")
Back to Journals » International Journal of Nanomedicine » Volume 16
Prostate-Specific Membrane Antigen and Esterase Dual Responsive Camptothecin–Oligopeptide Self-Assembled Nanoparticles for Efficient Anticancer Drug Delivery
Authors Xu B, Yan M, Zhou F , Cai D, Guo W, Jia X, Liu R, Ma T, Li T, Gao F, Wang P, Lei H
Received 28 July 2021
Accepted for publication 19 November 2021
Published 3 December 2021 Volume 2021:16 Pages 7959—7974
DOI https://doi.org/10.2147/IJN.S331060
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Dongwoo Khang
Bing Xu, Mengmeng Yan, Fei Zhou, Desheng Cai, Wenbo Guo, Xiaohui Jia, Runping Liu, Tao Ma, Tong Li, Feng Gao, Penglong Wang, Haimin Lei
School of Chinese Pharmacy, Beijing University of Chinese Medicine, Beijing, 102488, People’s Republic of China
Correspondence: Penglong Wang; Haimin Lei Email [email protected]; [email protected]
Background: The clinical utility of camptothecin (CPT) is restricted by poor aqueous solubility, high lipophilicity, active lactone ring instability, and off-targeted toxicities. We report here a prostate-specific membrane antigen (PSMA) and esterase dual responsive self-assembled nanoparticles (CPT-WT-H NPs) for highly efficient CPT delivery and effective cancer therapy.
Methods and Results: In this study, smart self-assembled nanoparticles CPT-WT-H NPs were elaborately designed and synthesized by combing hydrophobic CPT with hydrophilic PSMA-responsive penta-peptide via a cleavable ester bond. This dual responsive nanoparticle with negatively charged surface first respond to the extracellular PSMA and then to the intracellular esterase, achieving a programmable release of CPT at the tumor site and producing the byproducts of biocompatible glutamic acid and aspartic acid. Our data demonstrated that CPT-WT-H NPs exhibited greatly improved water solubility and stability. Results from MTT and flow cytometry showed CPT-WT-H NPs exhibited significantly higher cytotoxicity as well as apoptosis-inducing activity against PSMA-expressing LNCaP-FGC cells than the non-PSMA-expressing cancer cells, showing excellent cytotoxic selectivity. Moreover, the unique nanostructure provided the efficient transportation of CPT to tumor site, which resulted in the effective inhibition of tumor growth and low systemic toxicity in vivo.
Conclusion: CPT-WT-H NPs exhibited excellent in vitro PSMA-response ability and in vivo antitumor activity and safety, holding the promise to become a new and potent anticancer drug. The current research presents a promising strategy for efficient drug delivery.
Keywords: CPT prodrug, dual responsive, sequential drug release, prostate-specific membrane antigen, self-assembly
Introduction
Conventional chemotherapy is still a preferential cancer therapeutic strategy because of its high medical efficiency.1–3 As one of the DNA topoisomerases I inhibitors, camptothecin (CPT) has demonstrated remarkable antitumor properties against a variety of cancers in preclinical studies, but frequently failed in clinical uses due to its poor aqueous solubility, high lipophilicity, active lactone ring instability, and off-targeted toxicities.4–7 Great efforts have been devoted to increasing its tumor targeting and lowing the systemic toxicity by means of nano chemistry, their large-scale clinical application is still limited due to several major drawbacks, including low drug loading capacity (typically less than 10%), premature drug leakage, potential long-term toxicity of carrier materials, poor stability in vivo, and lack of active targeting.8–10
The self-assembling prodrugs with active targeting ability are emerging as a potential strategy for efficient chemo-therapeutic agents’ delivery due to their superior targeting ability, well-defined chemical structures, high drug loading capacities, and high stability.11–18 This drug self-delivery systems could not only take advantage of the enhanced permeability and retention (EPR) effect and the active targeting effect but also avoid the significant toxicity of drug carriers, showing excellent biocompatibility, safety and stability.19–21 Meanwhile previous researches of CPT derivative SN-38 (7-ethyl-10-hydroxycamptothecin) polymeric prodrugs confirmed the self-deliverable nano therapy was feasible, which maintained pharmacological activity while substantially alleviated the drug toxicity. The SN-38 polymeric prodrug nanoparticles exhibited higher stability and prolonged circulation, improved antitumor efficacy and lower toxicity compared to irinotecan (CPT-11), an approved drug for clinical applications.22–25 Prostate-specific membrane antigen (PSMA), a 750-amino acid type II membrane glycoprotein, has attracted significant research interests due to its unique glutamate carboxypeptidase II (GCPII) activity and selective expression in the neo vasculature of nearly all types of solid tumors.26–29 The expression level of PSMA is positively correlated with the tumor grade, pathological stage and tumor recurrence of prostate cancer.30,31 PSMA has been used as a biomarker for the cancer diagnosis as well as a therapeutic target for cancer treatments.28,29,32–36 Meanwhile, previous researches demonstrated that the PSMA-activated prodrug strategy produced a sufficient therapeutic index and excellent tumor targeting, which could be used to treat all solid tumors.29,37,38
The surface charges of the nanoparticles exert a great impact on their blood circulation, phagocytosis and nuclear localization functions.39–42 It is reported that the positive charge facilitated the binding of nanoparticles to the cell membrane and the uptake of nanoparticles by cells due to the electrostatic attraction with the anionic cell membrane. However, the positive charge might also strengthen the nonspecific binding of nanoparticles to the luminal surface of vascular wall and the clearance of nanoparticles by reticule-endothelial system (RES), thus shortening the blood circulation time.43–47 And the nanoparticles with neutral or negatively charged surface could reduce the undesirable clearance by RES and improve the blood compatibility, so that the anticancer drugs could be transported to the tumor site more efficiently.39,42–48
In this study, smart self-assembled nanoparticles CPT-WT-H NPs were elaborately designed and synthesized for highly efficient CPT delivery and effective cancer therapy, which could not only take advantage of the enhanced permeability and retention (EPR) effect and the active targeting effect but also avoid the significant toxicity of drug carriers, showing excellent biocompatibility, safety and stability. The prostate-specific membrane antigen (PSMA) and esterase dual responsive self-assembled nanoparticles CPT-WT-H NPs were prepared by combing hydrophobic CPT with hydrophilic PSMA-responsive penta-peptide via a cleavable ester bond. The relatively stable chemical linkage between PSMA-responsive penta-peptide and CPT, no expression of PSMA in blood and the negatively charged surface ensured that CPT-WT-H NPs were stable and inactive in blood circulation. Once they reached the tumor site, the dual responsive CPT-WT-H NPs were hydrolyzed by cell membrane antigen PSMA and the intracellular esterase successively, achieved a programmable release of CPT (Scheme 1). Our data demonstrated that CPT-WT-H NPs exhibited greatly improved water solubility and stability. Meanwhile, CPT-WT-H NPs exhibited excellent in vitro PSMA-targeting ability and in vivo antitumor activity and safety, holding the promise to become a new and potent anticancer drug.
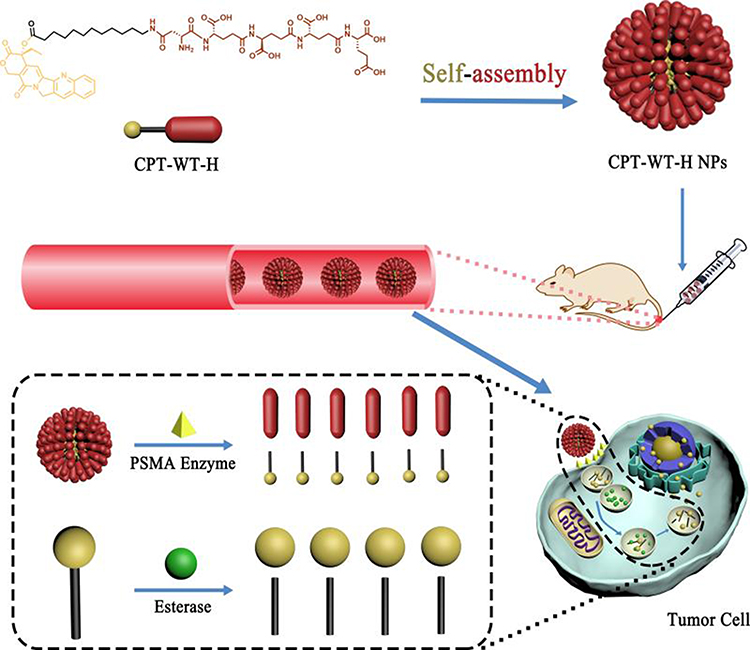 |
Scheme 1 Schematic illustration of the PSMA and esterase dual responsive CPT nanoparticles (CPT-WT-H NPs) with negatively charged surface, prolonged blood circulation, excellent PSMA-targeted delivery, enhanced tumor accumulation, potent antitumor efficacy and low side effects. |
Materials and Methods
More details of the experimental section and additional results are available in the Supporting Information.
Self-Assembly of the CPT-WT-H Prodrug
CPT-WT-H prodrug (10.0 mg) was dissolved in 10.0 mL phosphate-buffered saline (PBS, pH 7.4), and the solution was stirred at room temperature for 1 h. Finally, the concentration of CPT-WT-H NPs solution was adjusted to 0.1 mg·mL−1 for further experiments. For in vivo fluorescence imaging, DiR, a fluorescent probe, was loaded within the CPT-WT-H NPs. Briefly, 2.0 mg of DiR was dissolved in 1 mL of dichloromethane (DCM). Then, the DCM solution was injected dropwise into 10.0 mL PBS (pH 7.4) solution containing 10.0 mg CPT-WT-H prodrug while stirring slightly for 0.5 h. After that, the dispersion was frozen at −80°C overnight and lyophilized for 72 h, and DiR-loaded CPT-WT-H NPs was successfully prepared.
Characterization of CPT-WT-H NPs
The size distribution, polydispersity index (PDI), and zeta potential were evaluated using dynamic light scattering (DLS) (Zs90, Malvern, UK). The morphology was observed by transmission electron microscope (TEM) instrument (JEM2100, JEOL, Japan) operated at an accelerating voltage of 100 kV. The ultraviolet-visible (UV-vis) absorption spectrum between 200 and 400 nm was measured by a UV-vis spectrophotometer (Hitachi, Japan).
Characterization of in vitro PSMA Response of CPT-WT-H NPs
The in vitro PSMA response of the CPT-WT-H NPs was performed as described in the product catalog (Recombinant Human PSMA/FOLH1/NAALADase I, 4234-ZN, bio-techne, USA) with some modifications using LC-MS. CPT-WT-H NPs solutions (125 μL, 0.1 mg·mL−1 in HEPS buffer) were incubated with the rh-PSMA solution (125 μL, 1.0 μg·mL−1 in HEPS buffer) at 37°C for 2 h, 8 h, 12 h, 24 h and 48 h in a remonstrated oscillator (HZQ-QX, Harbin Donglian Instrument Co. Ltd., China). The sample incubating with the inactivated rh-PSMA (heating rh-PSMA at 95°C for 5 minutes) used as the control. After incubation, the reactions were stopped by heating at 95°C for 5 minutes and were centrifuged at 10,000 rpm for 10 min with high-speed centrifuge (Sorvall™ Legend™ Micro 21, Thermo Fisher Scientific, USA). A 100 μL sample of the supernatants was analyzed with LC-MS.
Characterization of Esterase Response of CPT-WT-H NPs
Esterase-mediated hydrolysis of CPT-WT-H NPs was performed by HPLC analysis in the presence of thermomyces lanuginosus lipase (Sigma-Aldrich, USA), a type of esterase, in PBS at 37 °C.49 Briefly, 0.6 mL of an aqueous solution of CPT-WT-H NPs (0.2 mg/mL) was incubated with 0.6 mL of thermomyces lanuginosus lipase (≥100,000 U/g, Sigma-Aldrich, USA). At predetermined time points (1, 2, 4 h), aliquot samples (0.2 mL) were collected and 200 μL of methanol was added to each sample, then it was centrifuged for 10 min at 10000r/min. Then, 200 μL of supernatant was subjected to HPLC analysis. UV detection was performed at a wavelength of 365 nm, and a C18 reverse-phase column (5 µm, 250 mm × 4.6 mm, Kromasil, Akzo Nobel Co., Ltd., Amsterdam, Netherlands) was utilized.
Cytotoxicity Assay
The in vitro cytotoxicity of CPT-WT-H NPs was assessed by the MTT method against PSMA-expressing LNCaP-FGC cells, non-PSMA-expressing cancer cells HepG2, HeLa, MCF-7, DU145, PC-3 cells and normal liver cells LO2 using CPT as the positive control. Briefly, the tumor cells growing in the logarithmic phase were seeded in 96-well plates at a density 4×103 cells/well and cultured for 24 h. Then, the cells were treated with serial dilutions of free CPT and CPT-WT-H NPs for 72 h. After incubation, 20 μL of 5.0 mg·mL−1 methylthiazol tetrazolium (MTT) was added to each well and incubated for another 4 h at 37°C. Following the incubation, 150 μL dimethyl sulfoxide (DMSO) was added to each well and absorbance was measured with a plate reader (BIORAD 550 spectrophotometer, Bio-Rad Life Science Development Ltd., Berkeley, CA, USA) at 490 nm. Experiments were performed in triplicates, and the values were the average of three (n = 3) independent experiments.
In vitro Cellular Uptake
The cellular uptake of the CPT-WT-H NPs was carried on LNCaP-FGC (PSMA+) and HepG2 (PSMA−) cell lines by confocal laser scanning microscopy. The LNCaP-FGC and HepG2 cells were seeded in 35 mm glass bottom dishes at a density of 1×104 cells per well with 1 mL complete 1640 medium. After incubation at 37°C for 24 h, the culture media was replaced with 2.0 mL of fresh medium containing 10.0 μM CPT-WT-H NPs. Then, the cells were incubated further for different incubation time periods (1 and 4 h). Following the removal of culture media, cells were washed with cold PBS, fixed with 70% ethanol for 30.0 min at 4°C. Subsequently, the cells were stained with 5 μg·mL−1 propidium iodide (excitation wavelength was 488 nm) for 5 min to visualize nucleus. The cells were observed by an Ultra View RS confocal laser scanning microscopy (PerkinElmer, Waltham, MA, USA).
Detection of Apoptosis Using Annexin V-FITC/PI Staining
Apoptosis of LNCaP-FGC (PSMA+) and HepG2 (PSMA−) cells was detected using the Annexin V-FITC Apoptosis Detection Kit (Beyotime Biotechnology, Jiangsu, China) as per-manufacturer instruction. The tumor cells were seeded into 6-well plates at a density of 1.0×105 cells per well and cultured for 24 h. Then, the cells were treated with different concentrations of CPT-WT-H NPs (2.5, 5.0, 20.0 μM for LNCaP-FGC; 20.0, 40.0, 80.0 μM for HepG2) for 72 h. Afterwards, the cells were harvested with trypsin (without EDTA) digestion, washed twice with cold PBS and centrifuged at 3000 g for 5 min. Then, the cells were re-suspended in 200 μL binding buffer and stained with Annexin V-FIT and PI according to the manufacturer’s instructions. Finally, the cells were immediately analyzed with flow cytometry (FACSCalibur, BD Biosciences, USA).
In vivo Near-Infrared Fluorescence (NIRF) Imaging
To investigate the bio-distribution of CPT-WT-H NPs, NIR fluorescence imaging experiments were carried out and analyzed using an IVIS Imaging Spectrum System in combination with IVIS Living Imaging 3.0 software (PerkinElmer, USA). DiR, a hydrophobic and lipophilic near-infrared fluorescence dye was encapsulated within CPT-WT-H NPs (designed as DiR-doped CPT-WT-H NPs). When the tumors reached to approximately 100–200 mm3, free DiR or DiR-doped CPT-WT-H NPs was intravenously injected via the tail vein into the MCF-7 tumor-bearing mice at an equivalent concentration of DiR (0.25 mg·mL−1, 100 μL). After the injection, the real-time bio-distribution and tumor accumulation of CPT-WT-H NPs in MCF-7 tumor-bearing mice were imaged by IVIS imaging system under certain parameters (λex = 740 nm, λem = 783 nm) at 0.5, 2.0, 4.0, 6.0, 8.0, 24.0, 32.0, and 48.0 h post-intravenous injection. The mice were euthanized after 48.0 h post-intravenous injection. The major organs (heart, liver, spleen, lung, kidney, and intestine) and tumor were harvested, and subjected for ex vivo fluorescence imaging. Their fluorescence intensities were analyzed by Living Image Software.
In vivo Antitumor Effect and Safety Studies
The antitumor efficacy of CPT-WT-H NPs was evaluated in MCF tumor-bearing BALB/c nude mice. The irinotecan group served as a positive control and the PBS group as the blank control. MCF tumor-bearing BALB/c nude mice were prepared by subcutaneously injecting 1×106 cells into the alar region. When the tumors reached to 100–200 mm3, the mice were randomly divided into five groups (n = 6/group). The tumor-bearing mice were intravenously injected via tail vein with PBS, irinotecan at a concentration of 10 mg·kg−1 and CPT-WT-H NPs at a dose of 10 mg·kg−1, 30 mg·kg−1 and 60 mg·kg−1 on days 0, 2, 4, 6, 8, 10, 12 and 14. The length and width of the tumor and the body weight of mice were measured every 2 days for 16 days. The tumor volume (V) was calculated as V =1/2 × length (mm) × width (mm)2. On the 16th day, the mice were euthanized, tumors were separated, weighted and photographed. Then, 0.5 mL of blood was collected from mice to investigate the blood biochemical analyses. Blood tests including alanine transaminase (ALT), aspartate transaminase (AST), blood urea nitrogen (BUN) and creatinine (CRE) were performed.
In addition, the main organs (liver, lungs, kidney, spleen and heart) were excised, washed with 0.9% NaCl, and fixed in the 10% neutral buffered formalin. For the hematoxylin and eosin (H&E) staining, the formalin-fixed tumors and main organs were embedded in paraffin, sectioned at 5 mm, stained with H&E, and examined by optical microscope (Axio Imager 2, ZESISS, Germany).
Statistical Analysis
Each experimental value is expressed as the mean ± SD. One-way analysis of variance (ANOVA) was performed to determine the significance between groups; p < 0.05 was considered as statistically significant and indicated by a single asterisk (*). p < 0.001 is considered as highly significant and indicated by three asterisks (***).
Results and Discussion
Synthesis of CPT-WT-H Conjugate
The amphiphilic CPT-WT-H conjugate was designed and synthesized as shown in Schemes S1 and 2 (Supplementary Materials). Firstly, WT-H (Asp-Glu*Glu*Glu*Glu) (“*” means the γ-glutamyl linkage; “-” means the α-glutamyl linkage), the penta-peptide employed as the hydrophilic part and tumor-targeting group, was prepared by liquid-phase peptide synthesis (LPPS) (Scheme S1). The successful synthesis of WT-H was confirmed by 1H NMR, 13C NMR and HRMS (Figures S1–S3, Supporting Materials). For the synthesis of PSMA-activated prodrug CPT-WT-H, the intermediates CPT-A-L12 and CPT-B-L12 were synthesized as shown in Scheme S2 according to our previous report.50 Then, reaction of CPT-B-L12 with WT-H in the presence of EDCI, N, N-Diisopropylethylamine (DIPEA), and 1-hydroxybenzotriazole (HOBt) in DCM afforded the corresponding intermediate CPT-WT-H-L12, which was further treated with TFA to remove the protective groups, yielding the final prodrug CPT-WT-H (Scheme S2). The structure of CPT-WT-H was confirmed by 1H NMR and 13C NMR, HRMS and UV absorbance (Figures S4–S7, Supplementary Materials). As shown in Figure S7A, the proton signals at ∼ 6.53 ppm related to the hydroxyl of CPT disappeared in CPT-WT-H conjugate, demonstrating the successful formation of ester linkage. Furthermore, the appearance of signals at ∼ 8.11 and 1.7–2.3 ppm belonging to imide protons and the methylene of penta-peptide, respectively, demonstrated the penta-peptide was successfully linked to CPT. High resolution mass spectrometry (HRMS) and analytical HPLC further verified the synthesis and purity of CPT-WT-H (Figure S7B and C). The determined molecular weight of CPT-WT-H conjugate (m/z, [M + H]+) was 1177.4950, which was consistent with the theoretical value (m/z, [M + H]+, 1177.4941). HPLC analysis indicated that the purity of CPT-WT-H was more than 96% (Figure S7C). The result of UV-vis absorption spectra also showed that CPT-WT-H exhibited the characteristic absorption peaks of CPT (~ 380 nm) in dimethyl sulfoxide (Figure S7D). Compared to the absorption peak of CPT, a 5 nm blue shift of the absorption peak from 382 nm to 378 nm was observed from the absorption of CPT-WT-H conjugate. Worth noting, the PSMA-activated prodrug CPT-WT-H contained as high as > 29.0% CPT. These experimental results further demonstrated the successful synthesis of CPT-WT-H conjugate.
Preparation and Characterization of CPT-WT-H NPs
Inherent amphiphilicity of CPT-WT-H molecule made it easy to self-assemble into nanoparticles spontaneously in aqueous solution such as PBS. When diluted into PBS, CPT-WT-H exhibited obvious Tyndall effect (Figure 1A), indicating the successful formation of abundant nanoparticles. To further confirm the formation of CPT-WT-H NPs from CPT-WT-H conjugate, the dynamic light scattering (DLS), transmission electron microscopy (TEM) was performed to determine and visualize the size and morphology of CPT-WT-H NPs. TEM suggested that CPT-WT-H NPs formed uniform spherical shapes in PBS (pH 7.4) with a mean diameter of around 100.0 nm (Figure 1A), which was in accordance with the result of DLS analysis. The DLS analysis revealed that CPT-WT-H NPs had average hydrodynamic diameter of 100.4 nm (PDI 0.247) (Figure 1B) and zeta potential of −22.3 ± 0.7 mV. The formation of stable nanoparticles also significantly enhanced the aqueous solubility of the prodrug CPT-WT-H (76 mg·mL−1 in PBS, pH = 7.4), which was much higher than CPT (0.003 mg·mL−1 in PBS, pH = 7.4).
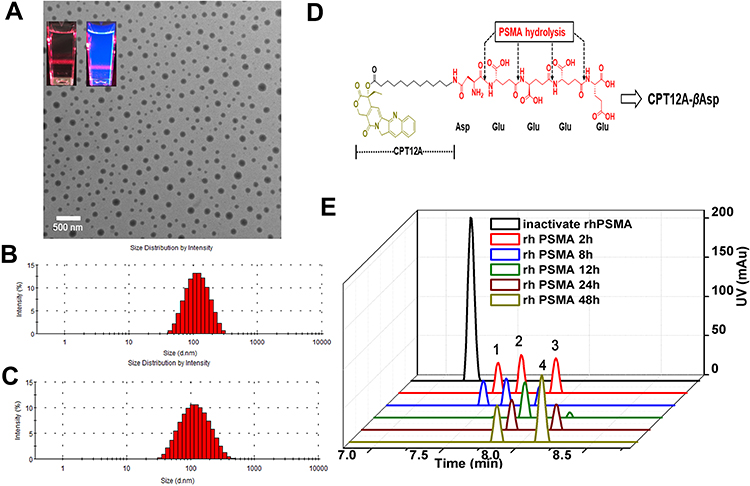 |
Figure 1 Characterization of CPT-WT-H NPs. (A) A TEM image of CPT-WT-H NPs dispersed in PBS (pH 7.4); inserts are the photographs of CPT-WT-H NPs in PBS in bright field (left) and under UV light at 365 nm with a red laser passing through (right). (B) DLS histograms of CPT-WT-H NPs in PBS. (C) Change of hydrodynamic diameter (Dh) of CPT-WT-H NPs in PBS for 120 h. (D) Schematic of sequential PSMA hydrolysis of CPT-WT-H (CPT12A-βAsp-GluγGluγGluγGlu) to yield the PSMA hydrolysate CPT12A-βAsp. (E) Characterization of in vitro PSMA response of CPT-WT-H NPs by LC-MS. |
In vitro Colloidal Stability
The colloid stability of nanoscale drug delivery system appeared to be one of the most important factors for their biomedical application.51,52 Previous studies have demonstrated that CPT lactone ring was hydrolyzed quickly in PBS, with a half-life of 24 min, and only 17% of CPT lactone form remained at equilibrium.53,54 It was reported that structural modifications at the 20-OH of CPT could enhance the stability of the lactone ring.50 In addition, the formation of stable nanoparticles not only enhanced the aqueous dispersity of CPT but also the stability of the lactone ring.55 The stability of CPT-WT-H NPs was estimated by DLS and HPLC analysis in this study. The DLS measurements revealed that the hydrodynamic diameter, PDI, and zeta potential of CPT-WT-H NPs showed almost no change in PBS buffer within 120 h, indicating high stability of CPT-WT-H NPs (Figure 1C). Furthermore, the HPLC analysis showed that no free CPT and degradation products were observed after incubating CPT-WT-H NPs in PBS buffer (pH 7.4) at 37°C for 24 h, further confirming their high stability (Figure S8, Supporting Information). All these results demonstrated that CPT-WT-H NPs showed excellent stability in aqueous environment without precipitation and phase separation, which were suitable for tumor-targeting delivery.
Characterization of in vitro PSMA and Esterase Response of CPT-WT-H NPs
One ideal prodrug should be inactive until activated by PSMA and be readily hydrolyzed by PSMA.29 The PSMA response of the CPT-WT-H NPs was evaluated by LC-MS in vitro. As shown in Figure 1D and E and Figure S9, after incubation with rh-PSMA for 2 h, CPT-WT-H (CPT12A-βAsp-GluγGluγGluγGlu) was sequentially hydrolyzed by rh-PSMA, the peak of the CPT-WT-H NPs was disappeared, the chromatographic peak of specific hydrolysate CPT12A-βAsp-GluγGluγGlu (peak 1), CPT12A-βAsp-GluγGlu (peak 2), CPT12A-βAsp-Glu (peak 3) generated. With the extension of incubation time, peak 1 and peak 2 disappeared gradually, while peak 4 (CPT12A-βAsp) gradually increased. This sequence hydrolysis process of CPT-WT-H was consistent with the PSMA activated prodrug G202.29 Above results that showed the prodrug CPT-WT-H exhibited excellent in vitro PSMA response.
To verify our proposed strategy of PSMA and esterase dual responsive, the CPT-WT-H NPs were incubated with thermomyces lanuginosus lipase (Sigma-Aldrich, USA), a type of esterase, in PBS at 37 °C. At predetermined time points (1, 2 and 4h), aliquot samples were collected and subjected to HPLC analysis. As shown in Figure S10, CPT-WT-H NPs could be hydrolyzed by thermomyces lanuginosus lipase easily in the beginning 1h. After 2 h and 4h incubation, CPT-WT-H NPs were almost completely hydrolyzed, which in principle validated our dual enzymatic strategy.
In vitro Cytotoxicity
The selective cytotoxic activities of CPT-WT-H NPs was investigated in PSMA-expressing (PSMA+) cancer cells LNCaP-FGC and non-PSMA-expressing (PSMA−) cancer cells (HepG2, MCF-7, HeLa, DU145, and PC-3) and the normal cell LO2 by MTT assay.56–60 The CPT was used as positive control. As presented in Figure 2A–F, the CPT-WT-H NPs exhibited moderate to potent cytotoxicity against all the tested tumor cell lines. But the cytotoxicity was much lower than that of the parent compound CPT at the same concentration. Meanwhile, CPT-WT-H NPs exhibited significantly higher cytotoxicity against the PSMA-expressing cell line LNCaP-FGC than the non-PSMA-expressing cell lines HepG2, HeLa, MCF-7, DU145, and PC-3, which could be attributed to the high expression of PSMA on the membrane of LNCaP-FGC cells. The cytotoxicity of CPT-WT-H NPs (IC50 = 1.12 ± 0.62 μM) against LNCaP-FGC (PSMA+) was 35-fold, 13-fold, 7-fold, 6-fold, and 10-fold, respectively, higher than that of the non-PSMA-expressing cells HepG2 (IC50 = 36.02 ± 4.46 µM), HeLa (IC50 = 13.47 ± 2.00 µM), MCF-7 (IC50 = 7.23 ± 1.23 µM), DU145 (IC50 = 6.87 ± 1.31 µM), and PC-3 (IC50 = 11.84 ± 2.14 µM) cells (Figure 2H). Moreover, CPT-WT-H NPs showed low toxicity to human normal liver cell LO2 (IC50 > 40.00 µM) (Figure 2G). Interestingly, among the non-PSMA-expressing cells, the CPT-WT-H NPs, showed much higher cytotoxicity against MCF-7, DU145, and PC-3 cell lines than other cell lines (HeLa and HepG2). Those significant difference cytotoxicity of CPT-WT-H NPs against the PSMA-expressing and non-PSMA-expressing cancer cells demonstrated that CPT-WT-H NPs could effectively target tumor cells overexpressing PSMA receptors, thereby confirming the successful construction of the targeted drug delivery system.
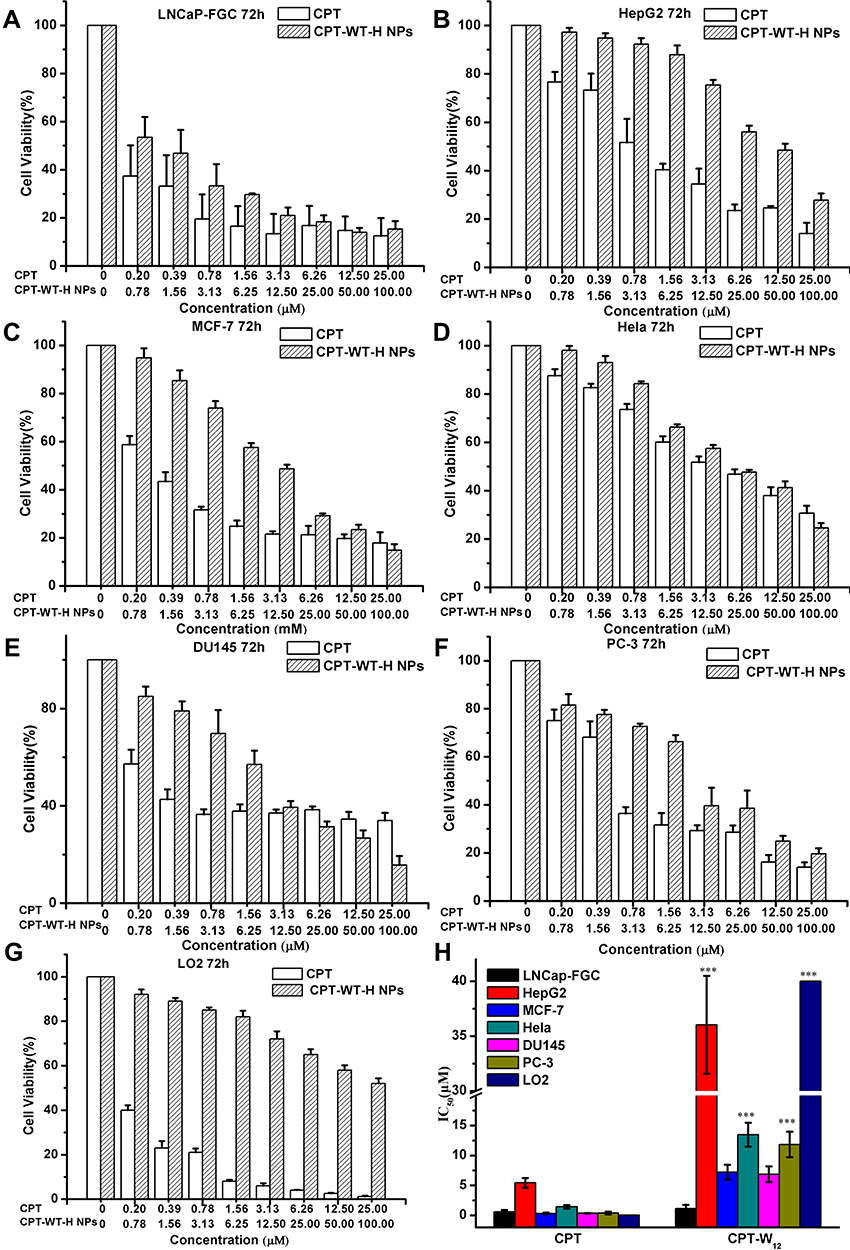 |
Figure 2 In vitro cytotoxicity of CPT-WT-H NPs and CPT against PSMA-expressing (PSMA+) cancer cell LNCaP-FGC (A) and non-PSMA-expressing (PSMA−) cancer cells HepG2 (B), MCF-7 (C), HeLa (D), DU145 (E), PC-3 (F) and normal liver cells LO2 (G), the IC50 values of CPT and CPT-WT-H NPs on different cell lines (H). ***p < 0.001, vs LNCaP-FGC cells. |
In vitro Cellular Uptake
In addition to solubilization, the penta-peptide serves to mask the cytotoxicity by preventing nonspecific uptake of drug into cells due to the presence of negatively charged amino acids. The cellular uptake behavior of CPT-WT-H NPs was investigated by confocal laser scanning microscope (CLSM) in the LNCAP-FGC (PSMA+) and HepG2 (PSMA−) cell lines. Propidium iodide (PI) was used to label the nuclei (red) and the fluorescence of CPT was pseudo-colored with blue. The cells were cultured with CPT-WT-H NPs for 1 and 4 h before observation. As shown in Figure 3A, the blue fluorescence generated from CPT was clearly observed inside LNCAP-FGC (PSMA+) cells and localized in the cytoplasm and nuclei after 1 h incubation, indicating that the active compound could enter the cancer cells successfully. In addition, the blue fluorescence intensity in LNCAP-FGC (PSMA+) increased with the prolonging incubation time (4 h), showing a time-dependent manner (Figure 3A). There was almost no blue fluorescence from CPT was detected in HepG2 (PSMA−) cells when treated with CPT-WT-H NPs for 1 h (Figure 3B). Although the fluorescence from CPT in HepG2 (PSMA−) cells increased with the prolonging incubation time (4 h) (Figure 3B), which was highly lower than that of LNCAP-FGC (PSMA+), demonstrating the poor uptake of CPT-WT-H NPs. It was mainly due to the hydrophilic and negative charge of the penta-peptide WT-H, which hampered the cellular uptake of CPT-WT-H NPs. Moreover, the mean fluorescence intensities of CPT at the above time points in each cell line were calculated using ImageJ software.61 As shown in Figure 3C, the blue fluorescence intensity detected in the LNCAP-FGC (PSMA+) cells was much higher than that of the HepG2 (PSMA−) cells (p < 0.001), demonstrating excellent PSMAresponse of CPT-WT-H NPs.
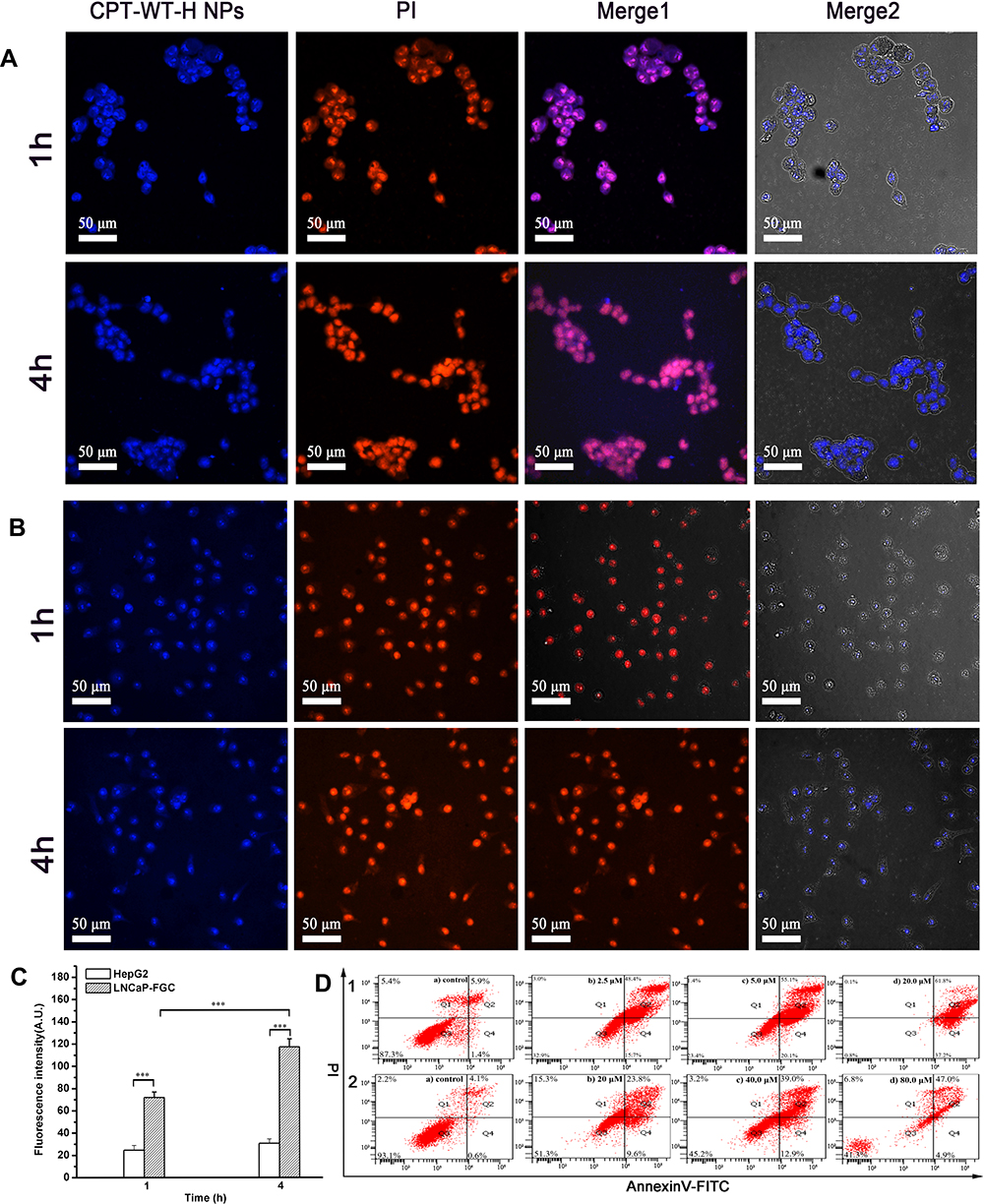 |
Figure 3 Cellular uptake of CPT-WT-H NPs in vitro. (A) CLSM images of LNCaP-FGC (PSMA+) cells incubated with CPT-WT-H NPs (10 μM) for 1 and 4 h. (B) CLSM images of HepG2 (PSMA−) cells incubated with CPT-WT-H NPs (10 μM) for 1 and 4 h. Nuclei were stained by PI (red), the blue color was indicative of CPT. (C) Mean fluorescence intensity of CPT-WT-H NPs internalized by LNCaP-FGC and HepG2 cells after incubation for 1, and 4 h. ***p < 0.001. (D) Flow cytometry analysis for apoptosis of LNCaP-FGC (PSMA+) (1) and HepG2 (PSMA−) (2) cells induced by CPT-WT-H NPs for 72 h. Q1, necrotic cells; Q2, late apoptotic cells; Q3, living cells; Q4, early apoptotic cells. Inserted numbers in the profiles indicate the percentage of the cells present in this area. |
In vitro Apoptosis
To further demonstrate the selective cytotoxicity of CPT-WT-H NPs against LNCaP-FGC (PSMA+) and HepG2 (PSMA−), the Annexin V-FITC/PI apoptosis detection was performed. The apoptotic effect was determined by counting the apoptosis ratios (including the early and late apoptosis ratios). It was observed that the LNCaP-FGC (PSMA+) cell demonstrated a remarkable response to the apoptotic effect of CPT-WT-H NPs in a dose-dependent manner. The percentage of apoptotic LNCaP-FGC cells was evaluated to be 64.10%, 75.20%, and 99.00% after treatment with 2.50, 5.00, and 20.00 μM of CPT-WT-H NPs, respectively (Figure 3D1). On the contrary, the drug-induced apoptosis for HepG2 (PSMA−) cell demonstrated a totally different trend. The percentage of apoptotic HepG2 (PSMA−) cell was evaluated to be 33.4%, 51.90%, and 51.90% after treatment with 20.00 μM, 40.00 μM and 80.00 μM of CPT-WT-H NPs, respectively (Figure 3D2). Thus, consistent with the in vitro cytotoxicity, the results demonstrated CPT-WT-H NPs showed PSMA-oriented targeting activity.
In vivo NIRF Imaging
Encouraged by the excellent selective toxicity and high cellular uptake to PSMA-producing prostate cancer cells, the in vivo tumor-targeting ability of CPT-WT-H NPs was next evaluated in MCF-7 human breast cancer xenograft (PSMA-producing).29 The lipophilic near-infrared fluorescence dye DiR was incorporated into CPT-WT-H NPs for in vivo NIRF imaging.2,5,61 After the injection of free DiR and DiR-doped CPT-WT-H NPs intravenously, the real-time bio-distribution and accumulation of CPT-WT-H NPs in MCF-7 tumor-bearing mice were monitored by IVIS imaging system. As shown in Figure 4A, in animals injected with free DiR into mice, the fluorescence signals mainly distributed within liver and no fluorescent signals could be observed in the tumor tissues. After injection of DiR-doped CPT-WT-H NPs, a strong fluorescence signal was observed in the whole body at 0.5 h, suggesting the rapid circulation of the DiR-doped CPT-WT-H NPs in the bloodstream. As time elapsed, the fluorescence signal was obviously accumulated at the tumor sites at 2 h post-injection. The average fluorescence signals in tumor tissue continued to increase gradually, reaching a maximum value at 24 h after post-injection (Figure 4A and B). The result indicated that the CPT-WT-H NPs could reduce the rapid capture by the reticuloendothelial system (RES) and accumulate in the tumor by the EPR effect and PSMA-mediated active targeting.
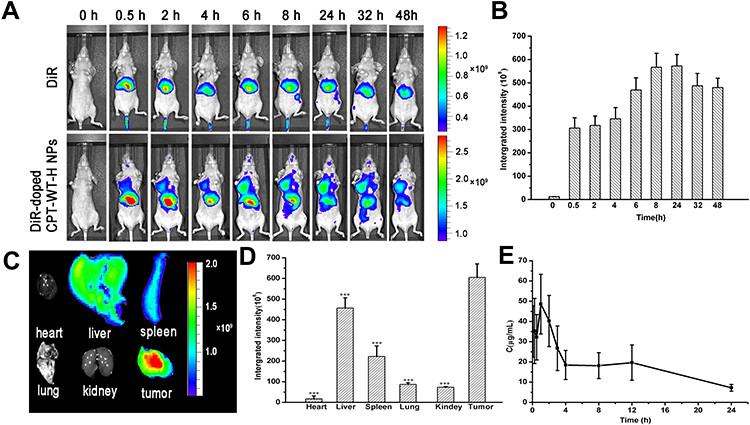 |
Figure 4 In vivo NIRF imaging of DiR-doped CPT-WT-H NPs. (A) In vivo NIRF images of the MCF-7 tumor-bearing nude mice (PSMA-producing) after intravenous injection of free DiR (0.25 mg·mL−1, 100 µL) or DiR-doped CPT-WT-H NPs (0.25 mg·mL−1, 100 µL). (B) Integrated fluorescence intensity of tumor obtained after intravenous injection of DiR-doped CPT-WT-H NPs (0.25 mg·mL−1, 100 µL). (C) Ex vivo NIRF images and (D) Integrated fluorescence intensity of the major organs and tumors excised at 48 h post intravenous injection of DiR-doped CPT-WT-H NPs, ***p < 0.001, vs tumor. (E) In vivo pharmacokinetics profiles of CPT-WT-H NPs s at the equivalent dose (30 mg·kg−1) in BALB/c mice. Error bars indicate SD (n = 6). |
To provide further evidence of the tumor targeting of the CPT-WT-H NPs, the tumor and major organs (heart, liver, spleen, lung, and kidney, respectively) were excised for ex vivo NIRF imaging after 48 h post-injection. As shown in Figure 4C and D, the brightest fluorescence was found in both the tumor and the liver in DiR-doped CPT-WT-H NPs group, which was significantly higher than the other organs. It should be noted that the relatively strong fluorescence of the important metabolic organs liver and spleen should be due to the macrophage clearance of CPT-WT-H NPs from the blood, which was a universal phenomenon for nano medicine. As confirmed by these data, the CPT-WT-H NPs could efficiently deliver CPT-WT-H NPs to the tumor site, which was likely attributed to both the EPR effect and PSMA-mediated active tumor targeting mechanisms. The result would contribute to improving the therapeutic efficiency as well as reducing the side effects.
In vivo Blood Circulation
Previous studies showed that NPs with a suitable size (smaller than 200 nm) had a longer retention time in the bloodstream than free small molecular drugs.2,62,63 In this study, the pharmacokinetic studies were performed in BALB/c mice (∼20 g) after intravenous injection of CPT-WT-H NPs (30 mg·kg−1). It is worth noting that CPT-WT-H NPs with negatively charged surface showed a significantly extended blood circulation half-life (t1/2 = 10.9 ± 1.86 h) (Figure 4E and Table S1) without the help of any drug carriers than free CPT (t1/2 = 0.165 ± 0.134 h).64 Thus, the significantly improved retention in bloodstream would provide the possibility of enhanced accumulation of CPT-WT-H NPs at the tumor site.
Potent Antitumor Efficacy and Reduced Side Effects of CPT-WT-H NPs
The in vivo antitumor efficacy of CPT-WT-H NPs was estimated in MCF-7 tumor xenograft models (PSMA-producing).29 When the tumors reached an average volume of 100–200 mm3, PBS, Irinotecan, and CPT-WT-H NPs were intravenously injected via the tail vein on days 0, 2, 4, 6, 8, 10, 12 and 14. Tumor size and body weight of MCF-7 tumor-bearing nude mice were monitored every 2 days for 16 days. As shown in Figure 5A, the tumor volume of mice receiving PBS as control rapidly increased at 3 weeks, while the growth of tumor was significantly suppressed by the treatment of CPT-WT-H NPs and IR. CPT-WT-H NPs and IR displayed potent antitumor effects and dramatically delayed tumor progression. On day 16, the tumor volumes in CPT-WT-H NPs-treated mice were remarkably smaller compared with those mice treated with PBS. By comparison, the antitumor efficacy of 30 mg·kg−1 CPT-WT-H NPs was similar to that of IR (10 mg·kg−1). Importantly, the body weights of mice receiving IR (10 mg·kg−1) treatments reduced observably compared to that of PBS (p < 0.05). However, mice treated with 30 mg·kg−1 CPT-WT-H NPs did not show any weight loss during the course of treatment (Figure 5B); Moreover, the antitumor efficacy of 60 mg·kg−1 CPT-WT-H NPs was comparable with IR.
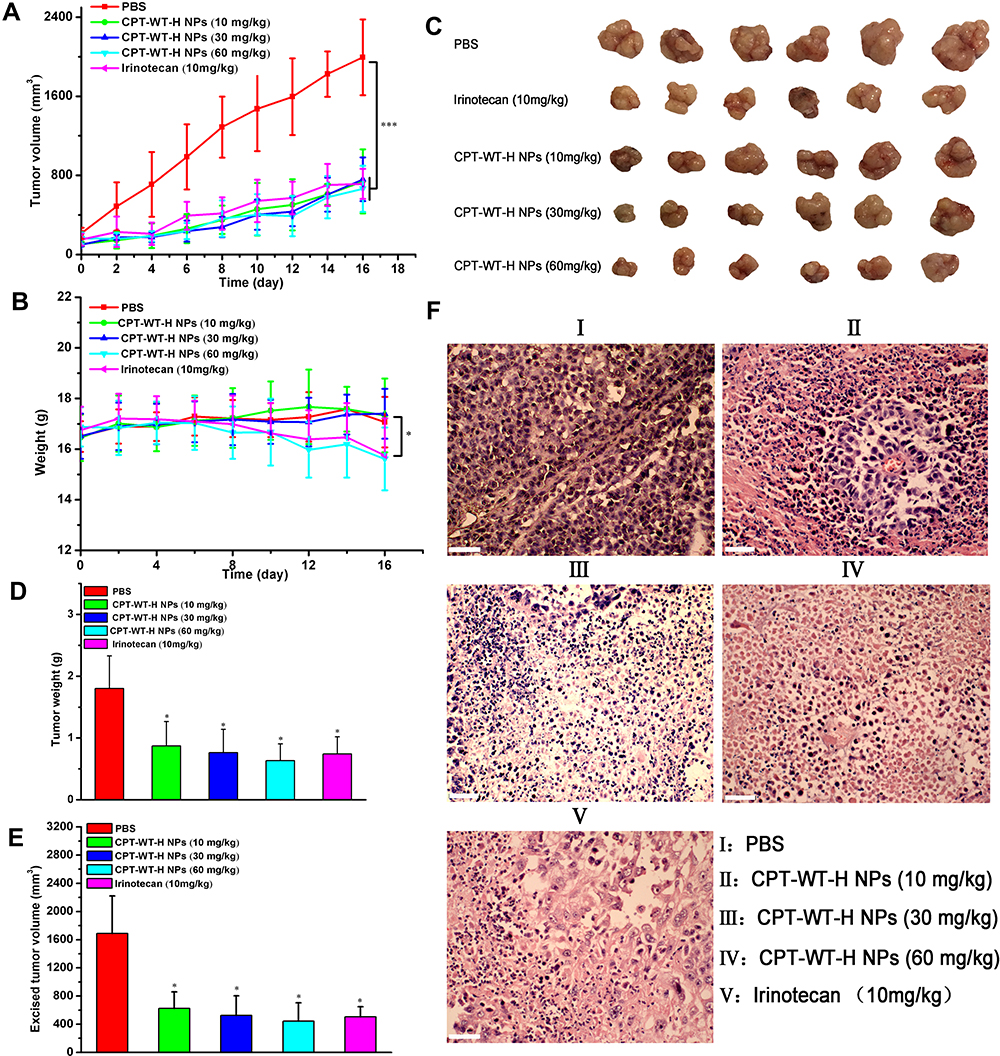 |
Figure 5 CPT-WT-H NPs significantly inhibited tumor progression and dramatically reduced side effects in an MCF-7 human breast cancer model. (A) The growth curves of MCF-7 tumor after treatment, which was initiated on day 13 post tumor inoculation, and drugs were intravenously injected every 2 days for 8 times (n = 6/group). (B) Changes of body weight in each group. (C) The tumor pictures in each group. (D) Excised tumor weight in each group. (E) Excised tumor volume in each group. *p < 0.05; ***p < 0.001. (F) Histological analysis of tumor sections stained with H&E. |
At the end of the experiments, the MCF-7 tumor-bearing nude mice were sacrificed, and tumors were dissected. The mice treated with CPT-WT-H NPs and IR had much smaller tumor size and tumor weight than that of the PBS group (Figure 5C–E). In addition, the inhibition rate of tumor growth of 30 mg·kg−1 CPT-WT-H NPs group was similar to the IR group, the tumor control rate of 60 mg·kg−1 CPT-WT-H NPs group was higher than the IR group. These results clearly demonstrated that CPT-WT-H NPs elicited potent therapeutic efficacy and reduced side effects.
Histological Analyses
Histological analyses of hematoxylin and eosin (H&E) staining of the tumor and major organs (heart, liver, spleen, lung, and kidney, respectively) were used to evaluate the antitumor effect and the toxicity of the CPT-WT-H NPs and IR. Histological examination of hematoxylin and eosin–stained tissue section (Figure 5F) indicated that the tumors treated with CPT-WT-H NPs and IR showed much irregularly shaped necrotic area with cellular shrinkage, nuclear fragmentation, and nucleus dissolution. These results demonstrated the potent in vivo therapeutic efficacy of CPT-WT-H NPs, showing great potential for cancer chemotherapy. Furthermore, the H&E staining of the main organs (heart, liver, spleen, lung, kidney) demonstrated that no significant organ damage was observed for the CPT-WT-H NPs-treated mice (Figures 6A and B, and S11). Notably, no significant organ damage was observed in the mice even treated with 60 mg·kg−1 of CPT-WT-H NPs. However, the mice treated with the positive drug IR (10 mg·kg−1) showed obvious interstitial inflammatory cell infiltration (Figure 6B), which was the characteristic pathologic changes of acute kidney injury.65,66 The H&E staining results further confirmed the superior in vivo antitumor efficacy of CPT-WT-H NPs without obvious systemic toxicity.
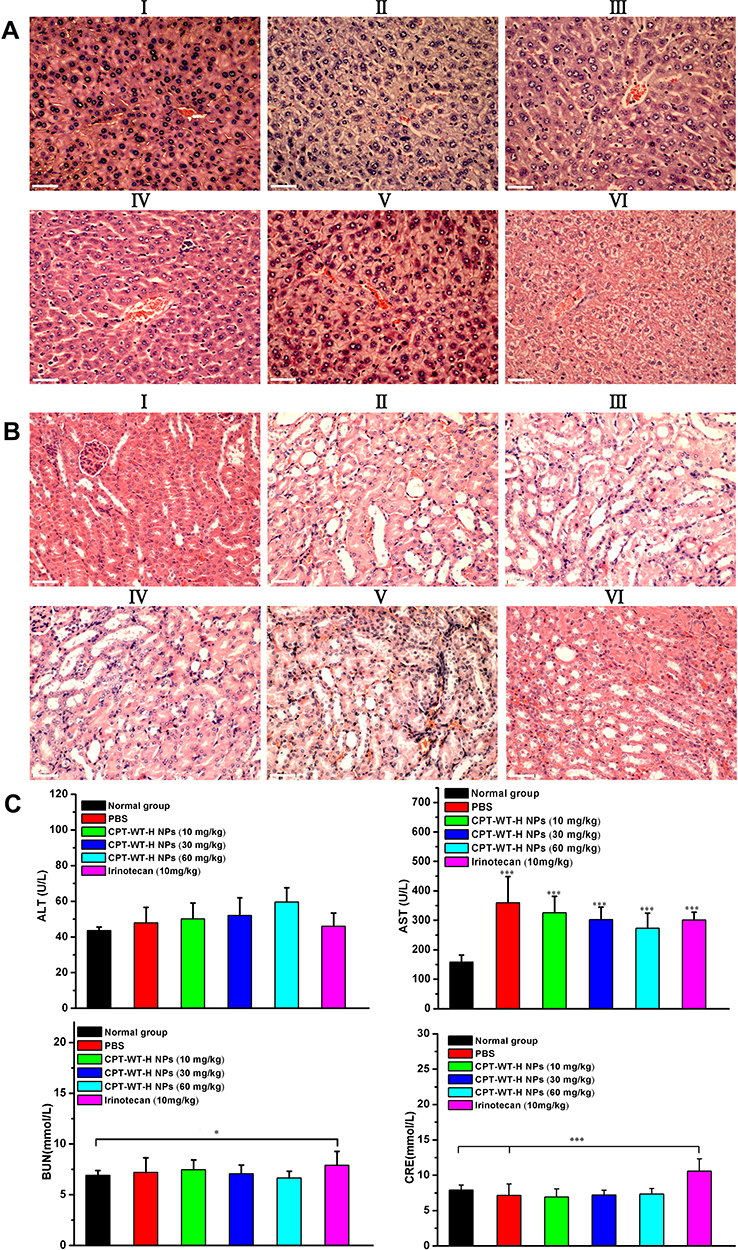 |
Figure 6 Representative H&E-stained histological images obtained from liver (A) and kidneys (B) after intravenous administration of irinotecan (10 mg·kg−1) and CPT-WT-H NPs (10, 30, 60 mg·kg−1). The PBS-treated group and the untreated group were used as the control. (I: PBS, II: CPT-WT-H NPs (10mg·kg−1), III: CPT-WT-H NPs (30mg·kg−1), IV: CPT-WT-H NPs (60mg·kg−1), V: Irinotecan (10 mg·kg−1), VI: Normal group). (C) Blood biochemical analysis of after the antitumor efficacy evaluation. The PBS-treated group and the untreated group were used as the control. The main parameters of hepatic and renal function are alanine transaminase (ALT), aspartate transaminase (AST), blood urea nitrogen (BUN), and creatinine (CRE). Error bars indicate SD (n = 6). *p < 0.05; ***p < 0.001. |
In vivo Safety Studies
In order to evaluate the hepatic and renal toxicity, the blood biochemical analyses were performed. The BALB/c nude mice of in vivo antitumor efficacy study were used to conduct the hematological and blood biochemical analyses before they were put to execution. As shown in Figure 6C, the measured factors including alanine transaminase (ALT), blood urea nitrogen (BUN), and creatinine (CRE) levels of CPT-WT-H NPs groups showed no statistically significant (p > 0.05) difference from those in PBS and the normal group. The aspartate transaminase (AST) levels of the model group and all the drug treatment group showed statistically significant (p < 0.001) difference from those in the normal group. However, the level of the blood urea nitrogen (BUN) and creatinine (CRE) in the irinotecan group increased obviously compared with the normal group (p < 0.05, p < 0.001 respectively). The results indicated that the CPT-WT-H NPs induced no obvious hepatic and renal dysfunction.
Conclusions
In this study, a novel kind of PSMA and esterase dual responsive actively targeted drug delivery system (CPT-WT-H NPs) has been successfully constructed for efficient delivery of CPT from a PSMA-activated CPT prodrug. The prodrug could be facilely synthesized and self-assemble into well-defined nanoparticles in an aqueous solution with defined chemical structures, high drug loading content and negatively charged surface, thus notably improving the solubility, stability and blood circulation time of CPT. Our data demonstrated that CPT-WT-H NPs exhibited high selective toxicity towards the PSMA-expressing, non-PSMA-expressing cancer cells and normal liver cells, high tumor accumulation via both EPR effect-mediated passive targeting and PSMA-mediated active targeting. More importantly, CPT-WT-H NPs exhibited excellent in vivo antitumor activity with low systemic toxicity compared to the positive drug IR. In summary, we demonstrated for the first time that this PSMA-triggered nanomedicine strategy could be a promising strategy for efficient delivery of CPT.
Supplementary Materials
The following files are available free of charge in the Supplementary materials file: 2 supporting schemes, 1 supporting table, 11 supporting figures, and some specific experimental details.
Ethics Statement
The animal study proposal was approved by the ethics committee of Beijing University of Chinese Medicine with the permit number 1115010900375. All animal experimental procedures were performed in accordance with the Regulations for the Administration of Affairs Concerning Experimental Animals approved by the State Council of People’s Republic of China.
Acknowledgments
This research was funded by the National Natural Science Foundation of China (Nos. 82104365, 81173519 and 81603256), the Fundamental Research Funds for the Central Universities (No. 2020-JYB-XJSJJ-007), Beijing “high-grade, precision and advanced” project, Beijing Key Laboratory for Basic and Development Research on Chinese Medicine (Beijing, 100102), and Young Chinese talent training project of the Chinese Academy of Chinese Medicine (CACM-2018-QNRC2-B08).
Author Contributions
All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article was submitted, gave final approval of the version to be published, and agreed to be accountable for all aspects of the work.
Disclosure
The authors declare no conflicts of interest for this work.
References
1. Zitvogel L, Galluzzi L, Smyth MJ, Kroemer G. Mechanism of action of conventional and targeted anticancer therapies: reinstating immune surveillance. Immunity. 2013;39:74–88. doi:10.1016/j.immuni.2013.06.014
2. Li Y, Lin J, Ma J, et al. Methotrexate–camptothecin prodrug nanoassemblies as a versatile nanoplatform for biomodal imaging-guided self-active targeted and synergistic chemotherapy. ACS Appl Mat Interfaces. 2017;9:34650–34665. doi:10.1021/acsami.7b10027
3. Baxevanis CN, Perez SA, Papamichail M. Combinatorial treatments including vaccines chemotherapy and monoclonal antibodies for cancer therapy. Cancer Immunol Immunother. 2009;58:317–324. doi:10.1007/s00262-008-0576-4
4. Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer. 2006;6:789. doi:10.1038/nrc1977
5. Liang X, Gao C, Cui L, Wang S, Wang J, Dai Z. Self‐assembly of an amphiphilic janus camptothecin–floxuridine conjugate into liposome‐like nanocapsules for more efficacious combination chemotherapy in cancer. Adv Mater. 2017;29:1703135. doi:10.1002/adma.201703135
6. Liu Y, Chen X, Ding J, Yu L, Ma D, Ding J. Improved solubility and bioactivity of camptothecin family antitumor drugs with supramolecular encapsulation by water-soluble pillar [6] arene. ACS Omega. 2017;2:5283–5288. doi:10.1021/acsomega.7b01032
7. Sawant RR, Sawant RM, Torchilin VP. Mixed PEG–PE/vitamin E tumor-targeted immunomicelles as carriers for poorly soluble anti-cancer drugs: improved drug solubilization and enhanced in vitro cytotoxicity. Eur J Pharm Biopharm. 2008;70:51–57. doi:10.1016/j.ejpb.2008.04.016
8. Park K. Facing the truth about nanotechnology in drug delivery. ACS Nano. 2013;7:7442–7447. doi:10.1021/nn404501g
9. Xue HY, Liu S, Wong HL. Nanotoxicity: a key obstacle to clinical translation of siRNA-based nanomedicine. Nanomedicine. 2014;9:295–312. doi:10.2217/nnm.13.204
10. Deng H, Zhang Y, Wang X, et al. Balancing the stability and drug release of polymer micelles by the coordination of dual-sensitive cleavable bonds in cross-linked core. Acta Biomater. 2015;11:126–136. doi:10.1016/j.actbio.2014.09.047
11. Shen Y, Jin E, Zhang B, et al. Prodrugs forming high drug loading multifunctional nanocapsules for intracellular cancer drug delivery. J Am Chem Soc. 2010;132:4259–4265. doi:10.1021/ja909475m
12. Huang P, Wang D, Su Y, et al. Combination of small molecule prodrug and nanodrug delivery: amphiphilic drug–drug conjugate for cancer therapy. J Am Chem Soc. 2014;136:11748–11756. doi:10.1021/ja505212y
13. Vemula PK, Cruikshank GA, Karp JM, John G. Self-assembled prodrugs: an enzymatically triggered drug-delivery platform. Biomaterials. 2009;30:383–393. doi:10.1016/j.biomaterials.2008.09.045
14. Vemula PK, Wiradharma N, Ankrum JA, Miranda OR, John G, Karp JM. Prodrugs as self-assembled hydrogels: a new paradigm for biomaterials. Curr Opin Biotechnol. 2013;24:1174–1182. doi:10.1016/j.copbio.2013.02.006
15. Luo C, Sun J, Sun B, He Z. Prodrug-based nanoparticulate drug delivery strategies for cancer therapy. Trends Pharmacol Sci. 2014;35:556–566. doi:10.1016/j.tips.2014.09.008
16. Cheng H, Zhu JY, Xu XD, et al. Activable cell-penetrating peptide conjugated prodrug for tumor targeted drug delivery. ACS Appl Mat Interfaces. 2015;7:16061–16069. doi:10.1021/acsami.5b04517
17. Liang Y, Li S, Wang X, et al. A nanosystem of amphiphilic oligopeptide-drug conjugate actualizing both αvβ3 targeting and reduction-triggered release for maytansinoid. Theranostics. 2017;7:3306. doi:10.7150/thno.20242
18. Liu Y, Zhou C, Wang W, et al. CD44 receptor targeting and endosomal pH-sensitive dual functional hyaluronic acid micelles for intracellular paclitaxel delivery. Mol Pharm. 2016;13:4209–4221. doi:10.1021/acs.molpharmaceut.6b00870
19. Wang Y, Cheetham AG, Angacian G, Su H, Xie L, Cui H. Peptide–drug conjugates as effective prodrug strategies for targeted delivery. Adv Drug Deliver Rev. 2017;110:112–126. doi:10.1016/j.addr.2016.06.015
20. Jiang Y, Wang X, Liu X, et al. Enhanced anti-glioma efficacy of ultrahigh loading capacity paclitaxel prodrug conjugate self-assembled targeted nanoparticles. ACS Appl Mat Interfaces. 2016;9:211–217. doi:10.1021/acsami.6b13805
21. Han X, Chen J, Jiang M, et al. Paclitaxel–paclitaxel prodrug nano assembly as a versatile nanoplatform for combinational cancer therapy. ACS Appl Mat Interfaces. 2016;8:33506–33513. doi:10.1021/acsami.6b13057
22. Wang H, Xie H, Wang J, et al. Self-assembling prodrugs by precise programming of molecular structures that contribute distinct stability, pharmacokinetics, and antitumor efficacy. Adv Funct Mater. 2015;25:4956–4965. doi:10.1002/adfm.201501953
23. Wang H, Zhou L, Xie K, et al. Polylactide-tethered prodrugs in polymeric nanoparticles as reliable nanomedicines for the efficient eradication of patient-derived hepatocellular carcinoma. Theranostics. 2018;8:3949–3963. doi:10.7150/thno.26161
24. Wang Y, Xie H, Ying K, et al. Tuning the efficacy of esterase-activatable prodrug nanoparticles for the treatment of colorectal malignancies. Biomaterials. 2021;270:120705–120718. doi:10.1016/j.biomaterials.2021.120705
25. Fang T, Ye Z, Chen X, Wang Y, Wan J, Wang H. Repurposing of camptothecin: an esterase-activatable prodrug delivered by a self-emulsifying formulation that improves efficacy in colorectal cancer. Int J Pharm. 2021;599:120399–120411. doi:10.1016/j.ijpharm.2021.120399
26. Liu T, Toriyabe Y, Kazak M, Berkman CE. Pseudoirreversible inhibition of prostate-specific membrane antigen by phosphoramidate peptidomimetics. Biochemistry. 2008;47:12658–12660. doi:10.1021/bi801883v
27. Davis MI, Bennett MJ, Thomas LM, Bjorkman PJ. Crystal structure of prostate-specific membrane antigen a tumor marker and peptidase. Proc Natl Acad Sci. 2005;102:5981–5986. doi:10.1073/pnas.0502101102
28. Afshar-Oromieh A, Haberkorn U, Schlemmer HP, et al. Comparison of PET/CT and PET/MRI hybrid systems using a 68Ga-labelled PSMA ligand for the diagnosis of recurrent prostate cancer: initial experience. Eur J Nucl Med Mol Imaging. 2014;41:887–897. doi:10.1007/s00259-013-2660-z
29. Denmeade SR, Mhaka AM, Rosen DM, et al. Engineering a prostate-specific membrane antigen–activated tumor endothelial cell prodrug for cancer therapy. Sci Transl Med. 2012;4:140ra86–140ra86. doi:10.1126/scitranslmed.3003886
30. Bravaccini S, Puccetti M, Bocchini M, et al. PSMA expression: a potential ally for the pathologist in prostate cancer diagnosis. Sci Rep. 2018;8:4254. doi:10.1038/s41598-018-22594-1
31. Kawatani M, Yamamoto K, Yamada D, et al. Fluorescence detection of prostate cancer by an activatable fluorescence probe for PSMA carboxypeptidase activity. J Am Chem Soc. 2019;141:26. doi:10.1021/jacs.9b04412
32. Isaacs JT. Prodrug Targeting Based Upon the NAALADase Activity to Prostate Specific Membrane Antigen by Prostate Cancer Cells. Johns Hopkins Univ Baltimore Md School of Medicine; 2001.
33. Kularatne SA, Venkatesh C, Santhapuram HKR, et al. Synthesis and biological analysis of prostate-specific membrane antigen-targeted anticancer prodrugs. J Med Chem. 2010;53:7767–7777. doi:10.1021/jm100729b
34. Lv Q, Yang J, Zhang R, et al. Prostate-specific membrane antigen targeted therapy of prostate cancer using a DUPA–paclitaxel conjugate. Mol Pharm. 2018;15:1842–1852. doi:10.1021/acs.molpharmaceut.8b00026
35. Wu X, Ding B, Gao J, et al. Second-generation aptamer-conjugated PSMA-targeted delivery system for prostate cancer therapy. Int J Nanomed. 2011;6:1747.
36. Nedrow‐Byers JR, Jabbes M, Jewett C, et al. A phosphoramidate‐based prostate‐specific membrane antigen‐targeted SPECT agent. Prostate. 2012;72:904–912. doi:10.1002/pros.21493
37. Hrkach J, Von Hoff D, Ali MM, et al. Preclinical development and clinical translation of a PSMA-targeted docetaxel nanoparticle with a differentiated pharmacological profile. Sci Transl Med. 2012;4:128ra39–128ra39. doi:10.1126/scitranslmed.3003651
38. Chen Z, Penet MF, Nimmagadda S, et al. PSMA-targeted theranostic nanoplex for prostate cancer therapy. ACS Nano. 2012;6:7752–7762. doi:10.1021/nn301725w
39. Xiao K, Li Y, Luo J, et al. The effect of surface charge on in vivo bio distribution of PEG-oligocholic acid based micellar nanoparticles. Biomaterials. 2011;32:3435–3446. doi:10.1016/j.biomaterials.2011.01.021
40. Albanese A, Tang PS, Chan WC. The effect of nanoparticle size shape and surface chemistry on biological systems. Annu Rev Biomed Eng. 2012;14:1–16. doi:10.1146/annurev-bioeng-071811-150124
41. He C, Hu Y, Yin L, Tang C, Yin C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials. 2010;31:3657–3666. doi:10.1016/j.biomaterials.2010.01.065
42. Dai L, Liu J, Luo Z, Li M, Cai K. Tumor therapy: targeted drug delivery systems. J Mater Chem B. 2016;4:6758–6772. doi:10.1039/C6TB01743F
43. Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Adv Drug Deliver Rev. 2014;66:2–25.
44. Cho EC, Xie J, Wurm PA, Xia Y. Understanding the role of surface charges in cellular adsorption versus internalization by selectively removing gold nanoparticles on the cell surface with a I2/KI etchant. Nano Lett. 2009;9:1080–1084. doi:10.1021/nl803487r
45. Yue ZG, Wei W, Lv PP, et al. Surface charge affects cellular uptake and intracellular trafficking of chitosan-based nanoparticles. Biomacromolecules. 2011;12:2440–2446. doi:10.1021/bm101482r
46. Bose T, Latawiec D, Mondal PP, Mandal S. Overview of nano-drugs characteristics for clinical application: the journey from the entry to the exit point. J Nanopart Res. 2014;16:2527. doi:10.1007/s11051-014-2527-7
47. Campbell RB, Fukumura D, Brown EB, et al. Cationic charge determines the distribution of liposomes between the vascular and extravascular compartments of tumors. Cancer Res. 2002;62:6831–6836.
48. Deng H, Liu J, Zhao X, et al. PEG-b-PCL copolymer micelles with the ability of pH-controlled negative-to-positive charge reversal for intracellular delivery of doxorubicin. Biomacromolecules. 2014;15:4281–4292. doi:10.1021/bm501290t
49. Tang W, Zhao ZB, Chong YY, et al. Tandem enzymatic self-assembly and slow release of dexamethasone enhances its antihepatic fibrosis effect[J]. ACS Nano. 2018;12:9966–9973. doi:10.1021/acsnano.8b04143
50. Xu B, Zhou F, Yan MM, et al. PSMA-oriented target delivery of novel anticancer prodrugs: design synthesis and biological evaluations of oligopeptide-camptothecin conjugates. Int J Mol Sci. 2018;19:3251. doi:10.3390/ijms19103251
51. Pei Q, Hu X, Liu S, Li Y, Xie Z, Jing X. Paclitaxel dimers assembling nanomedicines for treatment of cer Bertrandix carcinoma. J Control Release. 2017;254:23–33. doi:10.1016/j.jconrel.2017.03.391
52. Neuberger T, Schöpf B, Hofmann H, Hofmann M, Von Rechenberg B. Superparamagnetic nanoparticles for biomedical applications: possibilities and limitations of a new drug delivery system. J Magn Magn Mater. 2005;293:483–496. doi:10.1016/j.jmmm.2005.01.064
53. Burke TG, Mi Z. The structural basis of camptothecin interactions with human serum albumin: impact on drug stability. J Med Chem. 1994;37:40–46. doi:10.1021/jm00027a005
54. Zhao H, Lee C, Sai P, et al. 20-O-acylcamptothecin derivatives: evidence for lactone stabilization. J Org Chem. 2000;65:4601–4606. doi:10.1021/jo000221n
55. Zhang F, Zhu G, Jacobson O, et al. Transformative nanomedicine of an amphiphilic camptothecin prodrug for long circulation and high tumor uptake in cancer therapy. ACS Nano. 2017;11:8838–8848. doi:10.1021/acsnano.7b03003
56. Wullner U, Neef I, Eller A, Kleines M, Tur MK, Barth S. Cell-specific induction of apoptosis by rationally designed bivalent aptamer-siRNA transcripts silencing eukaryotic elongation factor 2. Curr Cancer Drug Targets. 2008;8:554–565. doi:10.2174/156800908786241078
57. Lee SJ, Kim H-S, Yu R, et al. Novel prostate-specific promoter derived from PSA and PSMA enhancers. Mol Ther. 2002;6:415–421. doi:10.1006/mthe.2002.0682
58. Wolf P, Gierschner D, Bühler P, Wetterauer U, Elsässer-Beile U. A recombinant PSMA-specific single-chain immunotoxin has potent and selective toxicity against prostate cancer cells. Cancer Immunol Immunother. 2006;55:1367–1373. doi:10.1007/s00262-006-0131-0
59. Fan Z, Liu G, Li Y, et al. Self-assembly of the active lactone form of a camptothecin–phospholipid complex for sustained nuclear drug delivery. RSC Adv. 2016;6:82949–82960. doi:10.1039/C6RA17714J
60. Liu X, Si J, Zhang Q, et al. Functionalized nanoparticles efficiently enhancing the targeted delivery tumor penetration and anticancer activity of 7‐ethyl‐10‐hydroxycamptothecin. Adv Healthc Mater. 2018;7:1701140. doi:10.1002/adhm.201701140
61. Kalchenko V, Shivtiel S, Malina V, et al. Use of lipophilic near-infrared dye in whole-body optical imaging of hematopoietic cell homing. J Biomed Opt. 2006;11:050507. doi:10.1117/1.2364903
62. Zhang R, Xing R, Jiao T, et al. Carrier-free chemophotodynamic dual nanodrugs via self-assembly for synergistic antitumor therapy. ACS Appl Mat Interfaces. 2016;8:13262–13269. doi:10.1021/acsami.6b02416
63. Wang G, Zhang F, Tian R, et al. Nanotubes-embedded indocyanine green–hyaluronic acid nanoparticles for photoacoustic-imaging-guided phototherapy. ACS Appl Mat Interfaces. 2016;8:5608–5617. doi:10.1021/acsami.5b12400
64. Ma P, Sun Y, Chen J, et al. Enhanced anti-hepatocarcinoma efficacy by GLUT1 targeting and cellular microenvironment-responsive PAMAM–camptothecin conjugate. Drug Deliv. 2018;25:153–165. doi:10.1080/10717544.2017.1419511
65. Furuichi K, Kaneko S, Wada T. Chemokine/chemokine receptor-mediated inflammation regulates pathologic changes from acute kidney injury to chronic kidney disease. Clin Exp Nephrol. 2009;13:9–14. doi:10.1007/s10157-008-0119-5
66. Ozkok A, Edelstein CL. Pathophysiology of cisplatin-induced acute kidney injury. Biomed Res Int. 2014;2014:967826. doi:10.1155/2014/967826
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.