and PDTC-ATO had significant body weight loss compared to the control (P < 0.01). However, the body weight loss difference between ATO group and PDTC-ATO group was not significant (P > 0.05), suggesting they might have similar system toxicity (Figure 4B). Mice treated with ATO and PDTC-ATO were less active than the other animal groups, but no other side effects were evident during the 4-week treatment.
Cleaved caspase-3 protein levels, associated with apoptosis in cells, were increased as judgement by IHC for tumor samples from animals treated with PDTC-ATO when compared with those from ATO group, PDTC, or the control. The result was consistent with tumor growth inhibition found in mouse models (Figure 4D and E, and supplementary Table S1). As shown by TUNEL assay, apoptosis in tumors of PDTC-ATO group was approximately 2.1-fold more than that of ATO group, 2.7-fold than PDTC, and 8.4-fold than the control (Figure 4D and Table S1). The result of H&E staining was consistent with that of the TUNEL assay, having less apparent apoptosis detected in tumor tissues of the control group, than those of any other group, and the most apoptosis was observed in PDTC-ATO group (Figure 4D).
PDTC-ATO Functioned to Perturb Ubiquitin-Proteasome Pathway
Previous reports demonstrated that arsenites and complexes comprised of PDTC as well as some other metals to induce apoptosis in cancer cells by inhibition of the ubiquitin-proteasome pathway.27,31 In the proteasome activity assay of this study though PDTC and ATO showed inhibiting effects on 20S proteasome activity in pancreatic cancer cell cultures, there was no significant difference when compared either of them with the control. Meanwhile, PDTC-ATO significantly inhibited 20S proteasome activity in SW 1990 cell cultures by 51%, and in PANC-1 by 44%, compared to the control, respectively (P < 0.001, Figure 5A).
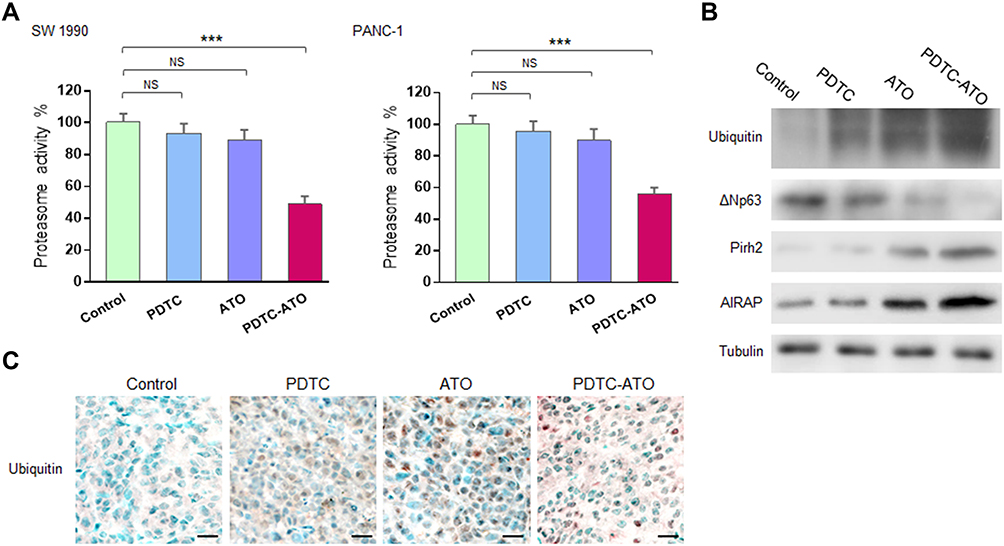 |
Figure 5 Proteasome inhibition induced by PDTC-ATO. Pancreatic cancer cells were cultured for 48 hours and tumor tissues were from mouse models receiving treatment described in the Figure 4 legend. (A) Cell pellet extracts were examined by the protocol supplied with the assay kit. Left, proteasome activity decreased by 51% in SW 1990 cells cultured with PDTC-ATO. Right, proteasome activity decreased by 44% in PANC-1 cells cultured with PDTC-ATO. Values are mean proteasome activities ± standard errors, determined for three independent tests. (B) Western blot of tumor tissue extracts using antibodies against ubiquitin, ΔNp63, Pirh2, AIRAP, and α1a-tubulin. Upregulation of ubiquitin, Pirh2, AIRAP, and downregulation of ΔNp63 in tumors from mice treated with PDTC-ATO were detected. (C) Proteasome inhibition induced by PDTC-ATO shown in IHC assay on tumor tissues. Scale bars, 20 μm. ***P < 0.001. Abbreviations: AIRAP, arsenite-inducible RNA-associated protein. NS, no significance. |
Emerging evidences demonstrated that arsenites were capable of perturbing ubiquitin-proteasome pathway in cancer cells by binding to the E3 ubiquitin ligase domain where contained zinc ions.10–12 Human Pirh2 (p53-induced RING-H2 protein), which contained zinc-binding sites and acted as an E3 ubiquitin ligase depending on the consensus RING-finger, was deregulated in various cancer cells.32 The results of Western blot assay showed that accumulation of ubiquitin, upregulations of Pirh2 and AIRAP which was an arsenite-inducible RNA-associated protein (namely ZFAND2A), as well as downregulation of ΔNp63 which was the target of Pirh2, was detected in tumor samples from mice treated with ATO or PDTC-ATO, and there were significant differences when compared them with the control (P < 0.01, Figure 5B, Table S1), meantime, the alteration of each protein caused by PDTC-ATO treatment was more potent than those by ATO monotherapy, indicating that the ubiquitin-proteasome pathway might be blocked owing to suppression of Pirh2 activity by ATO and PDTC-ATO. Moreover, the higher capacity of PDTC-ATO treatment than ATO alone suggested the synergy of PDTC for ATO against pancreatic cancer.
To verify the effects of PDTC-ATO on ubiquitin-proteasome pathway in vivo, the tumor samples were subjected IHC assay, and obvious accumulation of ubiquitin was observed in the tumor tissues of PDTC-ATO group, keeping consistent to the data of Western blot that ubiquitin was upregulated (Figure 5C, Table S1).
Discussion
ATO is used clinically in the management of APL, especially for the patients who carry the PML-RARα oncogene, and the curative outcome is attributed to its binding to the zinc finger domains of PML-RARα protein. The activity of ubiquitin-proteasome pathway requires three enzymes, E1s, E2s and E3s, to catalyze the ubiquitin-protein conjugates, respectively (Figure 6A).7 Among the E3 family, the majority of identified members contain a RING-finger domain with zinc sites where is conceived of as the target of arsenite treatment.12,31,33,34 Human Pirh2 contains a RING-H2 domain, charactered by a consensus sequence with six cysteine and two histidine residues that coordinate two zinc ions, besides, there is a CHY-zinc-finger domain conjugated to the RING finger (Figure 6B). As an E3 ubiquitin ligase being deregulated in various cancers, Pirh2 is considered an anticancer target of arsenite therapy due to the binding of arsenic ions like their action on PML-RARα.11,35
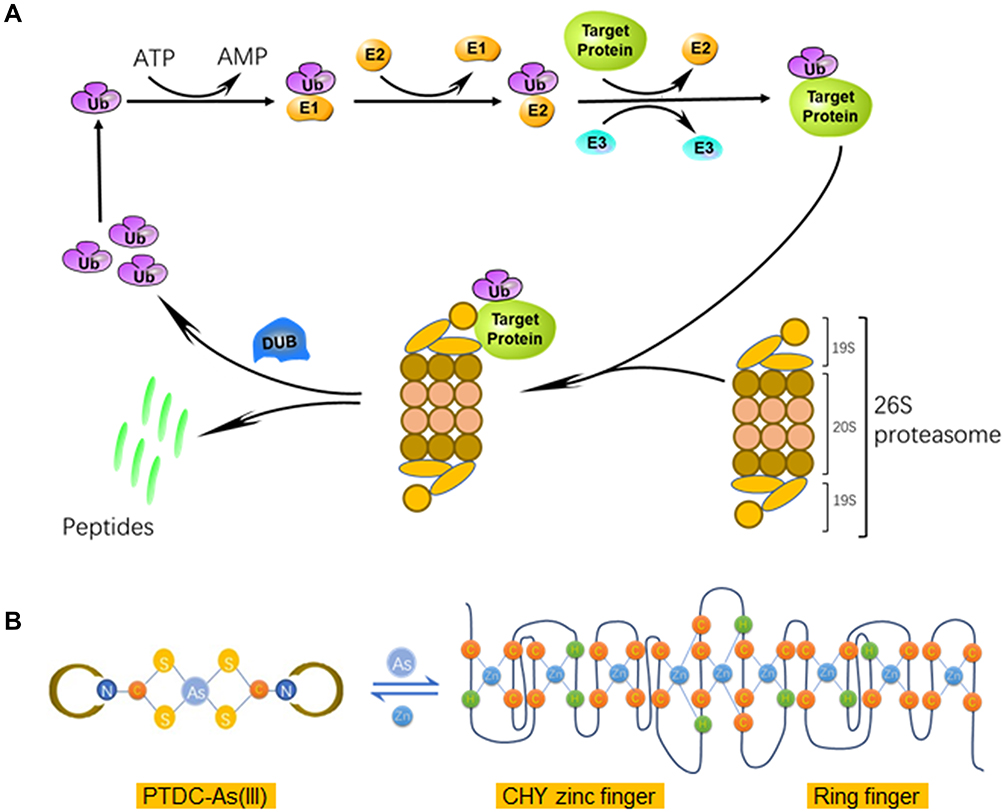 |
Figure 6 Schematic representation of ubiquitin-proteasome pathway and interaction between PTDC-ATO and Pirh2. (A) The human Ub conjugates to target protein by catalyzation of ubiquitin-protein ligases, and then subjects to degradation by 26S proteasomes. (B) The left, the structure of PDTC-As (III) complex; the right, the secondary sequence organization of the CHY-zinc-finger/RING-finger domain of Pirh2 protein; and the formula shows a dynamic balance between PDTC-As (III) and finger domain motifs via metal ion-exchange. Abbreviation: DUB, de-ubiquitinating enzyme. |
It is general in chemistry once complexes of different stabilities meet in solution, ion-exchange might happen between or among them to form new complexes having higher stability than the formers.36,37 Trivalent arsenic ions (As3+) are the effectors of ATO, and free arsenic ions could be captured by detoxication molecules such as glutathione (GSH) to reduce their chance to reach the target proteins within the body. Based on these, we hypothesized that a proper complexing agent might have the capacity to keep arsenic ions away from being captured by detoxication molecules, and then the complex could release the ions to bind to target proteins such as E3 ligases (Figure 6B).
The data from the Western blot assay on tumor samples showed that Pirh2 was detected in SW1990 pancreatic cancer cells, and its expression was suppressed by ATO treatment or PDTC-ATO, along with downregulation of ΔNp63 which was Pirh2-target protein, meantime, accumulation of ubiquitin as well as upregulation of AIRAP which was an arsenite-inducible subunit of the proteasome’s 19S cap was observed in the arenite-treated samples. These data collectively demonstrated that either ATO or PDTC-ATO had perturbed the ubiquitin-proteasome pathway in vivo, and the target might be Pirh2; meanwhile, the data suggested that the effect of PDTC-ATO was more potent and credible than ATO. As the hypothesis described above, the improved effect of PDTC-ATO therapy might be sourced from the greater number of arsenic ions arriving at the target proteins in a manner of the complex nuclei, and being released for the exchange with zinc ions contained by finger domains compared with ATO.
PDTC is currently investigated for its use as a metal remover, for the induction of cell cycle arrest, and for preventative induction of nitric oxide synthase.27,38–40 The data of CCK-8 test in this study demonstrated that though PDTC and ATO had modest anticancer effects on human pancreatic cancer cells, the complex constituted by their combination, PDTC-ATO, had an effective function to inhibit tumorigenesis (Figure 2A). A time-dependent cytotoxic effect for ATO on pancreatic cancer cells in culture has been demonstrated previously.15,16,41 As the previous reports and the data of this study showed, when treatment time was 24 hours, ATO usually increased pancreatic cancer cell viability at a concentration equal to or less than 5 μM in cultures, but PDTC-ATO was observed to inhibit pancreatic cancer cell viability effectively not only by 48-hour treatment but also by 24-hour (Figure 2A, and supplementary Figure S1).16
The previous studies suggested that the IC50 values of ATO to pancreatic cancer cells were above 10 μM when treatment time was 48 hours in vitro, and ATO had no obvious inhibiting effects if treatment time was 24 hours at concentrations less than 10 μM.15,16 Through the IC50 values of PDTC-ATO detected in this study (Table 1), together with the in vitro cytotoxic assay data (Figures 2A and S1), we had a judgement that PDTC-ATO was capable of inhibiting the viability of pancreatic cancer cell lines effectively not only by 48-hour treatment but also 24-hour, indicating that PDTC could change the time-dependent manner of ATO against pancreatic cancer cell proliferation in cultures.
Though body weights of mice treated with ATO and PDTC-ATO all decreased in this study (Figure 4B), adverse effects in mice treated with PDTC-ATO were less than those in mice treated with ATO in the experiment reported. The main adverse effects with ATO were physical inactivity and anorexia, meanwhile, these symptoms were seldom observed in animals treated with PDTC-ATO. Furthermore, from the IHC assay on the organs including brain, liver and kidney, less injury induced by PDTC-ATO than ATO could be observed (Supplementary Figure S2). These observed animal behaviors associating with treatment adverse effects indicated that PDTC-ATO might have slightly less system toxicity than ATO.
Conclusion
This study was designed according to the idea that arsenites were capable of perturbing ubiquitin-proteasome pathway by binding to RING-finger type E3 ligases.11,35 The results of Western blot assay on tumor samples demonstrated that either ATO or PDTC-ATO was capable of inducing alteration of protein expression led by Pirh2 activity suppression in pancreatic cancer cells. The data of anticancer capacity in cell cultures and xenografts indicated that PDTC-ATO had a higher capacity to inhibit the viability of pancreatic cancer cells than ATO; meanwhile, their system toxicity was similar.
Acknowledgments
The authors would like to thank Ms. Na Wang (Fudan University Shanghai Cancer Center) for the experimental assistance in the cellular study. This work was supported by the National Natural Science Foundation of China (81473498); Cross-disciplinary Research Fund of Shanghai Ninth People’s Hospital, Shanghai JiaoTong University School of Medicine (JYJC201910).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74(11):2913–2921.
2. Kamisawa T, Wood LD, Itoi T, Takaori K. Pancreatic cancer. Lancet. 2016;388(10039):73–85. doi:10.1016/S0140-6736(16)00141-0
3. Bax M, McKenna J, Do-Ha D, et al. The ubiquitin proteasome system is a key regulator of pluripotent stem cell survival and motor neuron differentiation. Cells. 2019;8(6):581. doi:10.3390/cells8060581
4. Mukhopadhyay D, Riezman H. Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science. 2007;315(5809):201–205. doi:10.1126/science.1127085
5. Han J, Liu L, Yue X, Chang J, Shi W, Hua Y. A binuclear complex constituted by diethyldithiocarbamate and copper(I) functions as a proteasome activity inhibitor in pancreatic cancer cultures and xenografts. Toxicol Appl Pharmacol. 2013;273(3):477–483. doi:10.1016/j.taap.2013.09.009
6. Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 2001;292(5521):1552–1555. doi:10.1126/science.292.5521.1552
7. Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78:399–434. doi:10.1146/annurev.biochem.78.101807.093809
8. Plechanovova A, Jaffray EG, Tatham MH, Naismith JH, Hay RT. Structure of a RING E3 ligase and ubiquitin-loaded E2 primed for catalysis. Nature. 2012;489(7414):115–120. doi:10.1038/nature11376
9. Budhidarmo R, Nakatani Y, Day CL. RINGs hold the key to ubiquitin transfer. Trends Biochem Sci. 2012;37(2):58–65. doi:10.1016/j.tibs.2011.11.001
10. Zhang F, Paramasivam M, Cai Q, et al. Arsenite binds to the RING finger domains of RNF20-RNF40 histone E3 ubiquitin ligase and inhibits DNA double-strand break repair. J Am Chem Soc. 2014;136(37):12884–12887. doi:10.1021/ja507863d
11. Yan W, Jung Y-S, Zhang Y, Chen X, Li Y. Arsenic trioxide reactivates proteasome-dependent degradation of mutant p53 protein in cancer cells in part via enhanced expression of Pirh2 E3 ligase. PLoS One. 2014;9(8):e103497. doi:10.1371/journal.pone.0103497
12. Jiang J, Tam LM, Wang P, Wang Y. Arsenite targets the RING finger domain of Rbx1 E3 ubiquitin ligase to inhibit proteasome-mediated degradation of Nrf2. Chem Res Toxicol. 2018;31(5):380–387. doi:10.1021/acs.chemrestox.8b00062
13. Zhang XW, Yan XJ, Zhou ZR, et al. Arsenic trioxide controls the fate of the PML-RARalpha oncoprotein by directly binding PML. Science. 2010;328(5975):240–243. doi:10.1126/science.1183424
14. Xu C, Wang X, Zhou Y, et al. Synergy between arsenic trioxide and JQ1 on autophagy in pancreatic cancer. Oncogene. 2019;38(47):7249–7265. doi:10.1038/s41388-019-0930-3
15. Beauchamp EM, Ringer L, Bulut G, et al. Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking hedgehog/GLI pathway. J Clin Invest. 2011;121(1):148–160. doi:10.1172/JCI42874
16. Han JB, Sang F, Chang JJ, et al. Arsenic trioxide inhibits viability of pancreatic cancer stem cells in culture and in a xenograft model via binding to SHH-Gli. Onco Targets Ther. 2013;6:1129–1138. doi:10.2147/OTT.S49148
17. Bhalla S, Gordon LI, David K, et al. Glutathione depletion enhances arsenic trioxide-induced apoptosis in lymphoma cells through mitochondrial-independent mechanisms. Br J Haematol. 2010;150(3):365–369. doi:10.1111/j.1365-2141.2010.08197.x
18. Dai J, Weinberg RS, Waxman S, Jing Y. Malignant cells can be sensitized to undergo growth inhibition and apoptosis by arsenic trioxide through modulation of the glutathione redox system. Blood. 1999;93(1):268–277. doi:10.1182/blood.V93.1.268
19. Zhang Y, Wade KL, Prestera T, Talalay P. Quantitative determination of isothiocyanates, dithiocarbamates, carbon disulfide, and related thiocarbonyl compounds by cyclocondensation with 1,2-benzenedithiol. Anal Biochem. 1996;239(2):160–167. doi:10.1006/abio.1996.0311
20. Francis P, Markman M, Hakes T, et al. Diethyldithiocarbamate chemoprotection of carboplatin–induced hematological toxicity. J Cancer Res Clin Oncol. 1993;119(6):360–362. doi:10.1007/BF01208846
21. Spivak AM, Andrade A, Eisele E, et al. A pilot study assessing the safety and latency-reversing activity of disulfiram in HIV-1-infected adults on antiretroviral therapy. Clinical infectious diseases: an official publication of the Infectious Diseases Society of America. 2014:58(6):883–890.
22. Pang H, Chen D, Cui QC, Dou QP. Sodium diethyldithiocarbamate, an AIDS progression inhibitor and a copper-binding compound, has proteasome-inhibitory and apoptosis-inducing activities in cancer cells. Int J Mol Med. 2007;19(5):809–816.
23. Hafberg ET. 50 years ago in the journal of pediatrics: hepatolenticular degeneration: the comparative effectiveness of D-penicillamine, potassium sulfide, and diethyldithiocarbamate as decoppering agents. J Pediatr. 2016;173:55. doi:10.1016/j.jpeds.2015.12.040
24. Kim KS, Oh DH, Choi HM, et al. Pyrrolidine dithiocarbamate, a NF-kappaB inhibitor, upregulates MMP-1 and MMP-13 in IL-1beta-stimulated rheumatoid arthritis fibroblast-like synoviocytes. Eur J Pharmacol. 2009;613(1–3):167–175. doi:10.1016/j.ejphar.2009.04.026
25. Milacic V, Chen D, Giovagnini L, Diez A, Fregona D, Dou QP. Pyrrolidine dithiocarbamate-zinc(II) and -copper(II) complexes induce apoptosis in tumor cells by inhibiting the proteasomal activity. Toxicol Appl Pharmacol. 2008;231(1):24–33. doi:10.1016/j.taap.2008.03.009
26. Chen J, Du C, Kang J, Wang J. Cu2+ is required for pyrrolidine dithiocarbamate to inhibit histone acetylation and induce human leukemia cell apoptosis. Chem Biol Interact. 2008;171(1):26–36. doi:10.1016/j.cbi.2007.09.004
27. Daniel KG, Chen D, Orlu S, Cui QC, Miller FR, Dou QP. Clioquinol and pyrrolidine dithiocarbamate complex with copper to form proteasome inhibitors and apoptosis inducers in human breast cancer cells. Breast Cancer Res. 2005;7(6):R897–R908. doi:10.1186/bcr1322
28. Ganesan S, Alex AA, Chendamarai E, et al. Rationale and efficacy of proteasome inhibitor combined with arsenic trioxide in the treatment of acute promyelocytic leukemia. Leukemia. 2016;30(11):2169–2178. doi:10.1038/leu.2016.227
29. Jimeno A, Feldmann G, Suarez-Gauthier A, et al. A direct pancreatic cancer xenograft model as a platform for cancer stem cell therapeutic development. Mol Cancer Ther. 2009;8(2):310–314. doi:10.1158/1535-7163.MCT-08-0924
30. Verma RJ, Vasu A, Saiyed AA. Arsenic toxicity in mice and its possible amelioration. J Environ Sci. 2004;16(3):447–453.
31. Chiu HW, Tseng YC, Hsu YH, et al. Arsenic trioxide induces programmed cell death through stimulation of ER stress and inhibition of the ubiquitin-proteasome system in human sarcoma cells. Cancer Lett. 2015;356(2 Pt B):762–772. doi:10.1016/j.canlet.2014.10.025
32. Jung Y-S, Qian Y, Chen X. Pirh2 RING-finger E3 ubiquitin ligase: its role in tumorigenesis and cancer therapy. FEBS Lett. 2012;586(10):1397–1402. doi:10.1016/j.febslet.2012.03.052
33. Stanhill A, Haynes CM, Zhang Y, et al. An arsenite-inducible 19S regulatory particle-associated protein adapts proteasomes to proteotoxicity. Mol Cell. 2006;23(6):875–885. doi:10.1016/j.molcel.2006.07.023
34. Tam LM, Jiang J, Wang P, et al. Arsenite binds to the zinc finger motif of TIP60 histone acetyltransferase and induces its degradation via the 26S proteasome. Chem Res Toxicol. 2017;30(9):1685–1693. doi:10.1021/acs.chemrestox.7b00146
35. Yan W, Chen X, Zhang Y, Zhang J, Jung YS, Chen X. Arsenic suppresses cell survival via Pirh2-mediated proteasomal degradation of DeltaNp63 protein. J Biol Chem. 2013;288(5):2907–2913. doi:10.1074/jbc.M112.428607
36. Piquette A, Cannon C, Apblett AW. Remediation of arsenic and lead with nanocrystalline zinc sulfide. Nanotechnology. 2012;23(29):294014. doi:10.1088/0957-4484/23/29/294014
37. Dawson JB, Bahreyni-Toosi MH, Ellis DJ, Hodgkinson A. Separation of protein-bound copper and zinc in human plasma by means of gel filtration -ion-exchange chromatography. Analyst (Lond). 1981;106(1259):153–159. doi:10.1039/an9810600153
38. Park SS, Lee DM, Lim JH, et al. Pyrrolidine dithiocarbamate reverses Bcl-xL-mediated apoptotic resistance to doxorubicin by inducing paraptosis. Carcinogenesis. 2018;39(3):458–470. doi:10.1093/carcin/bgy003
39. Chabicovsky M, Prieschl-Grassauer E, Seipelt J, et al. Pre-clinical safety evaluation of pyrrolidine dithiocarbamate. Basic Clin Pharmacol Toxicol. 2010;107(3):758–767. doi:10.1111/j.1742-7843.2010.00573.x
40. Cuzzocrea S, Chatterjee PK, Mazzon E, et al. Pyrrolidine dithiocarbamate attenuates the development of acute and chronic inflammation. Br J Pharmacol. 2002;135(2):496–510. doi:10.1038/sj.bjp.0704463
41. Zhang N, Wu ZM, McGowan E, et al. Arsenic trioxide and cisplatin synergism increase cytotoxicity in human ovarian cancer cells: therapeutic potential for ovarian cancer. Cancer Sci. 2009;100(12):2459–2464. doi:10.1111/j.1349-7006.2009.01340.x
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.