")
Back to Journals » International Journal of Nanomedicine » Volume 14
Psoralen-loaded lipid-polymer hybrid nanoparticles enhance doxorubicin efficacy in multidrug-resistant HepG2 cells
Authors Yuan Y, Cai T, Callaghan R, Li Q, Huang Y, Wang B, Huang Q , Du M, Ma Q, Chiba P , Cai Y
Received 6 October 2018
Accepted for publication 7 January 2019
Published 27 March 2019 Volume 2019:14 Pages 2207—2218
DOI https://doi.org/10.2147/IJN.S189924
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Linlin Sun
Yueling Yuan,1,* Tiange Cai,2,* Richard Callaghan,3,* Qianwen Li,4 Yinghong Huang,4 Bingyue Wang,1 Qingqing Huang,1 Manling Du,1 Qianqian Ma,1 Peter Chiba,5 Yu Cai1,6
1College of Pharmacy, Jinan University, Guangzhou, Guangdong 510632, China; 2College of Life Science, Liaoning University, Shenyang, Liaoning 110000, China; 3Research School of Biology, Australian National University, Canberra ACT 2601, Australia; 4Guangzhou Guoyu Pharmaceutical Technology Co., Ltd., Guangzhou, Guangdong 510663, China; 5Institute of Medical Chemistry, Medical University of Vienna, Vienna 1090, Austria; 6Cancer Research Institute, Jinan University, Guangzhou, Guangdong 510632, China
*These authors contributed equally to this work
Background: Psoralen (PSO), a major active component of Psoralea corylifolia, has been shown to overcome multidrug resistance in cancer. A drug carrier comprising a lipid-monolayer shell and a biodegradable polymer core for sustained delivery and improved efficacy of drug have exhibited great potential in efficient treatment of cancers.
Methods: The PSO-loaded lipid polymer hybrid nanoparticles were prepared and characterized. In vitro cytotoxicity assay, cellular uptake, cell cycle analysis, detection of ROS level and mitochondrial membrane potential (ΔΨm) and western blot were performed.
Results: The P-LPNs enhanced the cytotoxicity of doxorubicin (DOX) 17-fold compared to free DOX in multidrug resistant HepG2/ADR cells. Moreover, P-LPNs displayed pro-apoptotic activity, increased levels of ROS and depolarization of ΔΨm. In addition, there were no significant effects on cellular uptake of DOX, cell cycle arrest, or the expression of P-glycoprotein. Mechanistic studies suggested that P-LPNs enhanced DOX cytotoxicity by increased release of cytochrome c and enhanced caspase3 cleavage, causing apoptosis in HepG2/ADR cells.
Conclusion: The lipid-polymer hybrid nanoparticles can be considered a powerful and promising drug delivery system for effective cancer chemotherapy.
Keywords: lipid-polymer hybrid nanoparticles, psoralen, drug delivery, HepG2, ADR cells, apoptosis
Introduction
Hepatocellular carcinoma (HCC) is one of the most common types of cancer worldwide.1 Chemotherapy is frequently used in HCC treatment; however, its efficacy is limited by the development of multidrug resistance (MDR).2 Many studies have revealed that overexpression of drug efflux pumps, increased DNA damage repair, and hypoxia-inducible factor1-α (HIF1-α) signaling played important roles in the drug resistance of HCC.3–6 In recent years, reduced levels of ROS have also been implicated in cell resistance to chemotherapy drugs.7,8 In addition, evidence has been presented that Bcl-2 over expression resulted in the resistance of cells to doxorubicin (DOX) and paclitaxel.9,10 It is clear that multiple factors are involved in conferring multidrug resistance and that, regardless of the mechanism, strategies to overcome the phenotype are vital.
In China, Traditional Chinese Medicines have frequently been used for cancer treatment. For example, the naturally occurring coumarin compound psoralen (PSO) has anti-tumor, estrogen-like, and multidrug resistance modulating properties.11,12 Unfortunately, its use has been limited by pharmacokinetic issues, primarily due to its poor solubility. However, targeted nanoscale drug delivery systems have been developed to improve solubility and controlled drug release. More importantly, nanoparticles can bypass efflux pumps since they are internalized via endocytosis, which ensures higher intracellular accumulation of drugs.13–15
Lipid-polymer hybrid nanoparticles (LPNs) combine the advantages of liposome- and polymer-based nanoparticles.16,17 The LPNs comprise a polymer core coated with lipid layers, and the combination can improve stability, enhance cellular uptake, and increase encapsulation efficiency.18–20 Poly(D,L-lactide-co-glycolide) (PLGA), a biodegradable polymer approved by the US Food and Drug Administration, is commonly used as the polymer core to encapsulate poorly water soluble compounds.21 The lipid component contains natural (eg, crude soybean lipids) and synthetic phospholipids such as 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-carboxy (polyethylene glycol) 2000 (DSPE-PEG2000). The amphiphilic phospholipids form a monolayer around the hydrophobic polymer core, whereas the DSPE-PEG2000 embeds in the lipid monolayer to form a PEGylation stealth layer outside the lipid shell. This surface coating improves electrostatic properties and steric stability to prolong circulation half-life.22
In the present work, PSO-loaded lipid-polymer hybrid nanoparticles (P-LPNs) were prepared with an emulsification–solvent–evaporation method. A P-glycoprotein (P-gp) expressing DOX-resistant HepG2 (HepG2/ADR) cell was used to investigate whether the P-LPNs enhanced the efficiency of chemotherapy drug or showed a superior therapeutic effect to free PSO. A thorough understanding about the role of P-LPNs on HepG2/ADR cells is of great importance for a rational design of nanoparticle-based chemotherapy regimens.
Materials and methods
Materials
Psoralen (PSO) was purchased from Chengdu Pufeide Biotechnology Co., Ltd (Chengdu, Sichuang, China). DOX hydrochloride was obtained from Selleck Chemicals (Houston, TX, USA). Soybean phospholipids (injection grade) were produced by Shanghai Tywei Pharmaceutical Co., Ltd (Shanghai, China). 1,2-distearoyl-sn-glycero-3-phosphocholine-N-[methoxy (polyethylene glycol)-2000] (DSPE-PEG2000) was provided by Shanghai AVT Pharmaceutical Technology Co., Ltd. (Shanghai, China). Poly(D,L-lactide-co-glycolide) (PLGA, 50:50, Mw: 8,000–12,000) was obtained from Jinan Daigang Biomaterial Co., Ltd (Jinan, Shandong, China). Dimethylsulfoxide (DMSO) and MTT (3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide) were purchased from Sigma-Aldrich (St Louis, Missouri, USA). RPMI-1640 cell culture medium, penicillin–streptomycin, and trypsin-EDTA were purchased from Thermo Fisher Life Technologies (Guangzhou, Guangdong, China). FBS was provided by AusGeneX Pty Ltd (Molendinar, Queensland, Australia). Dialysis membranes with a molecular weight cut-off of 8,000–14,000 kDa were purchased from Sangon Biotech (Shanghai, China). AnnexinV-APC dye and SYTOX green dye were provided by KeyGEN BioTECH (Nanjing, Jiangsu, China). JC-1 (5,50,6,60-tetrachloro-1,10,3,30-tetraethylbenzimidazolyl-carbocyanine iodide) and DCFH-DA (2′,7′-dichlorofluorescein diacetate) were purchased from Beytime Biotechnology (Shanghai, China). Propidium iodide (PI) staining buffer was obtained from BD Biosciences Pharmingen (Shanghai, China). P-glycoprotein primary antibodies and horseradish peroxidase-conjugated goat anti-rabbit immunoglobulin were purchased from Abcam (Cambridge, UK). Other primary antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). HepG2 hepatoblastoma cell lines were provided by KeyGEN BioTECH (Nanjing, Jiangsu, China). Ethanol, acetonitrile, and all other chemicals were HPLC grade.
Methods
Preparation of the LPNs
LPNs were prepared according to a previously published method.22,23 Ester-terminated PLGA and PSO were dissolved in acetonitrile, whereas phospholipids and DSPE-PEG2000 dissolved in 4% ethanol solution. The PLGA drug solution was added dropwise to the surfactant solution with an oil-water ratio of 1:10 (v/v). This solution was stirred for 90 minutes at 70°C and formed the core-shell LPN by self-assembly. P-LPNs were filtered through a 0.22 μm membrane, then used immediately, stored at 4°C, or lyophilized for later use.
Size and zeta potential measurements
Particle size, zeta potential, and polydispersity were measured by dynamic light scattering using a Zetasizer Nano ZS90 (Malvern, UK). The dispersion of nanoparticle was diluted with ultrapure water and subsequently analyzed.20
Particle morphology
The shape and morphology of NPs was visualized by transmission electron microscopy (TEM, TECNAI 10, Philips, the Netherlands) using an accelerating voltage of 200 kV. One drop of the nanoparticle dispersion was deposited onto the carbon-coated copper grid, followed by air drying prior to imaging.24
Encapsulation efficiency and loading capacity
The quantity of PSO in the LPNs was measured by HPLC (Agilent, Santa Clara, CA, USA), equipped with a reverse-phase column (Agilent, 150 ×4.6 mm, 5 μm). Isocratic chromatography conditions included a mobile phase of acetonitrile and water (55:45 v/v) with a flow rate of 1.0 mL/min. Injection volume of all samples was 5 μL. The nanoparticle emulsion was centrifuged at 11,000 g for 30 minutes to collect free PSO in the supernatant. A fraction of the P-LPNs was suspended in the mobile phase to determine the total PSO for HPLC analysis.25
The drug loading (DL) and encapsulation efficiency (EE) were calculated using the following equations:18,26
|
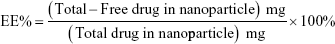
|
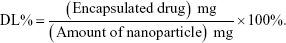
In vitro drug release study
In vitro release of PSO from P-LPNs was done using a standard dialysis procedure.18 Briefly, 30 mg of the drug loaded NPs were dispersed in PBS (0.01 M, pH 7.4) containing 0.5% (v/v) Tween-80; the latter added to improve the solubility of PSO in PBS. The dispersion, loaded in a dialysis bag, was placed into 150 mL PBS and stirred at 100 rpm at 37°C. At the indicated times, a 1 mL aliquot of the release medium (PBS) was removed for HPLC analysis and replaced with the same volume of fresh medium. The release medium was exchanged periodically during the dialysis process.
In vitro cell culture
HepG2/S cells were cultured in RPMI-1640 medium supplemented with 10% (v/v) FBS and 1% (v/v) penicillin-streptomycin under a humidified 5% CO2 atmosphere at 37°C. HepG2/ADR cell culture contained 1 μM DOX in RPMI-1640 medium to maintain P-gp expression every third passage. Cells were used for experiments at 80% confluency.27
In vitro cytotoxicity assay
Cells were incubated overnight in 96-well plates (Corning Inc., Corning, NY, USA) at a cell density of 5×103 cells/well. Spent medium was removed and replaced with fresh medium containing free DOX, free PSO, P-LPNs, DOX+PSO or DOX+P-LPNs, and incubated for 48 hours. Following incubation, 20 μL of MTT solution were added to each well, and the plates incubated for 4 hours. Absorbance was measured at a wavelength of 570 nm using a 96-well plate reader (BioTek, Winooski, VT, USA).
Cell toxicity was determined using the following equation:
|
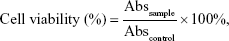
where Abssample is the absorbance of cells in the presence of different formulations, and Abscontrol is the absorbance of cells in the absence of drug. IC50 values were determined by nonlinear regression of the general dose–response equation.
Analysis of cell apoptosis
Cell apoptosis were measured by an AnnexinV-APC & SYTOX green apoptosis detection kit according to the manufacturer’s instructions. Briefly, HepG2 cells (2×105) were placed in 6-well plates (Corning Inc., Corning, NY, USA) and cultivated overnight. Medium was then removed and replaced by fresh medium containing free DOX, DOX+PSO, or DOX+P-LPNs, and incubated for 24 or 48 hours. Concentrations of DOX was 10 μM, and PSO, either in free form or nanoparticles, was 10 and 20 μM, respectively. Cells treated with only medium were used as a control. The cells were trypsinized, washed with PBS, resuspended in 400 μL of binding buffer containing 4 μL of APC-conjugated AnnexinV (AnnexinV-APC), and incubated in the dark for 10 minutes at ambient temperature. SYTOX green stain dye (10 μM) was added, and the samples were measured immediately with a flow cytometer (FACSVerse BD, San Jose, CA, USA). APC dye (fluorescence channel 4, FL4) and SYTOX green dye (fluorescence channel 2, FL2) were analyzed as FL2–FL4 dot plots. All observations were obtained from three independent experiments, done in triplicate.
Cellular uptake of DOX by flow cytometry
Cellular uptake of DOX from the different formulations was quantified by flow cytometry. Briefly, HepG2 cells (2×105) were placed in 6-well plates overnight. Cells were treated with 1 mL of fresh medium containing DOX (10 μM), DOX+PSO (20 μM), DOX+P-LPNs (20 μM) or DOX+Verapamil (3 μM), and incubated for 3 hours at 37°C. Cells treated with only medium were used as the respective control. The cells were trypsinized, washed twice in cold PBS, and analyzed by flow cytometery (FACSVerse BD).
Cell cycle analysis
The distribution of DNA in the cell cycle was studied by flow cytometry. HepG2 cells (2×105) were seeded in 6-well plates and were treated with different formulations of DOX (10 μM), DOX+PSO (10 and 20 μM), or DOX+P-LPNs (10 and 20 μM) for 48 hours. Cells treated with only medium were used as controls. The cells were trypsinized, washed with PBS, and fixed in cold 75% (v/v) ethanol solution at −20°C overnight. Samples were stained with cell cycle reagent containing PI/RNAse (BD Biosciences Pharmingen, Shanghai, China) and analyzed using a flow cytometer (FACSVerse BD).
ROS formation assay
The intracellular levels of ROS were evaluated using 2′,7′-dichlorodihydroluorescein diacetate (DCFH-DA). The HepG2 cells (2×105) were seeded in a 6-well plate and treated with different formulations of DOX (10 μM), DOX+PSO (20 μM), and DOX+P-LPNs (20 μM) for 48 hours. The cells were trypsinized, washed with PBS, and incubated with 0.5 mL of medium containing 5 μM of the ROS-sensitive probe DCFH-DA at 37°C for 20 minutes. Cells were subsequently collected by centrifugation and washed three times with cell culture medium without FBS. All samples were analyzed by the flow cytometer (FACSVerse BD).
Depolarization of mitochondrial membrane potential (ΔΨm)
Mitochondrial membrane potential (ΔΨm) was determined with a JC-1 reagent. JC-1 is a lipophilic cationic dye that selectively enters mitochondria. Depolarization of ΔΨm was quantified as an increase in the percentage of depolarized cells, which reflected a decrease in JC-1 aggregates and an increase of monomer when the inner mitochondrial membrane was depolarized.
HepG2 cells (2×105) were seeded in a 6-well plate and treated with different formulations of DOX (10 μM), DOX+PSO (10 μM or 20 μM), and DOX+P-LPNs (10 μM or 20 μM) for 48 hours. JC-1 staining solution (0.5 mL), at a concentration of 1 mg/mL, was added to each sample and incubated for 20 minutes at 37°C. Subsequently, the cells were washed three times with cell culture medium without FBS and prepared for flow cytometry (FACSVerse BD). Platelet bound JC-1 monomers (FL1) and JC-1 aggregates (FL2) were analyzed as FL1–FL2 dot plots.28,29
Western immunoblotting
HepG2 cells (2×106) were seeded in 100 mm2 dishes (Corning Inc., Corning, NY, USA) and treated with different formulations of DOX (10 μM), DOX+PSO (10 and 20 μM), and DOX+P-LPNs (10 and 20 μM) for 48 hours. Subsequently, the cells were harvested in RIPA lysis buffer, and protein quantitation was done using the BCA assay (Thermo Fisher Life Technologies, MA, USA). The protein samples (20–60 μg) were separated by 8%–15% SDS-PAGE and transferred to polyvinylidene fluoride membranes. Membranes were incubated with rabbit primary antibodies (as specified in figure legends) (1:1,000 dilutions) at 4°C overnight, subsequently washed with PBS containing 0.5% Tween-20, and probed with horseradish peroxidase-conjugated goat anti-rabbit immunoglobulin (Abcam, Cambridge, UK) (1:6,000 dilution) for 60 minutes at room temperature. The immune complexes were detected by chemiluminescence (4A Biotech Co., Ltd, Beijing, China) and visualized by ChemiDoc XRS (Bio-Rad, CA, USA). The intensity of the band for each protein was normalized to that of GAPDH.
Statistical analysis
Quantitative data were expressed as mean ± standard error of the mean (SEM). Statistical significance in different groups was determined by paired Student’s t-test and one-way ANOVA as appropriate using GraphPad Prism 6.0 software (GraphPad Software Inc, CA, USA). P<0.05 was considered to be statistically significant.
Results
Physicochemical characterization of P-LPNs
The lipid-polymer nanocarrier system was used to deliver PSO, and the properties of both unloaded LPNs and PSO-loaded LPNs were characterized as shown in Table 1 and Figure 1. The particle sizes were ~65 nm (Figure 1A), with zeta potentials of −28 mV, and polydispersity indices of 0.24. TEM images indicated that the nanoparticles show a well-defined spherical shape with a uniform particle diameter and no adhesion (Figure 1B). The PSO-loaded lipid-polymer nanocarrier system was designed to hold a hydrophobic core containing polymer and drug, as well as the hydrophilic lipid layer shell.30 Moreover, PSO was efficiently loaded into LPNs, with a drug loading of 2.08% and an encapsulation efficiency of 72.2%. The drug release profiles from P-LPNs are shown in Figure 1C. Non encapsulated PSO was completely released from the dialysis bag into surrounding PBS within 24 hours, the P-LPNs showed sustained release with a cumulative release of PSO exceeding 90% after 96 hours.
 | Table 1 Nanoparticle properties of unloaded LPNs and P-LPNs |
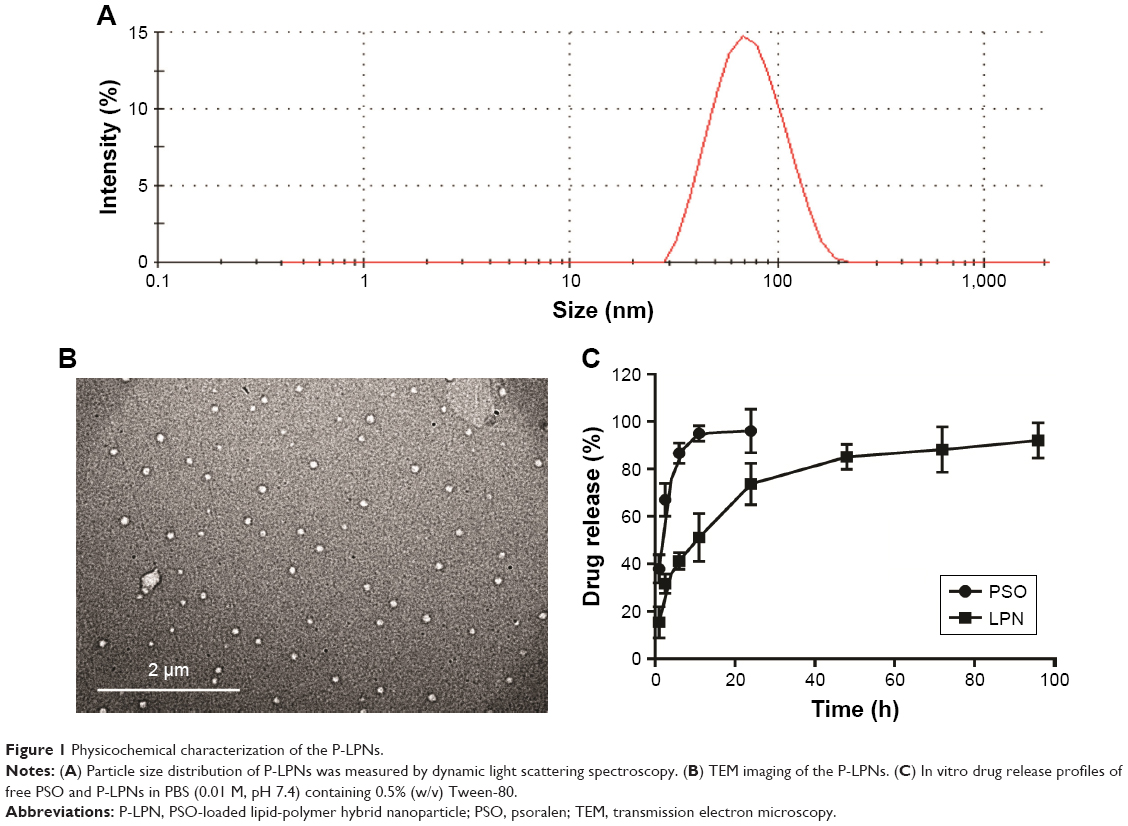 | Figure 1 Physicochemical characterization of the P-LPNs. |
In vitro cytotoxicity of PSO and LPNs
The cytotoxicity of free PSO and P-LPNs in HepG2 cells was assessed by MTT assays using a 48 hours exposure time. At concentrations below 40 μM, neither the modulator PSO nor P-LPNs produced a cytotoxic effect on HepG2/ADR cells (Figure 2A). The DOX concentration needed to kill 50% of the cells (IC50) was 0.32±0.06 μM for sensitive HepG2 (HepG2/S) cells, and 61.7±9.3 μM in HepG2/ADR cells. This indicates that HepG2/ADR cells were 192-fold resistant to DOX (Figure 2B and C). Co-administration of DOX and PSO (20 μM) on HepG2/S cells did not show a higher toxicity than DOX treatment alone (Figure 2B). Co-administration of DOX and P-LPNs (equivalent to 20 μM PSO) exhibited significantly higher cytotoxicity in HepG2/ADR cells compared to free DOX (P<0.01) or DOX+PSO (P<0.05) (Figure 2C). This demonstrates that co-administration of DOX and P-LPNs is superior to the individual components.
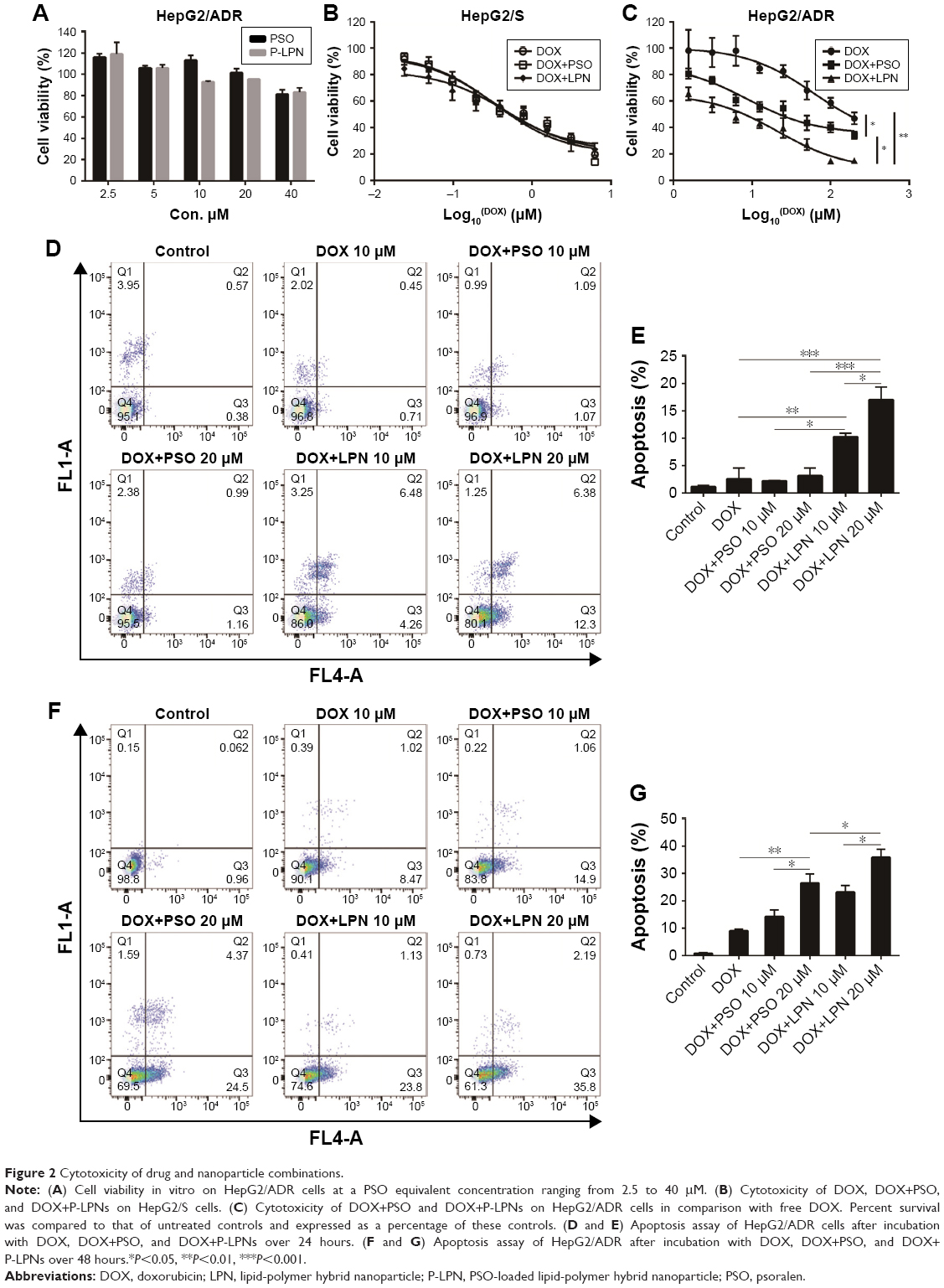 | Figure 2 Cytotoxicity of drug and nanoparticle combinations. |
Analysis of cell apoptosis
The ability of free drugs and the NP formulations to elicit an apoptotic response in vitro were measured with flow cytometry (Figure 2D–G). Co-administration of DOX and PSO on the HepG2/ADR cell resulted in extensive apoptosis after 48 hours; in contrast, there was no response with a shorter 24 hours incubation (Figure 2D and E). There was, however, a greater effect of DOX+P-LPNs compared to the free DOX or DOX+PSO treatment, at both 24 and 48 hours. This suggested that the nanoformulation enhanced the PSO-induced apoptosis in HepG2/ADR cells.
Cellular uptake of DOX
By taking advantage of the intrinsic fluorescence properties of DOX, quantitative analysis of cellular DOX uptake for the different formulations was measured in HepG2/ADR cells by flow cytometry. The activity of P-gp was inhibited with Verapamil to ascertain the effect of the drug efflux pump. Uptake of DOX is presented as mean fluorescence intensity (MFI) per cell. As shown in Figure 3A and B, the MFI for DOX+PSO in solution or in nanoparticle were not significantly different compared to DOX treatment alone. This was despite the significant effect of Verapamil (P<0.0001) on DOX accumulation. This suggests that PSO or P-LPNs do not potentiate the cytotoxic effect of DOX by increasing its cellular uptake.
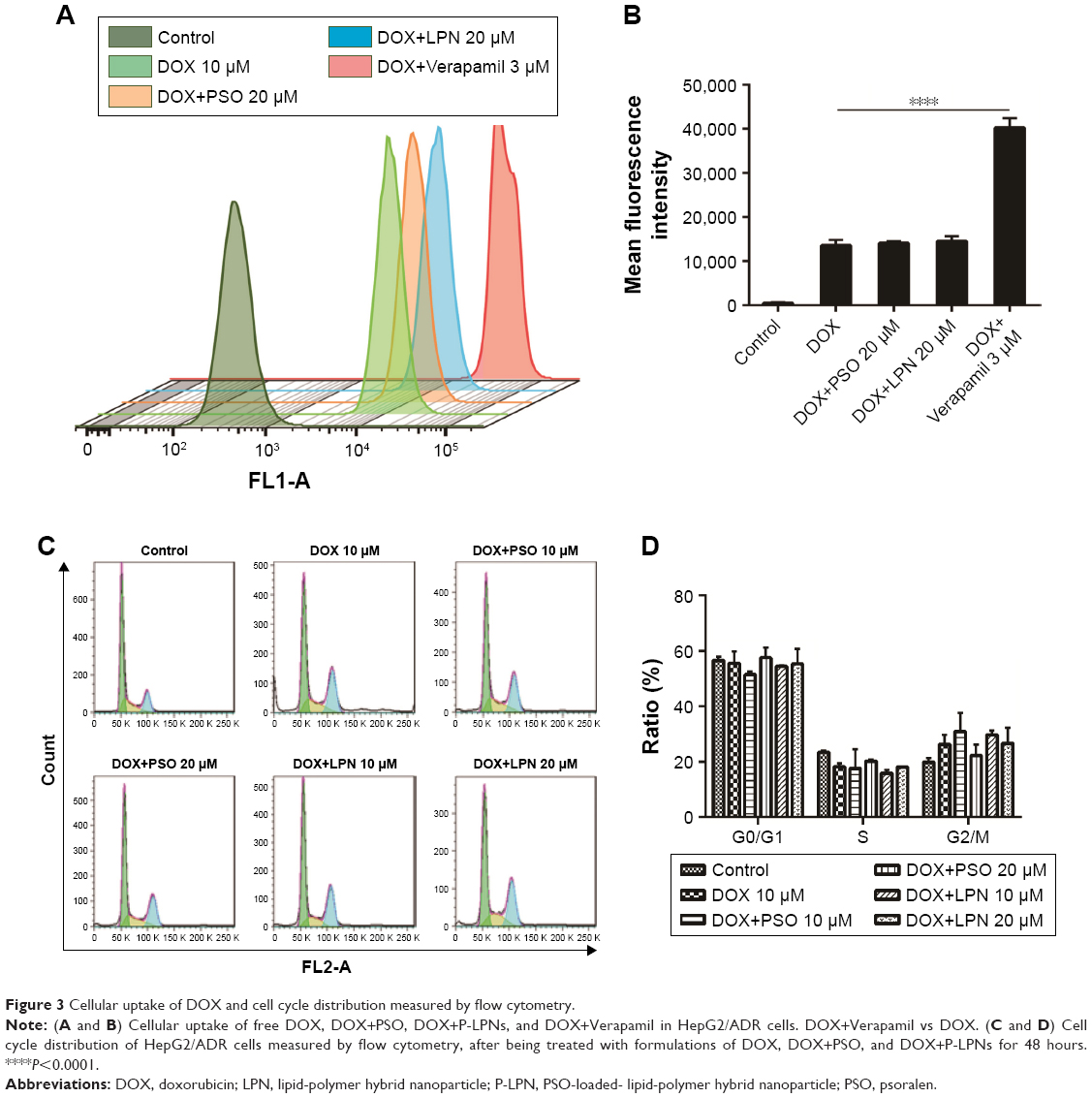 | Figure 3 Cellular uptake of DOX and cell cycle distribution measured by flow cytometry. |
Cell cycle analysis
To further investigate the mechanism of apoptosis, cell cycle studies were done on HepG2/ADR cells using flow cytometry (Figure 3C and D). The effects of treatment with DOX, a combination of DOX and PSO in solution, or in nanoparticle formulations, on the cell cycle were assessed. There was no significant difference in the cell cycle distribution between G0/G1, S, and G2/M phase of all treated groups compared to controls. The average percentage of cells in G0/G1 phase was 50%–55%, S phase cells 15%–20%, and G2/M phase cells were 20%–30%. These results indicate that PSO and the P-LPNs did not have any direct effect on cell cycle distribution. In addition, the PSO-enhanced apoptotic effect was not caused by cell cycle arrest in the HepG2/ADR cells.
ROS production
The levels of ROS in HepG2 and HepG2/ADR cells were assessed following incubation with combinations of DOX, PSO, and nanoparticles. Figure 4A and B demonstrated that levels of ROS in untreated HepG2/S cells were significantly (P<0.0001) higher than that in HepG2/ADR. A significant increase in ROS (ie, DCF fluorescence) in the resistant HepG2/ADR cells was observed after incubation with DOX, DOX+PSO, and DOX+P-LPNs compared to untreated cells. The greatest increase in ROS was observed for treatment with DOX+P-LPNs, wherein the levels were similar to that observed in the drug sensitive HepG2 cells.
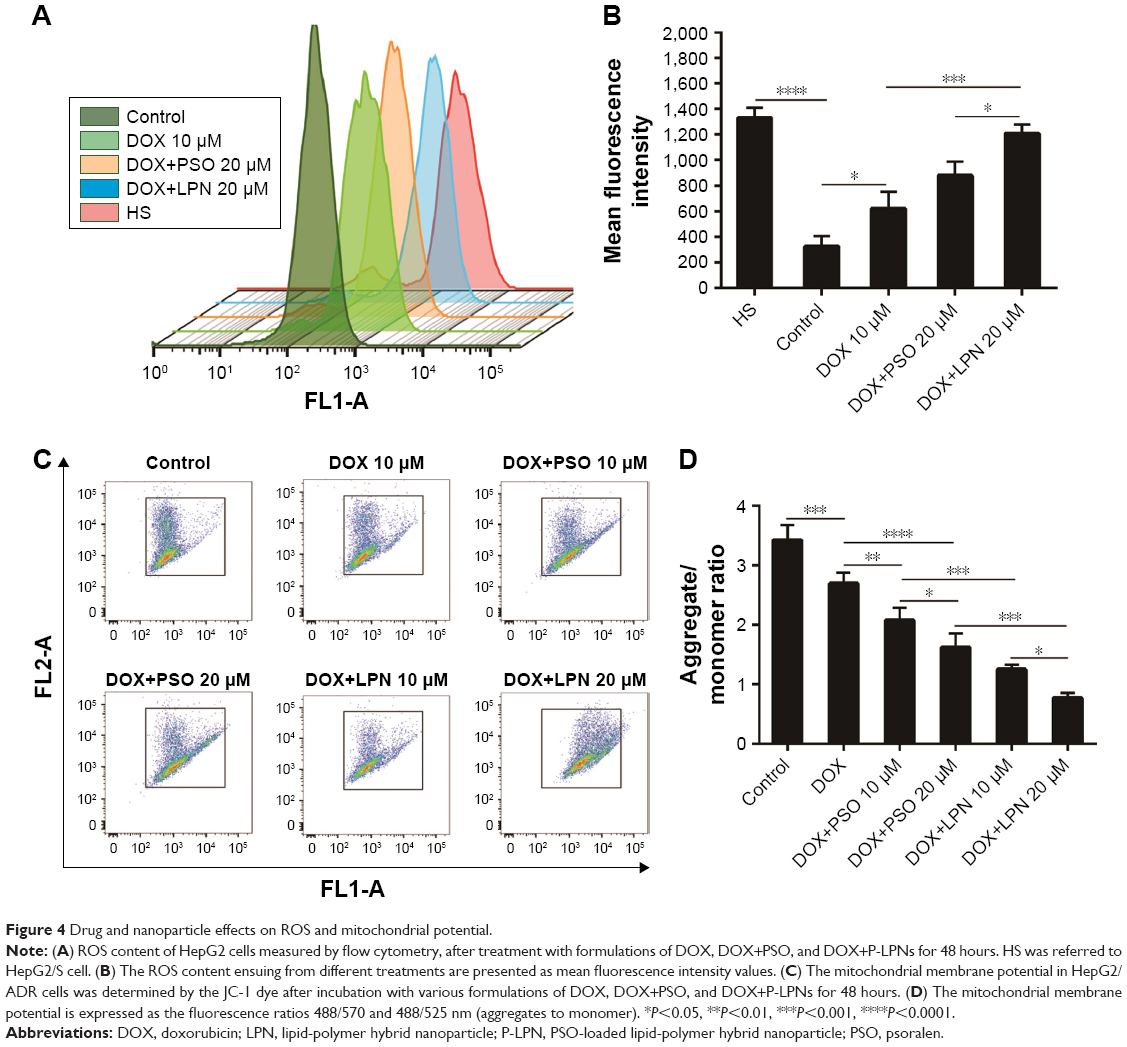 | Figure 4 Drug and nanoparticle effects on ROS and mitochondrial potential. |
Effects of P-LPNs on mitochondrial membrane potential
Mitochondria and the integrity of their inner membrane play a key role in the apoptotic pathway. An increased production of ROS results in depolarization of the mitochondrial membrane potential (ΔΨm).31,32 Due to the effects of DOX, PSO, and P-LPN formulations on ROS levels, their potential to alter ΔΨm in HepG2/ADR cells was measured with the fluorescent probe JC-1. As demonstrated in Figure 4C and D, treatment with the different formulations resulted in a significant loss in mitochondrial ΔΨm. The formulation of DOX+PSO caused a lower ΔΨm than DOX treatment alone. However, the largest collapse in mitochondrial membrane potential was produced by treatment with DOX+P-LPNs.
Analysis of P-LPNs on expression of P-gp
The resistant phenotype in HepG2/ADR cells was conferred by expression of P-gp, which was absent in the drug-sensitive parental line HepG2/S (Figure 5A and B). Moreover, as shown in lanes 3–7, treatment with combinations of DOX, DOX+PSO, and DOX+LPN did not alter the expression of P-gp in the HepG2/ADR cells.
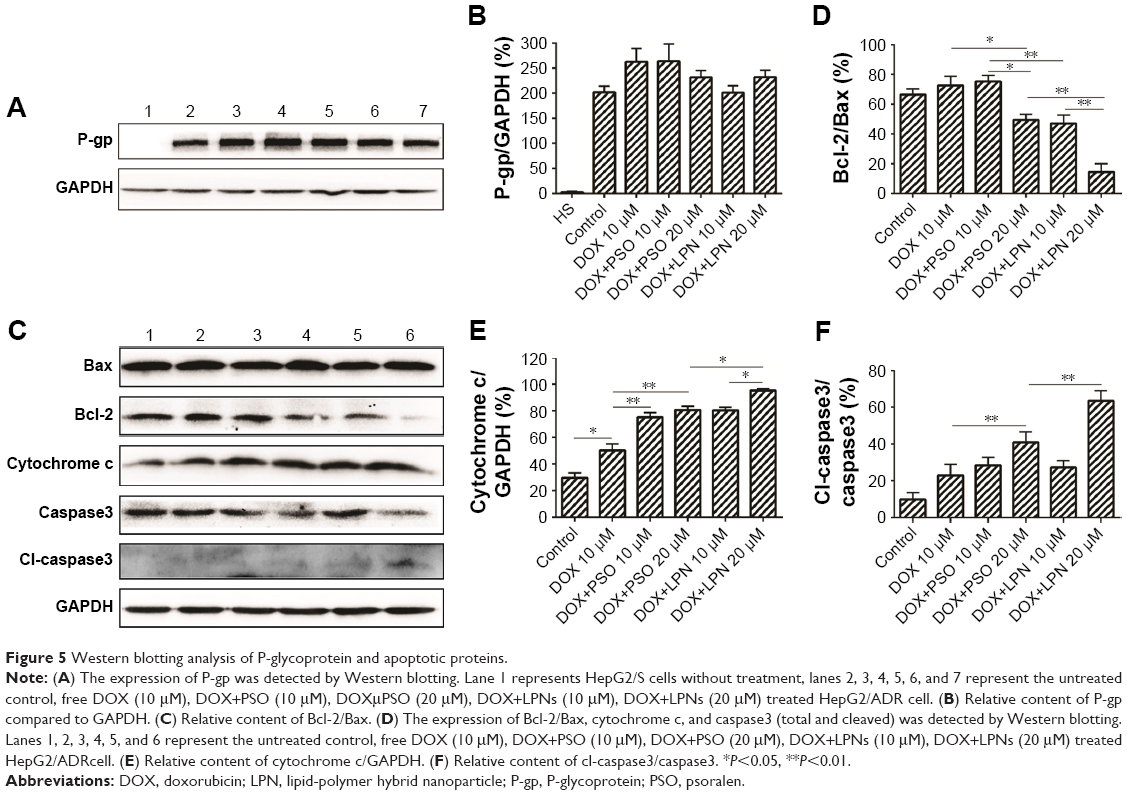 | Figure 5 Western blotting analysis of P-glycoprotein and apoptotic proteins. |
Analysis of P-LPNs on apoptosis pathways
Increased ROS and decreased mitochondrial ΔΨm are early indicators of an apoptotic effect. To determine whether the complete process was initiated, the levels of critical mitochondrial marker proteins were measured. As demonstrated in Figure 5C–F, PSO and the P-LPNs appeared to exert their cytotoxic activity by inducing mitochondrial apoptosis.33,34 Co-administration of DOX and P-LPNs decreased the ratio of Bcl-2 to Bax (Figure 5C and D) and was associated with elevated levels of cytochrome c (Figure 5C–E), compared to all other formulations. The results suggest that the combination of DOX with P-LPNs may induce apoptosis and was supported by the effects on the downstream apoptotic protein caspase3 (Figure 5C–F). Figure 5C demonstrates that the lower concentration of PSO (10 μM) in the DOX+PSO or DOX+P-LPNs treatments was less effective in causing cleavage of caspase3. In contrast, the inclusion of a higher PSO concentration (20 μM) induced significantly more extensive cleavage of caspase3 in HepG2/ADR cells.
Discussion
PSO displays anticancer activity in vitro; however, due to its low solubility, the compound has poor bioavailability. The objective of this investigation was to ascertain whether the effectiveness of PSO could be improved using lipid-polymer hybrid nanoparticles to encapsulate PSO. The P-LPNs were characterized with diameters of 65 nm and a negative zeta potential (−28 mV). TEM confirmed the spherical surface and the uniform distribution of nanoparticles and a delayed release of cargo.
Co-administration of DOX and P-LPNs was observed to decrease the viability of HepG2/ADR cells compared to treatment with free DOX or DOX+PSO. It has previously been shown that PSO induced KB cell death through apoptosis.35 P-LPNs elicited an apoptotic effect on HepG2/ADR cells without any alteration to the cell cycle. This was in contrast to observations of PSO arresting HaCaT keratinocyte cells in the S phase of the cell cycle and preventing cell progression from G1 into S phase in MCF-7/ADR breast cancer cells.36,37 These differences in PSO effects are likely explained by altered signaling pathways between the three cell types.
Over expression of P-gp confers resistance to DOX by mediating its active efflux. Despite the ability of PSO and P-LPNs to enhance the cytotoxicity of DOX in resistant HepG2/ADR cells, the formulations did not affect DOX accumulation. In addition, the level of P-gp expression was not altered in the presence of PSO or P-LPNs. However, Jiang et al38 demonstrated that the intracellular drug accumulation was significantly increased in MCF7/ADR cells after treatment with PSO, whereas the P-gp expression at the mRNA or protein level was unaffected.37,38 Hsieh et al11 revealed that PSO suppressed P-gp activity and decreased mRNA and proteins levels of P-gp in A549/D16 cell. However, the effects of P-gp on the extent of drug accumulation are highly dependent on its level of expression and the respective concentration of drug. Consequently, the cell phenotype provides a more accurate measure of its influence.
In the present study, DOX+P-LPNs treatment was also associated with elevated ROS levels and a concomitant depolarization of mitochondrial ΔΨm. Wang et al39 developed a lipid membrane-coated silica-carbon hybrid nanoparticle that targeted mitochondria to produce ROS and inhibited the production of ATP in NCI/AdrRES cells. The loss of ATP resulted in downregulation of efflux pumps and a loss of multidrug resistance. Although the DOX+P-LPNs treatment did not alter P-gp expression, there were marked effects on mitochondrial function in HepG2/ADR cell. Targeting mitochondria, which provided energy for tumor cells, may induce cancer cell death and overcome drug resistance produced by efflux pumps. In fact, delivering apoptotic agents to mitochondria has previously been shown as an effective strategy against the resistant phenotype.27,40,41
Increased generation of ROS causes oxidative stress, lipid peroxidation, loss of cytochrome c from permeabilized mitochondria, and activation of caspase cascade. This was also observed for DOX+P-LPNs and extended to downregulated bcl-2 expression, improved cytochrome c, and promoted the cleavage of caspase3. Bcl-2 and Bax are apoptosis regulators belonging to the Bcl-2 family and regulate apoptotic mediators, such as cytochrome c, by controlling the permeability of the mitochondrial membrane.42,43 Alternate lipid-polymer hybrid nanoparticles have also been demonstrated to induce a similar mitochondrial apoptotic pathway.44
Conclusion
In summary, we developed a lipid-polymer hybrid nanoparticle to encapsulate PSO, which could enhance the treatment efficiency of chemotherapy drugs in an HepG2/ADR cell, by increasing ROS level and improving the mitochondrial apoptotic pathway. Targeting the mitochondria, rather than modulating drug efflux pump activity, was responsible for the effectiveness of the nanoparticles in resistant cells. Based on these results, it is inferred that lipid-polymer hybrid nanoparticles hold promise as a drug delivery system for effective chemotherapy. Further studies on the in vivo efficiency of P-LPNs are required to ascertain how the encapsulation affects pharmacokinetic profiles.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81273707), the Ministry of Education in the New Century Excellent Talents (NECT-12-0677), the Natural Science Foundation of Guangdong (S2013010012880, 2016A030311037), the Science and Technology Program of Guangzhou (2014J4500005, 201704030141), the Science Program of the Department of Education of Guangdong (2013KJCX0021, 2015KGJHZ012), the Science and Technology Program of Guangdong (2015A050502027), and the Special Project of International Scientific and Technological Cooperation in Guangzhou Development District (2017GH16).
Disclosure
The authors report no conflicts of interest in this work.
References
Zhang Y, Qu C, Ren J, Zhang S, Wang Y, Dai M. [Liver cancer incidence and mortality data set in China]. Zhonghua Zhong Liu Za Zhi. 2015;37(9):705–720. Chinese. | ||
Chenivesse X, Franco D, Bréchot C. MDR1 (multidrug resistance) gene expression in human primary liver cancer and cirrhosis. J Hepatol. 1993;18(2):168–172. | ||
Gottesman MM, Pastan I. Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu Rev Biochem. 1993;62(1):385–427. | ||
Comerford KM, Wallace TJ, Karhausen J, Louis NA, Montalto MC, Colgan SP. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res. 2002;62(12):3387–3394. | ||
Park JG, Lee SK, Hong IG, et al. MDR1 gene expression: its effect on drug resistance to doxorubicin in human hepatocellular carcinoma cell lines. J Natl Cancer Inst. 1994;86(9):700–705. | ||
Wang L, Mosel AJ, Oakley GG, Peng A, Ling W. Deficient DNA damage signaling leads to chemoresistance to cisplatin in oral cancer. Mol Cancer Ther. 2012;11(11):2401–2409. | ||
Maiti AK. Genetic determinants of oxidative stress-mediated sensitization of drug-resistant cancer cells. Int J Cancer. 2012;130(1):1–9. | ||
Dahdouh F, Raane M, Thévenod F, Lee WK. Nickel-induced cell death and survival pathways in cultured renal proximal tubule cells: roles of reactive oxygen species, ceramide and ABCB1. Arch Toxicol. 2014;88(4):881–892. | ||
Dempsey CE, Roberts DL, Dive C. Bax, Bak and Bid: Key Mediators of Apoptosis. Wiley-VCH Verlag GmbH; 2008. | ||
Youle RJ, Strasser A, Rj Y. The Bcl-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9(1):47–59. | ||
Hsieh MJ, Chen MK, Yu YY, Sheu GT, Chiou HL. Psoralen reverses docetaxel-induced multidrug resistance in A549/D16 human lung cancer cells lines. Phytomedicine. 2014;21(7):970–977. | ||
Cai J, Chen S, Zhang W, et al. Paeonol reverses paclitaxel resistance in human breast cancer cells by regulating the expression of transgelin 2. Phytomedicine. 2014;21(7):984–991. | ||
Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7(9):771–782. | ||
Hillaireau H, Couvreur P. Nanocarriers’ entry into the cell: relevance to drug delivery. Cell Mol Life Sci. 2009;66(17):2873–2896. | ||
Bai F, Wang C, Lu Q, et al. Nanoparticle-mediated drug delivery to tumor neovasculature to combat P-gp expressing multidrug resistant cancer. Biomaterials. 2013;34(26):6163–6174. | ||
Wang Y, Kho K, Cheow WS, Hadinoto K. A comparison between spray drying and spray freeze drying for dry powder inhaler formulation of drug-loaded lipid-polymer hybrid nanoparticles. Int J Pharm. 2012;424(1–2):98–106. | ||
Gao J, Xia Y, Chen H, et al. Polymer-lipid hybrid nanoparticles conjugated with anti-EGF receptor antibody for targeted drug delivery to hepatocellular carcinoma. Nanomedicine. 2014;9(2):279–293. | ||
Liu Y, Pan J, Feng SS. Nanoparticles of lipid monolayer shell and biodegradable polymer core for controlled release of paclitaxel: effects of surfactants on particles size, characteristics and in vitro performance. Int J Pharm. 2010;395(1–2):243–250. | ||
Thevenot J, Troutier AL, David L, Delair T, Ladavière C. Steric stabilization of lipid/polymer particle assemblies by poly(ethylene glycol)-lipids. Biomacromolecules. 2007;8(11):3651–3660. | ||
Zhang L, Zhu D, Dong X, et al. Folate-modified lipid-polymer hybrid nanoparticles for targeted paclitaxel delivery. Int J Nanomedicine. 2015;10:2101–2114. | ||
Feng SS, Chien S. Chemotherapeutic engineering: application and further development of chemical engineering principles for chemotherapy of cancer and other diseases. Chem Eng Sci. 2003;58(18):4087–4114. | ||
Zhang L, Chan JM, Gu FX, et al. Self-assembled lipid–polymer hybrid nanoparticles: a robust drug delivery platform. ACS Nano. 2008;2(8):1696–1702. | ||
Wu XY, Xy W. Strategies for optimizing polymer-lipid hybrid nanoparticle-mediated drug delivery. Expert Opin Drug Deliv. 2016;13(5):609–612. | ||
Tang C, Zhang E, Li Y, Yang L. An innovative method for preparation of hydrophobic ion-pairing colistin entrapped poly(lactic acid) nanoparticles: loading and release mechanism study. Eur J Pharm Sci. 2017;102:63–70. | ||
Lu H, Zhang L, Liu D, Tang P, Song F. Isolation and purification of psoralen and isopsoralen and their efficacy and safety in the treatment of osteosarcoma in nude rats. Afr Health Sci. 2014;14(3):641–647. | ||
Cheow WS, Hadinoto K. Factors affecting drug encapsulation and stability of lipid–polymer hybrid nanoparticles. Colloids Surf B: Biointerfaces. 2011;85(2):214–220. | ||
Han M, Vakili MR, Soleymani Abyaneh H, Molavi O, Lai R, Lavasanifar A. Mitochondrial delivery of doxorubicin via triphenylphosphine modification for overcoming drug resistance in MDA-MB-435/DOX cells. Mol Pharm. 2014;11(8):2640–2649. | ||
Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med. 2000;6(5):513–519. | ||
Leytin V, Mykhaylov S, Starkey AF, et al. Intravenous immunoglobulin inhibits anti-glycoprotein IIb-induced platelet apoptosis in a murine model of immune thrombocytopenia. Br J Haematol. 2006;133(1):060207074859002–060207074859082. | ||
Yuan Y, Chiba P, Cai T, et al. Fabrication of psoralen-loaded lipid-polymer hybrid nanoparticles and their reversal effect on drug resistance of cancer cells. Oncol Rep. 2018;40(2):1055–1063. | ||
Skała E, Kowalczyk T, Toma M, et al. Induction of apoptosis in human glioma cell lines of various grades through the ROS-mediated mitochondrial pathway and caspase activation by Rhaponticum carthamoides transformed root extract. Mol Cell Biochem. 2018;445(1–2):89–97. | ||
Forbes-Hernández TY, Giampieri F, Gasparrini M, et al. The effects of bioactive compounds from plant foods on mitochondrial function: a focus on apoptotic mechanisms. Food Chem Toxicol. 2014;68:154–182. | ||
Jeffers M, Rong S, vande Woude GF. Hepatocyte growth factor/scatter factor-MET signaling in tumorigenicity and invasion/metastasis. J Mol Med. 1996;74(9):505–513. | ||
Jakubowska J, Wasowska-Lukawska M, Czyz M. STI571 and morpholine derivative of doxorubicin collaborate in inhibition of K562 cell proliferation by inducing differentiation and mitochondrial pathway of apoptosis. Eur J Pharmacol. 2008;596(1–3):41–49. | ||
Wang Y, Hong C, Zhou C, Xu D, Qu H-B. Screening antitumor compounds psoralen and isopsoralen from Psoralea corylifolia L. seeds. Evid Based Compl Alternative Med. 2011;2011(5):1–7. | ||
Ki JC, Joerges Christoph KI. Herzinge Thomas induction of a caffeine-sensitive S-phase cell cycle checkpoint by psoralen plus ultraviolet A radiation. Oncogene. 2003;22:9. | ||
Wang X, Cheng K, Han Y, et al. Effects of psoralen as an anti-tumor agent in human breast cancer MCF-7/ADR cells. Biol Pharm Bull. 2016;39(5):815–822. | ||
Jiang J, Wang X, Cheng K, et al. Psoralen reverses the P-glycoprotein-mediated multidrug resistance in human breast cancer MCF-7/ADR cells. Mol Med Rep. 2016;13(6):4745–4750. | ||
Wang H, Gao Z, Liu X, et al. Targeted production of reactive oxygen species in mitochondria to overcome cancer drug resistance. Nat Commun. 2018;9(1):562. | ||
Cui H, Huan ML, Ye WL, et al. Mitochondria and nucleus dual delivery system to overcome Dox resistance. Mol Pharm. 2017;14(3):746–756. | ||
Liu Y, Zhang X, Zhou M, Nan X, Chen X, Zhang X. Mitochondrial-targeting Lonidamine-Doxorubicin nanoparticles for synergistic chemotherapy to conquer drug resistance. ACS Appl Mater Interfaces. 2017;9(50):43498–43507. | ||
Los M, Mozoluk M, Ferrari D, et al. Activation and caspase-mediated inhibition of PARP: a molecular switch between fibroblast necrosis and apoptosis in death receptor signaling. Mol Biol Cell. 2002;13(3):978–988. | ||
Pawlowski J, Kraft AS. Bax-induced apoptotic cell death. Proc Natl Acad Sci. 2000;97(2):529–531. | ||
Zhang J, Hu J, Chan HF, Skibba M, Liang G, Chen M. iRGD decorated lipid-polymer hybrid nanoparticles for targeted co-delivery of doxorubicin and sorafenib to enhance anti-hepatocellular carcinoma efficacy. Nanomedicine. 2016;12(5):1303–1311. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.