")
Back to Journals » OncoTargets and Therapy » Volume 10
PUMA mediates the anti-cancer effect of osimertinib in colon cancer cells
Authors Guo L, Huang S, Wang X
Received 11 April 2017
Accepted for publication 15 August 2017
Published 3 November 2017 Volume 2017:10 Pages 5281—5288
DOI https://doi.org/10.2147/OTT.S139382
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Faris Farassati
Lingchuan Guo,1,* Shan Huang,1,* Xinwei Wang2
1Department of Pathology, Hospital of Suzhou University, Suzhou, 2Department of Oncology, Tumor Hospital of Jiangsu Province, Nanjing, Jiangsu, China
*These authors contributed equally to this work
Abstract: Osimertinib, an irreversible EGFR/HER2 inhibitor, has been found to be effective in the cancer cell with EGFR gene mutations in preclinical lung cancer models. However, the effect of osimertinib in colorectal cancer (CRC) cells is unclear. In the present study, we investigated how osimertinib suppresses CRC cells growth and potentiates effects of other chemotherapeutic drugs. We found that p73-mediated osimertinib-induced p53 upregulated modulator of apoptosis (PUMA) expression irrespective of p53 status following PI3K/AKT pathway inhibition in CRC cells. Furthermore, PUMA is required for osimertinib-induced apoptosis. In addition, osimertinib also synergized with 5-FU to induce significant apoptosis via PUMA in CRC cells. These results demonstrated a critical role of PUMA in mediating the anticancer effects of osimertinib and suggest that PUMA induction can be used as an indicator of osimertinib sensitivity.
Keywords: PUMA, osimertinib, apoptosis, p73, CRC
Corrigendum for this paper has been published
Introduction
Colorectal cancer (CRC) is the most common malignancy with the second largest incidence and mortality among all diagnosed cancers in the world.1 Right now, CRC accompanied with higher mortality is due to disease that is frequently diagnosed in the advanced stage without reliable biomarkers.2 The metastatic diseases are the main cause for the high mortality rates in CRC patients.3 Conventional chemotherapy for colorectal cancer treatment involves combinations of cytotoxic drugs such as 5-fluorouracil (5-FU), irinotecan and oxaliplatin, and has limited efficacy and substantial side effects because of lack of specificity. The use of anticancer-targeted therapy has significantly improved effectiveness of chemotherapy against colorectal cancer.4 Osimertinib through oral administration is the irreversible inhibitor against tyrosine kinase, an ErbB family protein, which is used for treating EGFR mutation-positive non-small cell lung cancer.5–7 However, the effect of osimertinib in CRC is poorly understood.
p53 upregulated modulator of apoptosis (PUMA), a Bcl-2 homology 3 (BH3)-only Bcl-2 family member, functions as a key regulator of apoptosis in cancer cells.8,9 PUMA is a critical mediator of p53-dependent and -independent apoptosis in multiple tissues and cell types.10–13 Usually, p53 gene initiating the transcription of PUMA is mainly dependent on DNA damage.8 The p53-independent manner of PUMA induction by the stimuli is mediated by the transcription factor such as p73, E2F1, STAT1, FoxO3a, or NF-κB.11,13–15 In previous study, PUMA successfully induces a proapoptotic effect in cancer cell via antagonizing the antiapoptotic Bcl-2 family proteins (Bcl-2 and Bcl-XL), thus activating the proapoptotic signaling members (Bak and Bax), subsequently leading to mitochondrial dysfunction and activation of the caspase cascade.16,17
In the present study, we found that osimertinib induced PUMA up-regulation through AKT/p73 pathway, and PUMA plays an important role in therapeutic response to osimertinib in CRC cell. Our results indicated that PUMA level is indicative of the therapeutic efficacy of osimertinib, and likely other targeted drugs as well.
Materials and methods
Cell culture and drug treatment
The human CRC cell lines HCT15 and SW48 were obtained from American Type Culture Collection. Cell lines were maintained in Roswell Park Memorial Institute (RPMI) 1640 culture medium with 2mM Glutamine, 10% fetal bovine serum, 100 units/mL penicillin, and 100 μg/mL streptomycin (Thermo Fisher Scientific, Waltham, MA, USA). The anticancer agents and chemicals used in this study including osimertinib (Selleckchem, Houston, TX, USA) and 5-FU (Sigma-Aldrich Co., St Louis, MO, USA) were diluted with dimethyl sulfoxide. Constitutively active AKT was obtained from Addgene (Cambridge, MA, USA).18
MTS assay
HCT15 or SW48 cells were seeded in 96-well plates at a density of 5×103 cells per well. After incubation for 24 hours, cells were treated with osimertinib for additional 72 hours. 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay was performed using the MTS assay kit (Promega Corporation, Fitchburg, WI, USA) according to the manufacturer’s instructions. Luminescence was measured with a Wallac Victor 1420 Multilabel Counter (PerkinElmer Inc., Waltham, MA, USA). Each assay was conducted in triplicate and repeated 3 times.
Real-time reverse transcription polymerase chain reaction (RT-PCR)
RNA 1 μg was used to generate cDNA using SuperScript II reverse transcriptase kit. PCR was performed in triplicate using SsoFasr™ Probes Supermix (Bio-Rad Laboratories Inc., Hercules, CA, USA) in a final reaction volume of 10 μL with gene-specific primer/probe sets, and a standard thermal cycling procedure (33 cycles) on a Bio-Rad CFX96™ Real-time PCR System. PUMA and β-actin levels were assessed using TaqMan Gene Expression Real-Time PCR assays. Results were expressed as the threshold cycle (Ct). The relative quantification of the target transcripts was determined by the comparative Ct method (ΔΔCt) according to the manufacturer’s protocol. The 2−ΔΔCt method was used to analyze the relative changes in gene expression. Control experiments were conducted without reverse transcription to confirm that the total RNA was not contaminated with genomic DNA.
Western blotting
Western blotting analysis was conducted as the previous study,19,20 with antibodies for cleaved-caspase 3, cleaved-caspase 9, p-p65, Bax, p-FoxO3a, FoxO3a, cytochrome oxidase subunit IV (Cox IV), E2F1, HA (Cell Signaling Technology, Boston, MA, USA), p73, PUMA (Abcam, Cambridge, MA, USA), cytochrome C and β-actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA).
Apoptosis assays
Apoptosis was analyzed by nuclear staining with Hoechst 33258 (Invitrogen). Annexin V/propidium iodide (PI) staining was performed using Annexin-Alexa 488 (Invitrogen) and PI staining. For cytochrome C release assay, cytosolic fractions were isolated by differential centrifugation, and probed by Western blotting for cytochrome C. Caspase activity was measured using the SensoLyte Homogeneous AMC Caspase-3/7 Assay Kit (Anaspec, Fremont, CA, USA) according to the manufacturer’s protocol.
shRNA knockdown
For stable transfection, HCT15 cells were transfected with the pLKO.1-puro lentiviral vector (Addgene) expressing shRNA containing the p53-targeting sequence (CACCATCCACTACAACTACAT), PUMA-targeting sequence (CCTGGAGGGTCATGTACAATCTCTT), or a vector containing a scrambled sequence. Lentiviral particles were generated by co-transfecting with lentiviral vector, pMD1.G, pMDLg/pRRE, and pRSV-REV (Addgene) into 293T cells. After lentiviral transduction, HCT15 cells were seeded in a 96-well plate with 2 μg/mL puromycin. The protein expression of puromycin-resistant clones was then confirmed by Western blotting.
Chromatin immunoprecipitation (ChIP) of p73 and PUMA promoter
ChIP was performed with p73 antibody and the ChIP Assay Kit (Sigma-Aldrich Co.) according to the manufacturer’s instruction. The precipitated DNA fragments (PUMA promoter fragment containing p73 binding site) were amplified by PCR using specific primers (5′-GTCGGTCTGTGTACGCATCG-3′, 5′-CCCGCGTGACGCTACGGCCC-3′).
Statistical analysis
Statistical analyses were performed using GraphPad Prism IV software (GraphPad Software, Inc., La Jolla, CA, USA). P-values were calculated using the Student t-test and were considered significant if P<0.05. The means ± SD were displayed in the figures.
Results
Osimertinib decreases the proliferation and induces apoptosis in CRC cells
To determine the effect of osimertinib on CRC cells, HCT15 and SW48 cells were treated with increasing concentrations of osimertinib for 72 hours, and the proliferation was analyzed by MTS assay. Based on the results, osimertinib effectively decreased the cell proliferation of these cell lines (Figure 1A). Next, we investigated if osimertinib induced apoptosis in CRC cells. In apoptotic assays, we found that 10 μg/mL osimertinib increased the population of Annexin V-positive cells in HCT15 cells (Figure 1B). Our findings also showed that osimertinib induced activation of the executioner caspases 3 and 7 in HCT15 cells (Figure 1C). The apoptotic response was also found to be attenuated upon pre-treatment with the pan-caspase inhibitor z-VAD-fmk (Figure 1B), suggesting that the apoptotic response is caspase-dependent. Also, osimertinib treatment increased caspase 3 and 9 activation in HCT15 cells (Figure 1D). The above data indicate that osimertinib inhibited proliferation and induced apoptosis in CRC cells.
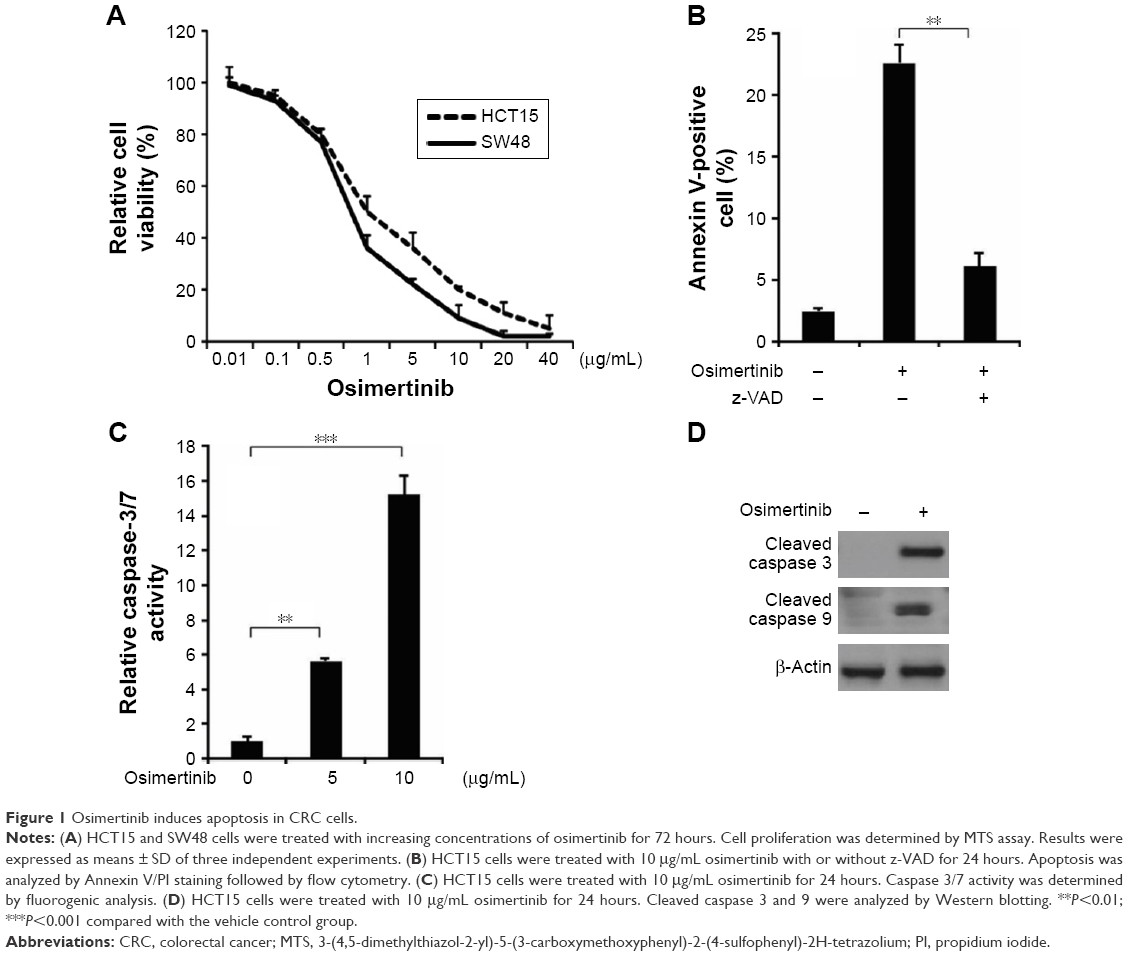 | Figure 1 Osimertinib induces apoptosis in CRC cells. |
Osimertinib induces p53-independent PUMA induction
Next, we investigated the mechanism of osimertinib-induced apoptosis in CRC cells. HCT15 cells were treated with 10 μg/mL osimertinib, and the induction of PUMA was examined. As shown in Figure 2A–C, treating HCT15 cells with osimertinib markedly up-regulated PUMA protein and mRNA level in a time-dependent manner. Osimertinib also induced PUMA mRNA expression in p53-Knockdown (p53-KD) HCT15 cells (Figure 2A). It is suggested that osimertinib induced PUMA expression via a p53-independent manner. Furthermore, osimertinib also induced PUMA expression in SW48 cells (Figure 2D). However, osimertinib treatment did not up-regulate other proapoptotic Bcl-2 family members, including Bax, Bid and Bim, but reduced the expression of the antiapoptotic proteins Mcl-1 (Figure 2E). These results suggest that PUMA is selectively induced by osimertinib regardless of p53 status and may mediate its anticancer effects.
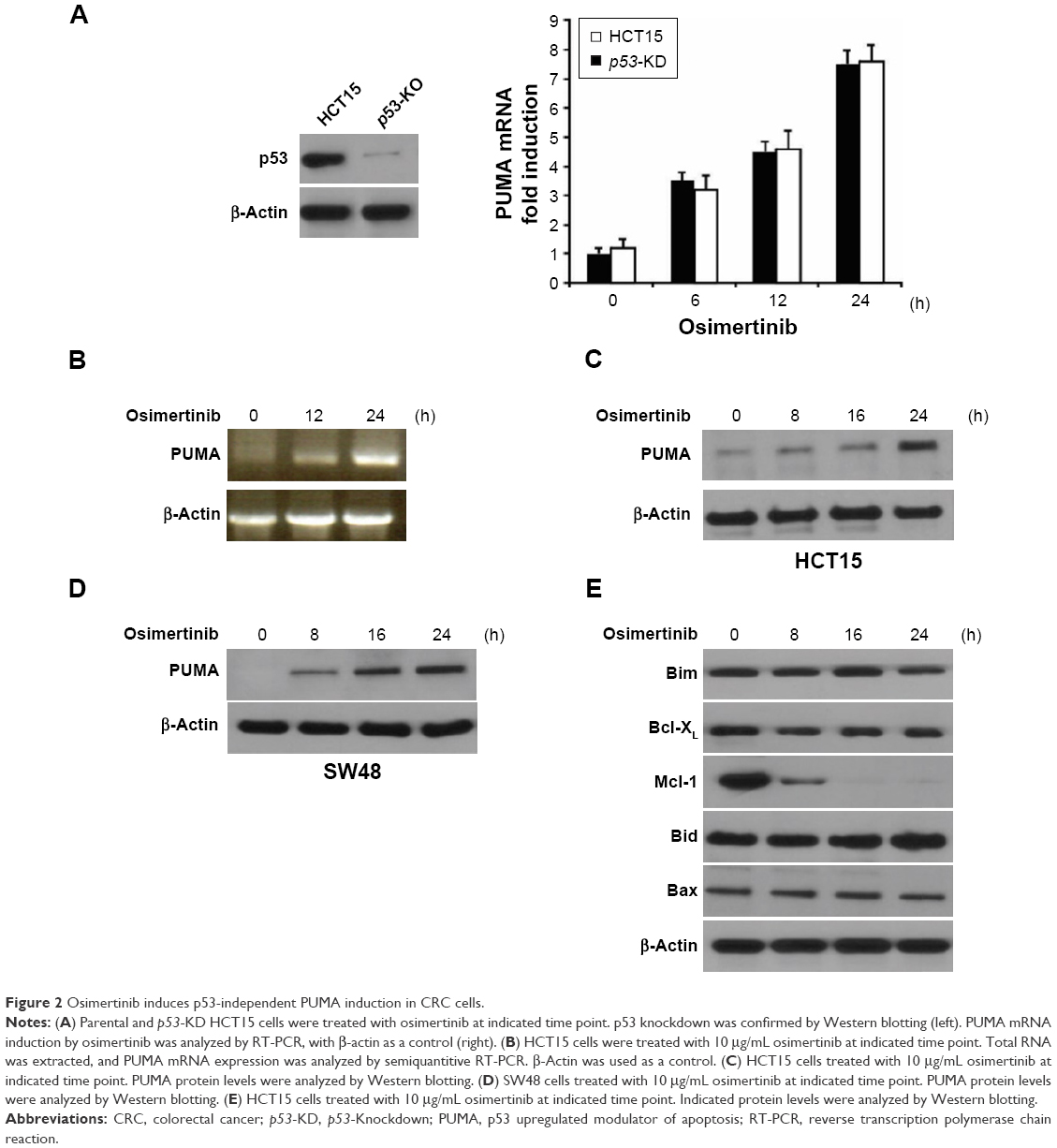 | Figure 2 Osimertinib induces p53-independent PUMA induction in CRC cells. |
PUMA mediates osimertinib-induced apoptosis
Next, we investigated whether PUMA is required for osimertinib-induced apoptosis. Apoptosis induced by osimertinib was significantly reduced in PUMA stable knockdown (PUMA-KD) HCT15 cells (Figure 3A). Annexin V/PI staining confirmed the reduction of osimertinib-induced apoptosis in PUMA-KD HCT15 cells (Figure 3B). Furthermore, our findings showed that osimertinib-induced caspases 3/7 activation was blocked in PUMA-KD HCT15 cells (Figure 3C). PUMA deficiency also blocked osimertinib-induced mitochondrial events, including activation of caspases 3, and 9 (Figure 3D) and cytochrome C release (Figure 3E). Our results suggest that PUMA is necessary for the anticancer effects of osimertinib in CRC cells.
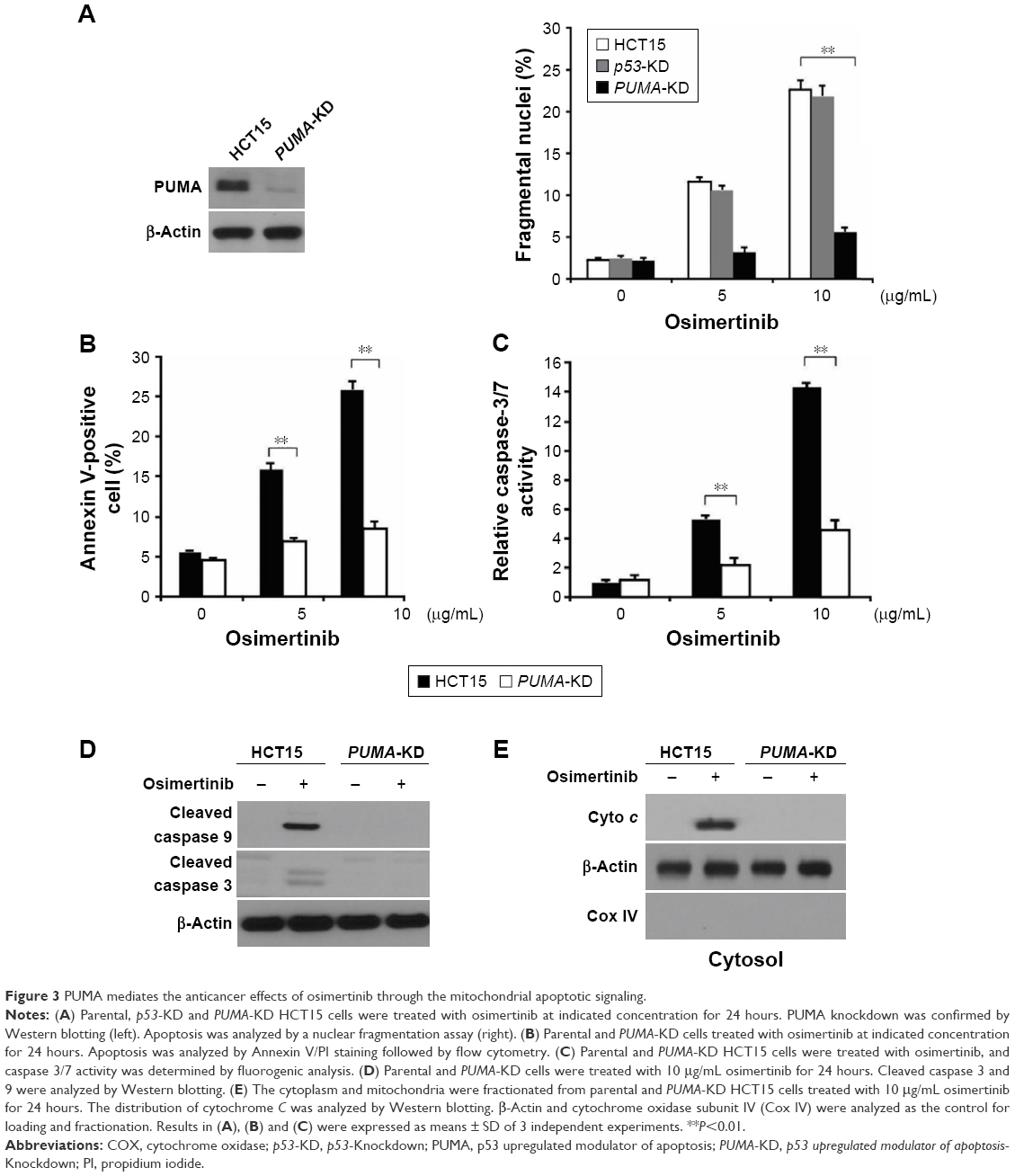 | Figure 3 PUMA mediates the anticancer effects of osimertinib through the mitochondrial apoptotic signaling. |
p73 is required for osimertinib-induced PUMA expression
Next, we analyzed the mechanisms of osimertinib-induced PUMA expression in CRC cells. Several transcription factors that can mediate PUMA induction in p53-independent manner were examined to further delineate the mechanism of PUMA induction. FoxO3a is not involved due to unchanged phosphorylation/activation following osimertinib treatment (Figure 4A). E2F1 and p65 were also ruled out due to lack of induction (Figure 4A). We found that osimertinib treatment induced p73 up-regulation in a time- and dose-dependent manner in HCT15 cells (Figure 4A and B). Knockdown of p73 by siRNA abrogated PUMA induction by osimertinib in HCT15 cells (Figure 4C). To determine whether p73 can directly activate PUMA transcription, p73 was found to be recruited to the genomic region containing the p73 binding sites following osimertinib treatment by ChIP (Figure 4D). These results indicate that p73 directly binds to PUMA promoter region to drive its transcriptional activation in response to osimertinib treatment.
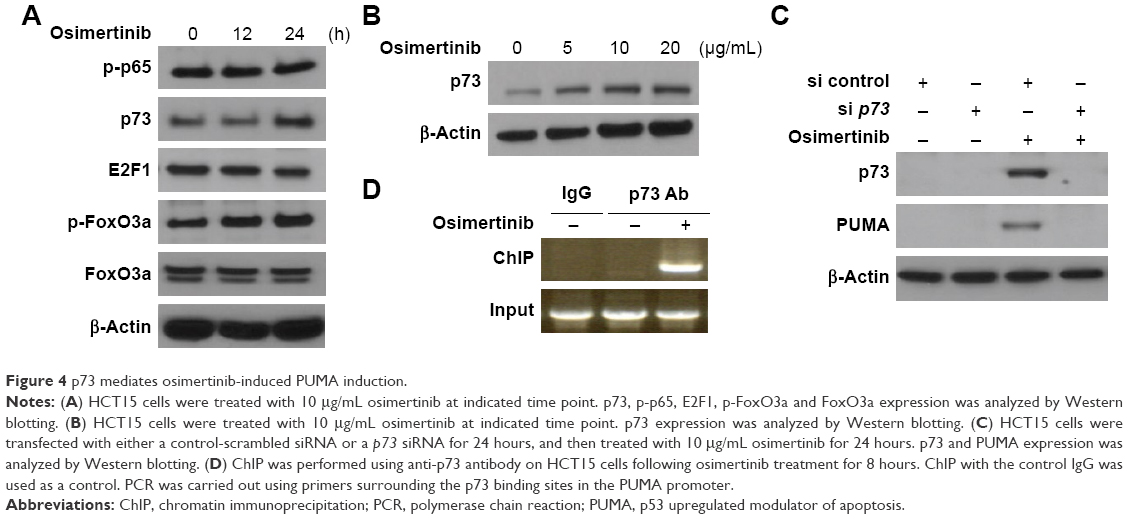 | Figure 4 p73 mediates osimertinib-induced PUMA induction. |
PI3K/AKT signaling is necessary for p73 induction after osimertinib treatment
The PI3K/AKT pathway promotes cell survival, and is a downstream effector of EGFR signaling pathway.21 Next, we investigated the effects of osimertinib on the PI3K/AKT signaling in relation to PUMA and p73. We found that osimertinib treatment strongly suppressed phosphorylation of AKT (Ser473) (Figure 5A). Exogenous expression of active AKT suppressed PUMA and p73 induction following osimertinib treatment (Figure 4B). These results suggest that PI3K/AKT pathway mediates PUMA expression through p73 by osimertinib.
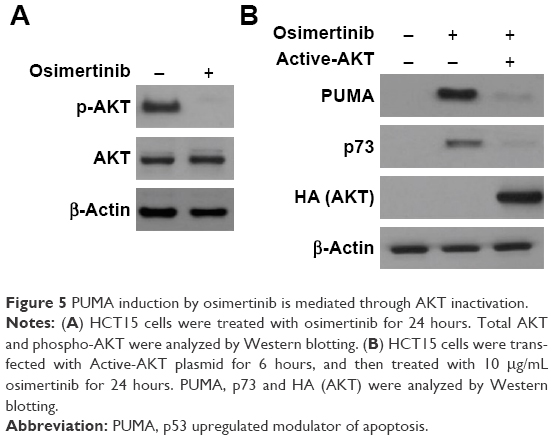 | Figure 5 PUMA induction by osimertinib is mediated through AKT inactivation. |
PUMA mediates the chemosensitization effects of osimertinib
Osimertinib has been used in combination with other chemotherapeutic agents in clinical studies.22,23 We reasoned that PUMA induction may mediate the chemosensitization effects of osimertinib, due to concurrent PUMA induction by osimertinib and other agents through different pathways. We found that osimertinib combined with 5-FU induced higher levels of PUMA, compared with single agent alone treatment (Figure 6A). Accordingly, the combination treatment induced higher level of apoptosis and caspase 3 activation in HCT15 cells. However, the combination induced apoptosis and caspase 3 activation were abolished in PUMA-KD HCT15 cells (Figure 6A and B). These findings indicate that PUMA mediates the chemosensitization effects of osimertinib.
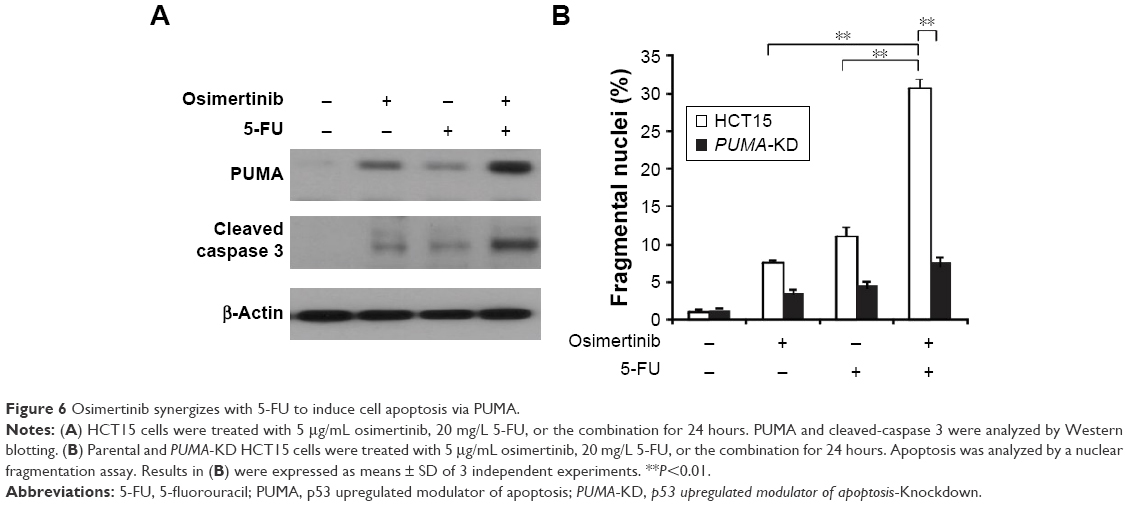 | Figure 6 Osimertinib synergizes with 5-FU to induce cell apoptosis via PUMA. |
Discussion
Osimertinib is a third generation of tyrosine kinase inhibitor that irreversibly inhibits ErbB family tyrosine kinase receptors.24 Some preclinical data suggested that the lung squamous cell carcinoma pathobiology has a strong dependency from the ErbB family pathway. In this study, we investigated the effect of osimertinib on CRC cells. Our results demonstrated for the first time that the therapeutic effect of osimertinib is at least in part mediated by the cell autonomous process of apoptosis induction, progressing from AKT inhibition, p73 induction leading to PUMA induction and activates mitochondria-dependent apoptosis. In addition to PUMA induction, depletion of Mcl-1 is an early event following osimertinib treatment, and may also contribute to anticancer action.
PUMA-mediated apoptotic event can be activated by oncogene and serves as a protective mechanism against the oncogenic signaling pathways. Our results showed that PUMA can be induced following AKT inhibition in response to osimertinib treatment and contributes to apoptosis initiation in CRC cells through the mitochondrial pathway. PUMA induction plays a key role in the proapoptotic effect of much chemotherapy, and is likely to be a useful indicator of chemosensitivity. Increased PUMA expression was associated with better prognosis in patients receiving 5-FU-based therapy in stage II and III colorectal cancer.25 Furthermore, a recent study demonstrated that response of isolated mitochondria from tumor cells to PUMA BH3 peptide correlates with chemotherapy response in patients with cancer.26 The results of the current study suggest that expression of PUMA can be a useful indicator for predicting response of colon cancer cells to osimertinib. In addition to PUMA induction, other Bcl-2 family members are also modulated following osimertinib treatment and contribute to apoptosis induction. Although it is hard to get biopsies from colorectal tumors treated with chemotherapy after surgery, it could be possible to detect PUMA induction using non-invasive approaches, such as analysis of circulating tumor cells.
In conclusion, this study demonstrates the inhibitory effect of osimertinib against tumorigenesis of human CRC cell regardless of p53 status. The subsequent experiments demonstrated that osimertinib significantly increased the cleavage of caspase 3 and 9 in HCT15 cells. Our results indicated that suppression of AKT and p73 induction may be responsible for osimertinib-induced PUMA expression. PUMA induction might be a useful biomarker for clinical trials testing osimertinib, and can have important implications for the future development and application.
Acknowledgments
We thank our lab members for critical reading. This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Disclosure
The authors report no conflicts of interest in this work.
References
Cunningham D, Atkin W, Lenz HJ, et al. Colorectal cancer. Lancet. 2010;375(9719):1030–1047. | ||
Osumi H, Shinozaki E, Suenaga M, et al. RAS mutation is a prognostic biomarker in colorectal cancer patients with metastasectomy. Int J Cancer. 2016;139(4):803–811. | ||
Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10(8):789–799. | ||
Sridharan M, Hubbard JM, Grothey A. Colorectal cancer: how emerging molecular understanding affects treatment decisions. Oncology (Williston Park). 2014;28(2):110–118. | ||
Yang JC, Ahn MJ, Kim DW, et al. Osimertinib in pretreated T790M-positive advanced non-small-cell lung cancer: AURA Study Phase II Extension Component. J Clin Oncol. 2017;35(12):1288–1296. | ||
Mok TS, Wu YL, Ahn MJ, et al. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N Engl J Med. 2017;376(7):629–640. | ||
Mayor S. Osimertinib effective in EGFR T790M-positive lung cancer. Lancet Oncol. 2017;18(1):e9. | ||
Yu J, Zhang L. No PUMA, no death: implications for p53-dependent apoptosis. Cancer Cell. 2003;4(4):248–249. | ||
Yu J, Wang Z, Kinzler KW, Vogelstein B, Zhang L. PUMA mediates the apoptotic response to p53 in colorectal cancer cells. Proc Natl Acad Sci U S A. 2003;100(4):1931–1936. | ||
Peng R, Tong JS, Li H, et al. Targeting bax interaction sites reveals that only homo-oligomerization sites are essential for its activation. Cell Death Differ. 2013;20(5):744–754. | ||
Ming L, Sakaida T, Yue W, Jha A, Zhang L, Yu J. Sp1 and p73 activate PUMA following serum starvation. Carcinogenesis. 2008;29(10):1878–1884. | ||
Yu J, Zhang L. PUMA, a potent killer with or without p53. Oncogene. 2008;(27 Suppl 1):S71–S83. | ||
Wang P, Qiu W, Dudgeon C, et al. PUMA is directly activated by NF-kappaB and contributes to TNF-alpha-induced apoptosis. Cell Death Differ. 2009;16(9):1192–1202. | ||
Sun J, Sun Q, Brown MF, et al. The multi-targeted kinase inhibitor sunitinib induces apoptosis in colon cancer cells via PUMA. PLoS One. 2012;7(8):e43158. | ||
Zhao Z, Wang J, Tang J, et al. JNK- and Akt-mediated puma expression in the apoptosis of cisplatin-resistant ovarian cancer cells. Biochem J. 2012;444(2):291–301. | ||
Sun J, Knickelbein K, He K, et al. Aurora kinase inhibition induces PUMA via NF-kappaB to kill colon cancer cells. Mol Cancer Ther. 2014;13(5):1298–1308. | ||
Ming L, Wang P, Bank A, Yu J, Zhang L. PUMA dissociates Bax and Bcl-X(L) to induce apoptosis in colon cancer cells. J Biol Chem. 2006;281(23):16034–16042. | ||
Xie R, Cheng M, Li M, et al. Akt isoforms differentially protect against stroke-induced neuronal injury by regulating mTOR activities. J Cereb Blood Flow Metab. 2013;33(12):1875–1885. | ||
Tong J, Tan S, Zou F, Yu J, Zhang L. FBW7 mutations mediate resistance of colorectal cancer to targeted therapies by blocking Mcl-1 degradation. Oncogene. 2017;36(6):787–796. | ||
Misale S, Bozic I, Tong J, et al. Vertical suppression of the EGFR pathway prevents onset of resistance in colorectal cancers. Nat Commun. 2015;6:8305. | ||
Berg M, Soreide K. EGFR and downstream genetic alterations in KRAS/BRAF and PI3K/AKT pathways in colorectal cancer: implications for targeted therapy. Discov Med. 2012;14(76):207–214. | ||
York ER, Varella-Garcia M, Bang TJ, Aisner DL, Camidge DR. Tolerable and effective combination of full dose crizotinib and osimertinib targeting MET amplification sequentially emerging after T790M positivity in EGFR mutant non-small cell lung cancer. J Thorac Oncol. 2017;12(7):e85–e88. | ||
Tanimoto A, Takeuchi S, Arai S, et al. Histone deacetylase 3 inhibition overcomes BIM deletion polymorphism-mediated osimertinib-resistance in EGFR-mutant lung cancer. Clin Cancer Res. 2017;23(12):3139–3149. | ||
Sullivan I, Planchard D. Osimertinib in the treatment of patients with epidermal growth factor receptor T790M mutation-positive metastatic non-small cell lung cancer: clinical trial evidence and experience. Ther Adv Respir Dis. 2016;10(6):549–565. | ||
Ni Chonghaile T, Sarosiek KA, Vo TT, et al. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science. 2011;334(6059):1129–1133. | ||
Misale S, Yaeger R, Hobor S, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486(7404):532–536. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.