")
Back to Journals » Journal of Blood Medicine » Volume 11
Psychosocial Impact and Disease Management in Patients with Congenital Factor VII Deficiency
Authors Peltier S, Kellum A , Brewer J, Duncan A, Cooper DL , Saad H
Received 25 April 2020
Accepted for publication 18 August 2020
Published 11 September 2020 Volume 2020:11 Pages 297—303
DOI https://doi.org/10.2147/JBM.S259909
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Skye Peltier,1 Angela Kellum,2 Janet Brewer,3 Alexander Duncan,4 David L Cooper,5 Hossam Saad5
1Center for Bleeding and Clotting Disorders, University of Minnesota Medical Center - Fairview, Minneapolis, MN, USA; 2Louisiana Center for Bleeding and Clotting Disorders, Tulane University, New Orleans, LA, USA; 3Comprehensive Health Education Services, Hanson, MA, USA; 4Department of Pathology & Laboratory Medicine, Emory University School of Medicine, Atlanta, GA, USA; 5Novo Nordisk Inc., Plainsboro, NJ, USA
Correspondence: Skye Peltier
Center for Bleeding and Clotting Disorders, University of Minnesota Medical Center – Fairview, Minneapolis, MN, USA
Tel +1 612-273-5047
Fax +1 612-273-5018
Email [email protected]
Purpose: Congenital factor VII (FVII) deficiency is a rare bleeding disorder of variable phenotype with predominantly mucocutaneous bleeding. The aim of this study was to identify the burden of FVII deficiency on patients and caregivers through a better understanding of the management and psychosocial impact of this disease.
Materials and Methods: A rare disease specialty recruiter from Comprehensive Health Education Services recruited participants for this online survey, which was conducted from January 31 to March 12, 2019. A moderator-assisted questionnaire was used to collect data on demographics, diagnosis, treatment, and psychosocial impact.
Results: Of the 45 respondents (25 patients and 20 caregivers), the majority were female (56%). Respondents reported a wide variety of initial bleeding symptoms, including bruising (58%), epistaxis (56%), and menorrhagia (36% of females). Because symptoms varied between individuals and were not always severe, diagnosis was often delayed. Mean time to obtain a diagnosis was 6.5 years and mean age at first diagnosis was 12.9 years. One-quarter (24%) of the respondents reported more than 100 bleeds of any severity over the previous year. When treating bleeds, 44% of patients reported using antifibrinolytics, and 42% reported using recombinant activated factor VII. Almost 31% of respondents reported missing schooldays as children, and 16% reported losing or resigning from a job in adulthood as a direct result of their disease. Notably, 29% of caregivers and 10% of their partners had also experienced issues with employment. Forty percent of respondents reported not participating in contact sports during childhood, and 22% continued to avoid contact sports in adulthood.
Conclusion: Overall, FVII deficiency has a substantial psychosocial impact, but most patients are satisfied with their disease management and are optimistic about their future. Patients desire additional educational, social, and financial support.
Keywords: bleeding disorder, psychosocial impact, recombinant activated factor VII, survey
Introduction
Congenital factor VII (FVII) deficiency is an autosomal recessive bleeding disorder caused by mutations in the gene encoding for FVII located in chromosome 13.1,2 More than 90% of identified mutational variants are point mutations and gene deletions. FVII is a vitamin K-dependent glycoprotein necessary for the initiation of coagulation and optimal hemostasis. Its normal plasma concentration is 10 nM (0.5 µg/mL). In plasma, FVII circulates mostly as an inactive zymogen, but it also occurs in active form.3
Estimated prevalence is 1:300,000–500,000 with a higher incidence in areas where consanguinity is common.2,3 The disease affects males and females equally, however, females tend to have more symptoms due to gynecologic bleeding.2
The European Network of Rare Bleeding Disorders (EN-RBD) registry reports a weak correlation between clinical severity and plasma FVII levels.4 Clinical phenotypes range from asymptomatic to severe life-threatening bleeding (central nervous system, gastrointestinal bleeding). Prediction of bleeding risk is based on multiple parameters, which poses a challenge to disease management.5 Despite the weak correlation between clinical severity and FVII levels, the International Society on Thrombosis and Haemostasis Scientific Standardization Committee (ISTH-SSC) classifies congenital FVII deficiency as follows: severe when FVII is <10% (highest risk of spontaneous major bleeding, including intracranial hemorrhage, gastrointestinal bleeding, and hemarthrosis), moderate when FVII is 10% to 20% (risk of mild spontaneous or triggered bleeding), and mild when FVII is 20% to 50% (often asymptomatic; comprising approximately 30%-40% of affected patients).2 Patients with severe deficiency are diagnosed in early childhood, whereas those with moderate deficiency present later in adolescence or at menarche in female patients.2 Overall, the most common symptoms include epistaxis, easy bruising, and mucocutaneous bleeding.2,6 Approximately 30% to 40% of affected individuals have asymptomatic disease.2
Laboratory findings include an increased prothrombin time (PT) and a normal activated partial thromboplastin time. Confirmatory diagnosis is possible with modified one-stage PT-based clotting assay, and differentiation of qualitative from quantitative FVII deficiency can be made by measuring plasma FVII antigen using immunoassay.1,7 Therapeutic and preventative options for congenital FVII deficiency include fresh frozen plasma, concentrates of FVII and prothrombin complex, and recombinant activated FVII (rFVIIa), although the availability of the agents varies between countries.1,6 While fresh frozen plasma and concentrates carry the risk of infection and volume overload, rFVIIa is generally well tolerated with a low risk (0.17%) of thromboembolic events8–10 and a half-life of 2.6 hours in patients with congenital FVII deficiency.11
There exists a continued need for more evidence-based studies to enhance our current understanding of congenital FVII deficiency. In this paper, we present the results from an online survey completed by patients with congenital FVII deficiency or their caregivers. The aim of the survey was to better understand the challenges that patients encounter in the diagnosis and treatment of the disease and the burden of congenital FVII deficiency on both patients and caregivers.
Materials and Methods
Study Design
A rare disease specialty recruiter from Comprehensive Health Education Services recruited participants for this online survey. Participants included patients with congenital FVII deficiency and caregivers of pediatric patients with the disease. This moderator-assisted survey was conducted from January 31 through March 12, 2019; average time to complete the 99-question survey was 45 minutes.
All participants provided informed consent. This survey was conducted in compliance with all national laws and guidelines protecting personal data. This survey was considered market research and as such did not require IRB or ethics committee approval.
Study Measures
Data were collected on patient demographics, initial symptoms, diagnosis, treatment history, and psychosocial impact. Bleeding assessment was conducted using the ISTH Bleeding Assessment Tool (BAT).12 To objectively evaluate the disease state of the patients, established cutoff values (≥4, adult males; ≥6, adult females; and ≥3, children)13 for this tool were used.
Statistical Analysis
Due to the small sample size, only descriptive statistics will be provided.
Results
Patient Demographics
Twenty-five patients and 20 caregivers completed the survey. Patient demographics are summarized in Table 1. Mean (range) age of the 25 adult patients was 33 (18–67) years; mean (range) age of children and adolescents whose caregivers completed the survey was 11 (5–17) years. Fifty-six percent of participants were female; 44% of participants were white, and 53% were employed full time. A family history of FVII deficiency was present in 58% of participants, and 50% were the first in their family to be diagnosed with FVII deficiency.
 |
Table 1 Demographic Characteristics |
Initial Symptoms
Owing to the wide variation in disease severity and the lack of clinical correlation between plasma levels of coagulation FVII and bleeds, neither patients nor their health care providers recognized initial symptoms as a bleeding disorder. Epistaxis was a common symptom at diagnosis, as 56% of respondents reported this as an initial symptom. Mouth bleeds (31%) and skin bruising (58%) were also reported. Forty-seven percent of females of reproductive age reported menorrhagia. The mean (range) BAT score was 7.67 (0–39) at diagnosis (Figure 1). A variety of bleeds contributed to the BAT total score, including epistaxis, bruising, bleeds from surgery or trauma, and joint bleeds.
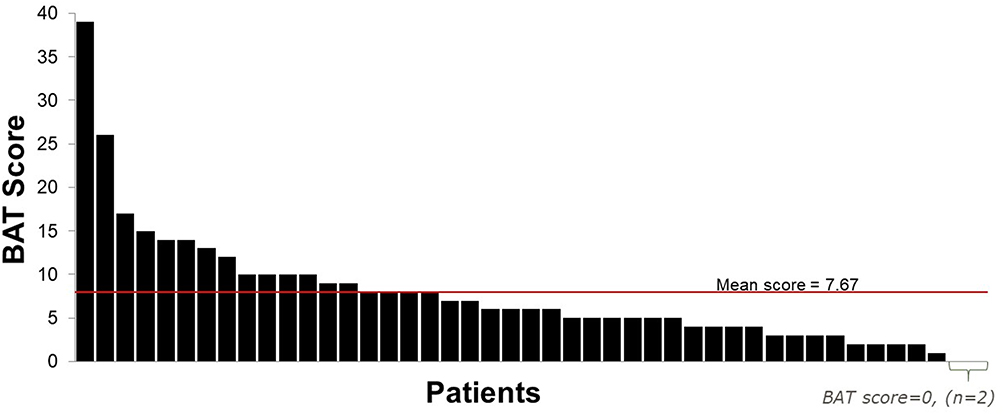 |
Figure 1 Distribution of ISTH-BAT scores. |
Diagnosis
The average age (range) at diagnosis was 12.9 (<1-47) years; 20% of the respondents were diagnosed after their 21st birthdays. The average time (range) from initial symptoms to a visit with a specialist was more than 4 (<1-37) years; the average time (range) from initial symptoms to diagnosis was 6.5 (<1-45) years.
Disease Management
Respondents reported that they prefer to see hematology specialists; they have observed that other health care providers have little understanding of their disease. Some patients and caregivers had difficulty finding a hematologist who had any knowledge of congenital FVII deficiency. Many respondents (89%) have regular follow-up visits; most of these follow-ups are with a private hematologist (51%) or at a hemophilia treatment center (49%). Many respondents were willing to travel long distances to see their hematologist; on average, respondents reported traveling an average of 64 minutes (each way) to their follow-up visits. The average travel time to the nearest location for treatment of an acute bleed was 26 minutes. Patients were comfortable with treating their own bleeds; 94% of bleeds were treated at home. Agents or products used to treat bleeds included antifibrinolytics (44% of respondents) and rFVIIa (42% of respondents). In addition, 18% of respondents reported using other methods (Figure 2).
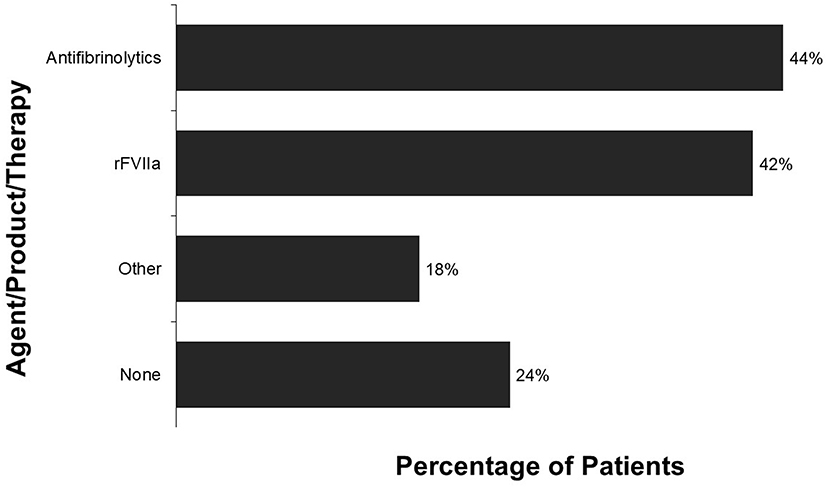 |
Figure 2 Treatments used to control recent bleeds (categories were not mutually exclusive). |
Despite regular follow-ups, many patients with FVII deficiency reported persistent symptoms. The most commonly reported symptoms were skin bruising and gum/tooth/mouth bleeds (Figure 3). Some (24%) respondents reported more than 100 bleeds of any severity over the previous year.
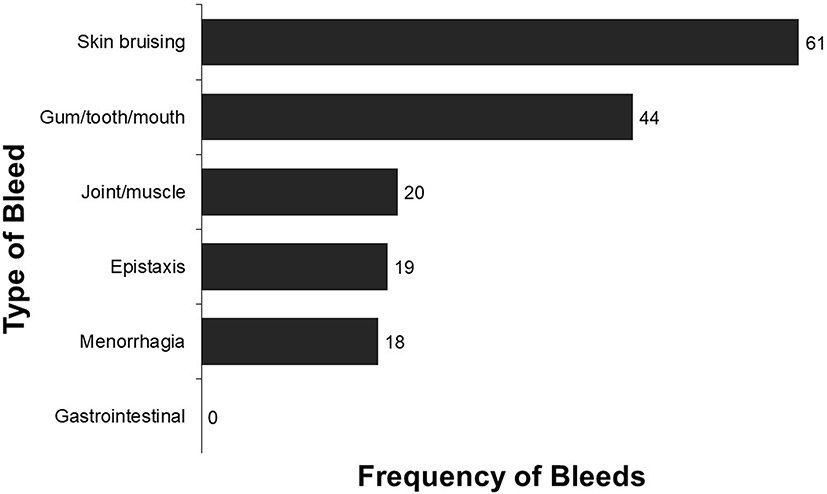 |
Figure 3 Average frequency of bleeds over the past year reported by all respondents. |
Many female respondents were disheartened by their gynecologist’s lack of knowledge and experience with FVII deficiency. Menorrhagia was a common issue among these women. Approximately 40% of these patients remain on continuous hormonal contraception to prevent regular menstruation. Childbearing is also a major area of concern for these patients; a few respondents reported that they were counseled by their health care providers to avoid having children.
Psychosocial Impact
Although many respondents reported consistent psychosocial impact, they expressed optimism about living with their disease in the future. Thirty-one percent of respondents missed school for reasons related to their disease; 33% reported experiencing excessive bleeding episodes at school. Some patients avoided activities that could put them at risk for a bleeding episode; 18% of respondents did not participate in gym class while they were in school, 40% reported not participating in contact sports during childhood, and 22% continued to avoid contact sports in adulthood. While many patients were not experiencing any issue with friends and peers at the time of the survey, 11% reported that their peers did not understand their disease, and 6% reported being bullied. Adult patients cited issues with employment; 16% lost or resigned from their job for reasons related to FVII deficiency. In addition, 12% stated that their employer was not understanding of their disease. Notably, 29% of caregivers and 10% of their partners had also experienced issues with employment. Respondents were satisfied with the level of support they receive from their significant other (74%) and family members (78%) (Figure 4).
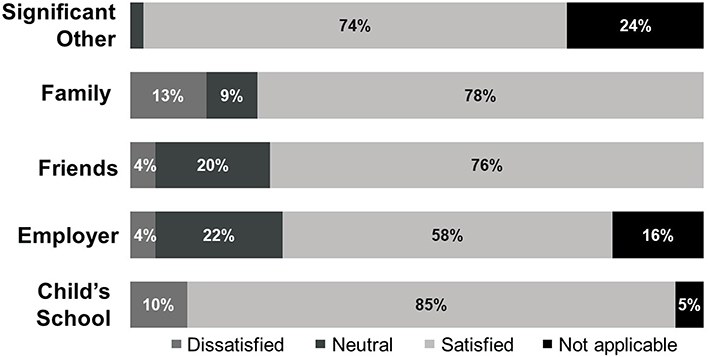 |
Figure 4 Proportion of patients satisfied with the level of support they receive. |
Patient Resources
Although patients were familiar with a wide variety of resources, they noted that disease-specific information was scarce. Top-rated resources were disease-specific and provided tangible support, such as retreats and conferences as well as opportunities to meet fellow patients, caregivers, and families, and leading health care professionals. The highest-rated organizations for support were Comprehensive Health Education Services, the Congenital FVII Deficiency Research Foundation, and the National Hemophilia Foundation (NHF). Patients reported that additional educational, social, and financial resources are still needed.
Discussion
The results of this online survey demonstrate the disease burden and challenges faced by patients with congenital FVII deficiency and their caregivers. Although the participants in the survey were satisfied with the support that they receive from their family and friends, most reported a continued need for additional educational resources for patients and caregivers and opportunities for networking within support groups. Comprehensive and integrative care involving hematologists, advanced practice providers, social workers, and caregivers can help better manage the psychosocial issues associated with congenital FVII deficiency.2,14 This approach could greatly benefit these patients and those with other rare bleeding disorders such as Glanzmann’s thrombasthenia and factor XIII deficiency, who likely have similar experiences with disease burden.
All physicians treating patients with congenital FVII deficiency should gather information from available resources such as the American Thrombosis and Hemostasis Network (ATHN) and multinational registries, including International Registry on Congenital FVII Deficiency and Seven Treatment Evaluation Registry (STER), to stay current with disease pathophysiology and treatment options.2,6 Given the highly variable clinical presentation and the long gap between symptom onset and disease diagnosis, knowledge of the ISTH-SSC classification is important for health care professionals to identify patients with higher risk of life-threatening bleeding, guide the choice of intervention, and improve patients’ quality of life.2
Menstruation and childbirth increase the risk of bleeding and are areas of challenge and confusion among female patients. Physiologic increases in FVII have been reported during pregnancy, but most patients with a bleeding history require peripartum prophylactic therapy with fresh frozen plasma, FVII concentrates, or rFVIIa.15 If possible, women with FVII deficiency should confirm that their chosen hospital for delivery is able to conduct FVII assays. Prophylaxis with rFVIIa, which has been reported in STER16 and other publications,17,18 may help reduce the development of iron deficiency anemia and unnecessary hysterectomies associated with menorrhagia in FVII-deficient patients. Although there is no consensus for the management of this rare condition, gynecologists and obstetricians treating patients with FVII deficiency should carefully assess the bleeding risk and need for prophylaxis.9,19 These health care providers should work closely with a hematologist to manage patients with FVII deficiency.
Periodic physical therapy is recommended for patients with arthropathy and those engaging in vigorous physical activities. Joint bleeding is not uncommon in congenital FVII deficiency, and arthropathy similar to that found in congenital hemophilia has been reported from childhood onwards.2,20 As one-half of the affected patients have a family history, genetic counseling and genotyping among known FVII-deficient patients can aid in the early diagnosis of family members and improve patient care.2
Among the various therapies for congenital FVII deficiency, rFVIIa is recommended by the National Hemophilia Foundation’s Medical and Scientific Advisory Council (NHF-MASAC) as a first-line treatment;21 plasma-derived FVII is not available in the United States.1,8 Although the formation of alloantibodies is a general concern with replacement therapies, the development of inhibitors against FVII is rare and inhibitors have a low affinity with rFVIIa when present.1,22 Of note, survey respondents reported having easy access to rFVIIa for prophylactic use or for acute bleeds and most patients were comfortable using it at home as needed.
Owing to the rareness of congenital FVII deficiency, there is a lack of homogenous epidemiologic studies and randomized clinical controlled studies. With evolving treatment options, more cross-sectional studies are needed to develop evidence-based management guidelines and to bridge the gap between our understanding and clinical practice.3,15 Some limitations of this qualitative research survey include small power and sample selection that limit the generalization of the reported results. In addition, reporting of disease severity is often inconsistent with the ISTH definitions, and FVII levels could not be collected in an Internet-based study; therefore, data could not be analyzed by disease severity. In addition, the survey collected data on the number of surgical procedures experienced by each patient; however, information on outcomes and adverse events was not collected. Nevertheless, the results highlight the current disease state of congenital FVII deficiency and can be used to guide management and optimize patient care. Given that the organization recruiting patients also offers disease-specific educational programs, there is a potential for recruitment bias; however, a recently published case-based discussion highlights the need for education about diagnosis, classification, and management.2
Conclusion
Congenital FVII deficiency is associated with high disease burden and psychosocial issues among affected patients and caregivers. Several resources, including networks and registries, provide useful information about disease state and management. Health care professionals should increase their awareness of disease classification and available treatment options to provide effective management of this rare condition.
Abbreviations
ATHN, American Thrombosis and Hemostasis Network; BAT, bleeding assessment tool; EN-RBD, European Network of Rare Bleeding Disorders; FVII, factor VII; ISTH, International Society on Thrombosis and Haemostasis; ISTH-SCC, International Society on Thrombosis and Haemostasis Scientific Standardization Committee; NHF, National Hemophilia Foundation; NHF-MASAC, National Hemophilia Foundation’s Medical and Scientific Advisory Council; PT, prothrombin time; rFVIIa, recombinant activated factor VII; STER, Seven Treatment Evaluation Registry.
Acknowledgments
This survey was sponsored by Novo Nordisk. Writing and editorial support were provided by PRECISIONscientia, Yardley, PA, in accordance with Good Publication Practice (GPP3) guidelines and were funded by Novo Nordisk. The authors would like to acknowledge Dr Shilpa Jain for her assistance with the data analysis.
Disclosure
SP is a consultant for Novo Nordisk. AK is a member of advisory boards for Novo Nordisk and Takeda and is a member of the speakers bureau for Takeda. JB is an employee of Comprehensive Health Education Services and reports grants from Novo Nordisk. AD is a consultant for Novo Nordisk. DLC and HS were employees of Novo Nordisk at the time of survey conception, drafting, and data collection and analysis. DLC is currently an employee of uniQure, Lexington, MA, USA. HS is currently an employee of Amgen, Thousand Oaks, CA, USA. The authors report no other conflicts of interest in this work.
References
1. Franchini M, Marano G, Mengoli C, et al. Inhibitors in patients with congenital bleeding disorders other than hemophilia. Semin Thromb Hemost.;44(6):595–603. doi:10.1055/s-0037-1607441
2. Jain S, Donkin J, Frey MJ, Peltier S, Gunawardena S, Cooper DL. Phenotypical variability in congenital FVII deficiency follows the ISTH-SSC severity classification guidelines: a review with illustrative examples from the clinic. J Blood Med. 2018;9:211–218. doi:10.2147/JBM.S157633
3. Shahbazi S, Mahdian R. Factor VII gene defects: review of functional studies and their clinical implications. Iran Biomed J. 2019;23(3):165–174. doi:10.29252/ibj.23.3.165
4. Peyvandi F, Palla R, Menegatti M, et al. Coagulation factor activity and clinical bleeding severity in rare bleeding disorders: results from the European Network of Rare Bleeding Disorders. J Thrombosis Haemostasis. 2012;10(4):615–621. doi:10.1111/j.1538-7836.2012.04653.x
5. Napolitano M, Siragusa S, Mariani G. Factor VII deficiency: clinical phenotype, genotype and therapy. J Clin Med. 2017;6:4. doi:10.3390/jcm6040038
6. de Moerloose P, Schved JF, Nugent D. Rare coagulation disorders: fibrinogen, factor VII and factor XIII. Haemophilia. 2016;22(Suppl 5):61–65. doi:10.1111/hae.12965
7. Mariani G, Bernardi F. Factor VII deficiency. Semin Thromb Hemost. 2009;35(4):400–406. doi:10.1055/s-0029-1225762
8. Hedner U. Recombinant activated factor VII: 30 years of research and innovation. Blood Rev. 2015;29(Suppl 1):S4–8. doi:10.1016/S0268-960X(15)30002-3
9. Loddo A, Cornacchia S, Cane FL, et al. Prophylaxis of peripartum haemorrhage using recombinant factor VIIa (rfVIIa) in pregnant women with congenital factor VII deficiency: a case report and literature review. Eur J Obstet Gynecol Reprod Biol. 2019;235:77–80. doi:10.1016/j.ejogrb.2019.02.017
10. Rajpurkar M, Croteau SE, Boggio L, Cooper DL. Thrombotic events with recombinant activated factor VII (rFVIIa) in approved indications are rare and associated with older age, cardiovascular disease, and concomitant use of activated prothrombin complex concentrates (aPCC). J Blood Med. 2019;10:335–340. doi:10.2147/JBM.S219573
11. Novo Nordisk Inc. NovoSeven®RT Package Insert. 2019.
12. Rodeghiero F, Tosetto A, Abshire T, et al. on behalf of the ISTH/SSC Joint VWF and Perinatal/Pediatric Hemostasis Subcommittees Working Group. ISTH/SSC Bleeding Assessment Tool: a standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J Thrombosis Haemostasis. 2010;8(9):2063–2065. doi:10.1111/j.1538-7836.2010.03975.x.
13. Elbatarny M, Mollah S, Grabell J, et al. Normal range of bleeding scores for the ISTH-BAT: adult and pediatric data from the merging project. Haemophilia. 2014;20(6):831–835. doi:10.1111/hae.12503
14. Page D. Comprehensive care for hemophilia and other inherited bleeding disorders. Transfusion Apheresis Sci. 2019;58(5):565–568. doi:10.1016/j.transci.2019.08.005
15. Bannow BS, Konkle BA. Inherited bleeding disorders in the obstetric patient. Transfus Med Rev. 2018;32(4):237–243. doi:10.1016/j.tmrv.2018.06.003
16. Napolitano M, Giansily-Blaizot M, Dolce A, et al. Prophylaxis in congenital factor VII deficiency: indications, efficacy and safety. Results from the Seven Treatment Evaluation Registry (STER). Haematologica. 2013;98(4):538–544. doi:10.3324/haematol.2012.074039
17. Holve S, Stuart R, Recht M. Prophylaxis of recurrent epistaxis and menorrhagia using recombinant factor VIIa in severe congenital factor VII deficiency. Pediatr Res. 2001;49(4):203A.
18. Huth-Kuhne A, Lages P, Zimmerman R. Regular prophylaxis with recombinant factor VIIa in a patient with severe congenital FVII deficiency. Hämostaseologie. 2008;28(Suppl S 01):S55. doi:10.1055/s-0037-1617127
19. Napolitano M, Di Minno MN, Batorova A, et al. Women with congenital factor VII deficiency: clinical phenotype and treatment options from two international studies. Haemophilia. 2016;22(5):752–759. doi:10.1111/hae.12978
20. Mariani G, Herrmann FH, Dolce A, et al. Clinical phenotypes and factor VII genotype in congenital factor VII deficiency. Thromb Haemost. 2005;93(3):481–487. doi:10.1160/TH04-10-0650
21. National Hemophilia Foundation. MASAC recommendations concerning products licensed for the treatment of hemophilia and other bleeding disorders; 2020. Available from: https://www.hemophilia.org/Researchers-Healthcare-Providers/Medical-and-Scientific-Advisory-Council-MASAC/MASAC-Recommendations/MASAC-Recommendations-Concerning-Products-Licensed-for-the-Treatment-of-Hemophilia-and-Other-Bleeding-Disorders.
22. Batorova A, Mariani G, Kavakli K, et al. Inhibitors to factor VII in congenital factor VII deficiency. Haemophilia. 2014;20(2):e188–191. doi:10.1111/hae.12376
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.