")
Back to Journals » International Journal of Nanomedicine » Volume 12
Protective effects on myocardial infarction model: delivery of schisandrin B using matrix metalloproteinase-sensitive peptide-modified, PEGylated lipid nanoparticles
Received 10 May 2017
Accepted for publication 8 August 2017
Published 26 September 2017 Volume 2017:12 Pages 7121—7130
DOI https://doi.org/10.2147/IJN.S141549
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Linlin Sun
Mingfeng Shao,1 Wenfang Yang,2 Guangying Han1
1Department of Cardiology, Linyi People’s Hospital, Linyi, Shandong, People’s Republic of China; 2Department of Internal Medicine, Linyi Hot Spring Hospital of Shandong Coal Mine, Linyi, Shandong, People’s Republic of China
Purpose: Schisandrin B (Sch B) is clinically applied for the treatment of hepatitis and ischemic disease. However, its clinical efficacy is limited due to the poor solubility and low bioavailability. This study aimed to develop matrix metalloproteinase (MMP)-sensitive peptide-modified, polyethylene glycol (PEG)-modified (PEGylated) solid lipid nanoparticles (SLNs) for loading Sch B (MMP-Sch B SLNs), and to evaluate the therapeutic effect in the myocardial infarction model.
Methods: PEG lipid and MMP-targeting peptide conjugate were synthesized. MMP-Sch B SLNs were prepared by solvent displacement technique. The physicochemical properties and pharmacokinetics of SLNs were investigated. In vivo effects on infarct size was evaluated in rats.
Results: The successful synthesis of lipid-peptide conjugate was confirmed. MMP-Sch B SLNs had a particle size of 130 nm, a zeta potential of 18.3 mV, and a sustained-release behavior. Higher heart drug concentration and longer blood circulation times were achieved by Sch B loaded SLNs than the drug solution according to the pharmacokinetic and biodistribution results. The best therapeutic efficacy was exhibited by MMP-Sch B SLNs by reducing the infarction size to the greatest extent.
Conclusion: The modified SLNs may be a good choice for delivery of Sch B for the treatment of myocardial infarction.
Keywords: cardiovascular diseases, CVDs, schisandrin B, matrix metalloproteinase, lipid nanoparticles
Introduction
Cardiovascular diseases (CVDs) continue to be one of the leading causes of death worldwide, with myocardial infarction (MI) contributing a large share of the deaths.1,2 In clinical practice, revascularization strategies followed by palliative care is the standard care for acute MI.3,4 However, these treatments only bring a short-term curative effect without eliminating disease. Therefore, there is an urgent need for improved MI treatments.
Schisandrin B (Sch B), isolated from the fruit of Schisandra chinensis, is clinically applied for the treatment of hepatitis and ischemic diseases, such as myocardial ischemia and cerebral ischemia.5–8 The potential mechanism underlying the cardioprotective effects of Sch B has been considered as the high antioxidant potential both in vitro and in vivo.9–11 Recent studies have further indicated that Sch B could improve cardiac function and attenuate myocardial remodeling in the MI mice model through down-regulating some inflammatory cytokines, activating eNOS pathway, inhibiting cell apoptosis, and enhancing cell proliferation.6 However, Sch B shows poor solubility and systemic delivery, and low bioavailability, which limit its clinical efficacy.12,13
Systemically administered nanoscale drug carriers could improve the solubility of hydrophobic drugs, prolong circulation half-life, and passively improve the accumulation of therapeutic agents in the target area.2,14 Several researches have proven that the bioactivity of Sch B was improved using nanotechnology such as phospholipid complex loaded nanoparticles, and folate-targeted and polyethylene glycol (PEG)-modified (PEGylated) TiO2 nanocarriers.12,13,15 Recent advances in nanoscale drug carriers for MI therapy include liposomes (containing PEGylated liposomes), core-shell hybrid liposomal vesicles, lipid nanoparticles, etc.16–19 In this study, we designed Sch B loaded PEG-modified solid lipid nanoparticles (SLNs) for MI treatment.
SLNs are the latest development in the arena of lipid nanoparticles after nanoemulsion and liposomes ever since their introduction in the early 1990s.20 SLNs are made from solid lipids with a mean diameter of 50 to 1,000 nm, which exhibit biocompatibility, physical stability, controlled release properties, and ease of manufacture.21 SLN systems represent a promising platform both for conjugating PEG, thus achieving long circulation in vivo, and for incorporation of active targeting ligand.22 For active targeting to the heart, multifunctional nanoparticles (matrix metalloproteinase [MMP]-sensitive peptide-modified, PEGylated SLNs) were successfully formulated.
MMPs are proteolytic enzymes which have been identified in the myocardium, and play an important role in myocardial remodeling and restructuring after MI.23,24 Among the MMPs, MMP-2 and MMP-9 are key factors in left ventricle remodeling.25 Since activated MMP-2 and MMP-9 were observed extracellularly in the infarction area, they have been reported as targets for drug delivery.23 Nguyen et al designed a micellar vehicle containing an MMP-targeting peptide (MMP-TP).23 Results suggest that MMP-TP micelles are candidates to target the infarcted myocardium in an MMP dependent manner. In the present study, we synthesized novel MMP-sensitive peptide-modified, PEGylated SLNs for loading Sch B (MMP-Sch B SLNs), thus delivering drugs to the heart.
The study was aimed at the development of novel MMP-Sch B SLNs, evaluation of SLNs’ effects on the parameters of pharmacokinetics, investigation of their biodistribution, and assessment of their effect in an MI model.
Materials and methods
Materials
Salvianolic acid B (purity ≥94%, HPLC), DMEM, MTT, DOTAP, and poloxamer 188 were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). MMP-TP (CPLGLAGG, molecular weight 300) was obtained from Shanghai Science Peptide Biotechnology Co., Ltd. (Shanghai, People’s Republic of China). COMPRITOL® 888 ATO (888 ATO) was provided by Gattefosse’ (Paramus, NJ, USA). Soybean lecithin (SL) in injection grade was purchased from Shanghai Taiwei Pharmaceutical Co., Ltd. (Shanghai, People’s Republic of China). Glyceryl monostearate (GMS) was purchased from Aladdin Industrial Corporation (Shanghai, People’s Republic of China). mPEG2000-NHS (molecular weight 2,000 Da) was purchased from Seebio Biochem Co., Ltd. (Shanghai, People’s Republic of China). All other reagents and chemicals were of analytical grade.
Synthesis of PEG and MMP-TP conjugate
PEG and MMP-TP conjugate (PEG-peptide, Figure 1) was synthesized as follows:26,27 mPEG2000-NHS (1 g) was dissolved in 10 mL of dimethyl formamide (DMF) and then 20 μL triethylamine was added. Peptide (0.3 g) was suspended in 2 mL of DMF (2 mL) and was added into the mPEG2000-NHS solution with an ice/water bath. The reaction was then continued for 24 h at room temperature under nitrogen protection. Unreacted peptide and chemical regents were removed by dialysis (molecular weight cut-off of 1,000 Da). The product was lyophilized before characterization by 1H nuclear magnetic resonance (1H NMR).
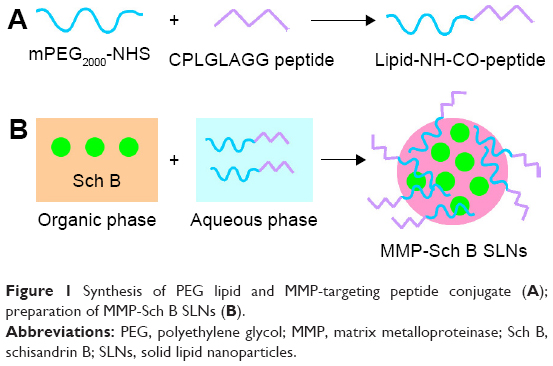 | Figure 1 Synthesis of PEG lipid and MMP-targeting peptide conjugate (A); preparation of MMP-Sch B SLNs (B). |
Preparation of MMP-Sch B SLNs
MMP-Sch B SLNs (Figure 1) were prepared by the solvent displacement technique with modifications.28 Sch B (50 mg), GMS (100 mg), 888 ATO (50 mg), and SL (100 mg) were accurately weighed and sonicated in 10 mL acetone to form the organic phase. The aqueous phase was prepared by dissolving PEG-peptide (200 mg), DOTAP (0.2%, w/v), and poloxamer 188 (0.1%, w/v) in Milli-Q water. The organic phase was injected into the aqueous phase under 600 rpm mechanical agitation at room temperature. The mixture was stirred at 600 rpm for 6 h until the organic solvent was removed. Free Sch B in the MMP-Sch B SLNs suspension was separated by ultrafiltration. Sch B loaded SLNs without MMP-TP (Sch B SLNs) were prepared by the same technique as MMP-Sch B SLNs using PEG instead of PEG-peptide. Blank SLNs not containing the drug were prepared by the same technique as Sch B SLNs without the presence of Sch B.
Particle size and zeta potential measurement
The MMP-Sch B SLNs, Sch B SLNs, and blank SLNs were diluted with Milli-Q water, separately.29 Their particle size and zeta potential were determined by dynamic light scattering in a Zetasizer (Nano ZS; Malvern Instruments, Malvern, UK). All measurements were taken at 25°C and an average of ten measurements were determined for each sample.
Drug entrapment efficiency (EE) and drug loading (DL) capacity
The drug EE and DL capacity of MMP-Sch B SLNs and Sch B SLNs were determined by the ultrafiltration method.30 The filtrate was collected and diluted with ethanol and the amount of drug was obtained by measuring the solution with high performance liquid chromatography (HPLC). EE and DL of Sch B loaded in SLNs were calculated by the following equations:
|
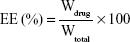
|
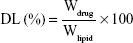
In vitro drug release study
The release of the drug was studied in PBS (pH 7.4) which was constantly shaking at 37°C.31,32 The drug release of Sch B solution (ethanol as solvent) was applied as control. At predetermined time points, the medium was taken out and replaced with fresh medium. The amount of Sch B was determined by HPLC.
Cells
Human cardiac myocytes (HCMs) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were cultured in 10% FBS (Fisher Chemicals, Fairlawn, NJ, USA) containing DMEM (Sigma-Aldrich Co.) in a 95% air/5% CO2 fully humidified atmosphere.
In vitro cytotoxicity in HCMs
HCMs were applied for the assessment of the in vitro cytotoxicity of Sch B loaded SLNs.33 Briefly, cells were seeded in 96-well plates at 104 cells/well and pre-incubated for 24 h. Different concentrations (1, 5, 10, 20, 50, 100 μM) of MMP-Sch B SLNs, Sch B SLNs, SLNs and 0.9% saline solution were added to the cells and incubated for 48 h. Then the cells were treated with 5 mg/mL of MTT solution and maintained for 4 h. The medium containing MTT was removed, 200 μL of DMSO was added to the wells. Plate was observed at 570 nm using a microplate reader. The relative cell viability was calculated by the following equation:
|
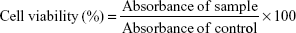
Animals and induction of MI model
Sprague-Dawley rats (SD rats, 220 to 250 g) were purchased from Nanjing Junke biological engineering Co., Ltd (Nanjing, People’s Republic of China) and housed under controlled conditions (temperature of 20°C±2°C and a 12 h light/12 h dark cycle). Experiments were performed according to the National Institutes of Health guide for the care and use of laboratory animals (NIH publication no 8023, revised 1978). An MI model was produced in male SD rats by partial ligation of the coronary artery.34 All the animal experiments were approved by the Medical Ethics Committee of Linyi People’s Hospital (reference no 201702121013).
In vivo pharmacokinetics evaluation
Sch B loaded SLNs and Sch B solution were injected through the tail vein of the MI rat models at a dose of 10 mg drug per kg body weight.35 Blood samples (200 μL) were taken via tail vein at 0.25, 0.5, 1, 1.5, 2, 4, 8, 12, 24, 48, and 72 h after injection and 15 μL of 1,000 U/mL heparin was added to each sample. The blood samples were immediately centrifuged at 5,000 rpm for 5 min at 4°C. The amount of Sch B was quantified by HPLC.
In vivo tissue biodistribution study
Sch B loaded SLNs and Sch B solution were administrated to the MI rat models as discussed in “In vivo pharmacokinetics evaluation” section. Rats were sacrificed after 2 h or 48 h and the heart, liver, spleen, lung, kidney, stomach, and colon were collected and washed of residual blood.36 Tissue samples were vortexed for 3 min and centrifuged at 20,000 rpm for 10 min. The supernatants were collected and the amount of Sch B was quantified by HPLC.
In vivo effects on infarct size
Sch B loaded SLNs and Sch B solution were administrated to the MI rat models as discussed in “In vivo pharmacokinetics evaluation” section. Rats were sacrificed at 48 h and the infarcted area was determined by TTC staining.37 Briefly, the hearts were excised and sliced into 2 mm thick sections. The slices were incubated in a solution of 1% TTC in PBS (pH 7.4) at 37°C for 15 min. The normal myocardium areas were stained brick red, and the infarct areas were unstained. The area of MI was calculated sing ImageJ (v 1.41; National Institutes of Health, Bethesda, MD, USA). The infarct size could be calculated by the following equation:
|

Statistical analysis
The results are reported as mean ± standard derivation. The difference between the groups was tested by two way ANOVA. The criterion for statistical significance was P<0.05.
Results
Characterization of lipid-peptide conjugate
The successful synthesis of lipid-peptide conjugate was confirmed by 1H NMR (Figure 2). The appearance of proton peaks corresponded to the structure of the lipid-peptide at 0.89 ppm (a, –NCO–C–CH3); 0.98 ppm (b, –CCH–N–); 1.31 ppm (c, –CO–CH2C–O–); 2.87 ppm (d, –C–C–OH); 3.21 ppm (e, –CO–CCH2–O–); 5.23 ppm (f, –CO–NH–); and 7.19 ppm (–COOH). The peaks of amide linkage, PEG and peptide proved the success of the synthesis.
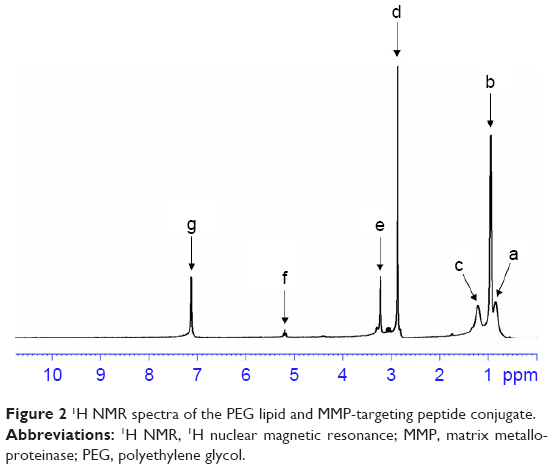 | Figure 2 1H NMR spectra of the PEG lipid and MMP-targeting peptide conjugate. |
Characterization of MMP-Sch B SLNs
The size of MMP-Sch B SLNs, Sch B SLNs, and SLNs were round 130 nm (Figure 3), with polydispersity indexes of 0.164, 0.122, and 0.109, respectively. The zeta potential of MMP-Sch B SLNs, Sch B SLNs, and SLNs were 18.3, 25.5, and 19.4 mV. EE of MMP-Sch B SLNs and Sch B SLNs were approximately 90%, with DL of 4.6% and 3.9%, separately.
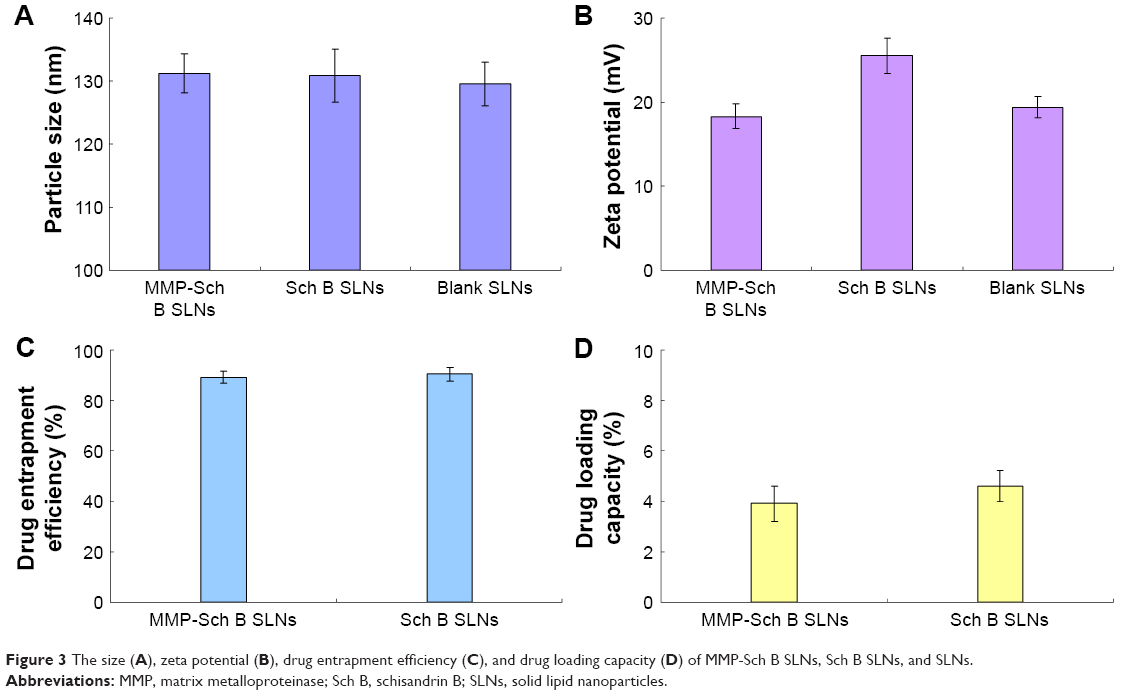 | Figure 3 The size (A), zeta potential (B), drug entrapment efficiency (C), and drug loading capacity (D) of MMP-Sch B SLNs, Sch B SLNs, and SLNs. |
In vitro drug release profile
In vitro release profile of Sch B loaded SLNs along with the Sch B solution were depicted in Figure 4. Sch B was released from the drug solution very fast, the release complete in the first 2 h. On the contrary, Sch B was released from MMP-Sch B SLNs and Sch B SLNs with a sustained-release behavior, with complete release after 48 h. The release profiles of MMP-Sch B SLNs and Sch B SLNs have significant differences.
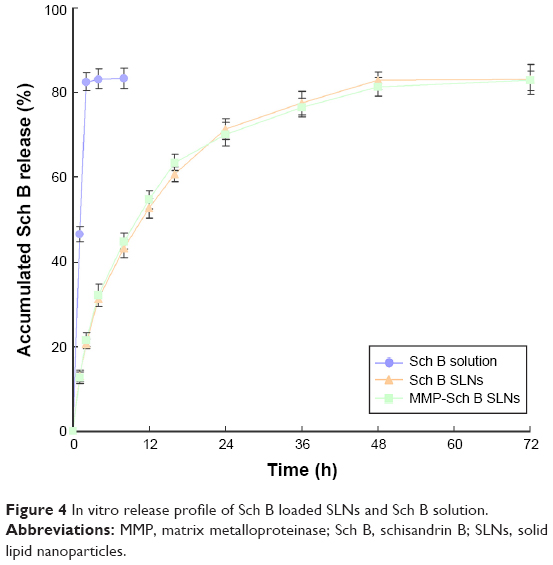 | Figure 4 In vitro release profile of Sch B loaded SLNs and Sch B solution. |
In vitro cytotoxicity
In vitro cytotoxicity of Sch B loaded SLNs and blank SLNs at various concentrations is shown in Figure 5. The HCM cell viabilities decreased slightly with the increased concentration of SLNs, while the average cell viabilities of different formulations at the studied concentrations were above 80% compared with control cells. All of the samples studied exhibited no obvious cytotoxicity, similar to the saline control group (P>0.05).
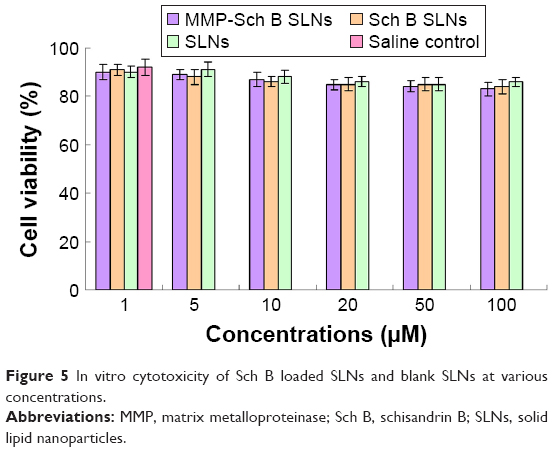 | Figure 5 In vitro cytotoxicity of Sch B loaded SLNs and blank SLNs at various concentrations. |
In vivo pharmacokinetics
The mean plasma drug concentration–time profile shown in Figure 6 and the main pharmacokinetic parameters are summarized in Table 1, respectively. The profile showed that the drug concentration of Sch B solution in plasma decreased rapidly and was cleared from the circulation within 4 h. On the contrary, Sch B loaded SLNs exhibited a prolonged plasma circulation time, up to more than 48 h. Table 1 showed Sch B-loaded SLNs exhibited higher AUC in comparison with Sch B solution (P<0.05). Sch B loaded SLNs exhibited longer t1/2 in comparison to Sch B solution (P<0.05). The volume of distribution (V) and plasma clearance (CL) of Sch B loaded SLNs were significantly lower than its injectable solution.
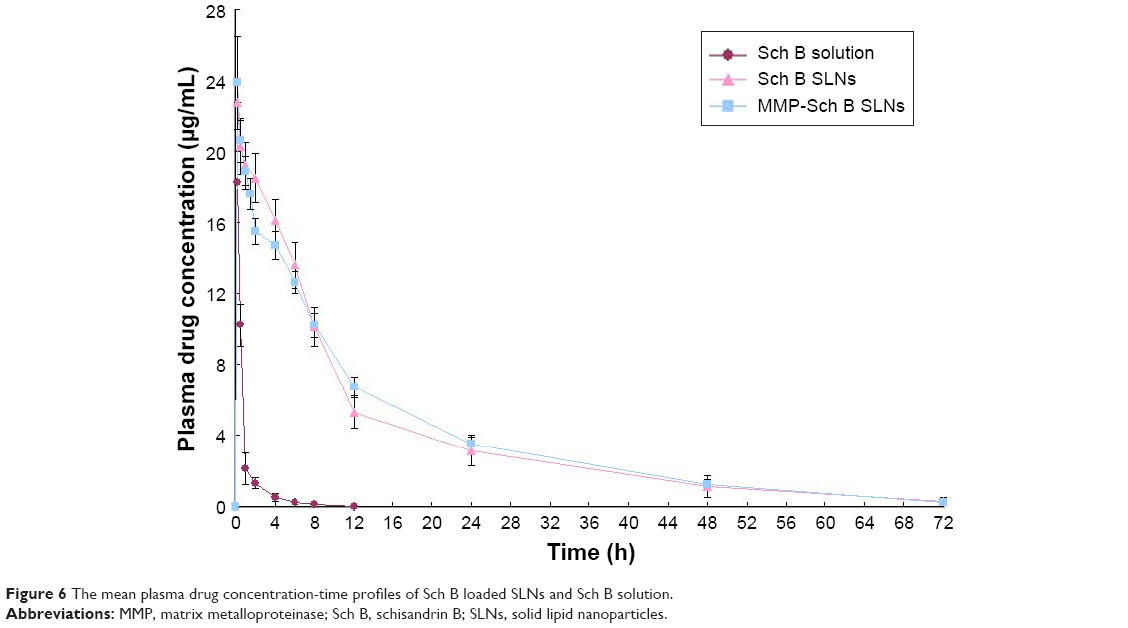 | Figure 6 The mean plasma drug concentration-time profiles of Sch B loaded SLNs and Sch B solution. |
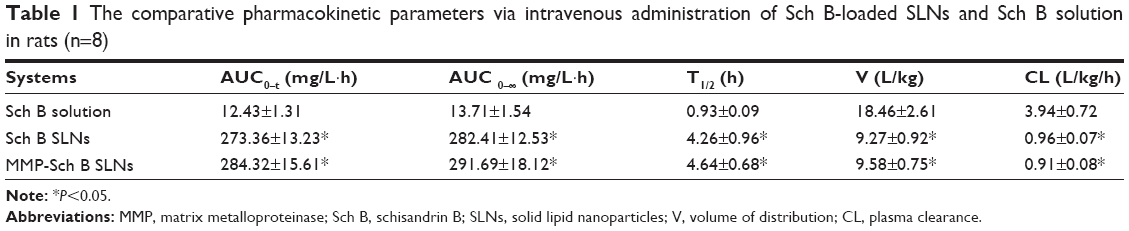 | Table 1 The comparative pharmacokinetic parameters via intravenous administration of Sch B-loaded SLNs and Sch B solution in rats (n=8) |
In vivo tissue biodistribution
In vivo tissue biodistribution behavior of Sch B loaded SLNs and Sch B solution were evaluated in the MI rat models after 2 and 48 h of administration (Figure 7). After both 2 h and 48 h of administration, the drug concentration of MMP-Sch B SLNs in the brain was the highest among all tissues, and also higher than Sch B SLNs and Sch B solution. At 2 h, Sch B SLNs did not show obvious heart accumulation than the solution. While at 48 h, significantly higher accumulation than Sch B solution was observed in the heart (P<0.05).
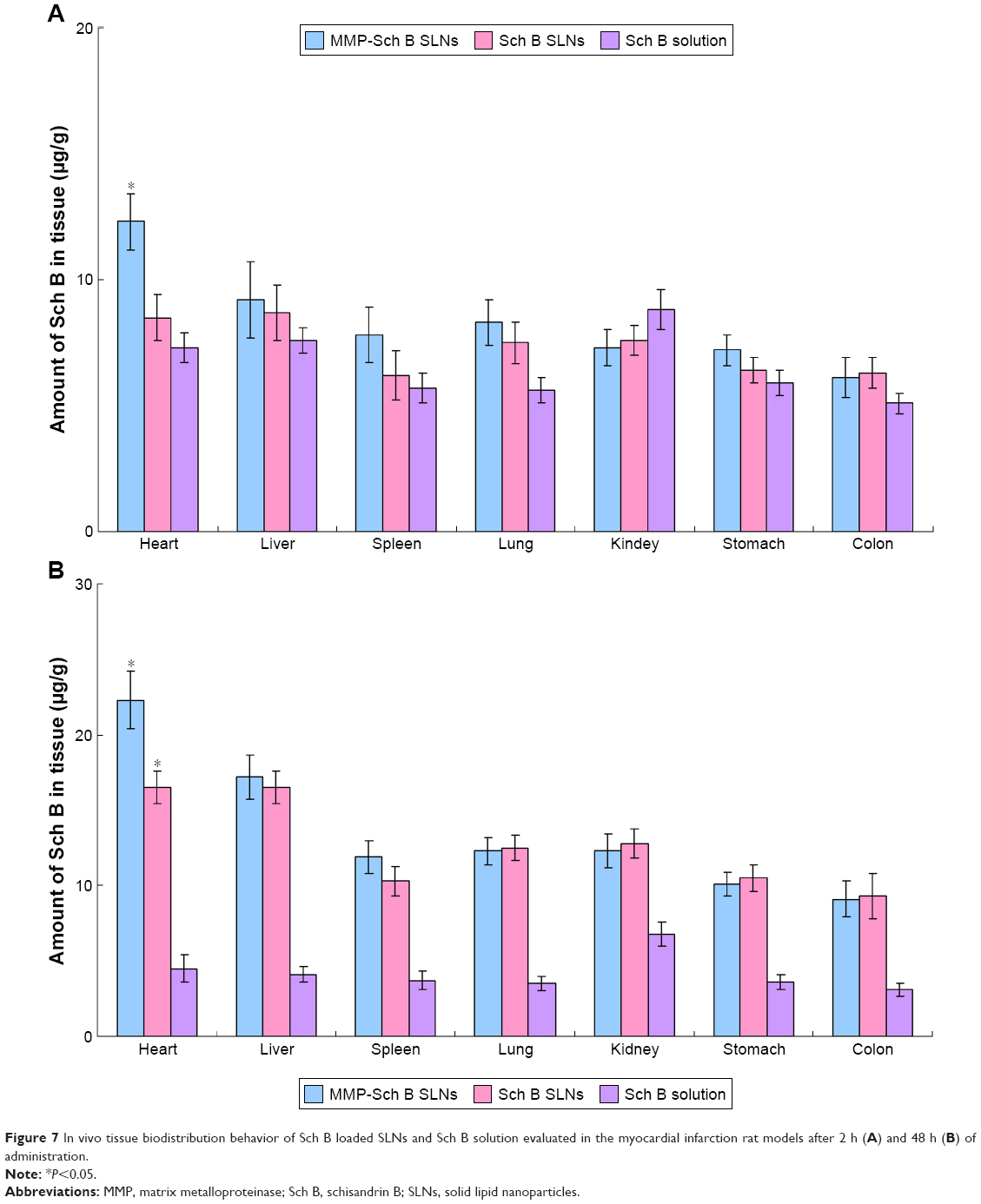 | Figure 7 In vivo tissue biodistribution behavior of Sch B loaded SLNs and Sch B solution evaluated in the myocardial infarction rat models after 2 h (A) and 48 h (B) of administration. |
In vivo infarct size
In vivo effects of Sch B loaded SLNs and Sch B solution on infarct size were illustrated in Figure 8. MMP-Sch B SLNs exhibited the most significant reduction in infarct size as compared with the other groups. The infarct size of MMP-Sch B SLNs, Sch B SLNs, Sch B solution, and saline groups were 19.2%, 27.3%, 36.5%, and 43.2%, respectively.
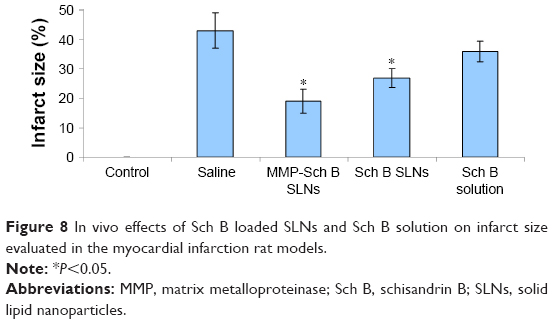 | Figure 8 In vivo effects of Sch B loaded SLNs and Sch B solution on infarct size evaluated in the myocardial infarction rat models. |
Discussion
In the present study, novel MMP-Sch B SLNs were applied for treatment of acute myocardial ischemia. At the beginning of this study, PEG and MMP-TP conjugate was synthesized. Several peptide sequences, such as GPLGIAGQ, GPLGV, GPLGVRG, and PVGLIG have been reported for the development of MMP-2/9-sensitive conjugate.26 These short peptides with a certain sequence can be cleaved by MMP-2/9 regardless of their secondary or tertiary structures. In this study, CPLGLAGG was used as MMP-TP.
In order to improve the pharmacological activity of Sch B in acute myocardial ischemia, MMP-Sch B SLNs were successfully prepared by the solvent displacement technique with GMS and 888 ATO as solid lipid. DOTAP and poloxamer 188 were used as emulsifier and stabilizer. PEG-peptide was applied as modification material to increase the circulation time and drug accumulation in the heart.21 The size of blank and Sch B SLNs was approximately 130 nm, with narrow polydispersity indexes lower than 0.2. This could be evidence that the loading of the drug has no obvious effect on the size of the SLNs. Particle size has a great impact on the in vitro and in vivo efficiency of the nanoparticles, including prolonging the blood circulation time and mediating the targeted effect.38 It has also been reported that zeta potential is a key factor to evaluate the stability of a colloidal dispersion. There are several approaches for measuring the drug EE and loading content of a nanoparticle system, including Sephadex column filtration, centrifugation, dialysis, ultrafiltration, and so on. Among these methods, centrifugation is frequently used due to its convenience. Hence, in this research this method was used with some modifications.
In vitro release of drugs from the lipid nanoparticles could include lipid matrix swelling, drug diffusion, and erosion or degradation process.39 Drugs located near the surface of SLNs may be delivered first, after which the inner drugs are diffused through the lipid matrix. The release profiles of Sch B loaded SLNs showed sustained-release behaviour. This behavior could maintain the drug concentration in the blood circulation and bring about a continuous therapeutic effect during the administration period.
Evaluation of the cytotoxicity of the cationic drug delivery systems is essential as cationic lipids may bring about a cytotoxic effect.40 Double-tailed cationic lipids, such as DOTMA and DDAB are reported to exhibit lower cytotoxicity than its single-tailed counterparts, such as CTAB.41 In this study, DOTAP was utilized and HCM cell viabilities for different formulations at various concentrations were evaluated. Over 80% cell viability of SLNs compared with control could prove this system has no obvious cytotoxicity, and it could be used as safe drug delivery system in CVDs.
In vivo pharmacokinetics evaluation showed that Sch B loaded SLNs exhibited higher AUC in comparison with Sch B solution. This could be explained by the PEGylated SLNs having a long circulation effect with the PEG chains on their surface.42 This confirms that the PEGylation of nanoparticles could improve the plasma half-life of the carriers and achieve sustained-release of the loaded drugs.16 PEG was reported to have the ability to improve the surface hydrophilicity of lipid particles and prevent the absorption of lipoproteins and opsonins effectively.22 Therefore, the PEG ligands present over the carriers could avoid the recognition of the reticuloendothelial system and prolong the circulation time, thus bringing about a persistent therapeutic effect.
In vivo biodistribution study results exhibited similar long-circulating characteristics of SLNs. MMP-Sch B SLNs exhibited higher drug biodistribution in the heart than that of Sch B SLNs and Sch B solution. MMP-TP modified carriers are candidates to target the infarcted myocardium in an MMP-dependent manner.23 The aim of the MMP-TP modification is to deliver more drugs to the infarct zone. MMP-Sch B SLNs exhibited higher heart Sch B concentration in comparison with Sch B SLNs in MI rats, which could be due to the targeted ability of MMP-sensitive peptide used for the modification.
Infarct size is an important factor for evaluating cardiac damage in the generation of ischemic heart diseases.43 In this study, cardiac protective effects of Sch B loaded SLNs were evaluated using an acute MI model. MMP-Sch B SLNs exhibited the most significant reduction in infarct size as compared with Sch B SLNs and other groups, showing the smallest infarct size. The infarct size was remarkably decreased after treatment with MMP-Sch B SLNs in acute MI-induced rats, suggesting the cardioprotective effect of the MMP-TP modified SLNs.
Conclusion
In summary, MMP-Sch B SLNs were successfully developed. Long-circulating and active targeting MMP-Sch B SLNs were used for heart-targeted drug delivery. The results of in vitro and in vivo studies revealed that the carriers could not only enhance drug penetration into heart tissues, but also improve heart protection efficacy from acute MI impairment by reducing the infarction size. The modified SLNs may be an effective therapeutic system for the treatment of MI.
Disclosure
The authors report no conflicts of interest in this work.
References
Esper SA, Subramaniam K. Heart failure and mechanical circulatory support. Best Pract Res Clin Anaesthesiol. 2012;26(2):91–104. | ||
Ho YT, Poinard B, Kah JC. Nanoparticle drug delivery systems and their use in cardiac tissue therapy. Nanomedicine (Lond). 2016;11(6):693–714. | ||
Adler ED, Goldfinger JZ, Kalman J, Park ME, Meier DE. Palliative care in the treatment of advanced heart failure. Circulation. 2009;120(25):2597–2606. | ||
Chang MY, Yang YJ, Chang CH, et al. Functionalized nanoparticles provide early cardioprotection after acute myocardial infarction. J Control Release. 2013;170(2):287–294. | ||
Chiu PY, Leung HY, Siu AH, Poon MK, Ko KM. Schisandrin B decreases the sensitivity of mitochondria to calcium ion-induced permeability transition and protects against ischemia-reperfusion injury in rat hearts. Acta Pharmacol Sin. 2007;28(10):1559–1565. | ||
Chen P, Pang S, Yang N, et al. Beneficial effects of schisandrin B on the cardiac function in mice model of myocardial infarction. PLoS One. 2013;8(11):e79418. | ||
Thandavarayan RA, Giridharan VV, Arumugam S, et al. Schisandrin B prevents doxorubicin induced cardiac dysfunction by modulation of DNA damage, oxidative stress and inflammation through inhibition of MAPK/p53 signaling. PLoS One. 2015;10(3):e0119214. | ||
Lee TH, Jung CH, Lee DH. Neuroprotective effects of Schisandrin B against transient focal cerebral ischemia in Sprague-Dawley rats. Food Chem Toxicol. 2012;50(12):4239–4245. | ||
Mak DH, Ip SP, Li PC, Poon MK, Ko KM. Effects of Schisandrin B and alpha-tocopherol on lipid peroxidation, in vitro and in vivo. Mol Cell Biochem. 1996;165(2):161–165. | ||
Yim TK, Ko KM. Schisandrin B protects against myocardial ischemia-reperfusion injury by enhancing myocardial glutathione antioxidant status. Mol Cell Biochem. 1999;196(1–2):151–156. | ||
Kim EY, Baek IH, Rhyu MR. Cardioprotective effects of aqueous Schizandra chinensis fruit extract on ovariectomized and balloon-induced carotid artery injury rat models: effects on serum lipid profiles and blood pressure. J Ethnopharmacol. 2011;134(3):668–675. | ||
Peng Q, Zhang ZR, Sun X, Zuo J, Zhao D, Gong T. Mechanisms of phospholipid complex loaded nanoparticles enhancing the oral bioavailability. Mol Pharm. 2010;7(2):565–575. | ||
Li H, Shi L, Wei J, et al. Cellular uptake and anticancer activity of salvianolic acid B phospholipid complex loaded nanoparticles in head and neck cancer and precancer cells. Colloids Surf B Biointerfaces. 2016;147:65–72. | ||
Mihardja SS, Gao D, Sievers RE, et al. Targeted in vivo extracellular matrix formation promotes neovascularization in a rodent model of myocardial infarction. PLoS One. 2010;5(4):e10384. | ||
Ding L, Li J, Huang R, et al. Salvianolic acid B protects against myocardial damage caused by nanocarrier TiO2; and synergistic anti-breast carcinoma effect with curcumin via codelivery system of folic acid-targeted and polyethylene glycol-modified TiO2 nanoparticles. Int J Nanomedicine. 2016;11:5709–5727. | ||
Zhang S, Wang J, Pan J. Baicalin-loaded PEGylated lipid nanoparticles: characterization, pharmacokinetics, and protective effects on acute myocardial ischemia in rats. Drug Deliv. 2016;23(9):3696–3703. | ||
Almer G, Frascione D, Pali-Schöll I, et al. Interleukin-10: an anti-inflammatory marker to target atherosclerotic lesions via PEGylated liposomes. Mol Pharm. 2013;10(1):175–186. | ||
Verma DD, Hartner WC, Levchenko TS, Bernstein EA, Torchilin VP. ATP-loaded liposomes effectively protect the myocardium in rabbits with an acute experimental myocardial infarction. Pharm Res. 2005;22(12):2115–2120. | ||
Zhang J, Han X, Li X, et al. Core-shell hybrid liposomal vesicles loaded with panax notoginsenoside: preparation, characterization and protective effects on global cerebral ischemia/reperfusion injury and acute myocardial ischemia in rats. Int J Nanomedicine. 2012;7:4299–4310. | ||
Thukral DK, Dumoga S, Mishra AK. Solid lipid nanoparticles: promising therapeutic nanocarriers for drug delivery. Curr Drug Deliv. 2014;11(6):771–791. | ||
Gao Y, Gu W, Chen L, Xu Z, Li Y. The role of daidzein-loaded sterically stabilized solid lipid nanoparticles in therapy for cardio-cerebrovascular diseases. Biomaterials. 2008;29(30):4129–4136. | ||
Pawar H, Surapaneni SK, Tikoo K, et al. Folic acid functionalized long-circulating co-encapsulated docetaxel and curcumin solid lipid nanoparticles: In vitro evaluation, pharmacokinetic and biodistribution in rats. Drug Deliv. 2016;23(4):1453–1468. | ||
Nguyen J, Sievers R, Motion JP, Kivimäe S, Fang Q, Lee RJ. Delivery of lipid micelles into infarcted myocardium using a lipid-linked matrix metalloproteinase targeting peptide. Mol Pharm. 2015;12(4):1150–1157. | ||
El-Aziz TA, Mohamed RH. Matrix metalloproteinase-9 polymorphism and outcome after acute myocardial infarction. Int J Cardiol. 2017;227:524–528. | ||
Phatharajaree W, Phrommintikul A, Chattipakorn N. Matrix metalloproteinases and myocardial infarction. Can J Cardiol. 2007;23(9):727–733. | ||
Zhang X, Wang X, Zhong W, Ren X, Sha X, Fang X. Matrix metalloproteinases-2/9-sensitive peptide-conjugated polymer micelles for site-specific release of drugs and enhancing tumor accumulation: preparation and in vitro and in vivo evaluation. Int J Nanomedicine. 2016;11:1643–1661. | ||
Shi L, Hu Y, Lin A, et al. Matrix metalloproteinase responsive nanoparticles for synergistic treatment of colorectal cancer via simultaneous anti-angiogenesis and chemotherapy. Bioconjug Chem. 2016;27(12):2943–2953. | ||
Qu J, Zhang L, Chen Z, et al. Nanostructured lipid carriers, solid lipid nanoparticles, and polymeric nanoparticles: which kind of drug delivery system is better for glioblastoma chemotherapy? Drug Deliv. 2016;23(9):3408–3416. | ||
Wang F, Li L, Liu B, Chen Z, Li C. Hyaluronic acid decorated pluronic P85 solid lipid nanoparticles as a potential carrier to overcome multidrug resistance in cervical and breast cancer. Biomed Pharmacother. 2017;86:595–604. | ||
Peng HS, Liu XJ, Lv GX, et al. Voriconazole into PLGA nanoparticles: improving agglomeration and antifungal efficacy. Int J Pharm. 2008;352(1–2):29–35. | ||
Chen W, Guo M, Wang S. Anti prostate cancer using PEGylated bombesin containing, cabazitaxel loading nano-sized drug delivery system. Drug Dev Ind Pharm. 2016;42(12):1968–1976. | ||
Saifullah B, Hussein MZ, Hussein-Al-Ali SH, Arulselvan P, Fakurazi S. Antituberculosis nanodelivery system with controlled-release properties based on para-amino salicylate-zinc aluminum-layered double-hydroxide nanocomposites. Drug Des Devel Ther. 2013;7:1365–1375. | ||
Wang C, Su L, Wu C, Wu J, Zhu C, Yuan G. RGD peptide targeted lipid-coated nanoparticles for combinatorial delivery of sorafenib and quercetin against hepatocellular carcinoma. Drug Dev Ind Pharm. 2016;42(12):1938–1944. | ||
Stanton LW, Garrard LJ, Damm D, et al. Altered patterns of gene expression in response to myocardial infarction. Circ Res. 2000;86(9):939–945. | ||
Jones AK, Bejugam NK, Nettey H, Addo R, D’Souza MJ. Spray-dried doxorubicin-albumin microparticulate systems for treatment of multidrug resistant melanomas. J Drug Target. 2011;19(6):427–433. | ||
Wang H, Sun G, Zhang Z, Ou Y. Transcription activator, hyaluronic acid and tocopheryl succinate multi-functionalized novel lipid carriers encapsulating etoposide for lymphoma therapy. Biomed Pharmacother. 2017;91:241–250. | ||
Yao C, Shi X, Lin X, Shen L, Xu D, Feng Y. Increased cardiac distribution of mono-PEGylated Radix Ophiopogonis polysaccharide in both myocardial infarction and ischemia/reperfusion rats. Int J Nanomedicine. 2015;10:409–418. | ||
Dong Z, Guo J, Xing X, Zhang X, Du Y, Lu Q. RGD modified and PEGylated lipid nanoparticles loaded with puerarin: Formulation, characterization and protective effects on acute myocardial ischemia model. Biomed Pharmacother. 2017;89:297–304. | ||
Huang J, Chen Z, Li Y, Li L, Zhang G. Rifapentine-linezolid-loaded PLGA microspheres for interventional therapy of cavitary pulmonary tuberculosis: preparation and in vitro characterization. Drug Des Devel Ther. 2017;11:585–592. | ||
Su Y, Hu J, Huang Z, et al. Paclitaxel-loaded star-shaped copolymer nanoparticles for enhanced malignant melanoma chemotherapy against multidrug resistance. Drug Des Devel Ther. 2017;11:659–668. | ||
Yu W, Liu C, Ye J, Zou W, Zhang N, Xu W. Novel cationic SLN containing a synthesized single-tailed lipid as a modifier for gene delivery. Nanotechnology. 2009;20(21):215102. | ||
Liu R, Wang Y, Li X, et al. Synthesis and characterization of tumor-targeted copolymer nanocarrier modified by transferrin. Drug Des Devel Ther. 2015;9:2705–2719. | ||
Liu X, Gu J, Fan Y, Shi H, Jiang M. Baicalin attenuates acute myocardial infarction of rats via mediating the mitogen-activated protein kinase pathway. Biol Pharm Bull. 2013;36(6):988–994. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.