")
Back to Journals » Journal of Inflammation Research » Volume 14
Pyroptosis: A New Regulating Mechanism in Cardiovascular Disease
Authors Ji N, Qi Z , Wang Y, Yang X, Yan Z , Li M, Ge Q, Zhang J
Received 24 February 2021
Accepted for publication 2 June 2021
Published 22 June 2021 Volume 2021:14 Pages 2647—2666
DOI https://doi.org/10.2147/JIR.S308177
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Nan Ji,1,2,* Zhongwen Qi,1,* Yueyao Wang,1 Xiaoya Yang,1 Zhipeng Yan,1 Meng Li,1 Qihui Ge,1 Junping Zhang1
1First Teaching Hospital of Tianjin University of Traditional Chinese Medicine, Tianjin, 300183, People’s Republic of China; 2National Clinical Research Center for Chinese Medicine Acupuncture and Moxibustion, Tianjin, 300193, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Junping Zhang Email [email protected]
Abstract: Pyroptosis is a kind of pro-inflammatory cell death. Compared with autophagy and apoptosis, pyroptosis has unique characteristics in morphology and mechanism. Specifically, pyroptosis is a kind of cell lysis mediated by the Gasdermin family, releases inflammatory cytokines IL-1β and IL-18. There are three different forms of mechanism, which are caspase-1-mediated, caspase-4/5/11-mediated and caspase-3-mediated. A large number of studies have proved that pyroptosis is closely related to cardiovascular disease. This paper reviewed the recent progress in the related research on pyroptosis and myocardial infarction, ischemia-reperfusion, atherosclerosis, diabetic cardiomyopathy, arrhythmia, heart failure hypertension and Kawasaki disease. Therefore, we believe that pyroptosis may be a new therapeutic target in the cardiovascular field.
Keywords: pyroptosis, cardiopathy, NLRP3, caspase-1, IL-1β
Introduction
Pyroptosis is a type of programmed cell death. It is characterized by the formation of holes in the cell membrane, the release of pro-inflammatory cytokines and cell lysis. Pyroptosis controls inflammation by releasing IL-1β, IL-18 and other inflammatory substances, which is an important innate immune mechanism. The occurrence of pyroptosis is closely related to the activation of NLRP3, the reactive oxygen species generated by oxidative stress activate NLRP3, and the structurally altered NLRP3 inflammasome induces caspase-1 activation. On the one hand, caspase-1 is activated and Gasdermin D is cleaved by activated caspase-1 to form a polypeptide containing the nitrogen-terminated active structural domain of Gasdermin D. This reaction can lead to myocardial perforation and rupture of cell membranes, resulting in the release of contents and causing an inflammatory response. On the other hand, the already activated caspase-1 cleaves the precursors of IL-1β and IL-18 to active IL-1β and IL-18 and releases them extracellularly, recruiting inflammatory cells and causing inflammatory cells to accumulate, thus amplifying the inflammatory response. Cholesterol destroys the lysosomal membrane structure, releases inflammatory contents, activates NLRP3, and induces pyroptosis. NLRP3 and caspase-1 are inseparable from the occurrence and development of many cardiovascular diseases. Pyroptosis and its pathological products can reduce angiogenesis, destroy the stability of blood vessel wall plaque, damage vascular endothelial cells, and cause myocardial hypertrophy and myocardial fibrosis. A large number of studies have proved that pyroptosis may be an endogenous regulator of cardiovascular disease and play an important role in cardiovascular disease.
Characteristics of Pyroptosis
Cell death is of considerable significance in both normal physiological and pathological states. In recent years, it has been discovered that a new type of programmed cell death, pyroptosis depends on the pore-forming activity of the Gasdermin protein family. In 1992, lytic death of macrophages was observed in Shigella flexneri, a gram-negative pathogen that was considered apoptosis at the time.1 It was not until 2001 that the study found that this form of death was caspase-1 activity-dependent. Unlike caspase-3 activity-dependent apoptosis, pyroptosis was officially defined as a new type of caspase-1-dependent cell programmability death.2 “Pyro” means fire, indicating that this programmed cell death causes an inflammatory response, while “ptosis” is fallout. Meaning, stating the nature of its programmed death. Cells that undergo pyroptosis lose membrane integrity, increase in size, and decrease in size of the nucleus.3–5
The Differences Between Pyroptosis, Apoptosis, Autophagy Necrosis and Necroptosis
In 1972, Kerr and his colleagues coined the term apoptosis to describe a particular form of cell death. Apoptosis is present in all life stages in multicellular organisms and allows for the timely removal of senescent, damaged, redundant, harmful cells, maintaining the organism’s stability. For a long time, apoptosis was thought to be the only programmed cell death, and it has been better understood. The onset of apoptosis is characterized by the gradual disappearance of specialized structures such as microvilli on the cell surface. The cell shrinks and separates from the surrounding cells. The cytoplasm and the cell membrane are separated from the surrounding cells. Nuclear chromatin sequestration, apoptosis-related cysteinases mediate the hydrolysis of ICAD and activated cysteinases activate DNAase (CAD), at this point, cleavage of the chromosomal DNA in the nucleus is induced. As a result, the DNA bands of apoptotic cells take on a stepped pattern and the cells that undergo apoptosis are subsequently engulfed by nearby mesenchymal cells or macrophages. The cell membrane remains intact during this period, so it does not trigger an inflammatory response. During this period, the cell membrane remains intact and does not trigger an inflammatory response.6
When the cell undergoes pyroptosis, the nucleus is concentrated, chromatin DNA randomly breaks and degrades, and the plasma membrane ruptures to form pores with a diameter of 1.1–2.4 nm. The cell permeability increases and inflammatory cytokines, lactate dehydrogenase, and other intracellular substances are released. The small pores in the cell membrane cause the cell to rapidly lose its ionic gradient, which allows a significant increase in osmotic pressure and leads to swelling of the cell; eventually, the cell membrane is destruction, the cellular contents are released into the extracellular environment, stimulating the body’s immune response, recruiting more inflammatory cells and expanding the Inflammatory response. The degradation of pyroptosis chromosomal DNA is mediated by an unknown endonuclease that undergoes random breaks, and now a small amount of ladder band DNA clips.7
The characteristics of pyroptosis and apoptosis are similar in that both are cell deaths that occur through the regulation of genes. Both scorch death and apoptosis are characterised by deoxynucleotidyl transferase-mediated dUTP nickel end-labelling (TUNEL) staining.
Autophagy occurs in most tissues at a basal level, where autophagy selectively removes excess cellular components, but in addition, autophagy is also induced to occur under different physiological and pathological conditions. The evolutionarily conserved autophagy-associated protein (ATG), however, plays an important role in all processes of autophagy.8
Ability of autophagy to degrade targets of different sizes: The process of autophagy is accompanied by the emergence of a small bilayer of membranes in the cytoplasm, called the separator membrane, which, as it extends, continues to encapsulate the surrounding intracellular material, eventually forming a bilayer of membrane structures called autophagosomes that enclose vesicles. Autophagosomes and lysosomes are fused by membranes to form a single membrane structure of autophagic lysosomes and degrade the intracellular material engulfed therein as well as the autophagic endosomal membrane. Autophagosomal surface transport proteins carry the degradation products to the autophagosome for cellular use.
Autophagosomes fuse with lysosomes, and all contents of autophagosomes are degraded by lysosomal hydrolases.9
Necrosis, long thought to be a passive death due to pathology, such as physical or chemical damage factors and hypoxia and malnutrition all lead to cell necrosis. The membrane permeability of the necrotic cells is increased, resulting in swelling of the cells, deformation or enlargement of the organelles, no obvious morphological changes in the early nuclei and finally cell rupture. Cell lysis releases inclusions and often causes an inflammatory response; the healing process is often accompanied by fibrosis of tissues and organs, resulting in scarring.10,11
Necroptosis is a newly discovered form of programmed cell death with morphological characteristics of necrotic cells and similar signalling mechanisms to those of apoptotic cells. Morphological manifestations include perforated cell membranes, increased intracellular osmotic pressure leading to rounding and swelling of cells, swelling of organelles, mitochondrial dysfunction, loss of mitochondrial membrane potential, loss of nuclear chromatin and explosion-like rupture of the plasma membrane. The cellular contents released after cell rupture exacerbate the surrounding inflammatory response. The difference with necrosis is that necroptosis strictly follows intracellular signalling and has an active energy-consuming character.12,13
Pyroptosis, autophagy, apoptosis, necrosis and necroptosis both have important implications for cardiovascular disease, with the onset of apoptosis generally causing the death of cardiomyocytes and leading to adverse cardiac outcomes. In contrast, autophagy can lead to very different consequences at different times in cardiac disease, with mild autophagy inhibiting apoptosis and reducing cellular damage. Severe autophagy can cause cellular damage (Table 1).
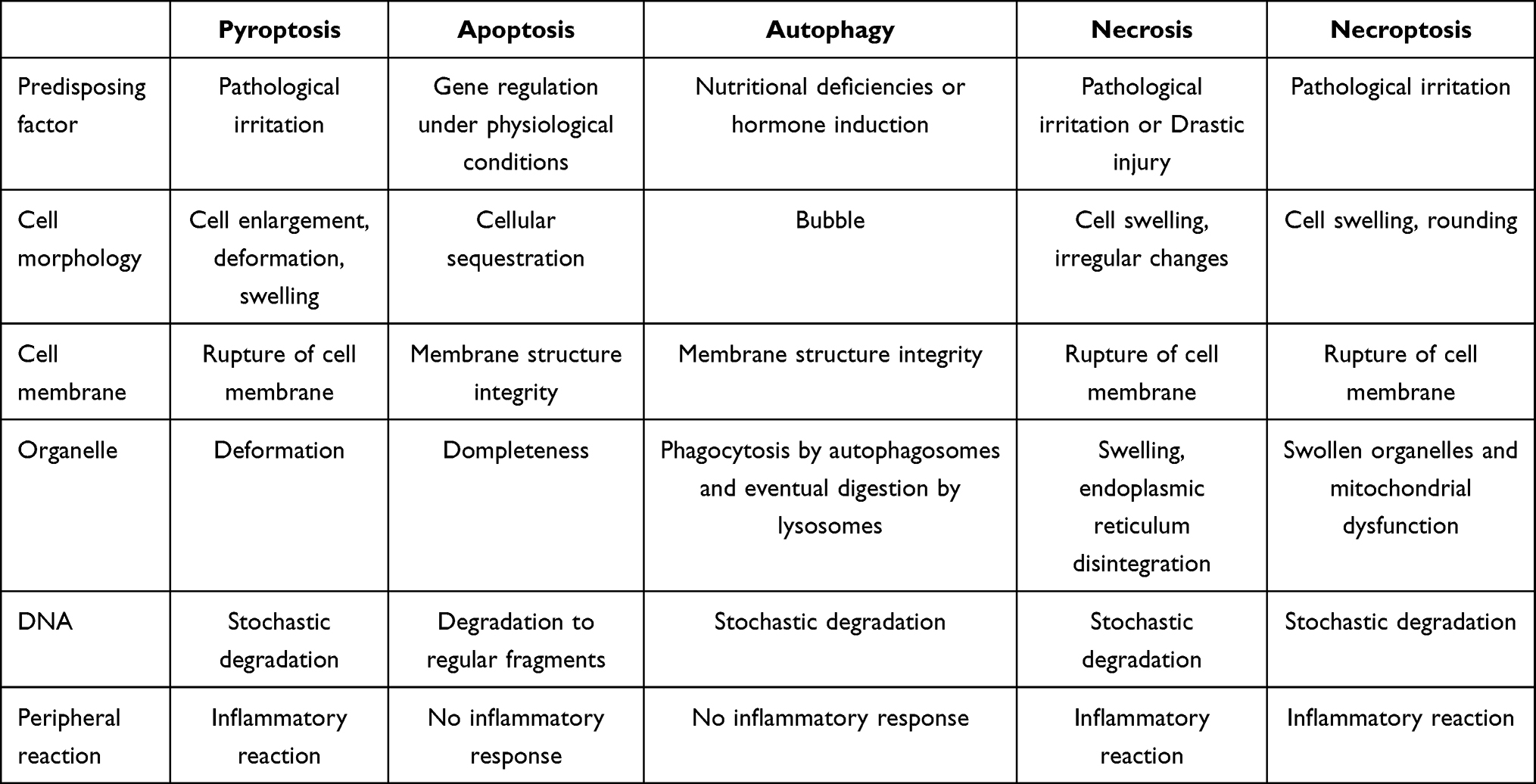 |
Table 1 The Differences Between Pyroptosis, Autophagy, Apoptosis, Necrosis and Necroptosis |
Three Molecular Pathways of Pyroptosis
Since the study, it has been generally accepted that there are two pathways to pyroptosis, one classical and the other non-classical pathways. However, in recent years, it has been discovered that there is a third new pathway for pyroptosis induced by caspase-3. After cells receive different stimuli, the pyroptosis process is initiated by different pathways, but is ultimately completed by the GSDMD protein.
The Canonical Pyroptosis Signaling Pathway
Activation of the canonical pyroptosis signaling pathway relies primarily on PRRs receiving stimulation by risk-signaling molecules, recruitment of pro-caspase-1 assembles to form inflammatory microsomes, activating caspase-1 molecule to cut further downstream GSDMD target proteins that promote pyroptosis.14 New studies show that GSDMD protein is a common substrate for inflammatory caspases and is an effector of pyroptosis. GSDMD proteins, known colloquially as “killer proteins”, play critical roles in both pyroptosis pathways. In the intracellular environment, GSDMD proteins are present in the cytoplasm and are subject to activation of the canonical pyroptosis pathway. Caspase-1 and non-canonical pyroptosis signaling pathway are activated by caspase-4/5/11 stimulation in the special. The locus divides the GSDMD protein into a lipophilic N-terminal domain and a hydrophilic C-terminal domain, with the N-terminal domain being the most important. Domains can bind to biological membranes and aggregate on the membrane’s inner side to form pores where water molecules can invade cells and trigger pyroptosis.15,16
The Non-Canonical Pyroptosis Signaling Pathway
It was found that there is also a non-caspase-1-dependent pyroptosis pathway in cells, which unlike the classical pathway, is dependent on the activation of caspase-4/5/11.17 Other experimental studies have shown that human caspase 4/5 and mouse caspase-11 can bind to bacterial LPS and thereby mediate inflammation leading to cellular necrosis. This caspase 4/5/11-dependent cell death mechanism constitutes atypical pyroptosis. Like caspase-1, activated caspase-11 also mediates the formation of cell membrane pores by cutting GSDMD, releasing mature IL-18 and IL-1β, and inducing the onset of pyroptosis. Besides, activated caspase-11 could promote K+ efflux not only by inducing GSDMD cell membrane pore formation, but also through the Pannexin-1/ATP/P2X7 pathway, mediating NLRP3/ASC/caspase-1 activation, promoting IL-1β maturation and release, and inducing cellular inflammatory responses.18–20
Caspase-3 - Dependent Pyroptosis Signaling Pathway
For a long time, pyroptosis was thought to occur in only two ways, in recent years, and it has been found that there is also a caspase-3-dependent pyroptosis pathway.21 In contrast to caspase-1/11/4/5, which induces GSDMD-dependent pyroptosis, caspase-3 induces cell pore formation by cutting GSDME and promoting recruitment of GSDME-N domains to the cell membrane, leading to pyroptosis. GSDME distribution and expression levels determine the mode of cell death by caspase-3 activation. Activated caspase-3 induces pyroptosis when cells overexpress GSDME, and for cells with low expression levels of GSDME, activation of caspase-3 triggers a subsequent rise after induction of apoptosis. This caspase-3-dependent mode of cell death is called apoptosis-like pyroptosis. In doxorubicin(DOX)-induced myocardial injury experiments, regulating the expression of caspase-3, GSDME by cell transfection. The experimental results showed that cardiomyocytes exposed to DOX exhibited the morphological characteristics of pyroptosis in vitro. Furthermore, DOX was found to induce caspase-3 activation, which ultimately triggers gsdme-dependent pyroptosis, while silencing or inhibiting caspase-3 reduced pyroptosis. We further found that the downregulation of GSDME inhibited dox-induced pyroptosis.22–24
Thus, inflammatory caspases, whether classical, non-classical, or a third focal pathway, ultimately transmit pyroptosis signals to perform GSDMD proteins.
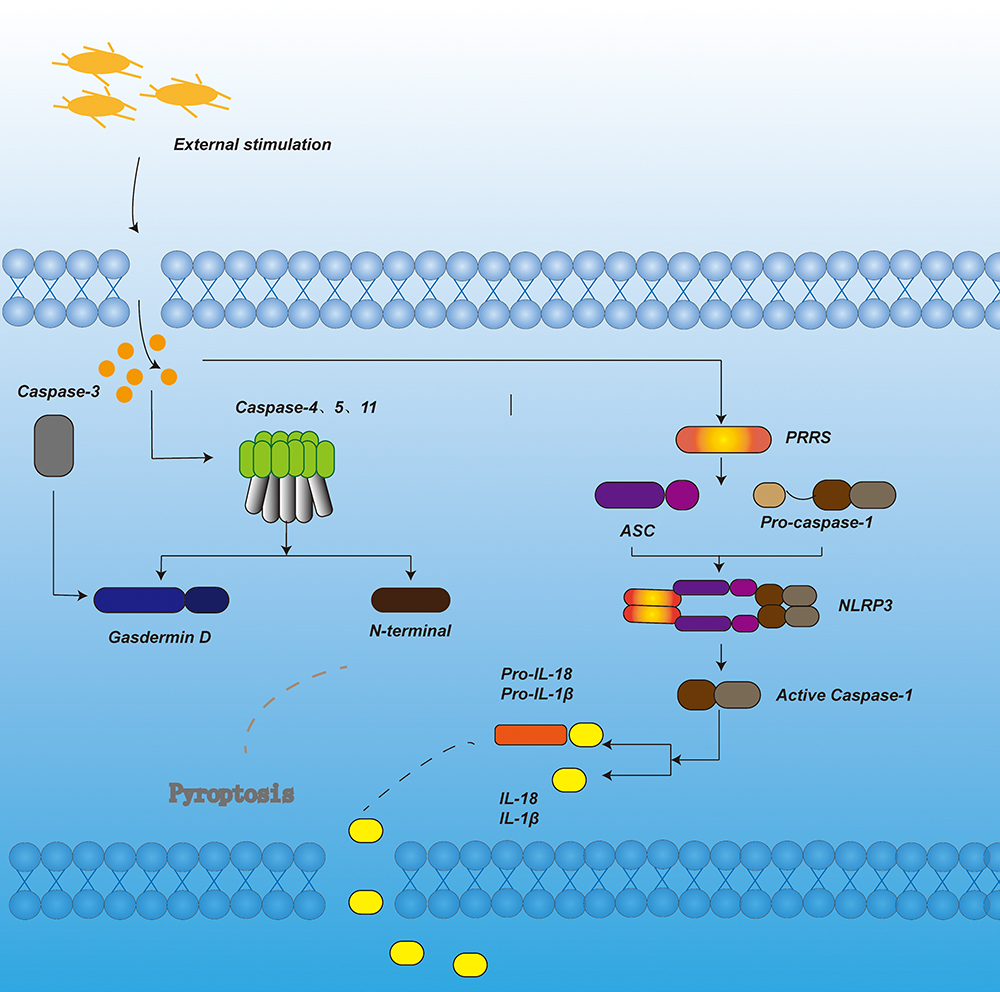 |
Figure 1 In the canonical pyroptosis signaling pathway, under the stimulation of bacteria, viruses and other signals, the pattern recognition receptor in the cell acts as a sensor to recognize these signals. Through the adaptor protein ASC, it binds to the precursor of Caspase-1 to form a multi-protein complex and activate Caspase-1. Activated Caspase-1 cleaves Gasdermin D to form peptides containing the nitrogen-terminal active domain of Gasdermin D, induce cell membrane perforation, cell rupture, release of contents, and cause inflammation. On the other hand, activated Caspase-1 cleaves the precursors of IL-1β and IL-18 to form active IL-1β and IL-18, which are released to the outside of the cell to recruit inflammatory cells to aggregate and expand the inflammatory response. In the non-canonical pyroptosis signaling pathway, under the stimulation of bacteria and other signals, Caspase-4, 5, and 11 are activated. The activated Caspase-4, 5, and 11 cleave Gasdermin D to form a peptide containing the nitrogen-terminal active domain of Gasdermin D. On the one hand, it induces cell membrane perforation and cell Rupture, release the contents and cause inflammation. On the other hand, it induces the activation of Caspase-1, cleaves the precursors of IL-1β and IL-18, forms active IL-1β and IL-18, and releases them to the outside of the cell, recruits inflammatory cells to aggregate, and expands inflammation reaction. In apoptotic-like pyrolysis, caspase-3 cuts GSDME, promotes the recruitment of GSDME-N domain to the cell membrane, induces the formation of cell membrane pores, and leads to pyroptosis. |
Pathological Factors Affect the Pyroptosis
Many pathological factors are involved in the development of pyroptosis, including oxidative stress, inflammatory cytokines, and cholesterol (Figure 1).
Oxidative Stress
Reactive oxygen species generated by oxidative stress can signal NLRP3 inflammasome activation, leading to an inflammatory response. In the ischemic brain, Bruton’s tyrosine kinase (BTK) is involved in the activation of NLRP3 inflammasomes. This process leads to the activation of caspase-1 and secretion of mature IL-1β.25
It was found that polychlorinated biphenyls (PCBs) are cytotoxic, especially for endothelial cells, leading to caspase-1 activation and cell membrane rupture, which are characteristics of pyroptosis. Experimental results show that PCB118 also induces excessive production of reactive oxygen species (ROS) in endothelial cells. The ROS scavenger (±)-α-tocopherol and the NF-κB inhibitor BAY11-7082 then prevented endothelial cells from being induced by PCB118. Increased expression of NLRP3 leads to a concomitant high activation of NLRP3 inflammatory vesicles. In addition to this, PCB 118-induced oxidative stress and focal death are dependent on the activation of the aromatic hydrocarbon receptor (AhR), which subsequently leads to a similar increase in the expression of cytochrome P450 1A1. All these evidences can prove that PCB 118 can lead to ex vivo endothelial pyroptosis by inducing NLRP3 inflammatory vesicle activation.And subsequently leads to endothelial cell pyroptosis in vitro and in vivo. AhR-mediated ROS production by triggering NF-κB-dependent NLRP3 expresses and promotes inflammasome activation and plays an essential role in PCB 118-induced pyroptosis.26
In a study by Yangshuo Tang, they used LPS and ATP stimulation to induce endothelial pyroptosis. To verify the involvement of ROS in the mechanism of inflammatory vesicle activation, they tested the pyroptosis process with respect to the oxidative stress component. ROS production is important for the activation of NLRP3 inflammatory vesicles because ROS scavenger (NAC) prevents the release of inflammatory cytokines and the activation of NLRP3 inflammatory vesicles. Through experiments, it was demonstrated that activated calpain can release caspase-1 that is separated by the cytoskeleton, thereby regulating NLRP3 activation and leading to a pyroptosis outcome.In summary, LPS-ATP is induced by regulation of the ROS/NLRP3/caspase-1 signaling pathway endothelial pyroptosis.27
Inflammatory Cytokines
Pyroptosis is a highly inflammatory pattern of cell death induced by inflammatory microsomes.28
The process of pyroptosis relies on the activation of caspase-1. Caspase-1 mediates the cleavage of interleukin-1β (IL-1β) precursors into active IL-1β, which is one of the essential functions of caspase-1. While active IL-1β recruits and activates other immune cells, induces chemokines (such as IL-18), inflammatory factors (such as IL-6) and adhesion factor synthesis. This ultimately leads to a “cascade effect”, amplifying the inflammatory response and thus inducing inflammation.But it is also for this reason that pyroptosis can tightly control the inflammatory response by controlling the release of inflammasomes and thereby orchestrating antimicrobial host defense and amplifying or maintaining inflammation. It is considered an essential innate immune effector mechanism against intracellular pathogens.29,30 In the mammalian inflammatory response, the apoptosis-associated spot-like protein cysteine-aspartate-containing protease recruitment domain (ASC) plays an important role. Studies have shown that ASC can regulate the activation of caspase-1 and induce the development of pyroptosis pairs and activate the production of cellular inflammatory factors. In addition to this, ASC interacts with receptor-interacting protein kinase 2 (RIPK2), which activates NF-κB. The scientists used the Japanese Aoki (Oryzias Layzias) fish model as the subject of their study, and used the CRISPR-CAS9 system to establish ASC knockout models (KO), ASC-KO and wild-type (WT) rice voles infected with Aeromonas aeruginosa, and observed the mortality rates of these models, and by comparing them, the scientists found that ASC-KO Aoki had a higher mortality rate than WT. In addition to this, ASC-KO and WT Medakas were analysed for the expression of immune-related genes in their kidneys and intestines following attack by Aeromonas aeruginosa. The results showed that the expression of NF-κB regulatory genes (eg IL-1β, IL-6, IL-8) and RIPK2 genes were significantly lower in WT after infection with Aeromonas aeruginosa. In addition, the bacterial load, superoxide anion production and lactate dehydrogenase release of ASC-KO cyanobacteria were measured in ASC-KO cyanobacteria kidney cells by the immune response of ASC to Aeromonas aerophilus, and it was found that these responses were significantly reduced in ASC-KO cyanobacteria compared to WT after infection. These results suggest that Ayahuasca ASC plays a key role in combating Aeromonas aerophilic infections by inducing inflammatory responses and cell death to eliminate bacteria. Thus, inflammatory cytokines play an important role in the process of pyroptosis.31
Cholesterol
In mammalian cell membranes, cholesterol is one of the important structural components. Cholesterol destroys the stability of lysosomal membrane structure, causes lysosomal damage, causes lysosomal contents to outflow, activates NLRP3, and causes pyroptosis. The lysosomal cathepsin B promotes the process. Type I interferon upregulates cholesterol-25-hydroxylase (Ch25h) and inhibits srebp transcription factors, and in macrophages this inhibits the inflammatory response caused by IL-1β. Macrophage production of 25-hydroxycholesterol (25-HC) prevents the activation of melanoma-deficient DNA sensor protein 2 (AIM2) by inflammatory vesicles, which is essential for macrophages. At the same time, we know that macrophages maintain inhibition of SREBP2 activation and cholesterol synthesis by upregulating CH25H, which alleviates the adverse effects of lipopolysaccharide (LPS) stimulation or bacterial infection. Upregulation of macrophage cholesterol levels results in the release of IL-1β. The secretion of this IL-1β is crystal-independent and dependent on angiotensin-converting enzyme 2. H25H deficiency then reduces cholesterol-dependent mitochondrial respiration and allows the release of mitochondrial DNA into the cytoplasm. In contrast, AIM2 deficiency decreases inflammatory vesicle activity in CH25H.Thus, activated macrophages utilize 25-HC for anti–inflammatory cycling to maintain mitochondrial integrity and prevent activation of false AIM2 inflammasomes.32 In an ABCA1/G1-deficient mouse model, we observed glomerulonephritis with lymph node swelling (LNS) and systemic lupus erythematosus (SLE). This lupus-like phenotype was once again seen in ABCA1/G1 knockout mouse dendritic cells (DCs), but not in macrophages or T cells. DC-ABCA1/G1 deficiency increases LN and splenic CD11b dendritic cells, as evidenced by the increasing accumulation of cholesterol, activation of inflammatory vesicles, increased levels of granulocyte-macrophage colony-stimulating factor receptors on the cell surface and increased secretion of inflammatory cytokines. And we know that systemic lupus erythematosus (SLE) is strongly associated with increased cardiovascular disease and decreased plasma high-density lipoprotein (HDL) levels. Thus, the efflux of cholesterol from immune cells is due to HDL via the ATP-binding cassette transporters A1 and G1 (ABCA1/G1). Thus, DC-ABCA1/G1 deficiency enhances T cell activation as well as T1 and T17 cell polarization. NLRP3 inflammatory microsomal defects can make the enlarged LNS smaller and enhance T1 cell polarization. These findings establish an important role for the DC cholesterol efflux pathway in the maintenance of immune tolerance and also reveal the involvement of cholesterol in the development of pyroptosis.33
During atherogenesis, cholesterol precipitates in the vessel wall as cholesterol crystals (CC), which trigger plaque inflammation by activating NACHT, LRR and PYD structural domain protein 3 (NLRP3) inflammatory vesicles.34 Pre-eclampsia is a hypertensive and inflammatory pregnancy disorder associated with cholesterol accumulation and inflammation at the maternal-fetal interface. Pre-eclampsia can be combined with fetal growth restriction (FGR) and shares risk factors and pathophysiological mechanisms with cardiovascular disease. Cholesterol crystal-mediated activation of NLRP3 inflammatory vesicles is essential for cardiovascular disease, and this pathway is associated with placental inflammation in pre-eclampsia.35
In an experimental study of the effect of inhaled particulate matter or quartz on NLRP3 inflammatory vesicle activation at a Swedish ironworks, flow cytometry was used to detect whole blood monocyte caspase-1 enzyme activity and real-time fluorescence quantitative PCR to detect the expression of inflammatory vesicle-associated genes. Multiple linear regression analysis was used to investigate the relationship between PM exposure and inflammatory markers. Significant exposure responses were found for respiratory dust and IL-18 and for inhaled dust and IL-1Ra. Whole blood drawn from study participants, stimulated by inflammatory vesicle initiation stimulation LPS or Pam3CSK4, resulted in increased caspase-1 enzyme activity in monocytes. The increase in caspase-1 activity was significantly attenuated in the high exposure group of the PM exposure measure. Thus, the level of PM exposure in the iron environment can influence NLRP3 inflammatory vesicles and the systemic inflammatory response.36
Mitochondrial Damage
It has been reported that NLRP3 is closely associated with mitochondria. In response to external changes, such as electrical stimulation, LPS or other stimuli, NLRP3 interacts with pro-caspase-1 via ASC, leading to activation of caspase-1. Activated caspase-1 promotes the cleavage and maturation of pro- IL-1β, pro-IL-18 and IL-33 in the cytoplasm and mature IL-1β is released.37 Mitochondrial localization of NLRP3 is critical for NLRP3 inflammatory vesicle activation. TLRs on the plasma membrane-associated and introns and RIG-like helicases (RIG-I and MDA-5) are recruited by the mitochondrial outer membrane protein MAVS to initiate type I interferon responses during viral infection. It was demonstrated that NLRP3 in the resting state was mainly localized to the endoplasmic reticulum, however, in the presence of NLRP3 inflammatory vesicle activators, NLRP3 and ASC were redistributed to the endoplasmic reticulum and mitochondria that were clustered in the nuclear periphery.38 A. baumannii is a Gram-negative bacterium, a conditional pathogen that causes hospital-acquired pneumonia and bacteraemia by infecting alveoli with epithelial cells and macrophages. There is evidence that the outer membrane protein 34 (Omp34) of A. baumannii induces a cellular immune response and an inflammatory response. In RAW264.7 mouse macrophages, Omp34 induced the expression of caspase-1-p10 and IL-1β, and NLRP3 gene silencing significantly reduced the expression of both proteins. Furthermore, Omp34 stimulated RAW264.7 mitochondria to produce ROS, while the ROS scavenger Mito-TEMPO inhibited Omp34-triggered expression of NLRP3 inflammatory vesicle-associated proteins and IL-1β synthesis. The above results suggest that mitochondrial-derived ROS play an important role in NLRP3 inflammatory vesicle activation.39
Lysosomal Rupture
As an organelle that breaks down various exogenous and endogenous macromolecules in eukaryotic cells, lysosomes are also involved in the regulation of NLRP3 inflammatory vesicle activation. When lysosomal histone B is released into the cytoplasm following stimulation by silica, alum or β-amyloid, which triggers destabilization of the cytophagocytic vesicles, NLRP3 activation can be induced. In addition, endocytic crystals or specific molecules may directly disrupt the lysosomal membrane, resulting in the diffusion of phagocytic particles into the cytoplasm, which may interact directly with inflammatory vesicle-associated proteins to promote NLRP3 activation.40 Studies have shown that NLRP3 inflammatory vesicle activation, lysosomal dysfunction and impaired autophagic flux play a key role in the pathophysiology of MI. Therapeutic strategies targeting NLRP3 activation, lysosomal enzyme release have shown beneficial effects in suppressing the early inflammatory response in cardiovascular disease. Therefore, inhibition of NLRP3 activation and correction of lysosomal dysfunction may be a new direction in the treatment of myocardial infarction.41,42
Other Pathological Factors
LPS-induced NLRP3 inflammatory vesicle activation, in which caspase-11 is involved, is prevalent in Gram-negative bacteria. Recent studies have demonstrated that IFN regulatory factor (IRF) 8 is essential for caspase-11-mediated NLRP3 inflammatory vesicle activation during LPS transfection, and that IRF8 promotes NLRP3 activation in bone marrow-derived macrophages (BMDMs) from mice infected with Gram-negative bacteria, and that BMDMs lacking IRF8 show substantially reduced caspase-11 activation and gasdermin D cleavage, which are required for NLRP3 inflammasome activation. Mechanistically, IRF8-mediated phosphorylation of IRF3 is required for transcription, which in turn triggers caspase-11-dependent NLRP3 inflammasome activation in infected BMDMs.43
Inflammatory mediators are important in the development of many RNA virus-infected diseases. Many RNA viruses and their component such as encephalomyocarditis virus (EMCV) 2B viroporin, the viral RNA of hepatitis C virus, the influenza virus M2 viroporin, the respiratory syncytial virus (RSV) small hydrophobic (SH) viroporin, and the human rhinovirus (HRV) 2B viroporin can activate the NLRP3 inflammasome to influence the inflammatory response. On the other hand, some viruses use virally encoded proteins to inhibit inflammatory activation, such as the influenza virus NS1 protein and the measles virus (MV) V protein.44
Bacterial, viral and environmental stimuli can all cause changes in NLRP3, which can lead to changes in the level of pyroptosis in the body.
Pathological Changes in Cardiovascular Disease by Pyroptosis
When pyroptosis occurs, it causes alterations to the ultrastructure of the cardiovascular system, resulting in more significant damage and a poor prognosis for cardiovascular disease (Figure 2).
Endothelial Damage
Vascular endothelial damage is the initiating factor for cardiovascular pathology. Endothelial cells are the most important protective barrier between blood and blood vessel wall. High-risk factors can accelerate endothelial cell death and lead to cardiovascular pathology. Different types of injuries, such as mechanical, immune-mediated and chemical, can lead to endothelial damage.45,46 Endothelial dysfunction induced by high glucose is a recognized cause of vascular complications of type 2 diabetes. Experiments have found that rutin may protect endothelial function and reduce vascular complications caused by diabetes by inhibiting NOX4-responsive oxidative stress and ROS-sensitive NLRP3/caspase-1 signaling pathway in vivo and in vitro.47 In the process of atherosclerosis, the deposition of low-density lipoprotein (LDL) plays an essential role in the damage of endothelial cells. Oxidized low-density lipoprotein (Ox-LDL) has also been confirmed to induce pyroptosis of endothelial cells, which plays a vital role in the course of the disease. In endothelial cells, Ox-LDL upregulates the expression of mixed-lineage kinase domain-like (MLKL) proteins at the mRNA and protein level, which are closely associated with the activation and secretion of caspase-1 and IL-1β, and it is also associated with the release of lactate depleting hydrogenase (LDH). Overexpression of MLKL increases the expression of caspase-1, IL-1β, pro-IL-1β and Ox-LDL-induced LDH. The NLRP3 specific inhibitor MCC950 abolished the activation of caspase-1 and the maturation of IL-1β induced by MLKL.48,49 There is also evidence that endothelial cell damage caused by pyroptosis in Kawasaki disease plays an important role in the disease process.50
Damage Arterial Wall Plaque Stability
Atherosclerosis usually begins insidiously and progresses slowly, and often does not show obvious clinical symptoms in the early stages: large and medium-sized musculoskeletal arteries, most commonly the aorta, coronary arteries, and cerebral arteries. With the continuous development of AS, the deposition of atheroma on the vessel wall gradually increases, and the vessel wall gradually becomes thicker and harder, making the lumen of the blood vessels becomes thinner, and the blood flow slows down, even eventually leading to blockages and impaired blood supply, causing a host of diseases.51
Although the death of macrophages in the early stages of atherosclerotic lesions has been reported to promote necrotic core formation and atherosclerotic plaque instability,52 during atherosclerosis, macrophage death is closely associated with inflammation. During the disease process, dying macrophages do not adequately clear atherosclerotic plaque progression. The accumulation of these dead cells leads to the formation of a necrotic core and the release of inflammatory cytokines, chemokines, proteases, which ultimately leads to inflammation, increased susceptibility to atherosclerotic plaque rupture, and the formation of thrombi.53 In diabetic atherosclerotic (DA) rats, small-dose, slow administration of erucic acid (SA) suppressed serum endothelin-1 (ET-1), and IL-1β in mice, inhibited the pyroptosis of bone marrow macrophages and inhibited the expression of eosinophils ASC, NRLP3, and caspase-1. The expression of LncRNA metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) was significantly upregulated in DA rat macrophages, and small doses of SA reduced MALAT1 expression. Gene overexpression and knockdown experiments indicate that MALAT1 promotes normal macrophage pyroptosis. Furthermore, in macrophages coincubated with high glucose and oxidized LDL, 1-μ-MSA inhibited pyroptosis in a manner similar to MALAT1 knockdown. Transfection of the pcDNA-MALAT1 expression vector antagonized the SA-induced decrease in MALAT1 expression and macrophage hypoplasia.54,55
Angiogenesis
Angiogenesis is a process by which new capillaries and blood vessels arise from the pre-existing vascular system and is tightly regulated by many anti-angiogenic factors.56 After the occurrence of ischemic diseases, the prognosis of dyslipidemia-related diseases such as myocardial ischemia, peripheral artery disease, ischemic stroke, may be related to insufficient angiogenesis.57 Studies have found that reduced angiogenesis can aggravate the progression of diseases such as myocardial ischemia, peripheral artery disease, and ischemic stroke.58 Dyslipidemia and the inflammatory environment will inhibit endothelial cell proliferation and angiogenesis and aggravate the prognosis of ischemia. Some scholars have prevented the occurrence of pyroptosis by inhibiting the activation of caspase-1. The results show that inhibiting caspase-1 can improve the tube formation of lysophosphatidylcholine-treated human aortic endothelial cells while reducing caspase-1 can improve mice angiogenesis and blood supply of hindlimb ischemic tissues, and confirmed that inhibiting endothelial cell atherosclerosis caspase-1 activation can improve angiogenesis and ischemic prognosis.59 In addition, H.A.MENA and his associates demonstrated experimentally that human recombinant histones trigger endothelial progenitor pyroptosis. This response can significantly inhibit angiogenesis, phenomena such as proliferation, migration/chemistry and the formation of rope-like structures.60 Therefore, pyroptosis is closely related to angiogenesis, but its specific mechanism needs to be further investigated.
Myocardial Hypertrophy
Myocardial hypertrophy is a compensatory response to stress or stimuli that can lead to arrhythmias and heart failure.61 Multiple molecular mechanisms have been found to be involved, but cardiac hypertrophy remains challenging to treat.62,63 IL-1β and IL-18 produced by pyroptosis can recruit other immune cells and induce the synthesis of IL-6, which acts as a ligand and binds to the relevant receptor, causing a homodimer to form with its linked GP130.64 At the same time, tyrosine kinases are activated, promoting increased transcriptional activity of cellular genes.A pathological model of cardiac hypertrophy in mice was developed by applying pressure overload to the mouse heart using aortic transverse contraction (TAC).65 Neonatal mouse cardiomyocytes were extracted, and cardiomyocyte angiotensin II (Ang II) was used to induce cardiomyocyte hypertrophy in vitro. Detection of caspase-1 and IL-1β expression revealed a significant increase in caspase-1 and IL-1β expression levels during the process of myocardial hypertrophy.66 Subsequently, we administered a caspase-1 inhibitor in conjunction with Ang II. The results showed that caspase-1 inhibitors attenuated the thickening effect of Ang II, which was associated with the down-regulation of caspase-1 and IL-1β expression. The experimental results provide new evidence that caspase-1-mediated pyroptosis is involved in the process of myocardial hypertrophy, and that inhibition of caspase-1 expression will alleviate the degree of myocardial hypertrophy, opening up new possibilities for its treatment,67 in diabetic cardiomyopathy (DCM) model in Sprague-Dawley rats treated with a high-fat diet plus low-dose streptozotocin. It was using siRNA (CMKLR1-siRNA) to down-regulate the expression of CMKLR1 (a G protein-coupled receptor that is an inducer of inflammation). The aim of the study was to evaluate the role of CMKLR1 in dilated cardiomyopathy. Enhanced expression of CMKLR1, NLRP3, caspase-1, activated caspase-1 and activated IL-1β in liver tissues of rats with enlarged cardiomyopathy as a model. DCM model rat cardiac hypertrophy, ultrastructural disorders, and functional impairment. By observing the pyroptosis process, it was found that silencing CMKLR1 suppressed NLRP3 expression and activated caspase-1 and IL-1β. CMKLR1-siRNA treatment alleviates cardiac inflammation, reduces cardiac hypertrophy and improves cardiac function. Silencing of either CMKLR1 or NLRP3 inhibited the expression of activated caspase-1, IL-1β and reduced the occurrence of pyroptosis. Thus, pyroptosis causes pathological changes in cardiac hypertrophy, and understanding the specific mechanisms of pyroptosis, and cardiac hypertrophy will help us to open up new ideas and new methods to inhibit cardiac hypertrophy.52,68
Myocardial Fibrosis
Myocardial fibrosis is an essential topic of modern medical research,69 heartbeat relies on the normal contraction and diastolic ability of the myocardium, and myocardial fibrosis leads to increased myocardial stiffness and decreased myocardial contraction and diastolic function, which in turn affects the heart’s pumping function.70–72 Myocardial fibrosis is a critical period in the progression of cardiac function from the compensated to the decompensated phase, which can lead to ventricular remodeling and eventually to heart failure. Therefore, a critical prognostic factor in cardiovascular disease.73–75 Activation of caspase-1 induces pyroptosis and release of the pro-inflammatory cytokines IL-1β and IL-18 initiates amplification of the inflammatory cascade, leading to endothelial dysfunction, which in turn produces or adds to the development of myocardial fibrosis. Myocardial fibrosis formation is associated with multiple complex mechanisms such as oxidative stress, chemokine families, NLRP3 inflammatory microsomes, pro-inflammatory cytokines, growth factors, and non-coding RNAs.76 Curcumin is the richest polyphenol in the dietary spice turmeric and is known to be the yellow curry pigment. Curcumin can reduce inflammation by inhibiting inflammatory cytokines, suppressing macrophage infiltration, and modulating immune cell activity, thereby slowing myocardial fibrosis.77
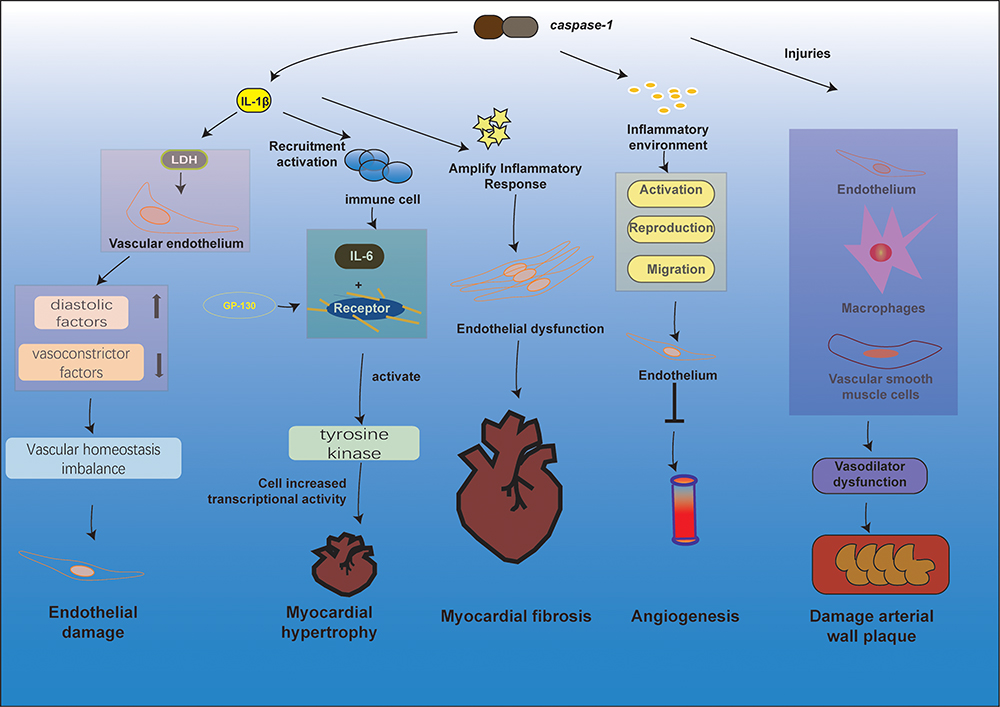 |
Figure 2 Pathological changes in the cardiovascular system through different pathways of pyroptosis. The release of IL-1β during anxiety is closely related to the release of LDH, and when the vascular endothelium is exposed to LDH and inflammatory substances, the release of diastolic factors decreases and vasoconstrictor factors increase, breaking the homeostasis of vascular homeostasis, leading to endothelial damage. In response to stimuli such as high blood lipids and oxidatively modified LDL, activation of Caspase-1 mediates the pyroptosis and inflammatory response of vascular endothelial cells, macrophages, and vascular smooth muscle cells, leading to vasodilatory dysfunction, formation of necrotic centers, stabilization of atherosclerotic plaques, and ultimately atherosclerosis. The inflammatory environment caused by inflammatory substances such as caspase-1 inhibits the activation, proliferation, and migration of endothelial cells and reduces angiogenesis. IL-1β and IL-18 produced by pyroptosis can recruit and activate other immune cells to induce the synthesis of the inflammatory factor IL-6, which acts as a ligand and, upon binding to the relevant receptor, causes the GP130 attached to it to form a homodimer, and tyrosine kinases are activated to promote increased cellular gene transcriptional activity. Activation of caspase-1 induces cellular pyroptosis and release of the pro-inflammatory factors IL-1β and IL-18 initiates amplification of the inflammatory cascade, leading to endothelial dysfunction, which in turn induces or adds to the development of myocardial fibrosis. |
Role of Pyroptosis in Cardiovascular Disease
The activation of NLRP3 inflammasome is strongly associated with the induction of pyroptosis, and NLRP3 is associated with hyperlipidemia, diabetes, and cardiovascular risk factors such as hypertension, obesity and hyperhomocysteinemia are associated. Pyroptosis has been found to be an essential factor in triggering cardiovascular inflammation, therefore, it may play a role in the pathogenesis of cardiovascular disease important role (Table 2).29
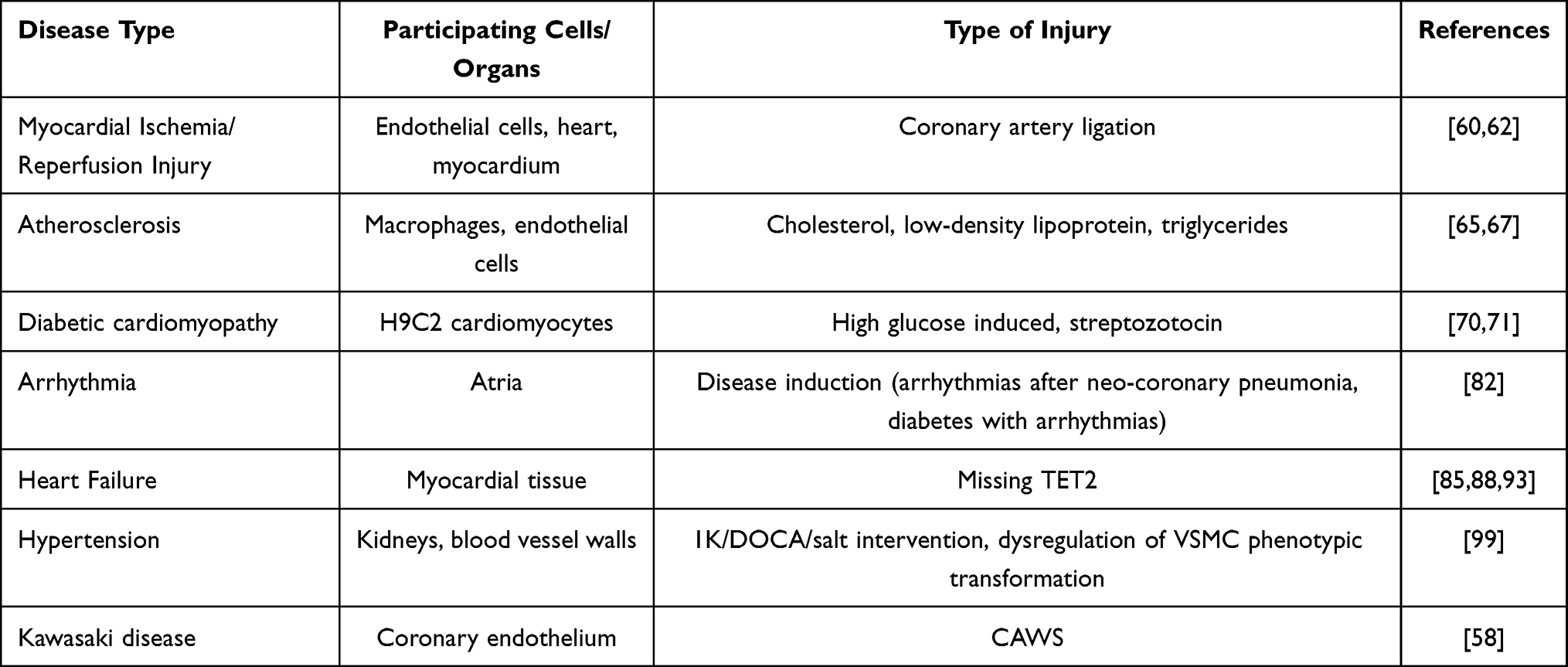 |
Table 2 Pyroptosis and NLRP3 Involvement of Organs and Modes of Injury in Various Cardiovascular Diseases |
Myocardial Ischemia/Reperfusion Injury
Myocardial infarction (MI) is a severe coronary artery-related disease, mainly caused by coronary artery atherosclerosis thrombosis or myocardial oxygen supply and demand imbalance, after the destruction of atherosclerosis, the released plaque can gather platelets, leading to coronary artery obstruction, resulting in myocardial ischemia and necrosis.78 There is some evidence that myocardial infarction is accompanied by a sterile inflammatory response, leading to leukocyte accumulation and subsequent wound resorption and scarring. It has been found that there is a general increase in the levels of inflammatory cytokines such as NLRP3 in myocardial cells that develop infarction and that aseptic inflammation after myocardial infarction is a significant factor in the development of aseptic inflammation. Responses are also generated primarily through inflammatory factor activation and release. It has been suggested that the inhibitory effect of Sirt1 on NLRP3 inflammasome is involved in a variety of diseases and that Sirt1 overexpression is a significant factor in the inflammatory response can effectively ameliorate MI-induced myocardial injury, so that Sirt1 can inhibit NLRP3 inflammasome activation, thereby reduces pyroptosis and myocardial infarction.79
Acute occlusion of coronary arteries can lead to myocardial infarction and is the leading cause of premature death. Timely recovery of myocardial blood flow can prevent excessive death of myocardial cells and improve clinical efficacy. It is now understood that in addition to ischemia-reperfusion injury, the process of reperfusion paradoxically leads to further damage known as myocardial ischemia-reperfusion (I/R) injury. Although necrosis is the primary mechanism of cell death after reperfusion, the pyroptosis pathway is currently considered to be involved in ischemia-reperfusion (I/R) injury.80 It has been suggested that cardiac troponin I-interacting kinase (TNNI3K) exacerbates ischemia-reperfusion (IR) through oxidative stress damage, thereby promoting cardiomyocyte death, by designing and synthesizing a novel TNNI3K inhibitor in a mouse model. TNNI3K inhibitor 6o inhibits cardiomyocyte pyroptosis and apoptosis and reduces circulation by interfering with p38MAPK activation Cardiac troponin I (cTnI) leakage decreased myocardial tissue damage and significantly reduced the area of myocardial infarction in rats.81
In the latest study, a model of acute myocardial infarction was established by electroacupuncture pretreatment of C57BL/6 mice for 3 days, followed by ligation of the left anterior descending coronary artery. Detection of myocardial injury revealed that it reduced infarct size, increased short fraction (FS) and ejection fraction (EF), and reduced the degree of inflammation after AMI injury. Meanwhile, EAP inhibited the expression of NLRP3, cleaved caspase-1 and IL-1β in ischaemic myocardial tissue and suppressed the expression of F4/80, CD11b, CD206 and activated M2 macrophages, and reduced the expression of Ly-6GCD11b neutrophils in ischaemic myocardial and splenic tissue.82 There is increasing evidence that artemisinin also exerts cardioprotective effects, with less myocardial damage, significant inhibition of myocardial autophagy, improved mitochondrial electron transport chain activity, and reduced NLRP3, ASC, cleaved caspase-1, and IL-1β in I/R rats treated with artemisinin.83
Atherosclerosis
Clinical practice has shown that atherosclerosis can be a serious health hazard, and it has become the leading cause of death among the sick population in developed countries.84 In recent years, the incidence of atherosclerosis has been rising rapidly and is characterized by fibrous tissue and lipid deposition in the intima of elastic arteries.85 Atherosclerosis can be triggered by endothelial damage and lipid deposition leading to thrombosis, thickening of the vessel wall, and hardening of the arteries. Subjected to physical (flow shear stress, tension, cramping or hypoxia) and chemical (oxidized LDL, free radicals, inflammatory response) Stimulated by (factor and infection) factors, endothelial cells release a large number of cytokines and adhesion molecules.86 Oxidised low-density lipoprotein (Ox-LDL) is a protein involved in oxidative stress damage and plays an important role in the disease process of atherosclerosis. This is because Ox-LDL is taken up by macrophages via scavenger receptors and Ox-LDL receptors, rather than by LDL receptors, leading to lipid deposition and macrophage foam formation. Recently, scientists have found that NLRP3 Inflammatory vesicles and caspase-1-mediated pyroptosis are involved in the formation and development of atherosclerosis, especially in the late stages of the disease. Caspase-1-mediated pyroptosis becomes a major mortality factor for Ox-LDL-processing macrophages in the atherosclerotic disease process. Activation of caspase-1 during death requires NLRP3 inflammatory vesicles, which can be activated by Ox-LDL and cholesterol crystals, which are most abundant in atherosclerotic lesions. As mentioned above, Caspase-1 mediates the breakdown of IL-1β precursors, activating IL-1β, which recruits and activates other immune cells and induces inflammatory cytokines such as IL-6, chemokines and adhesion molecules. This process promotes the development of atherosclerosis.87 In the high-fat diet (HFD)-induced (ApoE) mouse model of atherosclerosis, serum homocysteine, cholesterol, and triglyceride levels were elevated, atherosclerosis progression was accelerated, and macrophage infiltration into atherosclerotic lesions was increased, all of which were attenuated by piperonyl E (Peperomin E) treatment. Significantly elevated levels of inflammation-related regulators accompanied by increased expression of β-nuclear transcription factor B inhibitor B (κBα, IκBα) and nuclear factor-κB (NF-κB), a process that can be blocked by pepe, were found in arterial tissue of high-fat feed-fed ApoE mice with NLRP3 inflammatory microsomal activation. In vitro, silencing NLRP3 with small interfering RNA effectively inhibited oxidized LDL-induced ASC and caspase-1 expression, IL-1β and IL-18 production in human aortic endothelial cells. Further experiments showed that the NLRP3-ASC pathway was activated by reactive oxygen radicals (ROS) because the ROS scavenger of N-acetylcysteine (NAC) was blocked, and the addition of pepe further reduced the activity of the NLRP3-ASC pathway. However, NLRP3 overexpression eliminates the anti-inflammatory effect of pepe on Ox-LDL-incubated HAECs.88 Thus, caspase-1-dependent pyroptosis plays a vital role in the atherosclerotic disease process.
Statins are widely used in the treatment of hyperlipidaemia and cardiovascular disease. In addition to their lipid-lowering effects, statins have immunomodulatory, anti-inflammatory, antioxidant and anti-apoptotic functions. A growing body of research suggests that NLRP3 inflammatory vesicles and their downstream mediators are important targets for statins in the treatment of inflammatory diseases. Statins and hsCRP target LDL cholesterol by reducing cholesterol synthesis and increasing HDL cholesterol levels. Statins also act through immunomodulatory functions. Similar to atorvastatin, frequent administration of statins inhibited the TLR4/MyD88/NF-kB pathway associated with NLRP3 expression in human monocytes (THP-1) and induced a reduction in IL-1β.89
Diabetic Cardiomyopathy
Diabetic cardiomyopathy (DCM) is a common complication in the later stages of diabetes, leading to heart failure, arrhythmias and sudden death dengyixilie undesirable consequences. Metabolic alterations, mitochondrial dysfunction, oxidative stress, inflammation, cell death and extracellular matrix remodelling may all contribute to the pathogenesis of DCM. In diabetes, cleaved caspase-1 is activated following expression of NLRP3 inflammatory microsomes. In turn, it leads to the conversion of interleukin-1 beta precursors (pro-IL-1β) and pro-IL-18 to mature IL-1β and IL-18.90
Long non-coding RNAs (lncRNAs) are RNAs that are more than 200 nucleotides long and do not have a protein-coding function. The full name of lncRNA Kcnq1ot1 is KCNQ1 overlapping transcript 1, and lncRNA Kcnq1ot1 can regulate caspase-1 expression through miR-214-3p in the male C57BL/6 mouse DCM model. Using lentiviral transfection of the lncRNA Kcnq1ot1 revealed that silencing Kcnq1ot1 attenuated high glucose-induced cardiomyocyte pyroptosis in diabetic mice by affecting the expression of miR-214-3p and caspase-1.52,91
The study showed that the mRNA level and protein expression of caspase-1 was significantly increased in rat diabetic myocardium, and further investigations revealed that NLRP3 inflammasome activation of caspase-1-mediated pyroptosis plays an important role in the development of diabetic cardiomyopathy.92
In a study by Yebin Xie and his colleagues, it was found that chemerin may, through its receptor CMKLR1 induces inflammation NLRP3-mediated IL-1β caspase-1 activation and Cardiomyocyte pyroptosis. Increased expression of chemerin, CMKLR1, NLRP3, caspase-1, activated caspase-1 and mature IL-1β in DCM experiments using rats as animal models. The myocardium of the DCM model rats showed fibrosis, hypertrophy, ultrastructural disorders and impaired function, and the onset of pyroptosis was observed in both in vitro and in vivo experiments. Silencing CMKLR1 reduces NLRP3 expression and inhibits caspase-1 and IL-1β activation. CMKLR1-siRNA treatment reduces cardiac inflammation, fibrosis, hypertrophy and pyrosis, and improves cardiac function.93
To investigate whether ticagrelor, a P2Y12 receptor antagonist, and dapagliflozin, a sodium-glucose transporter-2 inhibitor, can inhibit NLRP3 inflammasome activation. Eight-week-old BTBR and wild-type mice were treated with drug-free, dapagliflozin, ticagrelor or a combination of them for 12 weeks. Their heart damage and inflammasome expression were tested. The results showed that both dapagliflozin and ticagrelor attenuated the progression of diabetic cardiomyopathy, NLRP3 inflammasome activation and fibrosis in BTBR mice, and that the combination had an additive effect.94
Arrhythmia
Arrhythmia can exist as a single or cardiomyopathy-associated disease, and myocardial damage associated with arrhythmia is often accompanied by temporary or permanent inflammatory reactions.
The novel coronavirus (CoV) causative virus Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) binds to cell surface ACE2 receptors and infects host cells, causing 2019 coronavirus disease (COVID −19).95,96 SARSCoV-2 has been shown to trigger an excessive systemic inflammatory response that can lead to cardiac damage, such as arrhythmias, in addition to acute lung injury and acute respiratory distress syndrome.97 Clinical results show that the impact of such diseases on the cardiovascular system of COVID-19 patients relies on inflammatory mediators, such as NLRP3, IL-1β, and TNF-.98,99 In vitro studies have demonstrated that SARS-CoV can open reading frame 3a (ORF3a) accessory protein, which acts as a potent activator of NLRP3 inflammatory microsomes, and thus SARS-CoV-2 may use the same mechanism of action to induce NLRP3-dependent cytokine storms and to lead to a severe inflammatory response. Although we have demonstrated that the arrhythmias caused by SARS-CoV-2 are associated with inflammatory microsomes such as NLRP3, there is no direct evidence that atrial fibrillation caused by SARS-CoV-2 is associated with pyroptosis, so further mechanisms remain to be explored.100
Atrial fibrillation is the most common arrhythmia, and its mechanism of action has not been elucidated to date, but there is considerable evidence that it is associated with inflammation.101 Elevated expression of NLRP3 inflammatory microsomes in cardiomyocytes from patients with persistent AF has been found to be increased by knockout techniques, and drug Inhibition of NLRP3 expression by inhibitors slows the progression of AF.102 In a rabbit model of diabetes with atrial fibrillation, atrial caspase-1 activity was found to be elevated, and serum IL-1β protein and IL-18 levels were elevated.103 To further confirm whether the activation and expression of NLRP3 inflammatory vesicles is altered in the diabetic rabbit model, we assessed NLRP3 protein levels and caspase-1 activity in atrial tissue lysates of different groups of rabbits.104 The overactive NLRP3 signaling pathway promotes the expression of RyR2 and caspase-1-mediated pyroptosis mechanism, resulting in increased RyR2-mediated arrhythmic SRCa2+ release events and increased inflammatory cytokine secretion. Thus, pyroptosis plays an essential role in the development of atrial fibrillation.105
In a study of the role of NLRP3 inflammatory vesicles in the pathophysiology of cardiac glycoside-induced cardiac inflammation and dysfunction, the cardiac glycoside vabain caused cardiac dysfunction and injury in wild-type mice stimulated with low doses of LPS, but no cardiac dysfunction was observed in mice treated with either vabain or LPS alone, thus suggesting that cardiac glycosides promote cardiac inflammation and dysfunction via NLRP3 inflammatory vesicles and providing new insights into the mechanisms of cardiac glycoside adverse effects.106
Heart Failure
IL-1β induces calcium overflow from the myocardial sarcoplasmic reticulum, which directly affects the calcium environment, myocardial contraction, and excitatory contraction IL-1β also stimulates the synthesis of inducible nitric oxide synthase (iNOS).107 Leading to cell death and tissue remodeling.108 Apoptosis-associated microproteins promote the inflammatory microsomal formation, which activates caspase-1, releases IL-1β and IL-18 promotes tumor necrosis factor-α (TNF-α) and INOS generation, which in turn reacts on caspase-1, thereby forming an inflammatory waterfall that together promotes pyroptosis and ultimately loss of cardiomyocytes and leading to heart failure.
The protein encoded by TET2 (10–11 translocation 2) is an epigenomic regulatory enzyme. A causal role of somatic TET2 mutations in atherosclerosis has been demonstrated. On this basis, using a mouse model of hyperlipidemia, bone marrow reconstituted in part by TET2-deficient hematopoietic cells resulted in their clonal expansion and a significant increase in atherosclerotic plaque size in mice.109 Mechanistically, TET2-deficient macrophages secrete more IL-1β and NLRP3, which leads to increased atherosclerotic plaque activity. However, recent studies have found that TET2 deficiency may have an even greater impact, as it may cause heart failure in MI patients.110,111 In both LAD permanent ligation and TAC models of heart failure, tet2-mediated clonal hematopoiesis results in the upregulation of IL-1β. IL-1β is activated and secreted upon lysis in the cytoplasm of NLRP3 inflammatory microsomal innate immune cells.112 The use of NLRP3 inhibitors, on the other hand, protects against heart failure caused by both models, and these data suggest that IL-1βblockade or the use of NLRP3 inflammatory microsomal inhibition may be particularly useful in the treatment of heart failure in individuals carrying these mutations, thus allowing us to explore new therapeutic ideas and preventive measures for heart failure in such patients from a pyroptosis perspective.113
Patients with heart failure often experience alterations in electrical remodeling that lead to cardiac arrhythmias.114 In cardiac macrophages, activation of TLR2 and NLRP3 inflammasome induces the production of IL-1β antioxidant protein in diabetic mice.IL-1β acetylase reduces the density of l-type Ca2+ (ICaL) and activates ROS signaling and protein kinase C, leading to arrhythmias and heart failure.115 IL-1β acetylase reduces the density of l-type Ca2+ (ICaL) and activates ROS signaling and protein kinase C, leading to arrhythmias and heart failure.IL-1β acetylase reduces the frequency of l-type Ca2+ (ICaL) and activates ROS signaling and protein kinase C, leading to arrhythmias and heart failure.116 During heart failure, sustained inflammatory stimuli lead to collagen accumulation and myocardial fibrosis, which is exacerbated by myocardial fibroblasts that then induce the production of inflammatory mediators. Anti-fibrotic drugs used to treat cardiac fibroblasts in mice inhibit the expression of NLRP3 and ASC and the assembly of inflammatory microsomes and block the NLRP3 transforming growth factor (TGF inhibitor 1)-Smad pathway.117
Administration of NaS after myocardial infarction in mice resulted in smaller left ventricular infarct scar areas, apoptotic signals measured by the Bcl-2/Bax ratio, and inhibition of cofilin-2 expression, a specific target of miR-21. NaS treatment after myocardial infarction maintained left ventricular function and improved survival by reducing inflammatory vesicle-mediated maladaptive remodeling and slowed the progression of heart failure.118
Hypertension
Chronic inflammation of the kidney and vascular wall is a major cause of hypertension, and dysregulation of VSMC phenotypic transformation is responsible for the development of hypertension and its associated vascular pathology. Clinical studies have also confirmed that the levels of pro-inflammatory cytokines IL-1β and IL-18 are elevated in the circulation and blood vessels of hypertensive patients. NLRP3 activation, inflammation, and phenotypic transformation were found in spontaneously hypertensive rats (SHR), which was attenuated by knockdown of NLRP3 in SHR-derived vascular smooth muscle cells (VSMCs) or by NLRP3 gene silencing in SHR aortas.119 A high-salt diet in rats induces activation of NF-κB, which further leads to activation of NLRP3 and caspase-1, and overload of ROS and PICs in the hypothalamic paraventricular nucleus. These changes and their interactions lead to sympathetic excitability, which ultimately accelerates the progression of hypertension. Thus, NF-κB plays a vital role in hypertension due to pyroptosis.120 Clinical studies have shown that the expression of aortic caspase-1 is positively correlated with TC, LDL-C, Lp (a), hypertension, and diabetes, but negatively correlated with HDL-C. However, the limitation of this study lies in the control group. Considering that 260 healthy people can rarely collect aortic tissue, the experiment had to select 261 renal arteries from kidney donors as the control group.121 By strictly regulating the relationship between pressure and urine sodium, the increase in blood pressure is accompanied by a compensatory decrease in proximal tubule sodium reabsorption, and the kidneys play an essential role in blood pressure homeostasis. In the 1K/DOCA/salt intervention of mouse kidney damage hypertension model, we observed that mouse hypertension is associated with the increase of NLRP3, ASC, pro-caspase-1 and pro-IL-1β mRNA levels, using Western blotting to measure caspase-1 p10 and IL-1β inhibitor p17 subunits, multiple immune response bands can be observed. In other environments, for example, gout and hyperuricemia are related to the formation of urate microcrystals in multiple sites, including the kidneys, which leads to the aggregation of inflammasomes. The application of uric acid-lowering compounds such as allopurinol can be used in many areas. Reduce blood pressure to a large extent.122 Inhibition of ASC expression provides protection against chronic pressurization, renal inflammation and pro-fibrosis of 1K/DOCA/salt, and hypertension of angiotensin II, which means that targeting inflammasome/IL-1β may be a new way to lower blood pressure123,124.
Kawasaki Disease
Kawasaki disease (KD) is more common in developed countries.125 It is a childhood heart disease that can cause permanent coronary artery damage and coronary artery aneurysms.126–128 At present, it is known that endothelial cell injury and inflammation are two important processes that lead to KD coronary endothelial dysfunction. In vivo experiments in KD patients showed that compared with healthy controls, the serum levels of ASC, caspase-1, IL-1β, IL-18, GSDMD and lactate dehydrogenase (LDH) in KD patients were significantly increased, the expression of NLRP3 mRNA in children with KD in the acute phase is positively correlated with the levels of C-reactive protein, IL-6, IL-1β, and prealbumin.129 Western blot analysis showed that the expression of GSDMD and IL-1β was significantly increased in the serum of KD patients.129 In cell experiments, in order to prove that the up-regulation of pyroptosis-related proteins is related to vascular endothelial damage, we used monocytes/macrophages (THP1 cell line) in KD-induced inflammatory conditions to simulate the impact of the inflammatory environment in vivo on endothelial cells. The results showed that in the in vitro model of KD treated ECs, the expression of pyroptosis-related proteins was upregulated. If GSDMD-derived inhibitor is added, the level of endothelial pyroptosis will decrease significantly, and the expression of pyroptosis-related protein will decrease. In addition, in the KD animal model, the inhibition of cathepsin B (CA074-Me) also rescued EC membrane rupture and reduced coronary artery inflammation. These evidences suggest that the EC pyroptosis of KD is mediated by cathepsin B, HMGB1/RAGE/cathepsin B signaling pathway may be one of the important pathways for endothelial cell pyroptosis activation. The KD mouse model induced by Candida albicans cell wall extract (CAWS) also validated the above viewpoint.130 Therefore, endothelial pyroptosis may play an important role in KD coronary artery endothelial injury.50
Genetic studies suggest that interleukin (IL)-1β may play a role in KD. Therefore, we explored the role of IL-1β in a mouse model of KD in which Lactobacillus casei cell wall extract (LCWE) was injected, and the results strongly suggested a key role for caspase-1 and IL-1β in the development of coronary lesions in a mouse model of KD blocked by IL-1 receptor antagonists. Thus, anti-lL-1β therapeutic strategies may be an effective and more targeted treatment for the prevention of coronary artery lesions in KD.131
Conclusion
Pyroptosis is a death method characterized by cell swelling, cell membrane rupture, and the release of inflammatory cytokines. At present, there are known caspase-1-dependent classic pathways, caspase-4/5/11-mediated non-classical pathways, and the newly discovered caspase-3-dependent apoptotic pathways in recent years. Among the traditional signaling pathways, NLRP3 and caspase-1 are the most widely involved in cardiovascular diseases. By affecting angiogenesis, myocardial hypertrophy, destabilizing plaques on the arterial wall, myocardial fibrosis, endothelial injury and other pathological processes, they play an important role in myocardial infarction/reperfusion, arrhythmia, Kawasaki disease and other cardiac diseases. Therefore, we can use our understanding of the molecular mechanisms of NLRP3 inflammatory vesicle activation to identify effective NLRP3 inhibitors or inhibitory pathways and assess their therapeutic potential. Scientists have identified many NLRP3 inhibitors, including those that directly inhibit NLRP3 or indirectly inhibit inflammatory vesicle components or related signalling events. However, it is worth noting that the mechanism or precise target of NLRP3 inhibition is not fully elucidated and there is a potential risk of off-targeting.
This review lists the latest progress and experimental studies of pyroptosis in cardiovascular diseases, studies have shown that lentivirus transfection, inhibitors and other means, by inhibiting the expression of NLRP3 inflammatory bodies or caspase-1 can reduce the degree of pyroptosis, so as to reduce cell damage and decrease the progress of the disease. This method may be a new target for the treatment of cardiac diseases. However, prospective clinical trials are needed to translate this into clinical practice. Future research needs to answer many unanswered questions. For example, can drugs or antibodies targeting the GSDMD inflammatory pathway enhance the therapeutic potential of a single NLRP3 inflammatory microsomal inhibitor for cardiovascular disease? Further exploration of new signaling pathways for pyroptosis will provide an important basis for the development of new therapeutic drugs. In addition, to promising research prospects in the cardiovascular field, pyroptosis is also involved in the process of inflammatory diseases such as tumors, neuroinflammation, and infectious diseases. We can extend the previous research on inflammatory pathways to pyroptosis. In this field, we can better explain and understand the molecular mechanism and treatment prospects of pyroptosis and open up a new situation in the treatment of inflammatory diseases.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Zychlinsky A, Prevost MC, Sansonetti PJ. Shigella flexneri induces apoptosis in infected macrophages. Nature. 1992;358(6382):167–169. doi:10.1038/358167a0
2. Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol. 2001;9(3):113–114. doi:10.1016/S0966-842X(00)01936-3
3. Zeng C, Wang R, Tan H. Role of Pyroptosis in Cardiovascular Diseases and its Therapeutic Implications. Int J Biol Sci. 2019;15(7):1345–1357.
4. Jia C, Chen H, Zhang J, et al. Role of pyroptosis in cardiovascular diseases. Int Immunopharmacol. 2019;67:311–318. doi:10.1016/j.intimp.2018.12.028
5. Zhaolin Z, Guohua L, Shiyuan W, Zuo W. Role of pyroptosis in cardiovascular disease. Cell Prolif. 2019;52(2):e12563. doi:10.1111/cpr.12563
6. Sl F, Bt C. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun. 2005;73(4):1907–1916.
7. Sl F, Bt C. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol. 2006;8(11):1812–1825. doi:10.1111/j.1462-5822.2006.00751.x
8. Li Z, Tian X, Ji X. ULK1-ATG13 and their mitotic phospho-regulation by CDK1 connect autophagy to cell cycle. PLoS Biol. 2020;18(6):e3000288. doi:10.1371/journal.pbio.3000288
9. Suzuki H, Osawa T, Fujioka Y, Noda NN. Structural biology of the core autophagy machinery. Curr Opin Struct Biol. 2017;43:10–17. doi:10.1016/j.sbi.2016.09.010
10. Tixeira R, Shi B, Parkes M, et al. Gasdermin E Does Not Limit Apoptotic Cell Disassembly by Promoting Early Onset of Secondary Necrosis in Jurkat T Cells and THP-1 Monocytes. Front Immunol. 2018;9:2842. doi:10.3389/fimmu.2018.02842
11. Chaudhary G, Yadav P, Yadav A, et al. Necrosis and necroptosis in germ cell depletion from mammalian ovary. J Cell Physiol. 2019;234(6):8019–8027. doi:10.1002/jcp.27562
12. Kishino A, Hayashi K, Maeda M, et al. Caspase-8 Regulates Endoplasmic Reticulum Stress-Induced Necroptosis Independent of the Apoptosis Pathway in Auditory Cells. Int J Mol Sci. 2019;20:23. doi:10.3390/ijms20235896
13. Wang M, Wan H, Wang S, et al. RSK3 mediates necroptosis by regulating phosphorylation of RIP3 in rat retinal ganglion cells. J Anat. 2020;237(1):29–47. doi:10.1111/joa.13185
14. Wang S. The role of Caspase-1/GSDMD-mediated pyroptosis in Taxol-induced cell death and a Taxol-resistant phenotype in nasopharyngeal carcinoma regulated by autophagy. Cell Biol Toxicol. 2020.
15. Liu Z, Wang C, Yang J, et al. Caspase-1 Engages Full-Length Gasdermin D through Two Distinct Interfaces That Mediate Caspase Recruitment and Substrate Cleavage. Immunity. 2020;53(1):106–114.e105. doi:10.1016/j.immuni.2020.06.007
16. Muendlein H, Jetton D, Connolly W, et al. cFLIP protects macrophages from LPS-induced pyroptosis via inhibition of complex II formation. Science. 2020;367(6484):1379–1384. doi:10.1126/science.aay3878
17. Shi J, Zhao Y, Wang Y, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514(7521):187–192. doi:10.1038/nature13683
18. Xia X, Wang X, Zheng Y, Jiang J, Hu J. What role does pyroptosis play in microbial infection? J Cell Physiol. 2019;234(6):7885–7892. doi:10.1002/jcp.27909
19. Wandel M, Kim B, Park E, et al. Guanylate-binding proteins convert cytosolic bacteria into caspase-4 signaling platforms. Nat Immunol. 2020;21(8):880–891. doi:10.1038/s41590-020-0697-2
20. Wang K, Sun Q, Zhong X, et al. Structural Mechanism for GSDMD Targeting by Autoprocessed Caspases in Pyroptosis. Cell. 2020;180(5):941–955.e920. doi:10.1016/j.cell.2020.02.002
21. Zhou B, Zhang J, Liu X, et al. Tom20 senses iron-activated ROS signaling to promote melanoma cell pyroptosis. Cell Res. 2018;28(12):1171–1185. doi:10.1038/s41422-018-0090-y
22. Zheng X, Zhong T, Ma Y, et al. Bnip3 mediates doxorubicin-induced cardiomyocyte pyroptosis via caspase-3/GSDME. Life Sci. 2020;242:117186. doi:10.1016/j.lfs.2019.117186
23. Zhang C, Li C, Wang Y, et al. Chemotherapeutic paclitaxel and cisplatin differentially induce pyroptosis in A549 lung cancer cells via caspase-3/GSDME activation. Apoptosis. 2019;24(3–4):312–325. doi:10.1007/s10495-019-01515-1
24. Wang Y, Gao W, Shi X, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547(7661):99–103. doi:10.1038/nature22393
25. Wang S. ROS-Mediated NLRP3 Inflammasome Activation in Brain, Heart, Kidney, and Testis Ischemia/Reperfusion Injury. Oxid Med Cell Longev. 2016;2016:2183026.
26. Long Y, Liu X, Tan X-Z. ROS-induced NLRP3 inflammasome priming and activation mediate PCB 118- induced pyroptosis in endothelial cells. Ecotoxicol Environ Saf. 2020;189:109937. doi:10.1016/j.ecoenv.2019.109937
27. Tang Y-S, Zhao Y-H, Zhong Y. Neferine inhibits LPS-ATP-induced endothelial cell pyroptosis via regulation of ROS/NLRP3/Caspase-1 signaling pathway. Inflammation Res. 2019;68(9):727–738. doi:10.1007/s00011-019-01256-6
28. Tavakoli Dargani Z, Singla R, Johnson T, Kukreja R, Singla DK. Exosomes derived from embryonic stem cells inhibit doxorubicin and inflammation-induced pyroptosis in muscle cells. Can J Physiol Pharmacol. 2018;96(3):304–307. doi:10.1139/cjpp-2017-0340
29. Zeng C, Wang R, Tan H. Role of Pyroptosis in Cardiovascular Diseases and its Therapeutic Implications. Int J Biol Sci. 2019;15(7):1345–1357. doi:10.7150/ijbs.33568
30. Que Y, Zhu T, Zhang F, Peng J. Neuroprotective effect of DUSP14 overexpression against isoflurane-induced inflammatory response, pyroptosis and cognitive impairment in aged rats through inhibiting the NLRP3 inflammasome. Eur Rev Med Pharmacol Sci. 2020;24(12):7101–7113. doi:10.26355/eurrev_202006_21704
31. Morimoto N, Okamura Y, Maekawa S, et al. ASC-deficiency impairs host defense against Aeromonas hydrophila infection in Japanese medaka, Oryzias latipes. Fish Shellfish Immunol. 2020;105:427–437. doi:10.1016/j.fsi.2020.07.027
32. Dang E, McDonald J, Russell D, Cyster J. Oxysterol Restraint of Cholesterol Synthesis Prevents AIM2 Inflammasome Activation. Cell. 2017;171(5):1057–1071.e1011. doi:10.1016/j.cell.2017.09.029
33. Westerterp M, Gautier E, Ganda A, et al. Cholesterol Accumulation in Dendritic Cells Links the Inflammasome to Acquired Immunity. Cell Metab. 2017;25(6):1294–1304.e1296. doi:10.1016/j.cmet.2017.04.005
34. Niyonzima N, Bakke S, Gregersen I, et al. Cholesterol crystals use complement to increase NLRP3 signaling pathways in coronary and carotid atherosclerosis. EBioMedicine. 2020;60:102985. doi:10.1016/j.ebiom.2020.102985
35. Silva G, Gierman L, Rakner J, et al. Cholesterol Crystals and NLRP3 Mediated Inflammation in the Uterine Wall Decidua in Normal and Preeclamptic Pregnancies. Front Immunol. 2020;11:564712. doi:10.3389/fimmu.2020.564712
36. Hedbrant A, Andersson L, Bryngelsson I, et al. Quartz Dust Exposure Affects NLRP3 Inflammasome Activation and Plasma Levels of IL-18 and IL-1Ra in Iron Foundry Workers. Mediators Inflamm. 2020;2020:8490908. doi:10.1155/2020/8490908
37. Liu G, Gu C, Liu M, et al. Protective role of p120-catenin on mitochondria by inhibiting NLRP3 in ventilator-induced lung injury. J Cell Mol Med. 2019;23(11):7360–7371. doi:10.1111/jcmm.14595
38. Yang S, Han Y, He J, et al. Mitochondria targeted peptide SS-31 prevent on cisplatin-induced acute kidney injury via regulating mitochondrial ROS-NLRP3 pathway. Biomed Pharmacother. 2020;130:110521. doi:10.1016/j.biopha.2020.110521
39. An Z, Su J. Acinetobacter baumannii outer membrane protein 34 elicits NLRP3 inflammasome activation via mitochondria-derived reactive oxygen species in RAW264.7 macrophages. Microbes Infection. 2019;21(3–4):143–153. doi:10.1016/j.micinf.2018.10.005
40. Campden R, Zhang Y. The role of lysosomal cysteine cathepsins in NLRP3 inflammasome activation. Arch Biochem Biophys. 2019;670:32–42. doi:10.1016/j.abb.2019.02.015
41. Nagoor Meeran M, Azimullah S, Laham F, et al. α-Bisabolol protects against β-adrenergic agonist-induced myocardial infarction in rats by attenuating inflammation, lysosomal dysfunction, NLRP3 inflammasome activation and modulating autophagic flux. Food Funct. 2020;11(1):965–976. doi:10.1039/C9FO00530G
42. Liu D, Zeng X, Li X, Mehta J, Wang X. Role of NLRP3 inflammasome in the pathogenesis of cardiovascular diseases. Basic Res Cardiol. 2018;113(1):5. doi:10.1007/s00395-017-0663-9
43. Karki R, Lee E, Sharma B, Banoth B, Kanneganti T. IRF8 Regulates Gram-Negative Bacteria-Mediated NLRP3 Inflammasome Activation and Cell Death. J Immunol. 2020;204(9):2514–2522. doi:10.4049/jimmunol.1901508
44. Choudhury S, Ma X, Abdullah S, Zheng H. Activation and Inhibition of the NLRP3 Inflammasome by RNA Viruses. J Inflamm Res. 2021;14:1145–1163. doi:10.2147/JIR.S295706
45. Ge X, Li W, Huang S, et al. The pathological role of NLRs and AIM2 inflammasome-mediated pyroptosis in damaged blood-brain barrier after traumatic brain injury. Brain Res. 2018;1697:10–20. doi:10.1016/j.brainres.2018.06.008
46. Romacho T, Valencia I, Ramos-González M, et al. Visfatin/eNampt induces endothelial dysfunction in vivo: a role for Toll-Like Receptor 4 and NLRP3 inflammasome. Sci Rep. 2020;10(1):5386. doi:10.1038/s41598-020-62190-w
47. Wang W, Wu Q, Sui Y, Wang Y, Qiu X. Rutin protects endothelial dysfunction by disturbing Nox4 and ROS-sensitive NLRP3 inflammasome. Biomed Pharmacother. 2017;86:32–40. doi:10.1016/j.biopha.2016.11.134
48. Wu Q, He X, Wu L, et al. MLKL Aggravates Ox-LDL-Induced Cell Pyroptosis via Activation of NLRP3 Inflammasome in Human Umbilical Vein Endothelial Cells. Inflammation. 2020;43(6):2222–2231. doi:10.1007/s10753-020-01289-8
49. Jin H, Ko Y, Park S, Kim H. P2YR activation by ATP induces oxLDL-mediated inflammasome activation through modulation of mitochondrial damage in human endothelial cells. Free Radic Biol Med. 2019;136:109–117. doi:10.1016/j.freeradbiomed.2019.04.004
50. Jia C, Zhang J, Chen H, et al. Endothelial cell pyroptosis plays an important role in Kawasaki disease via HMGB1/RAGE/cathespin B signaling pathway and NLRP3 inflammasome activation. Cell Death Dis. 2019;10(10):778. doi:10.1038/s41419-019-2021-3
51. Wu X, Zhang H, Qi W. Nicotine promotes atherosclerosis via ROS-NLRP3-mediated endothelial cell pyroptosis. Cell Death Dis. 2018;9(2):171. doi:10.1038/s41419-017-0257-3
52. Yang F, Qin Y, Lv J, et al. Silencing long non-coding RNA Kcnq1ot1 alleviates pyroptosis and fibrosis in diabetic cardiomyopathy. Cell Death Dis. 2018;9(10):1000. doi:10.1038/s41419-018-1029-4
53. Rashidi M, Wicks I, Vince J. Inflammasomes and Cell Death: common Pathways in Microparticle Diseases. Trends Mol Med. 2020;26(11):1003–1020. doi:10.1016/j.molmed.2020.06.005
54. Han Y, Qiu H, Pei X, Fan Y, Tian H, Geng J. Low-dose Sinapic Acid Abates the Pyroptosis of Macrophages by Downregulation of lncRNA-MALAT1 in Rats With Diabetic Atherosclerosis. J Cardiovasc Pharmacol. 2018;71(2):104–112. doi:10.1097/FJC.0000000000000550
55. Tumurkhuu G, Dagvadorj J, Porritt RA. Chlamydia pneumoniae Hijacks a Host Autoregulatory IL-1β Loop to Drive Foam Cell Formation and Accelerate Atherosclerosis. Cell Metab. 2018;28(3):432–448.e434. doi:10.1016/j.cmet.2018.05.027
56. Cosmai L, Gallieni M, Liguigli W, Porta C. Renal toxicity of anticancer agents targeting vascular endothelial growth factor (VEGF) and its receptors (VEGFRs). J Nephrol. 2017;30(2):171–180. doi:10.1007/s40620-016-0311-8
57. Ie S, Sa K. Vasculitis is an antiangiogenic state. J Am Soc Nephrology. 2012;23(1):8–10. doi:10.1681/ASN.2011111116
58. Jin F, Hagemann N, Brockmeier U, Schäfer ST, Zechariah A, Hermann DM. LDL attenuates VEGF-induced angiogenesis via mechanisms involving VEGFR2 internalization and degradation following endosome-trans-Golgi network trafficking. Angiogenesis. 2013;16(3):625–637. doi:10.1007/s10456-013-9340-2
59. L-p J, Lm F, Yf L, et al. Inhibition of Caspase-1 Activation in Endothelial Cells Improves Angiogenesis: a NOVEL THERAPEUTIC POTENTIAL FOR ISCHEMIA. J Biol Chem. 2015;290(28):17485–17494. doi:10.1074/jbc.M115.641191
60. Mena HA, Carestia A, Scotti L, Parborell F, Schattner M, Negrotto S. Extracellular histones reduce survival and angiogenic responses of late outgrowth progenitor and mature endothelial cells. J Thrombosis Haemostasis. 2016;14(2):397–410. doi:10.1111/jth.13223
61. Nakamura M, Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol. 2018;15(7):387–407.
62. Hou J, Kang Y. Regression of pathological cardiac hypertrophy: signaling pathways and therapeutic targets. Pharmacol Ther. 2012;135(3):337–354.
63. Tham Y, Bernardo B, Ooi J, Weeks K, McMullen J. Pathophysiology of cardiac hypertrophy and heart failure: signaling pathways and novel therapeutic targets. Arch Toxicol. 2015;89(9):1401–1438.
64. Peng X, Chen H, Li Y, Huang D, Huang B, Sun D. Effects of NIX-mediated mitophagy on ox-LDL-induced macrophage pyroptosis in atherosclerosis. Cell Biol Int. 2020;44(7):1481–1490. doi:10.1002/cbin.11343
65. Heger J, Schulz R, Euler G. Molecular switches under TGFβ signalling during progression from cardiac hypertrophy to heart failure. Br J Pharmacol. 2016;173(1):3–14. doi:10.1111/bph.13344
66. Man S, Karki R, Kanneganti T. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol Rev. 2017;277(1):61–75. doi:10.1111/imr.12534
67. Bai Y, Sun X, Chu Q, et al. Caspase-1 regulate AngII-induced cardiomyocyte hypertrophy via upregulation of IL-1β. Biosci Rep. 2018;38(2). doi:10.1042/BSR20171438
68. Li X, Du N, Zhang Q, et al. MicroRNA-30d regulates cardiomyocyte pyroptosis by directly targeting foxo3a in diabetic cardiomyopathy. Cell Death Dis. 2014;5(10):e1479. doi:10.1038/cddis.2014.430
69. Turner N. Inflammatory and fibrotic responses of cardiac fibroblasts to myocardial damage associated molecular patterns (DAMPs). J Mol Cell Cardiol. 2016;94:189–200. doi:10.1016/j.yjmcc.2015.11.002
70. Junttila M, Holmström L, Pylkäs K, et al. Primary Myocardial Fibrosis as an Alternative Phenotype Pathway of Inherited Cardiac Structural Disorders. Circulation. 2018;137(25):2716–2726. doi:10.1161/CIRCULATIONAHA.117.032175
71. González A, Schelbert E, Díez J, Butler J. Myocardial Interstitial Fibrosis in Heart Failure: biological and Translational Perspectives. J Am Coll Cardiol. 2018;71(15):1696–1706. doi:10.1016/j.jacc.2018.02.021
72. Liu C, Heckbert S, Lai S, et al. Association of Elevated NT-proBNP With Myocardial Fibrosis in the Multi-Ethnic Study of Atherosclerosis (MESA). J Am Coll Cardiol. 2017;70(25):3102–3109. doi:10.1016/j.jacc.2017.10.044
73. Senra T, Ianni B, Costa A, et al. Long-Term Prognostic Value of Myocardial Fibrosis in Patients With Chagas Cardiomyopathy. J Am Coll Cardiol. 2018;72(21):2577–2587. doi:10.1016/j.jacc.2018.08.2195
74. Henri O, Pouehe C, Houssari M, et al. Selective Stimulation of Cardiac Lymphangiogenesis Reduces Myocardial Edema and Fibrosis Leading to Improved Cardiac Function Following Myocardial Infarction. Circulation. 2016;133(15):1484–1497. doi:10.1161/CIRCULATIONAHA.115.020143
75. Ferreira V, Marcelino M, Piechnik S, et al. Pheochromocytoma Is Characterized by Catecholamine-Mediated Myocarditis, Focal and Diffuse Myocardial Fibrosis, and Myocardial Dysfunction. J Am Coll Cardiol. 2016;67(20):2364–2374. doi:10.1016/j.jacc.2016.03.543
76. Farris S, Don C, Helterline D, et al. Cell-Specific Pathways Supporting Persistent Fibrosis in Heart Failure. J Am Coll Cardiol. 2017;70(3):344–354. doi:10.1016/j.jacc.2017.05.040
77. Yu Y, Sun J, Wang R, Liu J, Wang P, Wang C. Curcumin Management of Myocardial Fibrosis and its Mechanisms of Action: a Review. Am J Chin Med. 2019;47(8):1675–1710. doi:10.1142/S0192415X19500861
78. Dreyer RP, Zheng X, Xu X. Sex differences in health outcomes at one year following acute myocardial infarction: a report from the China Patient-Centered Evaluative Assessment of Cardiac Events prospective acute myocardial infarction study. Eur Heart j Acute Cardiovascular Care. 2019;8(3):273–282. doi:10.1177/2048872618803726
79. Mao Q, Liang X-L, Zhang C-L, Pang Y-H, Lu Y-X. LncRNA KLF3-AS1 in human mesenchymal stem cell-derived exosomes ameliorates pyroptosis of cardiomyocytes and myocardial infarction through miR-138-5p/Sirt1 axis. Stem Cell Res Ther. 2019;10(1):393. doi:10.1186/s13287-019-1522-4
80. Rauf A, Shah M, Yellon DM, Davidson SM. Role of Caspase 1 in Ischemia/Reperfusion Injury of the Myocardium. J Cardiovasc Pharmacol. 2019;74(3):194–200. doi:10.1097/FJC.0000000000000694
81. Pang H, Wang N, Chai J. Discovery of novel TNNI3K inhibitor suppresses pyroptosis and apoptosis in murine myocardial infarction injury. Eur J Med Chem. 2020;197:112314. doi:10.1016/j.ejmech.2020.112314
82. Zhang T, Yang W, Wang Y, et al. Electroacupuncture preconditioning attenuates acute myocardial ischemia injury through inhibiting NLRP3 inflammasome activation in mice. Life Sci. 2020;248:117451. doi:10.1016/j.lfs.2020.117451
83. Wang F, Gao Q, Yang J, et al. Artemisinin suppresses myocardial ischemia-reperfusion injury via NLRP3 inflammasome mechanism. Mol Cell Biochem. 2020;474:171–180. doi:10.1007/s11010-020-03842-3
84. Xiao H, Lu M, Lin T, et al. Sterol regulatory element binding protein 2 activation of NLRP3 inflammasome in endothelium mediates hemodynamic-induced atherosclerosis susceptibility. Circulation. 2013;128(6):632–642. doi:10.1161/CIRCULATIONAHA.113.002714
85. Zhuang T, Liu J, Chen X, et al. Endothelial Foxp1 Suppresses Atherosclerosis via Modulation of Nlrp3 Inflammasome Activation. Circ Res. 2019;125(6):590–605. doi:10.1161/CIRCRESAHA.118.314402
86. Tumurkhuu G, Shimada K, Dagvadorj J, et al. Ogg1-Dependent DNA Repair Regulates NLRP3 Inflammasome and Prevents Atherosclerosis. Circ Res. 2016;119(6):e76–90. doi:10.1161/CIRCRESAHA.116.308362
87. Peng X, Chen H, Li Y, Huang D, Huang B, Sun D. Effects of NIX-mediated mitophagy on ox-LDL-induced macrophage pyroptosis in atherosclerosis. Cell Biol Int. 2020;44(7):1481–1490.
88. Yan J, Li M, Wang X, Lu Z, Ni X. Peperomin E (PepE) protects against high fat diet-induced atherosclerosis in Apolipoprotein E deficient (ApoE) mice through reducing inflammation via the suppression of NLRP3 signaling pathway. Biomed Pharmacother. 2018;105:862–869. doi:10.1016/j.biopha.2018.04.140
89. Parsamanesh N, Moossavi M, Bahrami A, Fereidouni M, Barreto G, Sahebkar A. NLRP3 inflammasome as a treatment target in atherosclerosis: a focus on statin therapy. Int Immunopharmacol. 2019;73:146–155. doi:10.1016/j.intimp.2019.05.006
90. Fink S, Cookson B. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun. 2005;73(4):1907–1916.
91. Yang F, Qin Y, Wang Y, et al. LncRNA KCNQ1OT1 Mediates Pyroptosis in Diabetic Cardiomyopathy. Cell Physiol Biochem. 2018;50(4):1230–1244. doi:10.1159/000494576
92. Li X, Du N, Zhang Q. MicroRNA-30d regulates cardiomyocyte pyroptosis by directly targeting foxo3a in diabetic cardiomyopathy. Cell Death Dis. 2014;5(10):e1479.
93. Xie Y, Huang Y, Ling X, Qin H, Wang M, Luo B. Chemerin/CMKLR1 Axis Promotes Inflammation and Pyroptosis by Activating NLRP3 Inflammasome in Diabetic Cardiomyopathy Rat. Front Physiol. 2020;11:381. doi:10.3389/fphys.2020.00381
94. Chen H, Tran D, Yang H, Nylander S, Birnbaum Y, Ye Y. Dapagliflozin and Ticagrelor Have Additive Effects on the Attenuation of the Activation of the NLRP3 Inflammasome and the Progression of Diabetic Cardiomyopathy: an AMPK-mTOR Interplay. Cardiovasc Drugs Therapy. 2020;34(4):443–461. doi:10.1007/s10557-020-06978-y
95. Dalamaga M, Karampela I, Mantzoros C. Commentary: could iron chelators prove to be useful as an adjunct to COVID-19 Treatment Regimens? Metabolism. 2020;108:154260. doi:10.1016/j.metabol.2020.154260
96. Klimke A, Hefner G, Will B, Voss U. Hydroxychloroquine as an aerosol might markedly reduce and even prevent severe clinical symptoms after SARS-CoV-2 infection. Med Hypotheses. 2020;142:109783. doi:10.1016/j.mehy.2020.109783
97. Liu J, Cao R, Xu M, et al. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discovery. 2020;6(1):16. doi:10.1038/s41421-020-0156-0
98. Jean S, Lee P, Hsueh P. Treatment options for COVID-19: the reality and challenges. J Microbiol Immunol Infect. 2020;53(3):436–443. doi:10.1016/j.jmii.2020.03.034
99. Maldonado V, Loza-Mejía M, Chávez-Alderete J. Repositioning of pentoxifylline as an immunomodulator and regulator of the renin-angiotensin system in the treatment of COVID-19. Med Hypotheses. 2020;144:109988. doi:10.1016/j.mehy.2020.109988
100. Moccia F, Gerbino A, Lionetti V, et al. COVID-19-associated cardiovascular morbidity in older adults: a position paper from the Italian Society of Cardiovascular Researches. GeroScience. 2020;42(4):1021–1049. doi:10.1007/s11357-020-00198-w
101. Wang S. Role of NLRP3-Inflammasome/Caspase-1/Galectin-3 Pathway on Atrial Remodeling in Diabetic Rabbits. J Cardiovasc Transl Res. 2020.
102. Chen G, Chelu MG, Dobrev D, Li N. Cardiomyocyte Inflammasome Signaling in Cardiomyopathies and Atrial Fibrillation: mechanisms and Potential Therapeutic Implications. Front Physiol. 2018;9:1115. doi:10.3389/fphys.2018.01115
103. Wan Y, Xu L, Wang Y, Tuerdi N, Ye M, Qi R. Preventive effects of astragaloside IV and its active sapogenin cycloastragenol on cardiac fibrosis of mice by inhibiting the NLRP3 inflammasome. Eur J Pharmacol. 2018;833:545–554. doi:10.1016/j.ejphar.2018.06.016
104. Yue L, Xie J, Nattel S. Molecular determinants of cardiac fibroblast electrical function and therapeutic implications for atrial fibrillation. Cardiovasc Res. 2011;89(4):744–753. doi:10.1093/cvr/cvq329
105. Chen G, Chelu M, Dobrev D, Li N. Cardiomyocyte Inflammasome Signaling in Cardiomyopathies and Atrial Fibrillation: mechanisms and Potential Therapeutic Implications. Front Physiol. 2018;9:1115.
106. Kobayashi M, Usui-Kawanishi F, Karasawa T, et al. The cardiac glycoside ouabain activates NLRP3 inflammasomes and promotes cardiac inflammation and dysfunction. PLoS One. 2017;12(5):e0176676. doi:10.1371/journal.pone.0176676
107. Cheng J, Akkerhuis K, Battes L, et al. Biomarkers of heart failure with normal ejection fraction: a systematic review. Eur J Heart Fail. 2013;15(12):1350–1362. doi:10.1093/eurjhf/hft106
108. Amit U, Kain D, Wagner A, et al. New Role for Interleukin-13 Receptor α1 in Myocardial Homeostasis and Heart Failure. J Am Heart Assoc. 2017;6(5):5. doi:10.1161/JAHA.116.005108
109. Maruyama S, Nakamura K, Papanicolaou K, et al. Follistatin-like 1 promotes cardiac fibroblast activation and protects the heart from rupture. EMBO Mol Med. 2016;8(8):949–966. doi:10.15252/emmm.201506151
110. Shibata R, Izumiya Y, Sato K, et al. Adiponectin protects against the development of systolic dysfunction following myocardial infarction. J Mol Cell Cardiol. 2007;42(6):1065–1074. doi:10.1016/j.yjmcc.2007.03.808
111. Shibata R, Ouchi N, Ito M, et al. Adiponectin-mediated modulation of hypertrophic signals in the heart. Nat Med. 2004;10(12):1384–1389. doi:10.1038/nm1137
112. Ørn S, Ueland T, Manhenke C, et al. Increased interleukin-1β levels are associated with left ventricular hypertrophy and remodelling following acute ST segment elevation myocardial infarction treated by primary percutaneous coronary intervention. J Intern Med. 2012;272(3):267–276. doi:10.1111/j.1365-2796.2012.02517.x
113. Sano S, Oshima K, Wang Y, et al. Tet2-Mediated Clonal Hematopoiesis Accelerates Heart Failure Through a Mechanism Involving the IL-1β/NLRP3 Inflammasome. J Am Coll Cardiol. 2018;71(8):875–886. doi:10.1016/j.jacc.2017.12.037
114. Monnerat G, Alarcón M, Vasconcellos L, et al. Macrophage-dependent IL-1β production induces cardiac arrhythmias in diabetic mice. Nat Commun. 2016;7(1):13344. doi:10.1038/ncomms13344
115. Long V, Bonilla I, Vargas-Pinto P, et al. Heart failure duration progressively modulates the arrhythmia substrate through structural and electrical remodeling. Life Sci. 2015;123:61–71. doi:10.1016/j.lfs.2014.12.024
116. El Khoury N, Mathieu S, Fiset C. Interleukin-1β reduces L-type Ca2+ current through protein kinase C activation in mouse heart. J Biol Chem. 2014;289(32):21896–21908. doi:10.1074/jbc.M114.549642
117. Westermann D, Lindner D, Kasner M, et al. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ Heart Fail. 2011;4(1):44–52. doi:10.1161/CIRCHEARTFAILURE.109.931451
118. Nguyen K, Chau V, Mauro A, et al. Hydrogen Sulfide Therapy Suppresses Cofilin-2 and Attenuates Ischemic Heart Failure in a Mouse Model of Myocardial Infarction. J Cardiovasc Pharmacol Ther. 2020;25(5):472–483. doi:10.1177/1074248420923542
119. Sun H, Ren X, Xiong X, et al. NLRP3 inflammasome activation contributes to VSMC phenotypic transformation and proliferation in hypertension. Cell Death Dis. 2017;8(10):e3074. doi:10.1038/cddis.2017.470
120. Qi J, Yu X, Shi X, et al. NF-κB Blockade in Hypothalamic Paraventricular Nucleus Inhibits High-Salt-Induced Hypertension Through NLRP3 and Caspase-1. Cardiovasc Toxicol. 2016;16(4):345–354. doi:10.1007/s12012-015-9344-9
121. Zheng F, Gong Z, Xing S, Xing Q. Overexpression of caspase-1 in aorta of patients with coronary atherosclerosis. Heart Lung Circ. 2014;23(11):1070–1074. doi:10.1016/j.hlc.2014.04.256
122. Doi T, Doi S, Nakashima A, et al. Mizoribine ameliorates renal injury and hypertension along with the attenuation of renal caspase-1 expression in aldosterone-salt-treated rats. PLoS One. 2014;9(4):e93513. doi:10.1371/journal.pone.0093513
123. Krishnan S, Dowling J, Ling Y, et al. Inflammasome activity is essential for one kidney/deoxycorticosterone acetate/salt-induced hypertension in mice. Br J Pharmacol. 2016;173(4):752–765. doi:10.1111/bph.13230
124. Krishnan S, Ling Y, Huuskes B, et al. Pharmacological inhibition of the NLRP3 inflammasome reduces blood pressure, renal damage, and dysfunction in salt-sensitive hypertension. Cardiovasc Res. 2019;115(4):776–787. doi:10.1093/cvr/cvy252
125. Zhang Y, Wang Y, Zhang L, et al. Reduced Platelet miR-223 Induction in Kawasaki Disease Leads To Severe Coronary Artery Pathology Through a miR-223/PDGFRβ Vascular Smooth Muscle Cell Axis. Circ Res. 2020.
126. Lee P, Day-Lewis M, Henderson L, et al. Distinct clinical and immunological features of SARS-COV-2-induced multisystem inflammatory syndrome in children. J Clin Invest. 2020;130(11):5942–5950. doi:10.1172/JCI141113
127. Pouletty M, Borocco C, Ouldali N, et al. Paediatric multisystem inflammatory syndrome temporally associated with SARS-CoV-2 mimicking Kawasaki disease (Kawa-COVID-19): a multicentre cohort. Ann Rheum Dis. 2020;79(8):999–1006. doi:10.1136/annrheumdis-2020-217960
128. Noval Rivas M, Arditi M. Kawasaki disease: pathophysiology and insights from mouse models. Nat Rev Rheumatol. 2020;16(7):391–405. doi:10.1038/s41584-020-0426-0
129. Liu L, Yuan Y, He X, et al. [Expression of Nod-like receptor protein 3 inflammasome in peripheral blood mononuclear cells of children with Kawasaki disease in the acute stage]. Zhongguo dang Dai Er Ke Za Zhi. 2019;21(10):992–997.
130. Anzai F, Watanabe S, Kimura H, et al. Crucial role of NLRP3 inflammasome in a murine model of Kawasaki disease. J Mol Cell Cardiol. 2020;138:185–196. doi:10.1016/j.yjmcc.2019.11.158
131. Lee Y, Schulte D, Shimada K, et al. Interleukin-1β is crucial for the induction of coronary artery inflammation in a mouse model of Kawasaki disease. Circulation. 2012;125(12):1542–1550. doi:10.1161/CIRCULATIONAHA.111.072769
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.