")
Back to Journals » Cancer Management and Research » Volume 11
Protective autophagy or autophagic death: effects of BEZ235 on chronic myelogenous leukemia
Authors Xin P, Xu W, Zhu X, Li C, Zheng Y, Zheng T, Cheng W, Peng Q
Received 7 February 2019
Accepted for publication 5 July 2019
Published 22 August 2019 Volume 2019:11 Pages 7933—7951
DOI https://doi.org/10.2147/CMAR.S204472
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Chien-Feng Li
Pengliang Xin,1 Wenqian Xu,1 Xiongpeng Zhu,1 Chuntuan Li,1 Yan Zheng,1 Tingjin Zheng,2 Wenzhao Cheng,3 Qunyi Peng1
1Department of Haematology, Quanzhou First Hospital Affiliated to Fujian Medical University, Quanzhou, People’s Republic of China; 2Central Laboratory, Quanzhou First Hospital Affiliated to Fujian Medical University, Quanzhou, People’s Republic of China; 3Stem Cell Translational Research Center, The Second Affiliated Hospital of Fujian Medical University, Quanzhou, People’s Republic of China
Correspondence: Xiongpeng Zhu
Quanzhou First Hospital Affiliated to Fujian Medical University, No. 250 East Street, Licheng District, Quanzhou City, People’s Republic of China
Tel +86 1 380 599 8581
Email [email protected]
Purpose: To investigate the effects of BEZ235 on chronic myeloid leukemia (CML) cells.
Methods: MTS assay was used to detect the proliferation of CML cells. The proteins expression were detected by Western blot assay. The effects of BEZ235 on autophagy in CML cells were verified through transmission electron microscopy and evaluated by laser confocal microscopy. Annexin V-FITC/PI double staining flow cytometry was used to detect apoptosis. A xenograft model was established to observe the therapeutic effect of BEZ235 in vivo.
Results: BEZ235 could inhibit the proliferation of CML cells; CQ and 3-MA could increase the proliferation inhibition and Z-VAD-FMK can reduce the proliferation inhibition of BEZ235 on CML cells (P<0.05). Results of TEM showed that the autophagosomes of CML cells treated with BEZ235 increased (P<0.05). The results by confocal microscopy showed that the autophagic activity of K562 cells increased with BEZ235 treatment. When BEZ235 combined with CQ, BEZ235-induced autophagic flow was blocked. FCM results showed that BEZ235 could induces apoptosis in CML cells. Z-VAD-FMK could decrease the apoptosis of CML cells induced by BEZ235. CQ increased the apoptosis of CML cells induced by BEZ235 (P<0.05). Western blot showed that BEZ235 inhibited the phosphorylation of AKT and S6K. BEZ235 alone could upregulate the expression of cleaved caspase-3 and LC3II. When combined with Z-VAD-FMK, the expression of cleaved caspase-3 was lower than that of BEZ235 alone. When combined with CQ, the expression of cleaved caspase-3 and LC3II were higher than those of BEZ235 alone (P<0.05). BEZ235 could inhibit the growth of xenografts of CML cell line.
Conclusion: BEZ235 can inhibit the proliferation of CML cells, induce apoptosis, and enhance autophagy activity. It induces protective autophagy. The combination of CQ can enhance the apoptosis and proliferation inhibition of CML cells induced by BEZ235.
Keywords: BEZ235, chronic myelogenous leukemia, proliferation, apoptosis, autophagy
Introduction
Chronic myelogenous leukemia (CML) is a malignant disease of the hematopoietic system characterized by Philadelphia chromosomes (Ph) and BCR-ABL fusion genes. The protein encoded by the fused gene is named BCR-ABL1 protein, and its tyrosine residue has strong phosphorylation activity, which can lead to phosphorylation of its own protein, and can also phosphorylate many important substrate proteins, thereby activating multiple downstream signaling pathways and developing disease.1–4 Tyrosine kinase inhibitors (TKI) are currently the backbone of CML treatment, but 15–20% of patients still have resistance to TKI.5–7 Therefore, people are still looking for new ways to treat CML.8–10 The PI3K/Akt/mTOR signaling pathway is located downstream of BCR-ABL and is an important signaling pathway in the pathogenesis of CML,4,9 so this study attempts to treat CML by inhibiting the activity of this pathway. It is known that the PI3K/AKT/mTOR pathway is closely related to various cell functional activities such as cell proliferation, apoptosis and autophagy activity. BEZ235 is a dual ATP-competitive PI3K and mTOR inhibitor, effectively inhibiting the activity of the pathway. It is a newly developed targeted anti-tumor drug that has therapeutic effects on a variety of tumors.11–13 In this study, BEZ235 was selected for research, and we tried to explore its effects on autophagy, proliferation and apoptosis of CML cells. Currently, PI3K and mTOR are key points in the autophagy signaling pathway, and BEZ235 is a PI3K and mTOR inhibitor, which may affect the autophagy activity of cells. Therefore, the focus of this study is on autophagy activity and autophagy activity of cells. Human CML cells K562 and KBM7R (T315I mutant) were used as the research object to investigate the effects of BEZ235 on autophagy, proliferation, and apoptosis of CML cells, and the effect of BEZ235-induced autophagy on cell proliferation and apoptosis. The efficacy and safety of BEZ235 on CML was further verified in vivo by establishing a subcutaneous tumor-forming animal model.
Materials and methods
Ethics
All procedures were conducted in accordance with the guidelines contained in the guide for the care and use of laboratory animals 8th edition 2011 (the guide). The protocol, the cell lines and experimental animals used in this study were approved by the Ethics Committee of Quanzhou First Hospital (No:2015–54), Quanzhou, China. The K562 cell line used in this study was from Fujian Provincial Institute of Hematology and KBM7R cell line was from Harbin Institute of Hematology. The animal experimental site was the teaching experimental building of Quanzhou Medical College. The experimental animals used were SCID mice.
Main reagents and instruments
BEZ235 was purchased from Selleck, dissolved in dimethyl sulfoxide (DMSO) and stored at −20 °C. Chloroquine (CQ) was purchased from Sigma, dissolved in sterile double distilled water, and stored at −20 °C. 3-methyladenine (3-MA) was purchased from Selleck, dissolved in PBS and stored at −20 °C. Z-VAD-FMK was purchased from Beyotime and stored at −20 °C. The human CML cell line K562 was obtained from the Fujian Institute of Hematology, and the KBM7R cell line was purchased from the Harbin Institute of Hematology. Fetal bovine serum and RPMI 1640 medium were purchased from Gibco. Annexin V-FITC/PI kit was purchased from Becton Dickinson. MTS kit was purchased from Promega. Primary antibody AKT, P-AKT, S6K, P-S6K, Cleaved casepase-3, LC3I/II and HRP-labeled goat anti-rabbit IgG were purchased from Abcam. The RFP-GFP-LC3 double-labeled adenovirus was purchased from Hanbio. Cell constant temperature incubator (Thermo, USA), Refrigerated centrifuge (Eppendorf, Germany), Infinite M200 microplate reader (Tecan, Switzerland), FACS Calibur TM flow cytometry (Becton Dickinson, USA). Western blot electrophoresis, Semi-dry meter and Gel imager (Bio-Rad, USA). Hu-12A Transmission electron microscope (Hitachi, Japan), Laser scanning confocal microscope (Zeiss, Germany).
Cell culture
K562 and KBM7R cells were cultured in RPMI 1640 medium containing 10% fetal bovine serum at 37 °C, 5% CO2. The logarithmic growth phase cells were taken for experiments.
MTS assay for cell proliferation
The desired cells were seeded in a 96-well culture plate, 200 μL per well, 2×104 cells/well. Cells were treated with different concentrations of drugs, and a control group was set up, with 3 replicate wells in each group, and complete medium was set for blank hole. The MTS assay detects cell viability at different times. The experiment was repeated 3 times. The 50% inhibitory concentration (IC50) value of each drug was calculated based on the cell survival rate, and the following experiments were performed according to the 50% infectious dose (ID50) value and the drug concentration of other researchers. The drug concentrations were selected from low to high.
Transmission electron microscopy to detect autophagy
The samples preparation and observation methods for the transmission electron microscopy experiments were as follows: Materials: After treatment of CML cells with BEZ235, cells were harvested for 3, 6, 12, and 24 h, centrifuged at 1200 rpm for 5 mins, and washed twice with 1×PBS. Pre-fixation: 1.5mL 1× PBS was suspended and transferred into 1.5mL EP tube, centrifuged at 1500 rpm for 10 mins, carefully discarded the supernatant, slowly added 3% glutaraldehyde-1.5% paraformaldehyde fixative to the wall and placed at 4 °C refrigerator for 2~24 hrs. Post-fixation: The specimen was placed in 1% citric acid-1.5% potassium ferrocyanide solution and fixed at 4 °C for 3 h. Rinse: 1 x PBS rinse 3 times for 10 mins each time. Dehydration: Dehydration of graded alcohol and acetone. Immersion: The specimen was placed in a dry acetone + epoxy resin 618 embedding agent (1:1) at room temperature for 1.5 h, and then transferred to a pure 618 embedding agent at 35 °C for 3 h. Embedding and polymerization: The conditions were 35 °C for 12 h, 45 °C for 12 h, and 60 °C for 72 h. Sectioning: Firstly cut a 1 μM thick semi-thin section with an ultra-thin slicer, bake it, and then drop it into a 1% basket-Azure II mixture for staining. After observation and positioning under a microscope, the samples were cut into 100 nm ultra-thin by the ultra-thin slicer. The samples were sectioned and stained with uranyl acetate and lead citrate for 5–10 mins, and rinsed with distilled water for 2–3 mins. Observation and filming: observation and filming under Hu-12A transmission electron microscopy.
Autophagy double-labeled adenovirus transfection, laser confocal microscopy for autophagy
1×106 cells were seeded into a 6-well plate, cultured in 1 mL of 1640 medium containing 10% fetal bovine serum, and the corresponding autophagy double-labeled adenovirus (mRFP-GFP-LC3) was added at a multiplicity of infection (MOI) value of 100 for 24 h. After changing the medium, the culture was continued for 24 h, and GFP and mRFP expression were observed by sampling in a fluorescence microscope. The cells were treated with different concentrations of BEZ235 alone or in combination with 40 μM of CQ. After 12 and 24 h of culture, the expression of GFP and mRFP were detected under laser confocal microscopy. The effect of BEZ235 on autophagy of CML cells was evaluated.
Annexin V-FITC/PI double staining method for detecting apoptosis rate of cells
The cells were seeded in a 6-well plate, 5×105 cells/well, the cells were treated with different concentrations of drugs, and the control group was set up. After 24 and 48 h of culture, all the cells in each group were collected by centrifugation; the cell concentration was adjusted using 1×binding buffer. 5×105/mL, 200 μL cells were pipetted into 1.5-mL EP tubes, 5 μL Annexin V-FITC and 5 μL of PI were added, and blank control and single staining control groups were set up. This was mixed well at room temperature for 15 min. Then, 300 μL of 1×binding buffer was added. Apoptosis was detected using a flow cytometer. The experiment was repeated 3 times.
Western blot detection of related protein expression
CML cells were treated with different concentrations of BEZ235 and cultured for 3–24 h respectively. The total protein of each group was extracted. The denatured protein was electrophoresed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). After transformation, the membrane was sealed at room temperature for 1–2 h, and then the primary antibody was stored at 4 °C and incubated overnight. The membrane was washed 3 times with phosphate buffered saline with Tween (PBST), and the second antibody was incubated at 32 °C for 2 h. After washing the membrane 33 times with PBST, color development was performed using electrochemiluminescence (ECL), and then exposed using a BIO-RAD gel imager to detect the gray value of the strip (protein relative expression = target strip gray scale/internal reference strip gray scale ×100%). The relative expression level of phosphorylated protein was measured as p-AKT = p-AKT band gray value/t-AKT band gray scale×100%.
Establish a subcutaneous xenograft model of CML SCID mice cells and administer BEZ235 intervention
KBM7R cells in logarithmic growth phase were collected at 1200 rpm × 5 min, washed twice in sterile 1 × PBS, and resuspended in serum-free RPMI-1640 to adjust the cell density to 4 × 106/0.2 mL. After disinfecting the skin of the mouse with 75% alcohol, 0.1 mL of the cell suspension was inoculated subcutaneously into the back. After successfully constructing the subcutaneous tumor formation model, a total of 12 experimental mice were randomly divided into 6 groups: of BEZ235 administration group and 6 control groups 24 h after inoculation of KBM7R cells. On the 10th day after inoculation, the tumor volume was approximately 100–150 mm3, and the administration was started. BEZ235 was intragastrically administered at 50 mg/kg/d once daily for 14 days, stopped for observation, and maintained until 31 days after inoculation. The control group was given an equal volume of NPM/polyethylene glycol 300 (10/90) mixture. The mental state, activity, food intake, drinking water, hair, signs of infection, urination, etc. of the mice were observed daily and recorded. The tumor formation and tumor growth of transplanted tumor in mice was also observed. The dosing day, the end of dosing day, and the end of the experimental day were measured, the long diameter (A) and the short diameter (B) of the tumor were measured, and the volume of the tumor was calculated according to the ellipsoid volume formula: volume = A×B2/2. The tumor inhibition rate was calculated using this formula: tumor inhibition rate = [(the average volume of tumors in the control group - the average volume of tumors in the treatment group)/the average volume of tumors in the control group]×100%. At the end of the experiment, the mice were sacrificed by cervical dislocation, the tumor was removed, and the liver and spleen were removed and weighed. The above tissues were fixed in 10% neutral formaldehyde for subsequent experiments. Hematoxylin and eosin (HE) and immunohistochemical staining were used to observe the pathological tissue morphology and related indicators.
Statistical processing
Statistical data were expressed as mean±standard deviation. Statistical analysis was performed using SPSS 17.0 software. The cell-value-added rate comparison between groups was measured using one-way analysis of variance (ANOVA). Comparison between apoptotic groups was done using paired t-test and Western blotting with quantity one software analysis. The comparison between the means was performed by randomized block design analysis of variance and rank sum test. P<0.05 was considered statistically significant.
Results
Effects of combined autophagy inhibitors and apoptosis inhibitors on the proliferation inhibition of BEZ235
As shown in Figure 1, the concentration of BEZ235 in the range of 0.01–10 μM significantly inhibited the proliferation of K562 and KBM7R cells. The effect was more obvious with and the increase of dose. Compared with the BEZ235 group alone, BEZ235 combined with CQ (40 μM) or 3-MA (5 mM) increased the proliferation inhibitory activity of CML cells and decreased the IC50 at 48 h (P<0.05). In contrast, when Z-VAD-FMK (20 μM) was used in combination with BEZ235, the proliferation inhibitory activity of BEZ235 on CML cells was reduced, and the IC50 of 48 h was increased (P<0.05). Among them, the IC50 of K562 cells alone was 0.39±0.14 μM for BEZ235, the IC50 of BEZ235 combined with CQ group was 0.05±0.01 μM, and the IC50 of BEZ235 combined with 3-MA group was 0.06±0.02 μM; the IC50 for BEZ235 combined with Z-VAD-FMK group was 1.61±0.38 μM. The IC50 of KBM7R cells alone was 0.89±0.23 μM for BEZ235, the IC50 for BEZ235 combined with CQ group was 0.08±0.02 μM, and the IC50 for BEZ235 combined with 3-MA group was 0.11±0.03 μM. The IC50 for BEZ235 combined with Z-VAD-FMK group was 2.21±0.45 μM.
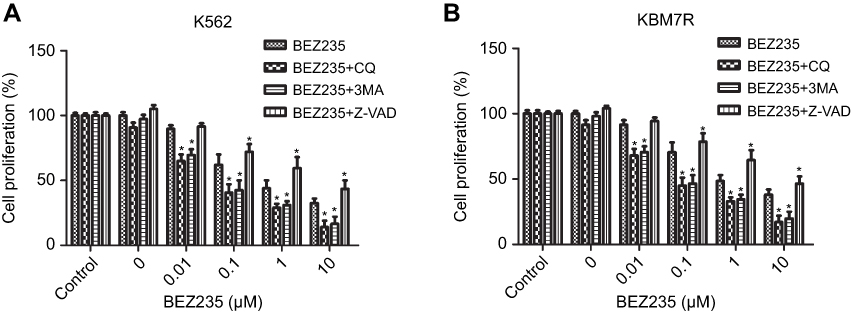 |
Figure 1 Effects of combined with autophagy inhibitor and apoptosis inhibitor on the proliferation of K562 or KBM7R cells. (A) K562 cells were treated with increasing concentrations of BEZ235 combined with autophagy inhibitor and apoptosis inhibitor and subjected to MTS assay. (B) KBM7R cells were treated with increasing concentrations of BEZ235 combined with autophagy inhibitor and apoptosis inhibitor and subjected to MTS assay. Mean ± SD. n=3. *P<0.05, compared with the same dose of BEZ235 group. |
Effect of BEZ235 on CML cell signaling pathway
As shown in Figure 2, the expression levels of P-AKT protein, and its downstream P-S6K protein decreased with the increase of BEZ235 concentration after different concentrations of BEZ235 were applied to the K562 and KBM7R cell lines for 12 h (P<0.05).
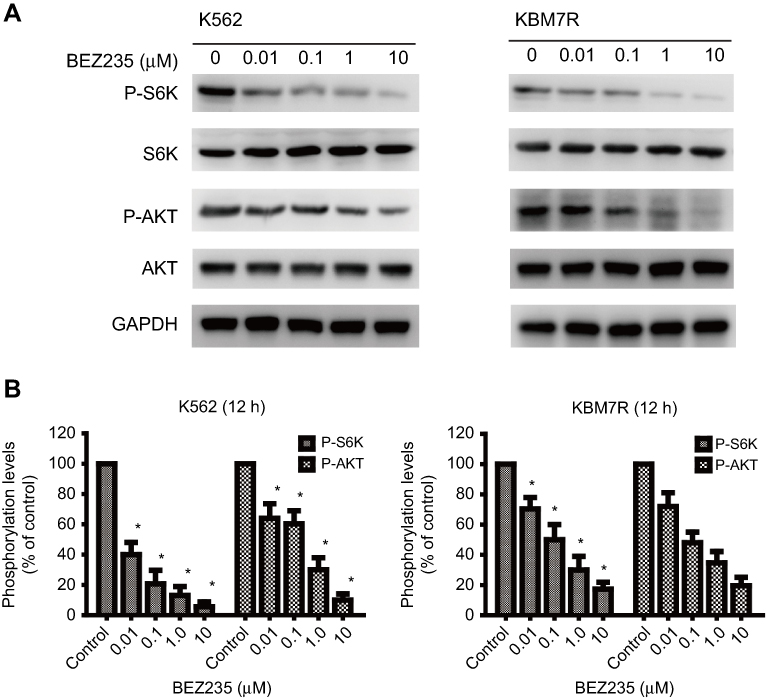 |
Figure 2 Dose dependent inhibition effect of BEZ235 on the phosphorylation levels of S6K and AKT (Ser473) in CML cells. (A) Results of Western blot; (B) Phosphorylationlevels of S6K, and AKT (Ser473) in cells treated with BEZ235 at concentrations of 0, 0.01, 0.1, 1.0, and 10 μM for 12 h. Mean ± SD. n=3. *P<0.05, compared with the control group. |
Expression of autophagy-related proteins
As shown in Figure 3, different concentrations of BEZ235 were administered to interfere with K562 and KBM7R cell lines for 24 h. With the increase of dose, the expression level of autophagy-related protein LC3II and LC3II/I ratio increased gradually (P<0.05). When 1 μM of BEZ235 was administered to K562 and KBM7R cell lines for 6, 12, 24, 48 h, the expression levels of LC3II and LC3II/I were gradually increased within 24 h in a time-dependent manner (P<0.05).
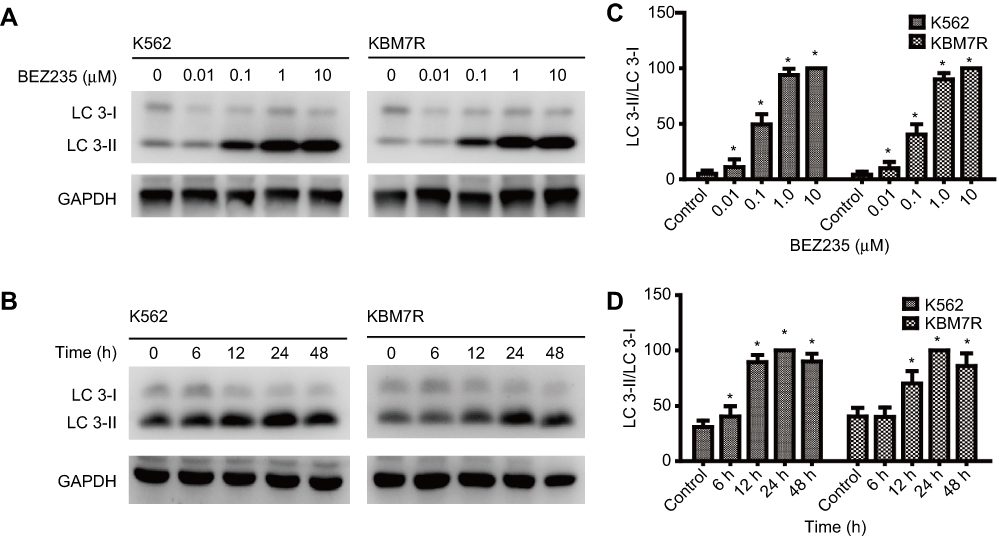 |
Figure 3 BEZ235 increases autophagy induction. (A) The cells were treated with BEZ235 at concentrations of 0, 0.01, 0.1, 1.0, and 10 μM for 24 h, and analyzed for LC3 expression by Western blot analysis. (B) The cells were treated with BEZ235 at concentrations of 1.0 μM for 0, 6, 12, 24, and 48 h, and analyzed for LC3 expression by Western blot analysis. (C) Quantification of LC3 protein expression of (A). (D) Quantification of LC3 protein expression of (B). Mean±SD. n=3. *P<0.05, compared with the control group. |
Transmission electron microscopy to detect autophagy in CML cells
The results of transmission electron microscopy showed that the autophagosomes in K562 cells and KBM7R cells increased gradually with time (P<0.05) within 24 h after treatment with BEZ235 (P<0.05). The effect was more obvious with the increase of dose (P<0.05). This confirms that BEZ235 does have an effect of promoting autophagy (see Figures 4 and 5).
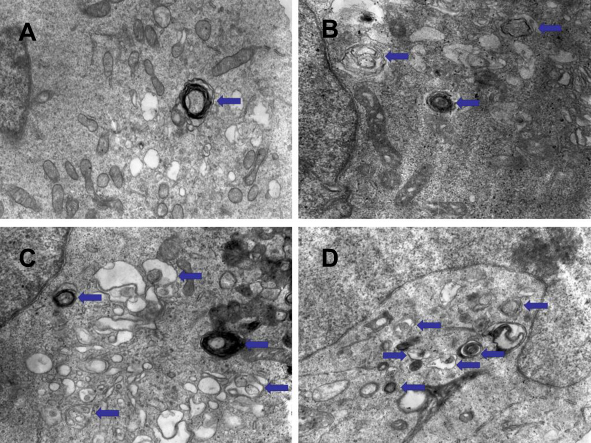 |
Figure 4 Electron microscopic results of BEZ235-induced autophagy in K562 cells (A panel is the control group, B panel is 1 μM for 12 h group, C panel is 1 μM for 24 h group, and D panel is for 5 μM for 24 h group, the arrows point to the autophogosomes). |
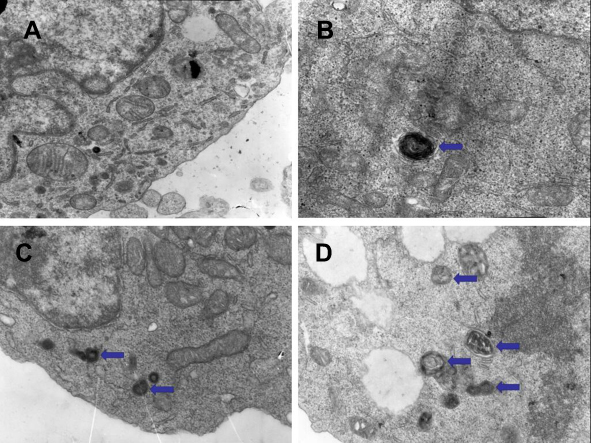 |
Figure 5 Electron microscopic results of BEZ235-induced autophagy in KBM7R cells (A panel is the control group, B panel is 1 μM for 12 h group, C panel is 1 μM for 24 h group, and D panel is for 5 μM for 24 h group, the arrows point to the autophogosomes). |
Autophagy detection by laser confocal microscopy
When autophagy is induced, both red and yellow fluorescent spots increase; when autophagy is inhibited, autophagosome production is reduced, both are reduced; when inhibition of autophagic flow leads to autophagosome degradation be blocked, yellow fluorescent spots increased, pure red fluorescent dots decrease or remain unchanged. In this way, it is possible to monitor whether the autophagic flow is unobstructed. After autophagy double-labeled adenovirus was transfected into K562 cells, GFP and mRFP expression were observed under laser confocal microscopy, suggesting that adenovirus transfection was successful. Compared with the control group, the mRFP red fluorescence and GFP green fluorescence of the BEZ235-treated group increased significantly, so the yellow fluorescent spots produced by the fusion of the 2 increased, and the effect was more obvious with the increase of the dose (P<0.05). Within the last 24 h, with the prolongation of time and the increase of dose, the simple mRFP red fluorescent bright spot of the BEZ235 treatment group was significantly enhanced compared with the yellow fluorescent bright spot (P<0.05) (see Figure 6). In the BEZ235 group 24 h after administration, the pure mRFP red fluorescence in combination with CQ (concentration 40 μM) was reduced compared with the BEZ235 single-use group, while the yellow fluorescence produced by mRFP fusion with GFP increased (P<0.05) (see Figure 7). Also attached is the WB map of the effect of BEZ235 alone or in combination with CQ on LC3 protein (see Figure 8).
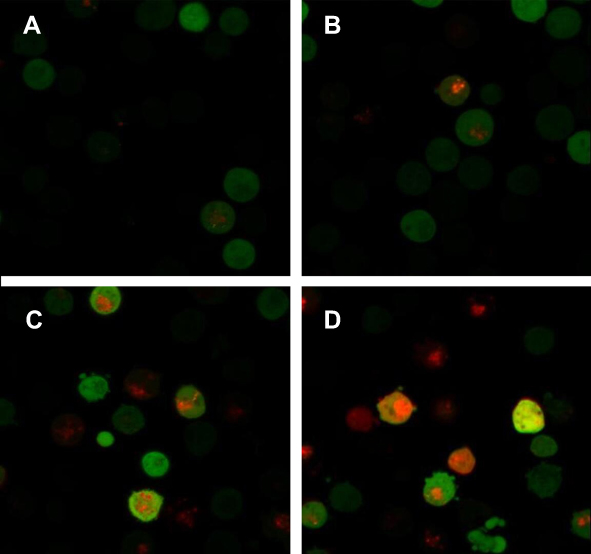 |
Figure 6 The autophagy induction of BEZ235 was observed under confocal microscopy (A panel is the control group, B panel is BEZ235 1 μM 12 h group, C panel is BEZ235 1 μM 24 h group, and D panel is BEZ235 5 μM 24 h group). |
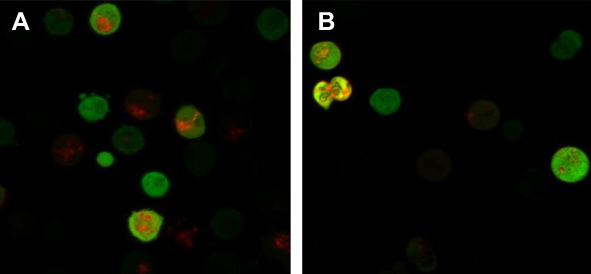 |
Figure 7 Confocal microscopy observation of CQ inhibition of autophagy induction of BEZ235 (A panel is BEZ235 1 μM 24 h group, B panel is CQ40 μM+BEZ235 1 μM 24 h group). |
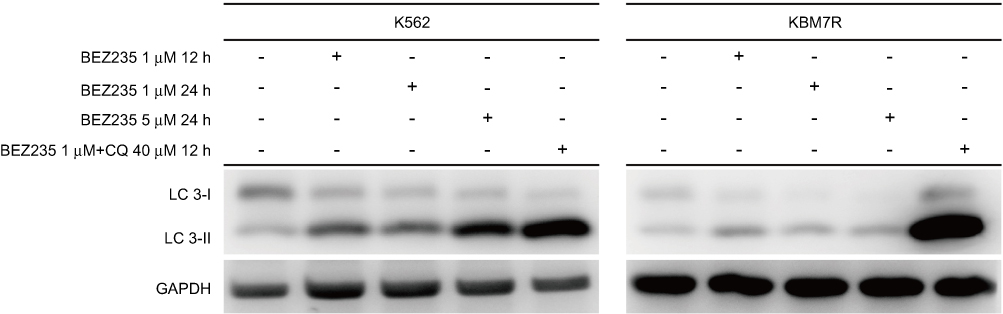 |
Figure 8 Effect of BEZ235 alone or in combination with CQ on LC3 protein. |
As can be seen from Figure 8, BEZ235 increased the expression level of LC3II and the ratio of LC3II/I in a time-and dose-dependent manner. When BEZ235 was combined with CQ, the expression level of LC3II and the ratio of LC3II/I increased more significantly. This result is consistent with the results of the confocal microscope.
Effect of BEZ235 alone and in combination with CQ, 3-MA, and Z-VAD-FMK on apoptosis of CML cells
The results of flow cytometry showed that the percentage of apoptosis of CML cells treated with BEZ235 was higher than that of the control group, and it was time- and dose-dependent (P<0.05). When Z-VAD-FMK was combined with BEZ235, the apoptosis of CML cells induced by the latter could be reduced (P<0.05), but the apoptotic rate was still higher than that of the blank control group (P<0.05). When CQ was combined with BEZ235, the apoptosis rate of CML cells induced by the latter was significantly increased (P<0.05). The combination of the 3-MA and BEZ235 group also increased the apoptosis induced by BEZ235, but there was no significant difference in the proportion of apoptosis between the 2 groups (P>0.05) (See Figures 9 and 10).
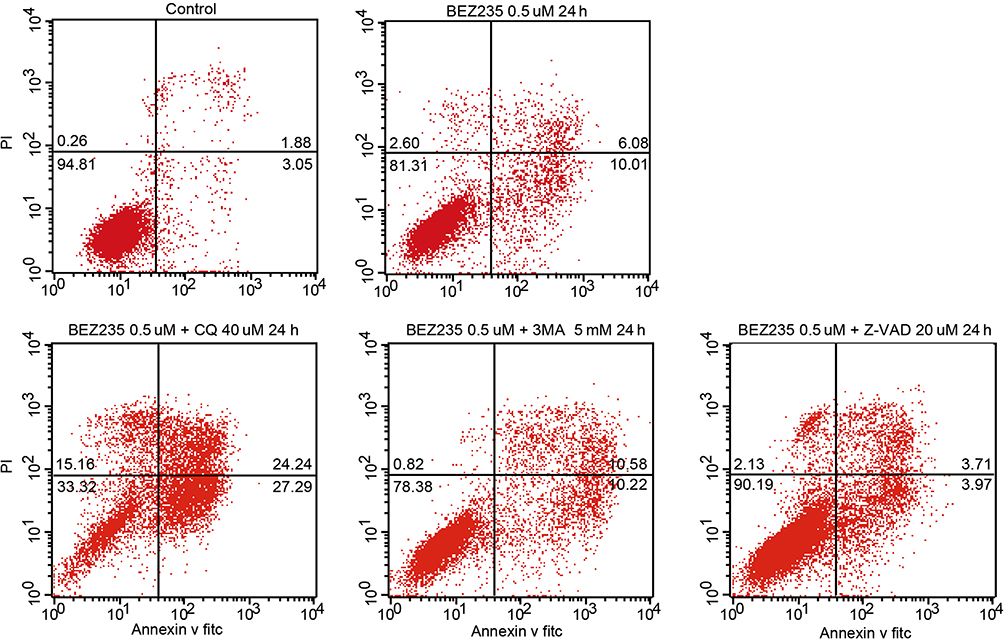 |
Figure 9 Effect of BEZ235 combined with autophagy inhibitor and apoptosis inhibitor on apoptosis of K562 cells. |
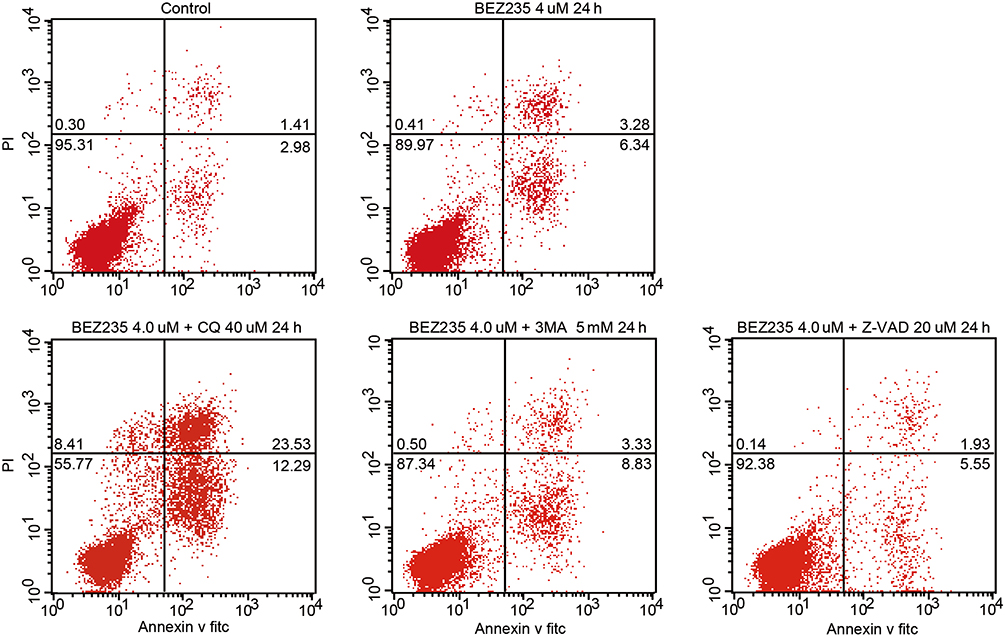 |
Figure 10 Effect of BEZ235 combined with autophagy inhibitor and apoptosis inhibitor on apoptosis of KBM7R cells. |
Additional WB maps of BEZ235 alone or in combination with CQ, 3-MA, and Z-VAD-FMK on Cleaved caspase-3 and LC3 proteins (see Figure 11).
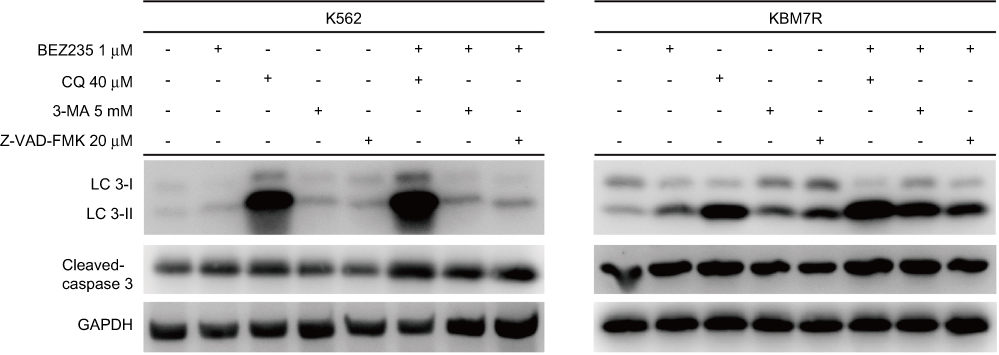 |
Figure 11 Effect of BEZ235 combined with autophagy and apoptosis inhibitor on the expression of autophagy and apoptosis-related proteins. |
As shown in Figure 11, BEZ235 alone could upregulate the expression of cleaved caspase-3 and LC3II, and increase the ratio of LC3II/I (P<0.05). When combined with Z-VAD-FMK, the expression level of cleaved caspase-3 was lower than that of BEZ235 alone (P<0.05). When combined with CQ, the expression levels of cleaved casepase-3 and LC3II, and the ratio of LC3II/I were higher than those of BEZ235 alone (P<0.05). When combined with 3-MA, the expression of cleaved caspase-3, LC3II, LC3II/I ratio and the BEZ235 group were not significantly different (P>0.05).
Animal experiment results
General condition of mice
After entering the sterile layer, the SCID mice were numbered, weighed, and caged, and then adapted to the new environment for one week. The daily observations included: mental state, activity, eating, drinking, hair, presence or absence of signs of infection, and defecation. Body weight and tumor volume (length and width) were recorded once every 5 days after inoculation and thereafter. On the 7th day after the inoculation, the tumor formation rate reached 100%, and the administration started on the 10th day after the inoculation. At this time, there was no significant difference in mental state, activity, diet, body weight, and tumor volume between the treatment group and the control group. In the first 10 days after the start of administration, the body weight and tumor volume of the treatment group were slightly lower than that of the control group, and there was no significant difference in general state of mind and diet. After that, the tumors in the control group increased significantly, the mental state gradually faded, and the activity decreased gradually. In the treatment group, the tumors were reduced or slowed, the mental state and diet were good, and the body weight gradually increased. By the end of the experiment, the weight of the control group was 28.50±2.62 g, and the weight of the treatment group was 28.41±2.23 g. The difference was not statistically significant (P>0.05) (Figures 12 and 13).
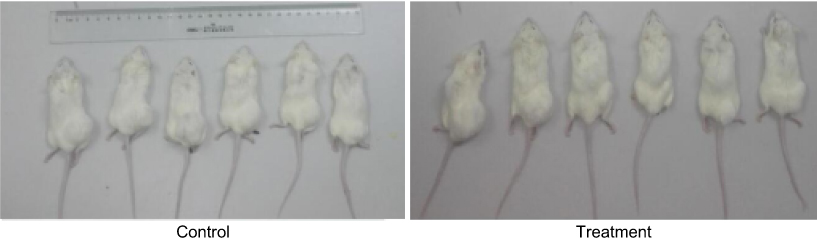 |
Figure 12 Control group and treatment group tumor-bearing mice 31 days after KBM7R cell inoculation. |
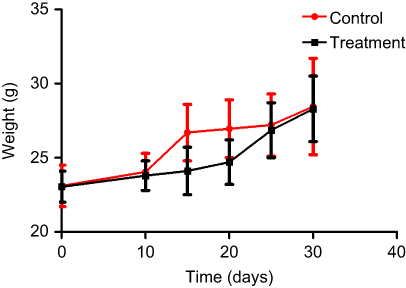 |
Figure 13 SCID mouse body weight curve. |
General situation of transplanted tumor tissue
The tumor tissue of the mice in the treatment group was easily peeled off, and the muscle infiltration was rare. The skin infiltration and subcutaneous muscle infiltration in the control group were obvious, and the tumor was difficult to peel off. Necrotic liquefaction can be seen in larger tumors. The tumor weight of the treatment group was 1.69±1.31 g, and the weight of the control group was 2.75±1.01 g. The difference was statistically significant (P<0.05). The tumor volume of the treatment group was 1720.2±1783.5 mm3, and the tumor volume of the control group was 2935.3±1662.4 mm3. The difference was statistically significant (P<0.05). The tumor inhibition rate was 89.57%, suggesting that BEZ235 can significantly inhibit the growth of subcutaneous xenografts of T315I mutant KBM7R cells in tumor-bearing mice (see Figures 14 and 15).
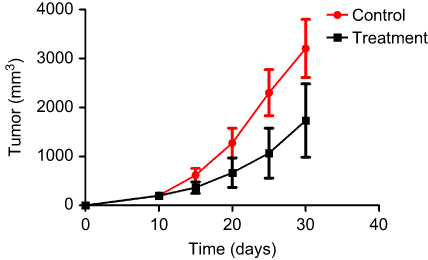 |
Figure 14 SCID mouse transplant tumor growth curve. |
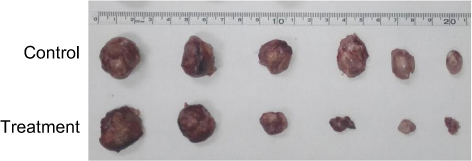 |
Figure 15 Tumors of the treatment group and the control group. |
HE staining of tumor in liver and spleen
Under the light microscope, the cells of the control group were compact, with many mitotic divisions, large tumor cell type, large cytoplasm of the nucleus, and the nucleus was on the cell side. The tumor cells grew actively and there was a small amount of necrotic tissue in the center of the tumor. While the treatment group tumor cells become smaller, mitotic figures are rare, hemorrhagic necrosis in the tumor tissue increased, and there is rare smooth muscle layer infiltration (Figure 16). There were no significant differences in the morphology of the liver and spleen between the 2 groups under light microscopy (Figure 17).
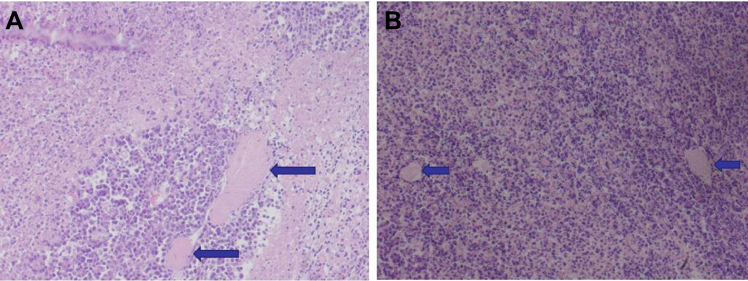 |
Figure 16 HE staining of the tumor (A is the control group, the long arrows point to muscle infiltration, B is the treatment group, the short arrows point to the ischemic necrosis, ×200). |
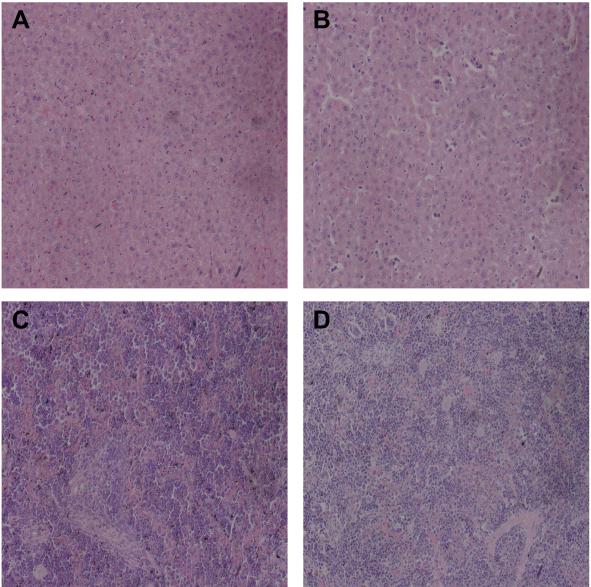 |
Figure 17 HE staining of mouse liver and spleen (A is the liver of the control group, B is the liver of the treatment group, C is the spleen of the control group, D is treatment group spleen, ×200). |
Immunohistochemistry results
In the present study, the immunohistochemical results of the pathological sections of the 2 groups of transplanted tumors showed strong positive expression of BCR-ABL, which was consistent with the pathological immunophenotype of human CML cells (Figure 18). In addition, we also detected the expression of PI3K/AKT/mTOR pathway-related proteins P-AKT, P-mTOR, P-S6K and apoptosis-related proteins BCL2 and caspase-3 downstream of BCR-ABL protein. P-AKT, P-mTOR, P-S6K, BCL2, caspase-3 and other proteins can be expressed in the cytoplasm and nucleus, according to the degree of positive expression from weak to strong. Histochemical staining can be light yellow, brownish yellow, or tan. P-AKT was moderately positive in the control group, while the positive rate of the treatment group was significantly lower (P<0.05) (Figure 18). P-mTOR was strongly positive in the control group, while the positive rate in the treatment group was decreased, and the difference was statistically significant (P<0.05) (Figure 18). P-S6K was strongly positive in the control group, while the positive rate of the treatment group was significantly lower, and the difference was statistically significant (P<0.05) (Figure 18). The apoptosis-related protein BCL2 was strongly positively expressed in the control group, while the positive rate of the treatment group was significantly lower, and the difference was statistically significant (Figure 19). Caspase-3 was moderately positive in the control group and strongly positive in the treatment group. The difference was statistically significant (P<0.05, P<0.05) (Figure 19). Immunohistochemistry showed that BEZ235 inhibited the phosphorylation of AKT, mTOR, and S6K in experimental mice, decreased the number of anti-apoptotic protein BCL2 and increased the expression of caspase-3. These results were consistent with the results of in vitro cell experiments. Therefore, the anti-tumor effect of BEZ235 may be related to PI3K/AKT/mTOR pathway blocking and promotion of apoptosis.
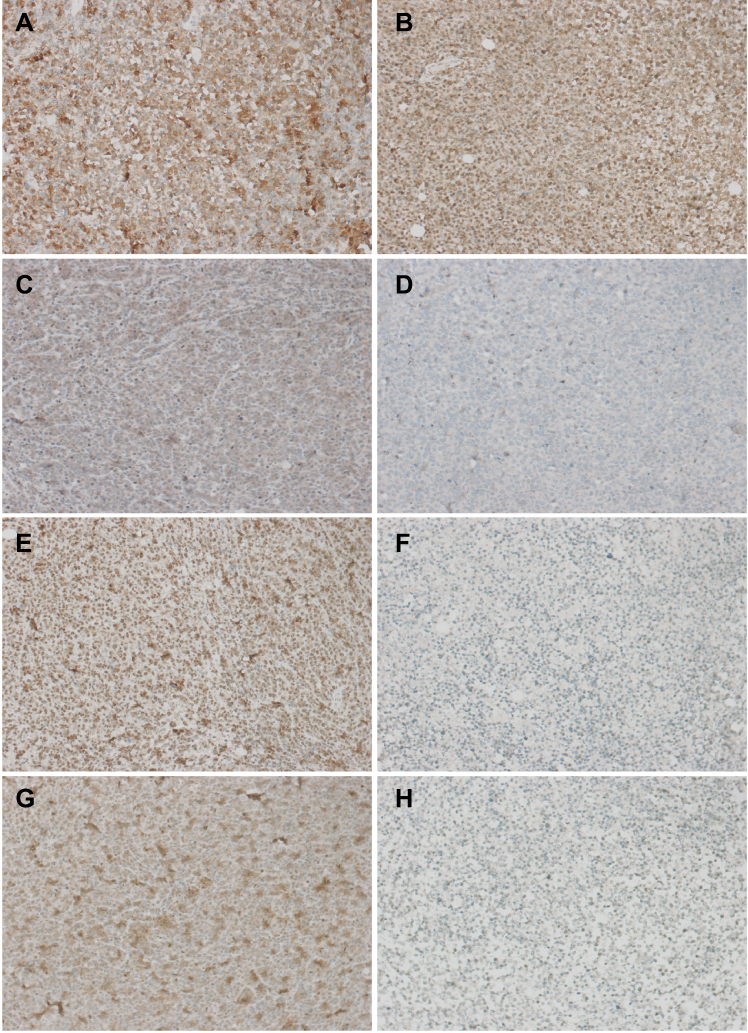 |
Figure 18 Immunohistochemical staining of tumor signaling pathway protein (A is the control group BCR-ABL, B is the experiment group, in which BCR-ABL was strongly positive; C was the control group, in which P-AKT was moderately positive; D was the treatment group, in which P-AKT was weakly positive; E was the control group, in which P-mTOR was strongly positive, and F was the treatment group in which P-mTOR was weakly positive; G was a control group, in which P-S6K was strongly positive, and H was a treatment group, in which P-S6K was weakly positive; ×200). |
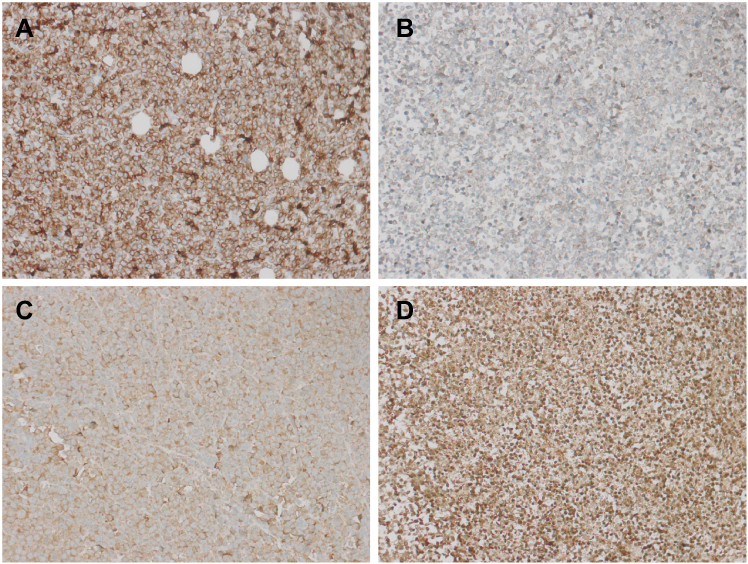 |
Figure 19 Immunohistochemical staining results of tumor apoptotic proteins (A is a control group BCL2, which is strongly positive; B was weakly positive in the treatment group BCL2; C was the control group caspase-3, which was moderately positive, and D was the treatment group caspase-3, which was strongly positive; ×200). |
Discussion
TKI resistance mechanisms in patients with CML include factors related to BCR-ABL and factors other than BCR-ABL. BCR-ABL related factors include new mutations in the BCR-ABL kinase domain,14 compound mutations involving T315I and P-loop mutations,15 and more research focuses on factors other than BCR-ABL. These factors include: high expression and resistance of Src family (such as Lyn kinase),16 epigenetic abnormalities,17 changes in telomerase activity,18 inhibition of apoptosis, induction of autophagy, external overexpression of the gene MDR1 and down-regulation of the inflow gene OCT1.19
We also reviewed some recent research advances. First, new developments in CML resistance mechanisms. Han SH studies have shown that activation of the autophagy regulatory axis of GCA-TRAF6-ULK1 is associated with imatinib resistance in CML.20 Mitchell R studies have shown that BCR-ABL-independent resistance in CML cells may be associated with activation of mTOR and activation of autophagy, and that combined with mTOR and autophagy inhibitors is expected to overcome BCR-ABL independent resistance.21 The study of Bedewy AML from the perspective of mRNA found that BMI1 mRNA expression is higher in the accelerated and blast phase of CML and is associated with shorter PFS.22 Multi-omics analysis of tyrosine kinase inhibitor-resistant K562 cells indicated that metabolic reprogramming promotes cell survival.23 Chandran RK study found that differential gene expression changes are associated with TKI resistance and disease progression in CML.24 Overexpression of c-MYC, down-regulation of ABCB1, BCL-2 and BAD, and complex interactions of several candidate genes such as C/EBPα/-β play an important role in the evolution and development of drug resistance in CML. In addition, other scholars explore the resistance and countermeasures of CML from the perspective of cytokines.25–27
Second, current research suggests that the degradation process of proteins, especially the degradation of apoptosis-related proteins, is associated with resistance to CML. R. Fahlman28 and others believe that the degradation of intracellular proteins is mainly mediated by the ubiquitin proteasome system (UPS). The first degradation signal was discovered and defined as the unstable N-terminal amino acid residue (or N-degron) of the protein substrate. The discovery of N-degrons produced a so-called N-terminal rule indicating that the half-life of the protein is determined by the nature of its N-terminal amino acid residue. The N-terminal regular pathway recognizes proteins containing N-terminally labile residues and mediates their polyubiquitination and subsequent degradation in the proteasome. Piatkov’s study found that the N-terminal regular pathway counteracts cell death by disrupting pro-apoptotic protein fragments, but caspase also inactivates components of the N-terminal regular pathway, indicating that there is a mutual inhibition between this pathway and the pro-apoptotic signal. Therefore, whether the N-terminal regular pathway also plays an anti-apoptotic role in TKI-resistant CML cells is worthy of study.29 Studies by Eldeeb et al have confirmed that the activity of pro-apoptotic protein fragments produced by most proteases can be inhibited by selective degradation of the N-terminal regular degradation pathway. Therefore, apoptosis of cancer cells is reduced and drug resistance occurs.30 Lyn is a non-receptor tyrosine kinase that plays an important role in normal cell growth, differentiation, survival, adhesion and migration. WU’s study found that Lyn protein may mediate imatinib resistance independent of BCR-ABL. Reduction of LYN expression by siRNA or inhibition of LYN activation by dasatinib promotes apoptosis in imatinib-resistant cells.31 Imatinib resistance of K562 cells caused by LyΔN expression can be reduced by N-terminal regular LynΔN degradation.32
In order to overcome TKI resistance, researchers have made various attempts and made some progress. Some scholars have used arsenic and interferon to treat drug-resistant CML,33 Some scholars believe that inhibition of autophagy can restore the sensitivity of drug-resistant tumors to chemotherapy, and even help eradicate drug-resistant cells in human leukemia, especially cancer stem cells.34,35 Other scholars have developed new drugs to overcome drug resistance,36 and some scholars have sought to overcome drug resistance from the perspective of gene therapy, such as silencing BCR-ABL gene by siRNA, making CML cells sensitive to tyrosine kinase inhibitor therapy,37 or using MicroRNA-221 to target STAT5 to make CML cells sensitive to imatinib.38 In addition, some scholars believe that due to the increase in surviving patients and the continuous improvement of transplantation effects, patients with drug-resistant CML may be treated with allogeneic stem cell transplantation.39
Our research is focused on finding ways to overcome TKI resistance in the downstream pathway of BCR-ABL, and found that the PI3K-mTOR dual-target inhibitor BEZ235 has good anti-CML activity, even effective for TKI-resistant cell lines.
As a dual-target inhibitor of PI3K and mTOR, BEZ235 affects CML cells by inhibiting the activation of the PI3K/AKT/mTOR pathway downstream of BCR-ABL.21,40 This study also demonstrates the decrease in the expression of P-AKT protein and its downstream P-S6K protein after BEZ235 intervention. The PI3K/AKT/mTOR pathway is closely related to cell proliferation, apoptosis, autophagy activity, and other cellular functions and is an important signal transduction pathway in cells. Current research suggests that PI3K and mTOR are key points in the autophagy signaling pathway.4,9 Among them, the pathway of type I PI3K and mTOR (especially mTOR complex 1, ie, mTORC1) inhibits the occurrence of autophagy. Type III PI3K promotes the occurrence of autophagy.41,42 As a dual-target inhibitor of type I PI3K and mTOR, BEZ235 may affect the autophagy activity of cells. Therefore, the focus of this study was on the effects of autophagy activity and autophagy activity on cell proliferation and apoptosis.
The results of MTS assay showed that BEZ235 inhibited the proliferation of CML cell line K562 and KBM7R cells. The autophagy inhibitors CQ and 3-MA increase the proliferation inhibitory activity of BEZ235 on CML cells. The apoptosis inhibitor Z-VAD-FMK can reduce the proliferation inhibitory activity of BEZ235 on CML cells. It is suggested that effectively inhibiting the autophagy activity induced by BEZ235 can help to enhance its proliferation inhibition.
In addition to proliferation inhibition, we found that BEZ235 also increased the expression level of autophagy-associated protein LC3II and increased the ratio of LC3II/I. This result suggests that BEZ235 may promote autophagy. However, although the number of autophagic vacuoles is directly proportional to the ratio of LC3-II or LC3-II/LC3-I, the increase in LC3-II, in addition to autophagy activation, can also result from inhibition of lysosomal function. Examples of this include accumulation of LC3-II, for example, when the enzyme activity in the lysosome is inhibited (such as CQ) or the fusion of autophagosomes with lysosomes is inhibited (such as vinblastine). To distinguish whether the increase in LC3-II is due to activation of autophagy or inhibition of degradation of autophagosomes,the concept of autophagic flow has been proposed by the academic community. Since autophagy is a continuous, multi-step process, the autophagic flow can better represent whether the process from autophagosome formation to autophagic substrate degradation is complete and smooth. Therefore, we know that when the autophagic flow is not smooth, it can affect the content of LC3-II and the ratio of LC3-II/LC3-I, so it is not easy to rely on increasing the content of LC3-II and the ratio of LC3-II/LC3-I to assert the enhancement of autophagy.43,44 For example, when CQ and BEZ235 were used in this experiment, the autophagy activity induced by BEZ235 was inhibited by an autophagy inhibitor CQ. However, the content of LC3-II and LC3-II/LC3-I in the 2-drug combination group at this time was higher than that of the BEZ235 group alone. To further observe the autophagy induction of BEZ235 and the autophagic flow, we used transmission electron microscopy(TEM), a gold standard for autophagy activity, to observe the autophagy induction of BEZ235 and used mRFP-GFP-LC3 adenovirus to study the real-time monitoring of autophagy/the autophagic flow.45
Scholars agree that the observation of autophagosomes under TEM is the gold standard for studying autophagy activity. Autophagy can be divided into early autophagosomes and late autophagosomes according to their morphology. The early autophagic corpuscle has a bilayer membrane structure, which is its most characteristic manifestation. In addition, early autophagy can also contain intact mitochondria, endoplasmic reticulum, ribosomes, and other organelles or cytoplasm. Since the inner membrane is degraded by lysosomal enzymes, the late autophagosomes have a monolayer membrane structure, which can contain organelles of different degradation stages because the electron density appears to be an amorphous state, which is relatively difficult to observe. The results of transmission electron microscopy showed that the autophagosomes in K562 cells and KBM7R cells gradually increased with the prolongation of time and the increasing of dose within 24 h after BEZ235 treatment. This result confirmed that BEZ235 does have an effect of promoting autophagy. To further observe the BEZ235-induced autophagic flow, we introduced mRFP-GFP-LC3 tandem fluorescent protein adenovirus, and used autophagy inhibitor CQ to block the autophagic flow for research.
mRFP-GFP-LC3 tandem fluorescent protein adenovirus can cause LC3 to carry both red fluorescence of mRFP and green fluorescence of GFP. When autophagy activity is enhanced, the accumulation of LC3 will occur on autophagy precursor and autophagosome membrane. It is a yellow fluorescent spot by the red-green synthesis function of laser confocal microscopy. When autophagosomes and lysosomes fuse to form autophagy lysosomes, it is easy to cause GFP green fluorescence quenching under acidic conditions, but mRFP red fluorescent is acid-resistant and not easy to degrade, so autophagy lysosomes appear to have red highlights. Therefore, LC3 in the whole process of autophagic flow can be marked by mRFP red fluorescence, while the decrease of late GFP represents the formation of autophagosomes. That is, a simple red fluorescent dot can be used to represent autophagosome lysosome, and a yellow fluorescence generated by red-green fusion represents an autophagosome. When autophagy is induced, both red and yellow fluorescent spots increase; when autophagy is inhibited, autophagosome production is reduced and both types of fluorescence are reduced. When autophagic flow is inhibited, autophagosome degradation is decreased, yellow fluorescent spots increased, and the simple red fluorescent dot decreases or does not change. In this way, it is possible to monitor whether the autophagic flow is unobstructed. Whether the autophagic flow is smooth can affect the physiological state of the cells. (It can also be understood that when both GFP and mRFP increase, suggesting that autophagy activity is enhanced, and GFP is relatively weakened over time and mRFP is enhanced, the unobstructed autophagic flow is further explained.)46
The results of laser confocal microscopy in this study showed that the yellow fluorescence produced by the fusion of mRFP and GFP in the BEZ235 treatment group was significantly increased in a dose-dependent manner compared with the control group. Within 24 h after administration, the simple mRFP red fluorescent bright spot of the BEZ235-treated group was significantly enhanced compared to the yellow fluorescent bright spot over time. When BEZ235 was combined with CQ, the simple mRFP red fluorescence was reduced compared with the BEZ235 single-use group, and the yellow fluorescence generated by the fusion of mRFP and GFP increased. This group of results indicates that BEZ235 can promote the autophagy activity of CML cells and facilitate the flow of autophagy. When CQ blocks the fusion of autophagosomes and lysosomes, resulting in poor autophagic flow, it can be seen that the yellow fluorescence generated by red-green fluorescence fusion is significantly enhanced. This result further confirms the autophagy induction of BEZ235.
After confirming the autophagy-inducing activity of BEZ235, in order to further understand the relationship between induced autophagy and cell death, autophagy inhibitor 3-MA, autophagy inhibitor CQ and apoptosis inhibitor Z-VAD-FMK were used in combination with BEZ235, respectively, and their ratio of induced apoptosis was compared to BEZ235 alone. The study found that there was no significant difference in the apoptosis rate of CML cells induced by autophagy inhibitor 3-MA combined with the BEZ235 group, and there was no significant expression of autophagy-related protein and expression of apoptosis-related protein. The other autophagy inhibitor, CQ, can significantly increase the proportion of apoptosis induced by BEZ235, promote the expression of apoptosis-related proteins, and also increase the expression of autophagy-related protein LC3II. We know that the type I PI3K and mTOR pathways inhibit the occurrence of autophagy. Type III PI3K promotes the occurrence of autophagy. BEZ235, a dual-target inhibitor of type I PI3K and mTOR, promotes autophagy activity derived from the inhibition of the type I PI3K/AKT/mTOR pathway, especially mTOR, without affecting the activity of type III PI3K. While 3-MA can slightly inhibit type I PI3K, it mainly inhibits autophagy by inhibiting the activity of type III PI3K. Therefore, when we combined 3-MA, we did not significantly enhance or attenuate BEZ235-induced autophagy activity. This explains why there is no significant difference in the proportion of apoptosis between the BEZ235-linked 3-MA group and the single-use BEZ235 group. Unlike 3-MA, CQ acts on the downstream part of autophagy by blocking the degradation of autophagosomes, resulting in poor autophagic flow and inhibition of autophagy, thus inhibiting BEZ235-induced autophagy. However, because its mechanism of action is not to inhibit the induction of autophagy, but to inhibit the degradation of autophagosomes, it does not affect the activation of LC3II, but inhibits the degradation of LC3II, which inevitably leads to the accumulation of LC3II in cells. This also explains how CQ inhibits autophagy, but it shows an increase in LC3II and LC3II/I. Apoptosis increased when CQ inhibited BEZ235-induced autophagy. This result suggests that BEZ235-induced autophagy may play a role in protecting cells and reducing apoptosis, rather than inducing autophagic death. The combination of CQ and BEZ235 can enhance the apoptosis of CML cells induced by the latter, which helps to improve the therapeutic effect. There is controversy regarding whether autophagy plays a protective role or induces autophagic death in the tumor among the academic community. Clarke P et al.47,48 support the discussion of autophagic death. Shen et al.49–51 prefer protective autophagy, for which our findings are consistent.
In addition, when Z-VAD-FMK was combined to inhibit the apoptosis induced by the caspase pathway in BEZ235, the percentage of apoptosis was significantly lower, but it was still higher than that of the control group. It seems that BEZ235 may pass other pathways than the caspase pathway to induce the apoptosis of CML cells,which warrants further study.
In the animal test, we found that after the start of administration of BEZ235, the body weight of the treatment group was once lower than that of the control group, but the mice had no hair loss, and the mental state, activity ability, and diet were generally good. Afterward, the volume of the tumor in the control group increased significantly. The mice in this group showed signs of wilting, disappointment, and less food consumption. The growth of the tumor in the treatment group was slow, the body weight continued to increase, and the activity, mental state, and diet were good. There was no significant difference in body weight between the 2 groups by the end of the experiment. However, because the tumor weight of the control group was significantly higher than that of the treatment group, the actual body weight treatment group of the mice was larger than the control group except the weight of the tumor. In addition, there was no significant difference in liver and spleen HE staining between the 2 groups, suggesting that there was no obvious liver and spleen damage in the treatment group. The above results indicate that BEZ235 is a relatively safe therapeutic amount at 50 mg/kg/d, the subjective discomfort is small, and there is no obvious toxic side effect on SCID mice.
There was no significant difference in tumor size between the 2 groups before the start of BEZ235 administration 10 days after inoculation. However, after the start of administration in the treatment group, the growth of the tumor in this group was slow or showed no significant increase, while the volume of the tumor in the control group increased rapidly. After statistical analysis, by the end of the experiment, the average volume of the tumors in the 2 groups and the average weight of the harvested tumors were significantly different. In addition, the skin infiltration and subcutaneous muscle infiltration of the tumor tissue of the control group were obvious, and the tumor was difficult to peel off. The tumor tissue of the treated group was easier to peel off, and the muscle infiltration was rare. The HE staining of the tumor tissue showed that the control group had more mitotic figures and tumor cell growth, and smooth muscle infiltration was more common. The mitotic figures in the treatment group are rare, the hemorrhage and necrosis of the tumor tissue is increased, and the smooth muscle infiltration is rare. These results indicate that BEZ235 can effectively inhibit the growth of CML cell xenografts and reduce the invasiveness of tumors in vivo, which is consistent with the previously reported BEZ235 in vivo study.12,52,53
To further clarify the effect of BEZ235 on KBM7R cell xenografts, the expression of P-AKT, P-mTOR, P-S6K, BCL2, and caspase-3 was detected in this experiment. The results showed that BEZ235 can inhibit the phosphorylation of AKT, mTOR, and S6K, decrease the expression of BCL2, and increase the expression of caspase-3. These results are consistent with the results of in vitro cell experiments. This indicates that BEZ235 also acts by downregulating the acidification level of the PI3K/Akt/mT0R pathway protein, thereby inhibiting the activity of this pathway, even for the IM-resistant T315I mutant KBM7R cell line54 and helping overcome TKI resistance. Moreover, HE staining results showed that hemorrhagic necrosis increased in the tumor tissue of the treatment group, suggesting that BEZ235 can also promote the death of CML cell xenografts in vivo, which may be related to the increased apoptosis induced by the decrease of BCL2 and the increase of caspase-3, consistent with the literature report.52,55
In this part of the study, we successfully constructed a mouse model of IM-resistant T315I mutant KBM7R cells in human chronic myeloid leukemia, further indicating that BEZ235 also inhibits CML cell proliferation, reduction of infiltration, induction of apoptosis, and other effects by inhibiting the PI3K/Akt/mT0R pathway in vivo. However, due to the limited number of experimental animals in this study, the indicators of drug safety are insufficient for observation. It is necessary to increase the sample size, increase the different dose groups, and conduct careful dose-safety evaluation.
Conclusion
BEZ235 can inhibit the proliferation of CML cells, induce apoptosis, and enhance autophagy activity. It induces protective autophagy. The combination of CQ can enhance the apoptosis and proliferation inhibition of CML cells induced by BEZ235.
Acknowledgments
The research was supported by Fujian Natural Science Foundation Program “Study on the effect of BCR-ABL downstream channel inhibitor on chronic myeloid leukemia cells and its resistance to TKI”, state contract No. 2016J01609.
Author contributions
All authors contributed toward writing, data analysis, discussion, drafting and revising the paper, gave final approval of the version to be published and agree to be accountable for all aspects of the work. All authors read and approved the final manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Ren R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat Rev Cancer. 2005;5(3):172–183.
2. Holyoake TL. Recent advances in the molecular and cellular biology of chronic myelogenous leukaemia: lessons to be learned from the laboratory. Br J Haematol. 2001;113(1):11–23.
3. Vakil E, Tefferi A. BCR-ABL1–negative myeloproliferative neoplasms: a review of molecular biology, diagnosis, and treatment. Clin Lymphoma Myeloma Leuk. 2011;11(Suppl 1):S37–S45.
4. Quintas-Cardama A, Cortes J. Molecular biology of bcr-abl1-positive chronic myelogenous leukemia. Blood. 2009;113(8):1619–1630.
5. Apperley JF. Chronic myelogenous leukaemia. Lancet. 2015;385(9976):1447–1459.
6. Apperley JF, Part I. mechanisms of resistance to imatinib in chronic myelogenous leukaemia. Lancet Oncol. 2007;8(11):1018–1029.
7. Konig H, Holyoake TL, Bhatia R. Effective and selective inhibition of chronic myelogenous leukemia primitive hematopoietic progenitors by the dual Src/Abl kinase inhibitor SKI-606. Blood. 2008;111(4):2329–2338.
8. Xu XH, Gan YC, Xu GB, et al. Tetrandrine citrate eliminates imatinib-resistant chronic myelogenous leukemia cells in vitro and in vivo by inhibiting Bcr-Abl/beta-catenin axis. J Zhejiang Univ Sci B. 2012;13(11):867–874.
9. Ahmed W, Van Etten RA. Signal transduction in the chronic leukemias: implications for targeted therapies. Curr Hematol Malig Rep. 2013;8(1):71–80.
10. Mak DH, Schober WD, Chen W, et al. Triptolide induces cell death independent of cellular responses to imatinib in blast crisis chronic myelogenous leukemia cells including quiescent CD34+ primitive progenitor cells. Mol Cancer Ther. 2009;8(9):2509–2516.
11. Kuger S, Corek E, Polat B, et al. Novel PI3K and mTOR inhibitor BEZ235 radiosensitizes breast cancer cell lines under normoxic and hypoxic conditions. Breast Cancer (Auckl). 2014;8:39–49.
12. Wang H, Zhang L, Yang X, et al. PUMA mediates the combinational therapy of 5-FU and BEZ235 in colon cancer. Oncotarget. 2015;6(16):14385–14398.
13. Hall CP, Reynolds CP, Kang MH. Modulation of glucocorticoid resistance in pediatric T-cell acute lymphoblastic leukemia by increasing BIM expression with the PI3K/mTOR inhibitor BEZ235. Clin Cancer Res. 2016;22(3):621–632.
14. Chandrasekhar C, Kumar PS, Sarma PVGK. Novel mutations in the kinase domain of BCR-ABL gene causing imatinib resistance in chronic myeloid leukemia patients. Sci Rep. 2019;9(1):2412.
15. Kang KH, Kim SH, Choi SY, et al. Compound mutations involving T315I and P-loop mutations are the major components of multiple mutations detected in tyrosine kinase inhibitor resistant chronic myeloid leukemia. Leuk Res. 2019;76:87–93.
16. Wu J, Meng F, Lu H, et al. Lyn regulates BCR-ABL and Gab2 tyrosine phosphorylation and c-Cbl protein stability in imatinib-resistant chronic myelogenous leukemia cells. Blood. 2008;111(7):3821–3829.
17. Elias MH, Baba AA, Husin A, et al. HOXA4 gene promoter hypermethylation as an epigenetic mechanism mediating resistance to imatinib mesylate in chronic myeloid leukemia patients. Biomed Res Int. 2013;1:129715.
18. Uziel O, Fenig E, Nordenberg J, et al. Imatinib mesylate (gleevec) downregulates telomerase activity and inhibits proliferation in telomerase-expressing cell lines. Br J Cancer. 2005;92(10):1881.
19. Hekmatshoar Y, Ozkan T, Altinok BG, et al. Characterization of imatinib-resistant K562 cell line displaying resistance mechanisms. Cell Mol Biol (noisy-le-grand). 2018;64(6):23–30.
20. Han SH, Korm S, Han YG, et al. GCA links TRAF6-ULK1-dependent autophagy activation in resistant chronic myeloid leukemia. Autophagy. 2019;1–15.
21. Mitchell R, Hopcroft LE, Baquero P, et al. Targeting BCR-ABL-independent TKI resistance in chronic myeloid leukemia by mTOR and autophagy inhibition. JNCI. 2017;110(5):467–478.
22. Bedewy AM, Elmaghraby SM, Kandil NS. ABCB1 and BMI1 mRNA expression in patients with chronic myeloid leukemia: impact on imatinib efficacy. Blood Res. 2019;54(1):57–62.
23. Noel BM, Ouellette SB, Marholz L, et al. Multi-omic profiling of TKI resistant K562 cells suggests metabolic reprogramming to promote cell survival. J Proteome Res. 2019;18(4):1842–1856.
24. Chandran RK, Geetha N, Sakthivel KM, et al. Differential gene expression changes and their implication on the disease progression in patients with chronic myeloid leukemia. Blood Cells Mol Dis. 2019;77:51–60.
25. Nievergall E, Ramshaw HS, Yong AS, et al. Monoclonal antibody targeting of IL-3 receptor α with CSL362 effectively depletes CML progenitor and stem cells. Blood. 2014;123(8):1218–1228.
26. Blank U, Karlsson S. TGF-β signaling in the control of hematopoietic stem cells. Blood. 2015;125(23):3542–3550.
27. Madapura HS, Nagy N, Ujvari D, et al. Interferon γ is a STAT1-dependent direct inducer of BCL6 expression in imatinib-treated chronic myeloid leukemia cells. Oncogene. 2017;36(32):4619.
28. Eldeeb M, Fahlman R. The-N-end rule: the beginning determines the end. Protein Pept Lett. 2016;23(4):343–348.
29. Piatkov KI, Brower CS, Varshavsky A. The N-end rule pathway counteracts cell death by destroying proapoptotic protein fragments. Proc Natl Acad Sci. 2012;109(27):E1839–E1847.
30. Eldeeb M, Fahlman R, Esmaili M, et al. Regulating apoptosis by degradation: the N-end rule-mediated regulation of apoptotic proteolytic fragments in mammalian cells. Int J Mol Sci. 2018;19(11):3414.
31. Wu J, Meng F, Kong LY, et al. Association between imatinib-resistant BCR-ABL mutation-negative leukemia and persistent activation of LYN kinase. JNCI. 2008;100(13):926–939.
32. Eldeeb MA, Fahlman RP. The anti-apoptotic form of tyrosine kinase Lyn that is generated by proteolysis is degraded by the N-end rule pathway. Oncotarget. 2014;5(9):2714.
33. El Eit R, Itani AR, Nassar F, et al. Antitumor efficacy of arsenic/interferon in preclinical models of chronic myeloid leukemia resistant to tyrosine kinase inhibitors. Cancer. 2019; 125:2818–2828.
34. Jia X, Zheng Y, Guo Y, et al. Sodium butyrate and panobinostat induce apoptosis of chronic myeloid leukemia cells via multiple pathways. Mol Genet Genomic Med. 2019;7:e613.
35. Rothe K, Porter V, Jiang X. Current outlook on autophagy in human leukemia: foe in cancer stem cells and drug resistance, friend in new therapeutic interventions. Int J Mol Sci. 2019;20(3):461.
36. Arken N. Schizandrol A reverses multidrug resistance in resistant chronic myeloid leukemia cells K562/A02. Cell Mol Biol (noisy-le-grand). 2019;65(1):78–83.
37. KC R, Thapa B, Ubeda A, Jiang X, Uludağ H. BCR-Abl silencing by short interfering RNA: a potent approach to sensitize chronic myeloid leukemia cells to tyrosine kinase inhibitor therapy. Stem Cells Dev. 2019;28(11):734–744.
38. Jiang X, Cheng Y, Hu C, et al. MicroRNA-221 sensitizes chronic myeloid leukemia cells to imatinib by targeting STAT5. Leuk Lymphoma. 2019;60(7):1709–1720.
39. Craddock CF. We do still transplant CML, don’t we? Hematology Am Soc Hematol Educ Program. 2018;2018(1):177–184.
40. Xin P, Li C, Zhu X, et al. Efficacy of the dual PI3K and mTOR inhibitor NVP-BEZ235 in combination with imatinib mesylate against chronic myelogenous leukemia cell lines. Drug Des Devel Ther. 2017;11:1115–1126.
41. Wu YT, Tan HL, Huang Q, et al. Activation of the PI3K-Akt-mTOR signaling pathway promotes necrotic cell death via suppression of autophagy. Autophagy. 2009;5(6):824–834.
42. Qin L, Wang Z, Tao L, et al. ER stress negatively regulates AKT/TSC/mTOR pathway to enhance autophagy. Autophagy. 2010;6(2):239–247.
43. Tanida I, Minematsu-Ikeguchi N, Ueno T, et al. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy. 2005;1(2):84–91.
44. Ju JS, Varadhachary AS, Miller SE, et al. Quantitation of “autophagic flux” in mature skeletal muscle. Autophagy. 2010;6(7):929–935.
45. Klionsky DJ, Abdalla FC, Abeliovich H, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8(4):445–544.
46. Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12(1):1–222.
47. Liu TJ, Koul D, LaFortune T, et al. BEZ235, a novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor, elicits multifaceted antitumor activities in human gliomas. Mol Cancer Ther. 2009;8(8):2204–2210.
48. Clarke PGH, Puyal J. Autophagic cell death exists. Autophagy. 2012;8(6):867–869.
49. Cerniglia GJ, Karar J, Tyagi S, et al. Inhibition of autophagy as a strategy to augment radiosensitization by the dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor BEZ235. Mol Pharmacol. 2012;82(6):1230–1240.
50. Shen SS, Kepp O, Kroemer G. The end of autophagic cell death? Autophagy. 2012;8(1):1–3.
51. Ji YH, Di WY, Yang QH, et al. Inhibition of autophagy increases proliferation inhibition and apoptosis induced by the PI3K/mTOR inhibitor BEZ235 in breast cancer cells. Clin Lab. 2015;61(8):1043–1051.
52. Yu Z, Xie G, Zhou G, et al. BEZ235, a novel dual PI3K-mTOR inhibitor displays anti-glioma activity and reduces chemoresistance to temozolomide in human glioma cells. Cancer Lett. 2015;367(1):58–68.
53. Schrauwen S, Depreeuw J, Coenegrachts L, et al. Dual blockade of PI3K/AKT/mTOR (BEZ235) and Ras/Raf/MEK (AZD6244) pathways synergistically inhibit growth of primary endometrioid endometrial carcinoma cultures, whereas BEZ235 reduces tumor growth in the corresponding xenograft models. Gynecol Oncol. 2015;138(1):165–173.
54. Ribback S, Cigliano A, Kroeger N, et al. PI3K/AKT/mTOR pathway plays a major pathogenetic role in glycogen accumulation and tumor development in renal distal tubules of rats and men. Oncotarget. 2015;6(15):13036–13048.
55. Fraveto A, Cardinale V, Bragazzi MC, et al. Sensitivity of human intrahepatic cholangiocarcinoma subtypes to chemotherapeutics and molecular targeted agents: a study on primary cell cultures. PLoS One. 2015;10(11):e0142124.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.