")
Back to Journals » Clinical Interventions in Aging » Volume 11
PSEN1 L226F mutation in a patient with early-onset Alzheimer’s disease in Korea
Authors Bagyinszky E, Park SA, Kim HJ, Choi SH , An SSA , Kim SY
Received 2 May 2016
Accepted for publication 30 June 2016
Published 12 October 2016 Volume 2016:11 Pages 1433—1440
DOI https://doi.org/10.2147/CIA.S111821
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Walker
Eva Bagyinszky,1,* Sun Ah Park,2,* Hyung Jun Kim,2 Seong Hye Choi,3 Seong Soo A An,1 SangYun Kim4
1Department of BioNano Technology, Gachon University, Seongnam-si, 2Department of Neurology, Soonchunhyang University Bucheon Hospital, Bucheon, 3Department of Neurology, Inha University School of Medicine, Incheon, 4Department of Neurology, Seoul National University College of Medicine & Neurocognitive Behavior Center, Seoul National University Bundang Hospital, Seongnam-si, Republic of Korea
*These authors contributed equally to this work
Abstract: In this study, we report a first 226leucine (Leu) mutation to phenylalanine (Phe) in (PSEN1, CTC>TTC, L226F) in Asia from a Korean early-onset Alzheimer’s disease (EOAD) patient. Polymerase chain reaction (PCR)–single strand conformation polymorphism, sequencing, and in silico predictions were performed. Previously, L226F was reported in EOAD patients by Zekanowski et al and Gómez-Tortosa et al. Disease phenotypes appeared in their thirties, and family history was positive in both cases. In our patient, age of onset was similar (37 years of age), but the mutation seemed to be de novo, since no affected family member was found. This leucine to phenylalanine substitution may cause additional stresses inside the transmembrane region due to large aromatic side chain and increased hydrophobic interactions with hydrocarbon chains in the membrane and its binding partners. Clinical phenotype of the mutation was aggressive progression into neurodegeneration, resulting in rapid cognitive decline. One of the patients was initially diagnosed with frontotemporal dementia, but the diagnosis was revised to AD upon postmortem studies in which Aβ plaques were seen. A second mutation, L226R, was found for the L226 residue. Similar to L226F, the patient with L226R also developed the first symptoms in his 30s, but EOAD was diagnosed in his 40s. These findings suggested that L226 might be an important residue in PSEN1, since mutations could result in neurodegenerative disease phenotypes at relatively young ages. There are mutations, such as L226F, which may not present clear clinical symptoms for the definitive diagnosis between frontotemporal dementia and AD. In addition, the similarities in the phenotypes could also be possible between AD and frontotemporal dementia, suggesting difficulties in differential diagnosis of various neurodegenerative diseases.
Keywords: Alzheimer’s disease, PSEN1 mutation, sequencing, frontotemporal dementia
Introduction
Alzheimer’s disease (AD) is the most common form of senile dementia, especially in the elder individuals, older than 65 years. Two main pathological phenotypes could be associated with AD: the amyloid β (Aβ) accumulation in senile plaques and the neurofibrillary tangle formation, outside and inside of the nerve cells, respectively1 AD could be distinguished as early-onset AD (EOAD) and late-onset AD, which occur under and above 65 years of age, respectively. Majority of AD cases occurred over 65 years of age. EOAD was reported quite rarely (5%–10% of all AD cases), but their genetic backgrounds were well understood, involving three main genes for AD, APP, PSEN1, and PSEN2. Mutations in the abovementioned genes could be validated as causative factors for AD, especially EOAD, and their inheritance pattern is autosomal dominant. However, there are several cases of genetic EOAD, in which no family history of disease has been observed, called de novo case of AD. The APP gene encodes a 770-amino acid long protein, which is cleaved by two enzymes, the beta- and gamma-secretases, resulting in 39–43 bp Aβ peptides, which are the main inclusions of amyloid plaques. Insoluble Aβ, located in plaques, or fibrillar Aβ may not be toxic. However, Aβ in soluble oligomer form could result in neuronal loss and reduced synaptic activity. Soluble Aβ dimers could be released from the plaques, resulting in neurotoxicity.2 In APP, >30 mutations have been described, which were established as pathogenic variants. PSEN1 and PSEN2 genes were suggested to be involved in gamma secretase activity. Most of the PSEN1 mutations have been reported as gain-of-function mutations, which could increase the gamma secretase cleavage. However, additional reports revealed that some mutations such as P264L, P267S, L435F, and C410Y may interfere with enzyme function, suggesting that they could affect through loss-of-function activities.3,4 Majority of genetic EOAD cases correlated with mutations in PSEN1.5,6 PSEN2 was described as a rare causative gene, for EOAD, but emerging studies have reported novel, possibly or probably pathogenic variants. Several clinical phenotypes, such as late-onset AD, amyloid angiopathy, hemorrhagic stroke, and dementia with Lewy Bodies, were described in PSEN2 carriers.7,8
Based on two main databases for AD mutations, the Alzheimer Research Forum or Alzforum (http://www.alzforum.org/mutations)9 and AD and frontotemporal dementia (FTD) mutations database (http://www.molgen.ua.ac.be/admutations/),10 >200 mutations were found in PSEN1 for EOAD. The age of disease onset could be mostly 40–50 years, but several mutations were associated with young-onset AD in their 30s.6,11 The associated disease phenotype with PSEN1 mutations could be aggressive and rapid progression of AD occurs. Alternative phenotypes, such as motor neuron symptoms, FTD, and spastic paraparesis, also appeared in patients with PSEN1 mutations.12
In this article, we report a case of a female patient with EOAD, who developed EOAD at the age of 37 years. Family history was negative, since no additional affected family member or relative was found. Hence, this patient may belong to a known de novo mutation of PSEN1 L226F, which was previously discovered in European EOAD patients.
Patients and methods
Patient information
The Ethical Committee of the Soonchunhyang University Bucheon Hospital approved the study. The patient and her parents gave consent to publish this case report and the accompanying images. The proband patient developed anxiety and paranoid ideation about her husband at the age of 37 years. The family members thought that it might be due to her recent moving to an unfamiliar town. But the progressive memory deficits and difficulties in daily household chores became more evident.
When she was thoroughly assessed on cognitions at the age of 39 years, the score on mini-mental state examination was 10, and she demonstrated severe deficits in multiple cognitive domains. Her speech was fluent but showed poor performances on Boston naming test. Her calculation, praxis, visual function on Rey complex figure copy, and frontal executive functions on stroop test (both word and color) were impaired. The most distinct deficits were observed in memory tests. She could not remember any items on delayed recall of Rey complex figure test and Seoul verbal learning test.13
Frontal releasing signs were noted on neurological test, but there were not any other focal deficits. On brain magnetic resonance imaging, suspicious bilateral hippocampal and distinct bilateral parietal cortical atrophy were noted. Subsequently, 18F-fluorodeoxyglucose positron emission tomography was taken, which demonstrated severe hypometabolism in bilateral parietal regions (Figure 1). Thereafter, she has taken cholinesterase inhibitors and memantine, but her cognitive declines were rapidly progressed. Her speech became nonfluent and finally mutic. And the slowness of the movements with increased muscle rigidity became evident with time. She was bedridden for one and a half years before her death at age 44 years. The follow-up brain CT was taken 1 year before her death, which demonstrated diffuse severe brain atrophy (Figure 1). No detailed family tree is available on the patient, since we have information only of her first-degree relatives. This case might be a de novo case of AD, since no additional affected family member was found.
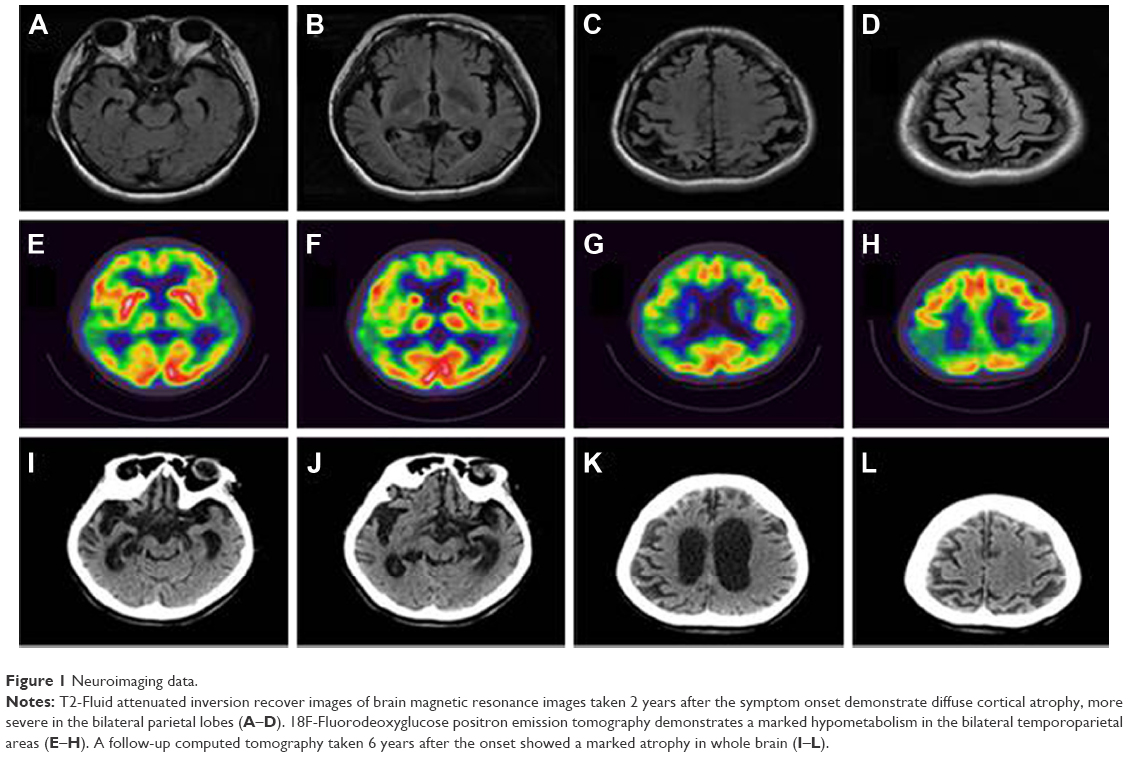 | Figure 1 Neuroimaging data. |
Genetic screening
Buffy coat was isolated after centrifugation at 800× g for half an hour. The genomic DNA was purified by following the protocol of GeneAll blood kit (Seoul, Korea). DNA samples were stored at -20°C before the analysis. In this project, a genetic analysis was performed with specific PCR primers of APP,14,15 PSEN1, and PSEN2.16,17 PCR products were subjected to single strand conformation polymorphism analysis (Figure 2A). Formamide was added to the PCR products, and these mixtures were incubated at 98°C for 10 minutes, to denature the double-stranded DNA bands. Native polyacrylamide gel electrophoresis (10%–12%) was performed for 20–22 hours.18 The ssDNAs could have different mobility in the gel, depending on the variants in the DNA. SYBR Gold staining (Invitrogen, Waltham, MA, USA) was used for visualization of DNA bands. To confirm and identify the mutation, all PCR products were duplicated and sequenced at both directions (Figure 2B). Sequencing was performed by the BioNeer Inc. (Daejeon, Korea). Prior to sequencing, PCR products were purified by GeneAll PCR kit, by following the protocol. Big Dye Terminator Cyclic sequencing was performed, and ABI 3730XL DNA Analyzer (http://eng.bioneer.com/home.aspx; Bioneer Inc.) was used for the sequencing. Sequencing results were aligned by NCBI Blast (http://blast.ncbi.nlm.nih.gov/Blast.cgi), and the chromatograms were analyzed by DNA BASER (http://www.dnabaser.com) software. Mutations and sequence variants were identified by NCBI Gene (http://www.ncbi.nlm.nih.gov/gene) and UniProt (http://www.uniprot.org) databases. Since pathological overlap may occur between EOAD and other early onset forms of neurodegenerative disorders,1,5 such as prion diseases and FTD, patients have also been screened for PRNP,19 progranulin (PGRN), and microtubule-associated protein tau (MAPT) genes.20,21
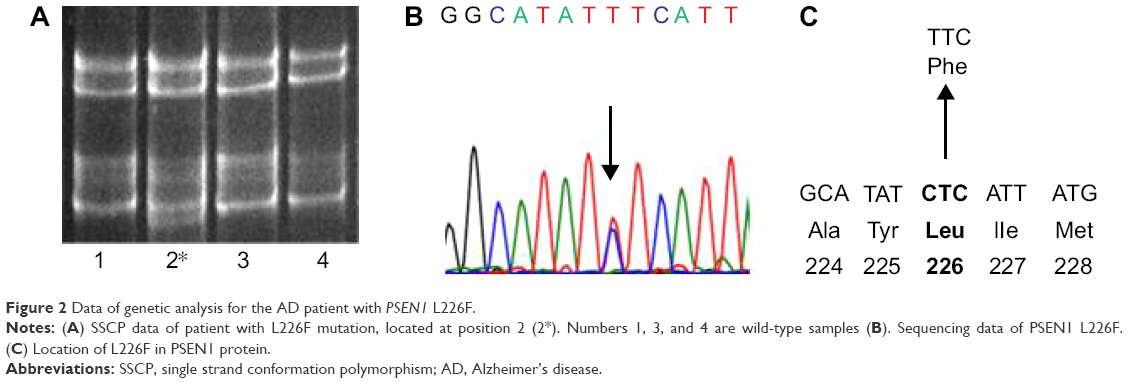 | Figure 2 Data of genetic analysis for the AD patient with PSEN1 L226F. |
In silico modeling
Mutation was analyzed by two kinds of in silico online tools: PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) and Sorting Intolerant from Tolerant (SIFT; http://sift.jcvi.org/), which make predictions on the pathogenic properties of missense mutations. PolyPhen-2 compares the normal and mutant proteins using different categories, such as homology searching and multiple sequence alignment. It also performs identity- and structure-based searches, such as accessible surface area and hydrophobicity features. These predictions might define the potential role of amino acid alterations in the protein structure. Three categories of mutations could be distinguished: probably, possibly damaging, and benign variants, depending on the scores of prediction. PolyPhen2 also provides a multiple sequence alignment, which compared the homologous sequences from different vertebrate as well as nonvertebrate species. In addition, if the three-dimensional (3D) protein structure might be available, it could provide a structural comparison between the wild-type and mutant protein. Two types of data sets are available: the HumDiv and HumVar scores. HumDiv could be used for the prediction of pathogenic nature of rare alleles. In HumDiv scores, the mildly damaging protein exchanges should be treated as possibly damaging alleles. HumVar would be used in the case of Mendelian disorder diagnosis, where highly damaging phenotypes should be distinguished from the alleles with less damaging phenotypes.22 SIFT algorithm can predict the potential effects of exchanges missense point mutations on protein function. In SIFT, different databases, such as SWISS-PROT, SWISS-PROT/TrEMBL, and protein databases, of NCBI are used. The software calculates the possibility on the deleterious properties of variants by comparing the alleles with mutation and the normal ones. SIFT scores the amino acid exchanges, which can be defined as deleterious or tolerated, depending on the scores. Under and over the score of 0.05, mutations could be damaging and tolerated, respectively.23
Structure prediction
3D structures of PSEN1 with mutation(s) were conducted by Raptor X, which is an online software (http://raptorx.uchicago.edu/). Protein structure prediction server used the full amino acid sequence of PSEN1 (1–467 amino acids). Discovery Studio 3.5 Visualizer from Accelrys (Seoul, Korea) was used to display superimposed images.24
Results
Single strand conformation polymorphism gel mobility revealed the presence of potential mutation in PSEN1 exon 7 (Figure 2A). After DNA sequencing PCR product of exon 7, a missense variant, L226F (CTC→TTC), was identified (Figure 2B and C), which was already verified as pathogenic mutation.25 Family members of patient were also screened for the possible mutation, but all of them were negative for L226F. Hence, this mutation might be a de novo. Patient was negative for any pathogenic or possibly pathogenic mutations in PRNP, PGRN, and MAPT genes.
PSEN1 (chromosome 14, 73,603,142-73,690,399) L226F was checked in the Korean Centers for Disease Control and Prevention (KCDC) database (http://www.cdc.go.kr/). In the KCDC, whole genome sequencing was performed in 622 healthy control individuals. PSEN1 L226F did not appear in these controls. Mutation was also checked in the ExAC Data set (Exome Aggregation Consortium; http://exac.broadinstitute.org/about), which screened the sequence of 60,706 unrelated individuals. This database is useful in disease-associated analyses and population genetic studies. PSEN1 L226F was also missing in ExAC database, which additionally supports its pathogenicity.
PolyPhen-2 prediction suggested this mutation as “probably damaging” variant with both 1.00 HumDiv and HumVar scores. Multiple sequence alignment suggested that L226 might be conservative among the presenilin-or presenilin-like proteins in different vertebrate species. Leucine was located in the same position of homolog proteins in sequences from different mammalian species, such as in mouse, horse, marmoset, hamster, and rat. Leucine was also located at the same residue in the presenilin- or presenilin-like sequences of other vertebrate species, such as chicken, toad, and zebrafish. Nonvertebrate sequences were not available in the multiple sequence alignments. SIFT also revealed the L226F as pathogenic (damaging) mutation with the score of 0.
The 3D modeling might confirm the data performed by Zekanowski et al25 before 2012. Leucine and phenylalanine are both nonpolar and hydrophobic amino acids. Phenylalanine is one of the biggest amino acids with higher hydrophobicity, which is usually located inside the protein core. Since the L226F mutation site was located at the outside of the 3D structure, its analysis suggested that this mutation might result in extra stress to the helix structure by increasing hydrophobic interactions between the PS1 and its binding partners. The benzene ring of phenylalanine could also result in disturbances inside the helix structure (Figure 3).
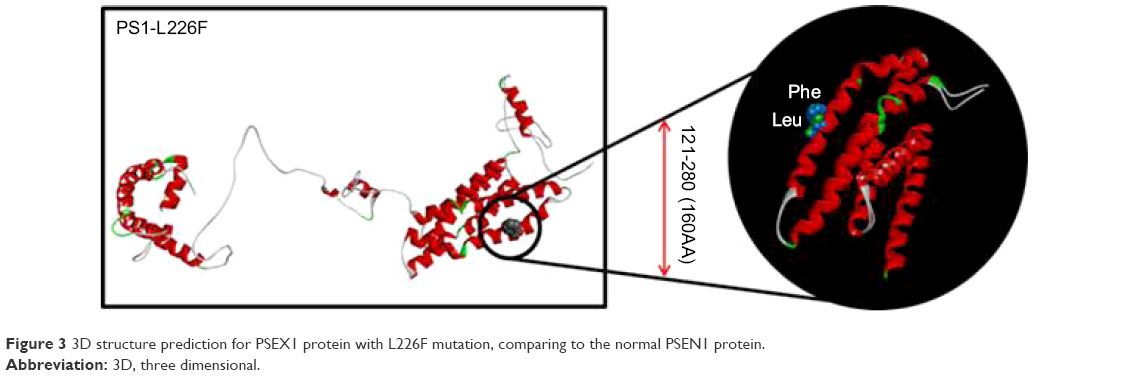 | Figure 3 3D structure prediction for PSEX1 protein with L226F mutation, comparing to the normal PSEN1 protein. |
Discussion
PSEN1 L226F is a known pathogenic mutation, which was discovered in European EOAD patients earlier (Table 1). Clinical phenotypes of FTD were also reported in patients with this mutation. This is the third case report of L226F mutation in AD patients. In addition, this is the first case of reporting PSEN1 L226F in a probably de novo case of AD. L226F was revealed in a Polish patient for the first time, who was clinically diagnosed with FTD. However, his symptoms were not typical FTD, and the autopsy and neuropathological examinations, based on Braak staining, confirmed the AD diagnosis according to the CERAD and NIA-Reagan criteria. In this case, the family history was positive, since the mother of the patient was also diagnosed with an unknown form of dementia at the age of 33 years. Mean age of death was 41 years. In silico modeling was performed by the same study, and they suggested that L226F exchange might promote increased hydrophobic interactions between the benzene rings of F226 and Y225 due to increased surface of transmembrane area. In addition, L226F might increase the hydrophobic interactions within domains of PSEN1 protein.25 PSEN1 L226F was expressed in HEK293 cells, and it was coexpressed in APP with the Swedish mutation. Elevated Aβ42, Aβ40, and Aβ42/Aβ40 were seen, compared with the cells, which expressed wild-type PSEN1.26
 | Table 1 Comparisons of dominant features of patients with PSEN1 L226F |
The second case of L226F was described in a female patient and in her father from a Spanish (Caucasian) family. The proband’s father died 12 years after the diagnosis with progressive form of dementia. In the proband patient, the first symptoms appeared at the age of 33 years, which were depression, dysarthria with nonfluent speech, tremor, and memory problems. Later, her symptoms became worse with increased level of memory deficit, and dysgraphia and aphasia appeared in the language.27
An additional mutation for codon 226, L226R, was described in four members of a Spanish family (Table 2).28 The proband patient had encephalopathy at the age of 33 years, followed up at 49 years with AD diagnosis. This patient had also motor skill delay in his childhood. Additional affected family members were also found, since the mother and the aunt of the patient were diagnosed with EOAD. Interestingly, the son of the affected aunt was clinically diagnosed with Pick’s disease.28
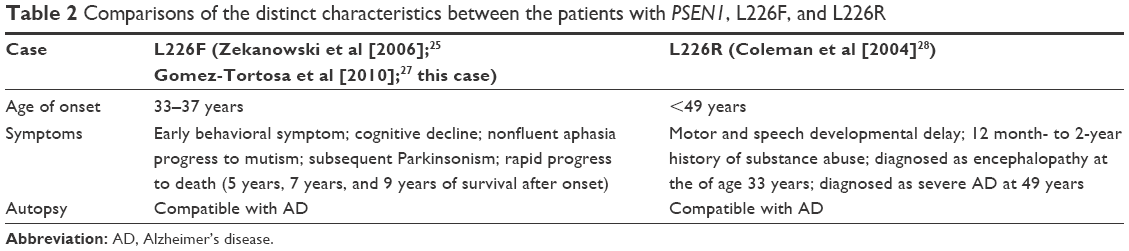 | Table 2 Comparisons of the distinct characteristics between the patients with PSEN1, L226F, and L226R |
Our data were the first case of PSEN1 L226F mutation in an Asian patient without any family history of dementia. Our patient revealed similar age of onset as that of previously diagnosed PSEN1 L226F EOAD cases, since the clinical symptoms appeared before 40 years (between 33 years and 36 years). However, both European cases were associated with positive familial EOAD (Table 1), but our patient did not have any family history of dementia. Hence, this Korean patient with PSEN1 L226F might be a de novo case of mutation, since no affected family members were found. Variants in PSEN1 were reported as the most common causative mutations for EOAD, and several PSEN1 mutations, such as L85P,29 L113Q,30 and H163P,31 were identified with young-onset dementia that occurred under 40 years of age.6
PSEN1-associated disease was usually reported in classical EOAD, but alternative phenotypes, such as spastic paraparesis and myoclonus and seizures, may be possible. In these cases, patient could develop FTD-like syndrome.32 L226F was discovered first in a patient, who was initially diagnosed with FTD, and the diagnosis was later suggested as AD from neurological tests. A few additional mutations in PSEN1, such as L113P, insR352 and G183V, M233L, were reported, where FTD phenotypes or Pick-type tauopathy appeared (Table 3).25 L113P was found in a French family, with the age of onset of 38–50 years. Patients developed personality and behavioral changes and spatial orientation. CT and single-photon emission computed tomography scans showed frontotemporal atrophy and hypoperfusion in the frontal lobes, respectively. Mutation appeared only in the affected family members and was absent in the asymptomatic ones.33 G183V was discovered in a Belgian individual, who developed FTD-like dementia at the age of 52 years. Patient developed personality changes, such as apathy, overeating, and frontal disinhibition. Pick bodies, tau-positive neuronal inclusions appeared in the patient’s brain, but Aβ deposits were missing.34 The M233L carrier patient developed the first symptoms at the age of 39 years, which included becoming detached and disengaged in daily activities, impairment in communications, altered eating habits (overeating), and becoming paranoid. Significant drop was observed in her mini-mental state examination scores, and impairment was found in her clock test drawing. PET screening revealed hypoperfusion and hypometabolism in the posterior parietal areas and in prefrontal, parietal, and temporal cortex, respectively.32 R352ins (ins353R) was discovered in an FTD case, but it coexisted with a MAPT mutation (A239T) and with a nonsense PGRN mutation (R493X). This case was associated with an ubiquitin-positive but Tau-negative form of FTD (which mostly occurs in PGRN-mutant patients). Currently, it is unclear whether the PSEN1 mutation could be associated with pathogenic disease phenotype.35 These findings revealed a possible clinical overlap between AD and FTD, suggesting that PSEN1 may be a candidate gene for FTD-type dementia.34 All patients who are suspected with EOAD or FTD should be screened for APP, PSEN1, PSEN2, PGRN, and MAPT genes.36 In our future research, more complex genetic profiling will be performed on our samples. We are planning to perform a gene panel analysis with neurodegenerative disease-causing and risk factor genes, as well as candidate genes. We have designed primers for these genes, and next-generation sequencing approaches are planned with them. We believe that a complex genetic screening could improve the disease diagnosis as well as the therapeutic approaches.37
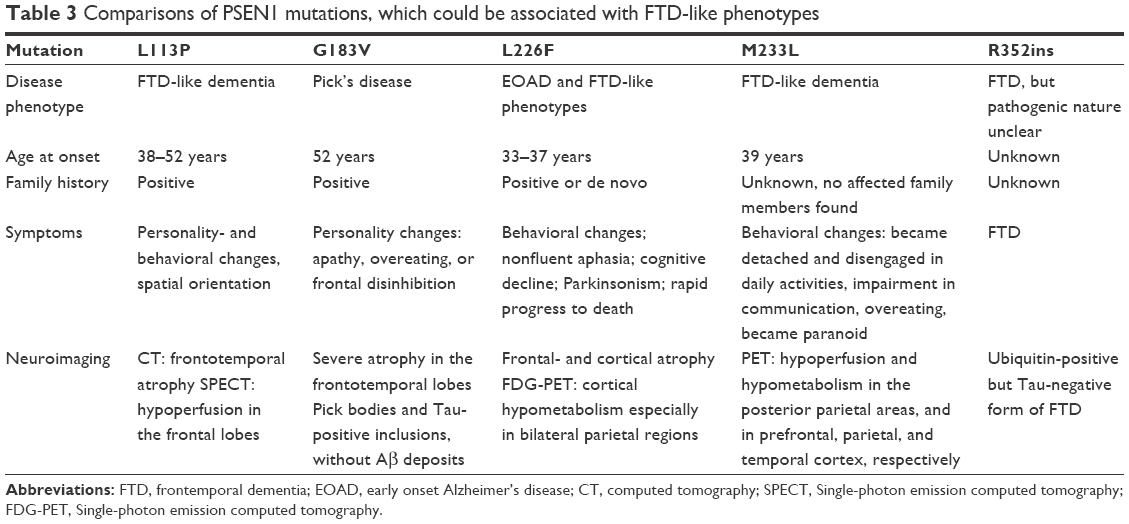 | Table 3 Comparisons of PSEN1 mutations, which could be associated with FTD-like phenotypes |
Acknowledgments
Neurological analyses were performed by Doctor Sun Ah Park and Doctor SangYun Kim. Genetic screening and analyses were performed by Doctor Eva Bagyinszky. Three-dimensional modeling was performed by SunOh Bae. We thank the patients and their families for agreeing the genetic test and for the CREDOS study for Alzheimer samples. This work was supported by grants from the Korea Healthcare technology R&D Project, Ministry of Health and Welfare, Republic of Korea (HI10C2020 to CSH & HI14C3331).
Disclosure
The authors report no conflicts of interest in this work.
References
Zou Z, Liu C, Che C, Huapin H. Clinical genetics of Alzheimer’s disease. Biomed Res Int. 2014;2014:291862. | ||
Shankar GM, Li S, Mehta TH, et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14(8):837–842. doi:10.1038/nm1782. | ||
Xia D, Watanabe H, Wu B, et al. Presenilin-1 knockin mice reveal loss-of-function mechanism for familial Alzheimer’s disease. Neuron. 2015;85(5):967–981. doi:10.1016/j.neuron.2015.02.010. | ||
Ben-Gedalya T, Moll L, Bejerano-Sagie M, et al. Alzheimer’s disease-causing proline substitutions lead to presenilin 1 aggregation and malfunction. EMBO J. 2015;34(22):2820–2839. doi:10.15252/embj.201592042. | ||
Rademakers R, Cruts M, Van Broeckhoven C. Genetics of early-onset Alzheimer dementia. ScientificWorldJournal. 2003;3:497–519. | ||
Bagyinszky E, Youn YC, An SS, Kim S. The genetics of Alzheimer’s disease. Clin Interv Aging. 2010;9:535–551. | ||
Jayadev S, Leverenz JB, Steinbart E, et al. Alzheimer’s disease phenotypes and genotypes associated with mutations in presenilin 2. Brain. 2010;133(4):1143–1154. doi:10.1093/brain/awq033. | ||
Canevelli M, Piscopo P, Talarico G, et al. Familial Alzheimer’s disease sustained by presenilin 2 mutations: systematic review of literature and genotype-phenotype correlation. Neurosci Biobehav Rev. 2014;42:170–179. doi:10.1016/j.neubiorev.2014.02.010. | ||
Alzforum Networking for a cure [homepage on the Internet]. Mutations. Available from: http://www.alzforum.org/mutations. Accessed September 18, 2016. | ||
Cruts M, Theuns J, Van Broeckhoven C. Locus-specific mutation databases for neurodegenerative brain diseases. Human Mutation. 2012; 33:1340–1344. | ||
Youn YC, Bagyinszky E, Kim H, Choi BO, An SS, Kim S. Probable novel PSEN2 Val214Leu mutation in Alzheimer’s disease supported by structural prediction. BMC Neurol. 2014;14:105. | ||
Hattori S, Sakuma K, Wakutani Y, et al. A novel presenilin 1 mutation (Y154N) in a patient with early onset Alzheimer’s disease with spastic paraparesis. Neurosci Lett. 2004;368(3):319–322. | ||
Ahn HJ, Chin J, Park A, et al. Seoul neuropsychological screening battery-dementia version (SNSB-D): a useful tool for assessing and monitoring cognitive impairments in dementia patients. J Korean Med Sci. 2010;25(7):1071–1076. | ||
Tanzi RE, Vaula G, Romano DM, et al. Assessment of amyloid β-protein precursor gene mutations in a large set of familial and sporadic Alzheimer disease cases. Am J Hum Genet. 1992;51(2):273–282. | ||
Schellenberg GD, Pericak-Vance MA, Wijsman EM, et al. Linkage analysis of familial Alzheimer disease, using chromosome 21 markers. Am J Hum Genet. 1991;48(3):563–583. | ||
Cruts M, van Duijn CM, Backhovens H, et al. Estimation of the genetic contribution of presenilin-1 and -2 mutations in a population-based study of presenile Alzheimer disease. Hum Mol Genet. 1998;7(1):43–51. | ||
Kamimura K, Tanahashi H, Yamanaka H, Takahashi K, Asada T, Tabira T. Familial Alzheimer’s disease genes in Japanese. J Neurol Sci. 1998;160(1):76–81. | ||
Hayashi K. PCR-SSCP: a simple and sensitive method for detection of mutations in the genomic DNA. PCR Methods Appl. 1991;1(1):34–38. | ||
Jeong BH, Ju WK, Huh K, et al. Molecular analysis of prion protein gene (PRNP) in Korean patients with Creutzfeldt-Jakob disease. J Korean Med Sci. 1998;13(3):234–240. | ||
Rizzu P, Van Swieten JC, Joosse M, et al. High prevalence of mutations in the microtubule-associated protein tau in a population study of frontotemporal dementia in the Netherlands. Am J Hum Genet. 1999;64(2):414–421. | ||
Cruts M, Gijselinck I, van der Zee J, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442(7105):920–924. | ||
Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. | ||
Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31(13):3812–3814. | ||
Källberg M, Wang H, Wang S, et al. Template-based protein structure modeling using the Raptor X web server. Nat Protoc. 2012;7(8):1511–1522. | ||
Zekanowski C, Golan MP, Krzyśko KA, et al. Two novel presenilin 1 gene mutations connected with frontotemporal dementia-like clinical phenotype: genetic and bioinformatic assessment. Exp Neurol. 2006;200(1):82–88. | ||
Bialopiotrowicz E, Szybinska A, Kuzniewska B, et al. Highly pathogenic Alzheimer’s disease presenilin 1 P117R mutation causes a specific increase in p53 and p21 protein levels and cell cycle dysregulation in human lymphocytes. J Alzheimers Dis. 2012;32(2):397–415. | ||
Gómez-Tortosa E, Barquero S, Barón M, et al. Clinical-genetic correlations in familial Alzheimer’s disease caused by presenilin 1 mutations. J Alzheimers Dis. 2010;19(3):873–884. | ||
Coleman P, Kurlan R, Crook R, Werner J, Hardy J. A new presenilin Alzheimer’s disease case confirms the helical alignment of pathogenic mutations in transmembrane domain 5. Neurosci Lett. 2004;364(3):139–140. | ||
Ataka S, Tomiyama T, Takuma H, et al. A novel presenilin-1 mutation (Leu85Pro) in early-onset Alzheimer disease with spastic paraparesis. Arch Neurol. 2004;61(11):1773–1776. | ||
Finckh U, Kuschel C, Anagnostouli M, et al. Novel mutations and repeated findings of mutations in familial Alzheimer disease. Neurogenetics. 2005;6(2):85–89. | ||
Kim J, Bagyinszky E, Chang YH, et al. A novel PSEN1 H163P mutation in a patient with early-onset Alzheimer’s disease: clinical, neuroimaging, and neuropathological findings. Neurosci Lett. 2012;530(2):109–114. | ||
Mendez MF, McMurtray A. Frontotemporal dementia-like phenotypes associated with presenilin-1 mutations. Am J Alzheimers Dis Other Demen. 2006;21(4):281–286. | ||
Raux G, Gantier R, Thomas-Anterion C, et al. Dementia with prominent frontotemporal features associated with L113P presenilin 1 mutation. Neurology. 2000;55(10):1577–1578. | ||
Dermaut B, Kumar-Singh S, Engelborghs S, et al. A novel presenilin 1 mutation associated with Pick’s disease but not beta-amyloid plaques. Ann Neurol. 2004;55(5):617–626. | ||
Pickering-Brown SM, Baker M, Gass J, et al. Mutations in progranulin explain atypical phenotypes with variants in MAPT. Brain. 2006;129(11):3124–3126. | ||
Shen L, Bagyinszky E, Youn YC, An SS, Kim SY. Genetic factors in frontotemporal dementia: a review. Toxicol Environ Health Sci. 2013;5(3):113–130. | ||
Giau VV, An SS, Bagyinszky E, Kim SY. Gene panels and primers for next generation sequencing studies on neurodegenerative disorders. Mol Cell Toxicol. 2015;11(2):89–143. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.