")
Back to Journals » Drug Design, Development and Therapy » Volume 15
PTBP1 Targets ILK to Regulate the Hypoxia-Induced Phenotypic Transformation of Pulmonary Artery Smooth Muscle Cells
Authors Yan G, Sun R, Chen Z, Pan X, Sheng Z, Tang C
Received 7 October 2020
Accepted for publication 17 March 2021
Published 13 May 2021 Volume 2021:15 Pages 2025—2033
DOI https://doi.org/10.2147/DDDT.S275000
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Georgios Panos
Gaoliang Yan, Renhua Sun, Zhongpu Chen, Xiaodong Pan, Zulong Sheng, Chengchun Tang
Department of Cardiology, Zhongda Hospital of Southeast University Medical School, Nanjing, 210009, People’s Republic of China
Correspondence: Gaoliang Yan
Department of Cardiology, Zhongda Hospital of Southeast University Medical School, Nanjing, 210009, People’s Republic of China
Tel +86 18761890380
Email [email protected]
Purpose: Pulmonary hypertension (PH) is a pathological process mainly characterized by the progressive increase in pulmonary vascular resistance. The degradation of pulmonary artery smooth muscle cells (PASMCs) from contractile/differentiated phenotype to synthetic/dedifferentiated phenotype is a key factor for hypoxic pulmonary hypertension.
Materials and Methods: In this study, qPCR was performed to evaluate the gene expression of mRNAs. Western blot, immunofluorescence and RNA pull down were used to detect gene expression levels.
Results: We found that the gene expression of polypyrimidine tract-binding protein1 (PTBP1) was increased significantly in a time-dependent manner in rats PA tissues and PASMCs after hypoxia. PTBP1 knockdown can inhibit the phenotypic transition of PASMCs. PTBP1 inhibits the phenotypic transition of PASMCs. In addition, PTBP1 inhibits the integrin-linked kinase (ILK) expression under hypoxic conditions, thereby down-regulating the expression of downstream proteins. It inhibits the phenotypic transition of PASMCs and alleviates pulmonary hypertension.
Conclusion: In conclusion, PTBP1/ILK axis promotes the development of PH via inducing phenotypic transition of PASMCs. This may provide a novel therapy for PH.
Keywords: pulmonary hypertension, polypyrimidine tract-binding protein 1, integrin-linked kinase, pulmonary artery smooth muscle cells
Introduction
Pulmonary hypertension (PH) is a pathological process mainly characterized by the progressive increase in pulmonary vascular resistance and progressive failure of right heart function.1,2 PH is a common complication of many clinical diseases. Although extensive researches have been carried out to study the initiation and progression of pulmonary hypertension, the potential mechanisms are still unclear. Studies suggested PH may be related to several factors such as hypoxia, neurohumoral, congenital, genetic and others.3,4 Its pathogenesis has not been fully understood.
Polypyrimidine tract-binding protein1 (PTBP1) is a member of heterogeneous nuclear ribonucleoproteins (hnRNPs) (also named hnRNPI),5 which is involved in the transcriptional regulation of many genes. PTBP1 modulates tumor metastasis by regulating metastasis-related genes. In tumor cells, PTBP1 promotes the expression of PKM2 isoforms by regulating the alternative splicing of the pyruvate kinase PKM gene, thereby promoting the glycolysis process of tumor cells and thus inducing tumorigenesis.6 Integrin-linked kinase (ILK) is a serine/threonine kinase that regulates cell survival, proliferation, and apoptosis through glycogen synthesis kinase 3β (GSK-3β).7 Hypoxia suppresses the expression of myocardin in smooth muscle cells and may be an essential mechanism involved in the formation of hypoxic PH.8,9 ILK is upstream to many intracellular molecules involved in hypoxic stress. Investigating the role of the ILK signaling pathway in the molecular mechanism of PH may provide theoretical and experimental evidence for clinical drug treatment of pulmonary hypertension.
In this study, we explored the role of PTBP1 and ILK in the hypoxia-induced PASMC phenotypic transition. We speculate that PTBP1 regulates PASMC by inhibiting the expression of ILK. To test this hypothesis, we identified the role of PTBP1 in the phenotypic transformation of PASMCs through silencing and overexpressing the PTBP1 gene in vivo and in vitro. Our results may provide a new therapeutic strategy for the prevention and treatment of PH.
Materials and Methods
Pulmonary Hypertension Animal Model
Thirty male SD rats, weighing 280–320g, were provided by the Animal Center of Zhongda Hospital of Southeast University Medical School. The rats were divided into control group and hypoxia treatment group. To establish the PH model, the rats were housed in a hypobaric hypoxia chamber depressurized to 380 mmHg (PO2 was reduced to about 79.6 mmHg accordingly) for 4 weeks. All the rats were housed in a condition of 12:12 hour light-dark cycle with free access to food and water. The Ethics Committee in Zhongda Hospital of Southeast University Medical School approved the animal study. All animal procedures were performed following the institutional guidelines and approved by the Zhongda Hospital of Southeast University Medical School.
Relative Ventricular Weight
The relative ventricular weight was assessed after sacrificing of the rats and clearing the blood from isolated hearts. The moisture over the hearts was absorbed with filter paper, and we determined the hearts’ weight. Then we calculated the right ventricular (RV)/left ventricle + septum (LV + S), RV weight-to-body weight (RV/BW), and (LV + S)/BW.
Hemodynamics Measurements
After hypoxia, the rats were injected with pentobarbital sodium (60 mg/k). A polyethylene microcatheter was inserted into the right ventricle and pulmonary artery and the mean pulmonary arterial pressure (mPAP).
The Detection of Wall Thickness (WT) and Wall Area (WA)
The right lower lung from the sacrificed animals were immersed in 10% formalin fixation. Then, specimens were dissected and stained with Hematoxylin. Ten pulmonary arterioles, each having a tube diameter of 50 to 100 μm, were randomly selected. The wall thickness (WT) and external diameter (ED) were determined to calculate the percentage of pulmonary arteriolar wall thickness (WT%) to the diameter of the tube using this equation (WT% = 2 WT/ED ×100%). The wall area (WA) % of the blood vessel was calculated using this equation, WA% [WA% = (TA-LA)/TA × 100%].
Primary Culture of Pulmonary Artery Smooth Muscle Cells
Wister rats were anaesthetized intraperitoneally with 1% sodium pentobarbital (40mg/kg). Rat heart and lung tissues were removed under aseptic conditions, pulmonary arteries were separated, and washed with Hanks’ solution to remove blood stains on the tissue surface. Then, the connective tissue and blood vessels were removed and the vascular endothelial cells were scraped. The tissues were divided into 0.5–1 mm3 size pieces and rinsed with Hanks solution until it became clear. Next, Hanks solution was removed, and digestion was performed using 0.2 mL type II collagenase solution at 37°C for 15 min. The digested samples were filtered and centrifuged at 1000 r/min for 5 min, and the supernatant was discarded. The pellet was washed with Hanks solution, followed by centrifugation; then the cells were resuspended in DMEM culture medium containing 10% fetal bovine serum and seeded into a 24-well culture plate.
Inducing Cellular Hypoxia
PASMCs were cultured medium containing 0.1% FBS for 24 hours. Then, PASMCs were treated with CoCl2 (100 μM) under the hypoxic condition for 1 h, 6 h, 24 h, 48 h, 72 h. The cells were placed in a hypoxia-specific incubator (XBS-08, Aipu, Hangzhou, China) containing 5% CO2 at 37°C. The control PASMCs were maintained under normoxic conditions.
Plasmid Transfection
siRNA for PTBP1 were synthesized by Genewiz (Beijing, China). The overexpressing vector of PTBP1 and ILK were purchased from Hanbio (Shanghai, China). Plasmids were transfected using Lipofectamine 3000 (Invitrogen, USA) kit following the manufacturer’s instructions. Adeno virus silencing PTBP1 was purchased from Hanbio (Beijing, China). Ad-sh-PTBP1 (0.5 mL of 4×108 plaque-forming units) was injected into the rats via tail vein.
Immunoprecipitation (IP)
Total protein was extracted from PASMCs with RIPA buffer. The protein sample was incubated with 25 μL protein A/G agarose and 25 μL immunoglobulin for 1 h at 4°C followed by centrifugation at 2000 × g at 4°C for 5 min. Primary antibody (4 μg) was added and incubated overnight at 4°C. Immunoglobulin (4 μg) was added to the protein sample as a negative control. Then, 25 μL of protein/G agarose was added, followed by centrifuging at 4°C for 2 h. After washing, the immunoprecipitation complex was boiled in 2 × protein loading buffer for 10 min for further Western blot analysis.10
Immunofluorescence
The changes in phenotypic markers of PASMCs were detected by immunofluorescence staining. According to the referenced method, cells grown over coverslips were washed with PBS buffer, fixed with formaldehyde for 10min, and further washed with PBS. The primary antibody was added (1:1000 dilution) and incubated for 1 h at 37°C on a shaker. Coverslips were washed three times with PBS and incubated with a FITC-labeled secondary antibody diluted 1:80 at 37°C for 30 min on a shaker. We used an imaging analysis system at 400x magnification power for each specimen. Images were taken to observe the intensity of F-actin staining and cell morphology. PTPB1 and ILK co-localization was determined by double staining.11
Western Blot
Proteins from tissue samples were isolated and quantified using BCA kit according to the instructions. Then, standard Western blotting was performed as following: SDS-PAGE electrophoresis, membrane transfer, primary antibody, and secondary antibody addition, development, and fixation. GAPDH was used as an internal reference. Band analysis was performed using image analysis software. The ratio of the grayscale of the target protein to the grayscale of the GAPDH color represents the relative expression of the target protein.12
Statistical Analysis
SPSS 19.0 software package was used for statistical data analysis Each index is expressed as a mean value ± standard deviation. Differences between different groups were analyzed using the Mann–Whitney non-parametric tests. P<0.05 was considered as statistically significant.13
Results
Identification PH Animal Model
In order to study the pathogenesis of PH, we constructed the PH animal model. After establishing a PH model, we measured the RV/(LV + S), mPAP, WT, and WA (n=6). Compared to the control group, these indexes started to increase 2 weeks after establishing the model and reached their maximum at 4 weeks. These findings indicated the successful establishment of the PH model (Figure 1A). We examined the expression of PTBP1 in pulmonary arterial tissue of rats with PH by Western blot and immunofluorescence assays. The results showed that the expression of PTBP1 was significantly increased 3 weeks after hypoxia (Figure 1B). Then, we evaluated the expression of PTBP1 of PASMCs using Western blot and immunofluorescence. The results indicated that hypoxia significantly promoted the expression PTBP1 after 24h (Figure 1C and D).
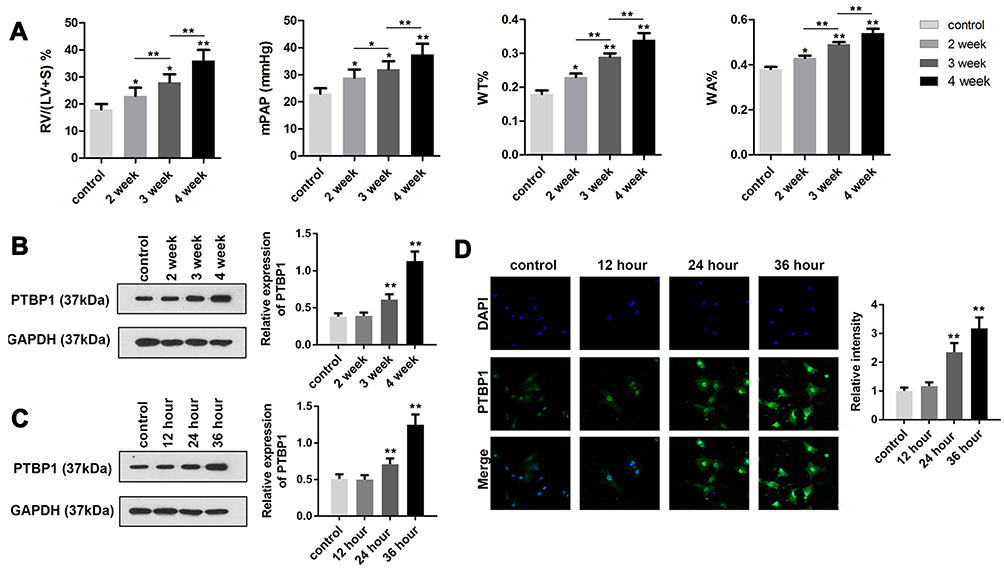 |
Figure 1 Establishment of an animal model of pulmonary hypertension. (A) Detection of RV/(LV + S)%, mPAP, WT% and WA% in rats. The results showed that the model was successfully prepared. (B) The western method was used to detect the expression of PTBP1 in the pulmonary artery tissue of rats with pulmonary hypertension. (C and D) The Western blot and immunofluorescence method were used to detect the expression of PTBP1 in the PASMCs. n=6, *p<0.05, **p<0.01. |
PTBP1 Silencing Inhibits Phenotype Transition of PASMCs
Since the expression of PTBP1 was significantly increased in rats with HP, we speculated that it might play a role in the transformation of PASMCs, so we established a hypoxia model of PASMCs. We found that knockdown of PTBP1 inhibited the phenotype switching of PASMCs induced by hypoxia (Figure 2A), and the PASMCs length and area were significantly reduced (Figure 2B and C). The experimental results show that PTBP1 plays an essential role in the phenotypic transition of PASMCs.
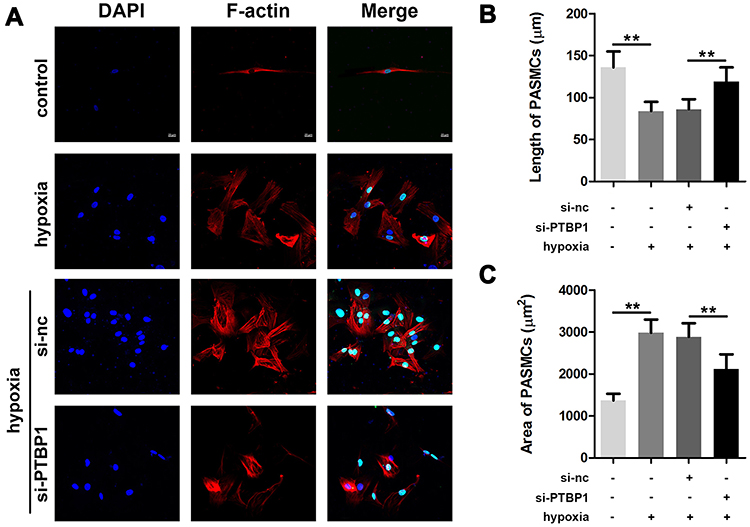 |
Figure 2 PTBP1 interference inhibits phenotypic switching of pulmonary artery smooth muscle cells. (A) F-actin staining was used to detect phenotypic changes in smooth muscle cells. (B) The cell length of PASMCs was calculated. (C) Results of the area of PASMCs were detected. n=6, **p<0.01. |
PTBP1 Inhibits Expression of ILK and Downstream Signaling Pathway
In order to further confirm the regulation of PTBP1 on the deformation and transformation of PASMCs, we examined the expression of PTBP1-related proteins. The results showed that knockdown of PTBP1 significantly decreased the protein level of ILK, meantime, it can inhibit the expression of Myocardin, SM a-actin, Calponin, and increase the expression of osteopontin (Figure 3A). ILK overexpression can reverse the PTBP1 regulatory effect. This finding indicates that PTBP1 can regulate ILK and affect the expression of several downstream signaling proteins (Figure 3B).
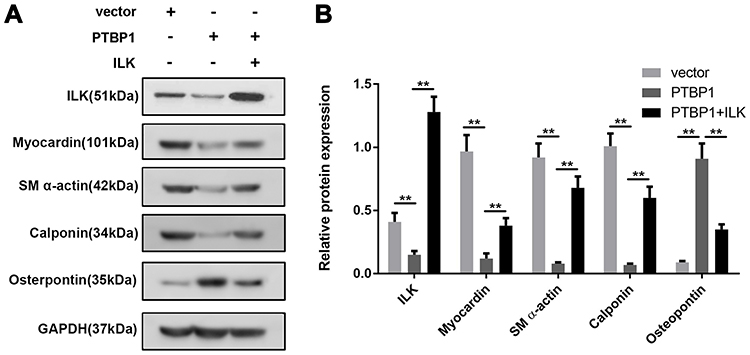 |
Figure 3 PTBP1 inhibits ILK and downstream signaling pathway protein expression. (A) The expressions of ILK, Myocardin, SM a-actin, Calponin, and osteopontin were detected by Western blot. (B) Statistical results of expression changes of ILK, Myocardin, SM a-actin, Calponin, and osteopontin. n=6, **p<0.01. |
PTBP1 Binds to ILK’s RNA and Maintains the RNA Stability of ILK
To clarify the precise mechanism of action of PTBP1, we performed RNA pulled down and mass spectrometry experiments (Figure 4A) also, RIP experiments (Figure 4B). The results showed that PTBP1 and ILK had direct interaction (Figure 4C). Bioinformatics analysis proves that these two molecules can interact with each other (Figure 4D). After the addition of transcription suppressor actinomycin D to the PASMCs, we found that PTBP1 overexpression inhibited the stability of ILK mRNA, and PTBP1 silencing can stabilize ILK’s mRNA (Figure 4E). These results were further confirmed by Western blotting and immunofluorescence (Figure 4F). The FISH analysis demonstrated the co-localization of PTBP1 and ILK, which verified the interaction of PTBP1 and ILK (Figure 4G).
 |
Figure 4 PTBP1 interact with ILK and regulates its RNA stability. (A) RNA pulled down + silver staining were performed to identify that ILK mRNA interact with PTBP1 protein. (B) RIP experiments along with the PCR was used to confirm the interaction between PTBP1 and ILK. (C) RNA pull down was performed to detect the interaction between PTBP1 and ILK. (D) Bioinformatics analysis proves that PTBP1 and ILK can be combined. (E) qPCR was used to evaluate the mRNA expression of ILK at different time point in the PASMCs after actinomycin D treatment. (F) Western blot was performed to confirm that overexpression of PTBP1 could inhibit ILK protein expression while PTBP1 knockdown promoted ILK expression. (G) Fish experiment was conducted to detect the co-localization of PTBP1 and ILK. n=6, **p<0.01. |
PTBP1 Silencing Modulates the Expression of Phenotypic Transition Related Genes
Next, based on the above results, we detected the expression of phenotypic transition-related proteins. The results showed that ILK and osteopontin expression was significantly increased in PASMCs after hypoxia while ILK, myocardin, SM α-actin and Calponin were downregulated. However, this was reversed by PTBP1 knockdown (Figure 5A and B).
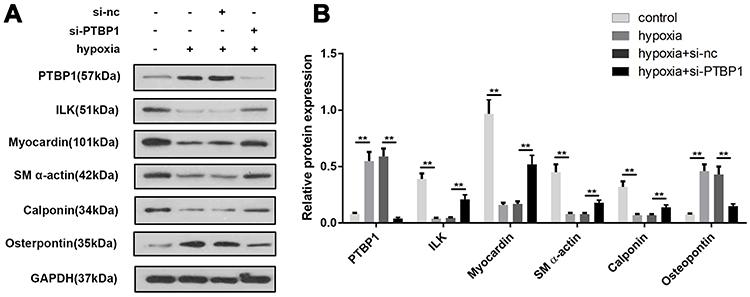 |
Figure 5 PTBP1 silencing modulates the expression of phenotypic transition-related genes. (A) Western blot was used to detect the expression of phenotypic transition-related genes in PASMCs including PTBP1, ILK, Myocardin, SM a-actin, Calponin, and osteopontin. (B) Statistical results of protein expression was calculated. n=6, **p<0.01. |
PTBP1 Silencing Alleviated the Pulmonary Hypertension of Rats
In order to further confirm the effect of PTBP1 in pulmonary hypertension, we carried out in vivo study (n=6). Adeno virus silencing PTBP1 was injected into rats via tail vein. We found that compared to the control group, RV/(LV + S)%, mPAP, WT%, and WA% increased 4 weeks after establishing the model, and PTBP1 silencing notably reduced these index compared to the Ad-sh-nc treatment group (Figure 6A–D).
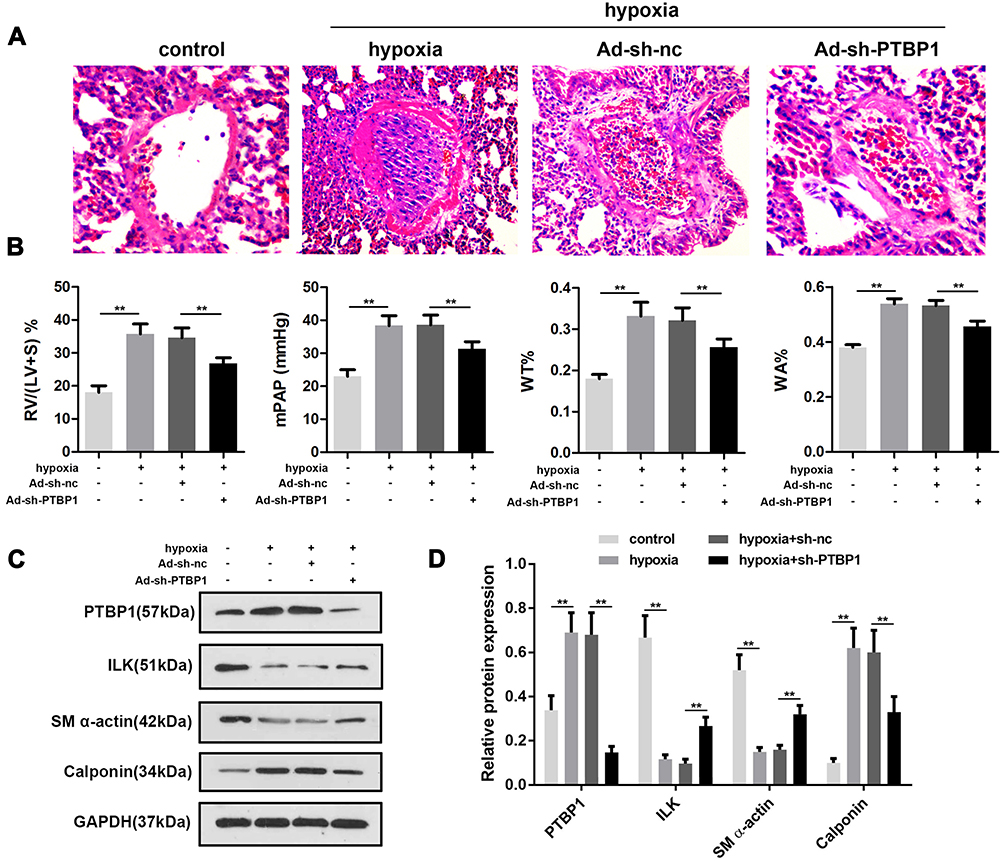 |
Figure 6 PTBP1 silencing alleviated pulmonary hypertension in rats. (A–D) After administration of Ad-sh-PTPB1 or Ad-sh-nc, RV/(LV + S)%, mPAP, WT% and WA% of the pulmonary hypertension or control rats were detected. n=6, **p<0.01. |
Discussion
Pulmonary hypertension (PH) is a group of diseases characterized by a progressive increase in the circulation resistance of the pulmonary artery system.14–17 Its pathological changes include pulmonary vasoconstriction and remodeling, abnormal proliferation of pulmonary vascular smooth muscle and endothelial cells. However, the cause of hypertension causes is still unclear. Several reasons could be involved in pulmonary hypertension diagnosis difficulty. Therefore, it is crucial to improve the understanding of pulmonary hypertension, effective early screening, and early treatment.18–20
PASMCs exist in two forms: synthetic phenotype and contractile phenotype. The contractile phenotype of PASMCs is rich in Myosin and α-actin, which are the main components of contractile proteins.21–23 Therefore, observing the expression of α-actin in PASMCs is an important method to determine the phenotypic transformation of cells. Previous reports showed that after 4 weeks of hypoxia, the content of α-actin in the cytoplasm of PASMCs is significantly reduced, and the degree of cell proliferation is increased. The previous results are consistent with the results of pulmonary hypertension modeling in the literature, providing a basis for better follow-up research.
PASMCs are the main components of the pulmonary artery vascular media, and play a significant role in pulmonary vascular remodeling. The abnormal proliferation of PASMCs manifested as the contraction, proliferation, differentiation, and matrix secretion of smooth muscle cells, which caused the thickening of the pulmonary artery vascular medial membrane. A variety of factors (such as hypoxia, inflammatory response, abnormal mechanical forces in blood vessels) induce PASMCs proliferation. It has been reported that PTBP1 knockout in glioma cells attenuates cell proliferation, and migration, and enhances cell adhesion via regulating the alternative splicing of the transmembrane factor RNT4.24 PTBP1 has been indicated to mediate the miR-124’s effect on regulating the endothelial cell glycolysis in pulmonary arterial hypertension. In PH, miR-124, through the alternative splicing factor PTBP1, regulates the PKM2/PKM1 ratio to modulate the proliferative, and inflammatory state of cells.
In the present study, we confirmed that PTBP1 was increased significantly in PASMC after hypoxia which indicate the critical role of PTBP1 in PH progression. PTBP1 knockdown inhibited the phenotypic transition of PASMC. As PTBP1 is a RNA binding protein, to elucidate the mechanism of PTBP1, we performed RIP and mass spectrum assay. ILK was found to be bound with PTBP1. ILK was first discovered by Hannigan et al7. ILK is a central regulatory protein of cell signaling; it also depends on phosphatidylinositol-3 kinase (PI3K) activation, which in turn acts on the downstream GSK-3β. Shen et al25 reveal that ILK is one of the necessary genes to maintain the aortic vasoconstriction phenotype. ILK has a wide range of regulatory roles in various pathophysiological processes, such as cell differentiation, development, and tumor growth. In this study, PTBP1 bound with ILK and inhibited the mRNA stability of the ILK, thus lead to the dysregulation of ILK. Moreover, ILK was involved in the modulation of phenotypic transition-related proteins including myocardin, SM α-actin, Calponin and osteopontin. However, more investigations such as rescue experiments will make this conclusion more credible.
Conclusion
This study found that PTBP1 was overexpressed in PASMCs after hypoxia. Silencing of PTBP1 inhibited the transformation of PASMCs by regulating ILK and its downstream signaling. This study suggests that PTBP1 is involved in regulating the occurrence and development of PH and may become a new biomarker for the diagnosis and prognosis of pulmonary hypertension.
Acknowledgment
This study is supported by National Natural Science Foundation of China (Research Grant no. 81600227).
Author Contributions
All authors made substantial contributions to the design and conception of the study, and acquisition, analysis and interpretation of data, and took part in either drafting or revising the manuscript. All authors gave final approval of the version to be published, have agreed on the journal to which the article has been submitted, and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Disclosure
There is no interest conflict of all the authors.
References
1. Zhao L, Mason NA, Morrell NW, et al. Sildenafil inhibits hypoxia-induced pulmonary hypertension. Circulation. 2001;104(4):424–428. doi:10.1161/hc2901.093117
2. Olschewski H, Simonneau G, Galiè N, et al. Inhaled iloprost for severe pulmonary hypertension. N Engl J Med. 2002;11(6):19.
3. Walsh-Sukys MC, Tyson JE, Wright LL, Bauer CR. Persistent pulmonary hypertension of the newborn in the era before nitric oxide: practice variation and outcomes. Pediatrics. 2000;105(1 Pt 1):14–20. doi:10.1542/peds.105.1.14
4. Olschewski H, Ghofrani HA, Walmrath D, et al. Inhaled prostacyclin and iloprost in severe pulmonary hypertension secondary to lung fibrosis. Am J Respir Crit Care Med. 1999;160(2):600–607. doi:10.1164/ajrccm.160.2.9810008
5. Kornblihtt AR, Schor IE, Alló M, Dujardin G, Petrillo E, Muñoz MJ. Alternative splicing: a pivotal step between eukaryotic transcription and translation. Nat Rev Mol Cell Biol. 2013;14(3):153–165. doi:10.1038/nrm3525
6. Christofk HR, Vander Heiden MG, Harris MH, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452(7184):230–233. doi:10.1038/nature06734
7. Hannigan GE, Leunghagesteijn C, Fitzgibbon L, et al. Regulation of cell adhesion and anchorage-dependent growth by a new |[beta]|1-integrin-linked protein kinase. Nature. 1996;379(6560):91–96. doi:10.1038/379091a0
8. Jie W, Guo J, Shen Z, et al. Contribution of myocardin in the hypoxia-induced phenotypic switching of rat pulmonary arterial smooth muscle cells. Exp Mol Pathol. 2010;89(3):301–306. doi:10.1016/j.yexmp.2010.06.010
9. Chettimada S, Gupte R, Rawat D, Gebb SA, Gupte SA, Physiology M. Hypoxia-induced glucose-6-phosphate dehydrogenase overexpression and activation in pulmonary artery smooth muscle cells: implication in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2014;308(3):
10. Meng J, Ai X, Lei Y, et al. USP5 promotes epithelial-mesenchymal transition by stabilizing SLUG in hepatocellular carcinoma. Theranostics. 2019;9(2):573–587. doi:10.7150/thno.27654
11. Xi X, Liu N, Wang Q, et al. ACT001, a novel PAI-1 inhibitor, exerts synergistic effects in combination with cisplatin by inhibiting PI3K/AKT pathway in glioma. Cell Death Dis Oct. 2019;10(10):757. doi:10.1038/s41419-019-1986-2
12. Zhong W, Yang W, Qin Y, et al. 6-Gingerol stabilized the p-VEGFR2/VE-cadherin/beta-catenin/actin complex promotes microvessel normalization and suppresses tumor progression. J Exp Clin Cancer Res. 2019;38(1):285. doi:10.1186/s13046-019-1291-z
13. Zhong W, Sun B, Gao W, et al. Salvianolic acid A targeting the transgelin-actin complex to enhance vasoconstriction. E Bio Med. 2018;37:246–258. doi:10.1016/j.ebiom.2018.10.041
14. Rich S, Dantzker DR, Ayres SM, Bergofsky EH, Koerner SK. Primary pulmonary hypertension: a national prospective study. Ann Intern Med. 1987;107(2):216–223. doi:10.7326/0003-4819-107-2-216
15. Park YM, Chung WJ, Lee SP, et al. Efficacy of inhaled iloprost in cor pulmonale and severe pulmonary hypertension associated with tuberculous destroyed lung. J Cardiovasc Ultrasound. 2014;22(2):95–97. doi:10.4250/jcu.2014.22.2.95
16. McLaughlin VV, Shillington A, Rich S. Survival in primary pulmonary hypertension: the impact of epoprostenol therapy. Circulation. 2002;106(12):1477–1482. doi:10.1161/01.CIR.0000029100.82385.58
17. Herbay AV, Illes A, Waldherr R, Otto HF. Pulmonary tumor thrombotic microangiopathy with pulmonary hypertension. Cancer. 1990;66(3):587–592. doi:10.1002/1097-0142(19900801)66:3<587::AID-CNCR2820660330>3.0.CO;2-J
18. Galiè N, Hoeper MM, Humbert M, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the Internati. Eur Heart J. 2015;37(1):67–119. doi:10.1093/eurheartj/ehv317
19. D’Alonzo GE, Barst RJ, Ayres SM, et al. Survival in patients with primary pulmonary hypertension: results from a national prospective registry. Ann Intern Med. 1991;115(5):343–349. doi:10.7326/0003-4819-115-5-343
20. Fishman AP. Clinical classification of pulmonary hypertension. Clin Chest Med. 2001;22(3):385–391. doi:10.1016/S0272-5231(05)70278-1
21. Abe K, Shimokawa H, Morikawa K, et al. Long-term treatment with a Rho-kinase inhibitor improves monocrotaline-induced fatal pulmonary hypertension in rats. Circ Res. 2004;94(3):385–393. doi:10.1161/01.RES.0000111804.34509.94
22. Baber SR, Deng W, Master RG, et al. Intratracheal mesenchymal stem cell administration attenuates monocrotaline-induced pulmonary hypertension and endothelial dysfunction. Am J Physiol Heart Circ Physiol. 2007;292(2):H1120–H1128. doi:10.1152/ajpheart.00173.2006
23. Rosenberg HC, Rabinovitch M. Endothelial injury and vascular reactivity in monocrotaline pulmonary hypertension. Am J Physiol. 1988;255(6 Pt 2):H1484–H1491. doi:10.1152/ajpheart.1988.255.6.H1484
24. Cobbold LC, Wilson LA, Sawicka K, et al. Upregulated c-myc expression in multiple myeloma by internal ribosome entry results from increased interactions with and expression of PTB-1 and YB-1. Oncogene. 2010;29(19):2884–2891. doi:10.1038/onc.2010.31
25. Shen D, Li J, Lepore JJ, Anderson T. Aortic aneurysm generation in mice with targeted deletion of integrin-linked kinase in vascular smooth muscle cells. Circ Res. 2011;109(6):616–628. doi:10.1161/CIRCRESAHA.110.239343
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.