")
Back to Journals » Journal of Experimental Pharmacology » Volume 10
Propranolol for the treatment of vascular sarcomas
Authors Wagner MJ, Cranmer LD , Loggers ET, Pollack SM
Received 5 April 2018
Accepted for publication 31 May 2018
Published 6 September 2018 Volume 2018:10 Pages 51—58
DOI https://doi.org/10.2147/JEP.S146211
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Bal Lokeshwar
Michael J Wagner,1,2 Lee D Cranmer,1,2 Elizabeth T Loggers,1,2 Seth M Pollack1,2
1Division of Medical Oncology, 2Clinical Research Division University of Washington and Fred Hutchinson Cancer Research Center, Seattle, WA, USA
Abstract: Vascular sarcomas are abnormal proliferations of endothelial cells. They range from benign hemangioma to aggressive angiosarcoma, and are characterized by dysregulated angiogenic signaling. Propranolol is a β-adrenergic receptor inhibitor that has demonstrated clinical efficacy in benign infantile hemangioma, and is now being used experimentally for more aggressive vascular sarcomas and other cancers. In this review, we discuss the use of propranolol in targeting these receptors in vascular tumors and other cancers.
Keywords: propranolol, vascular sarcoma, angiosarcoma, β-blocker, cancer
Introduction
Vascular sarcomas are abnormal proliferations of endothelial cells (ECs). They range from benign hemangioma to aggressive angiosarcoma, and are characterized by dysregulated angiogenic signaling.1 Benign infantile hemangiomas (IHs) are among the most common vascular tumors, with an incidence of approximately 3%.2 The natural history of IH is to first expand during a proliferative phase, and then regress during an involuting phase. Lesions that are symptomatic or otherwise problematic can be treated with topical or systemic agents, including corticosteroids or β-adrenergic receptor inhibitors.3 Propranolol is a nonselective β-adrenergic receptor blocker that has been implicated in several cancers and has had success in treating IH.4
Angiosarcoma is an aggressive cancer of ECs. It can occur anywhere in the body, with the most common sites being cutaneous lesions in the head and neck, breast, and extremities. They can be further subclassified into primary and secondary angiosarcoma, with the latter as a result of chronic lymphedema or radiation exposure. Outcomes for patients with angiosarcoma, even those who present with localized disease, are poor. For patients who develop metastatic disease, median survival is about 1 year.5–7 Primary treatment usually includes a combination of cytotoxic chemotherapy, surgery, and radiation. The advent of drugs targeting angiogenesis pathways were theoretically promising for treating tumors of ECs, but clinical results have been disappointing. Response rates to drugs targeting the VEGF/VEGFR axis range from 9%–20%.1 Combining bevacizumab, an anti-VEGF antibody, with paclitaxel yielded no clinical benefit.8 Drugs targeting other angiogenesis pathways such as the angiopoietin–TIE2 axis have also thus far been unsuccessful.9 The PI3K/AKT/mTOR pathway has also been implicated in both benign and aggressive vascular tumors.10,11
Molecular and genomic characterization has yielded some insights into the drivers of angiosarcoma, but to date no targeted agents have demonstrated a clear benefit for most patients. Some angiosarcomas harbor activating mutations in KDR12 or PLCG1,13 and others have CIC mutations or rearrangements14 which serve as potential driver events. Secondary angiosarcomas are characterized by MYC and FLT4 amplification.15 Even with this improved understanding as a result of high-throughput sequencing of several cohorts of angiosarcomas, driver events for most cases remain unknown. Recently, focus has sharpened on the β-adrenergic receptors that play a key role in normal EC function and may play a role in supporting angiosarcoma growth. In this review, we will discuss the use of propranolol in targeting these receptors in angiosarcoma and other vascular tumors.
β-adrenergic signaling and cancer
Adrenergic receptors are 7-transmembrane G-protein coupled receptors that consist of α, β, and γ subunit subtypes.16 β-adrenergic receptors play a vital role in several physiologic processes and are key mediators of the physiologic stress response. Drugs have been developed to inhibit the receptors with varying levels of affinity. Modulators of adrenergic signaling are some of the oldest drugs in clinical use, with clinical benefit particularly for cardiovascular disease and prevention of esophageal varix bleeding in advanced hepatic cirrhosis.
Recently, β-adrenergic signaling is gaining attention as a potential therapeutic target in cancer.17 Several mechanisms by which β-blockers improve outcomes in cancers have been proposed, including both direct anticancer effects and effects on multiple cell types in the cancer microenvironment (Figure 1).
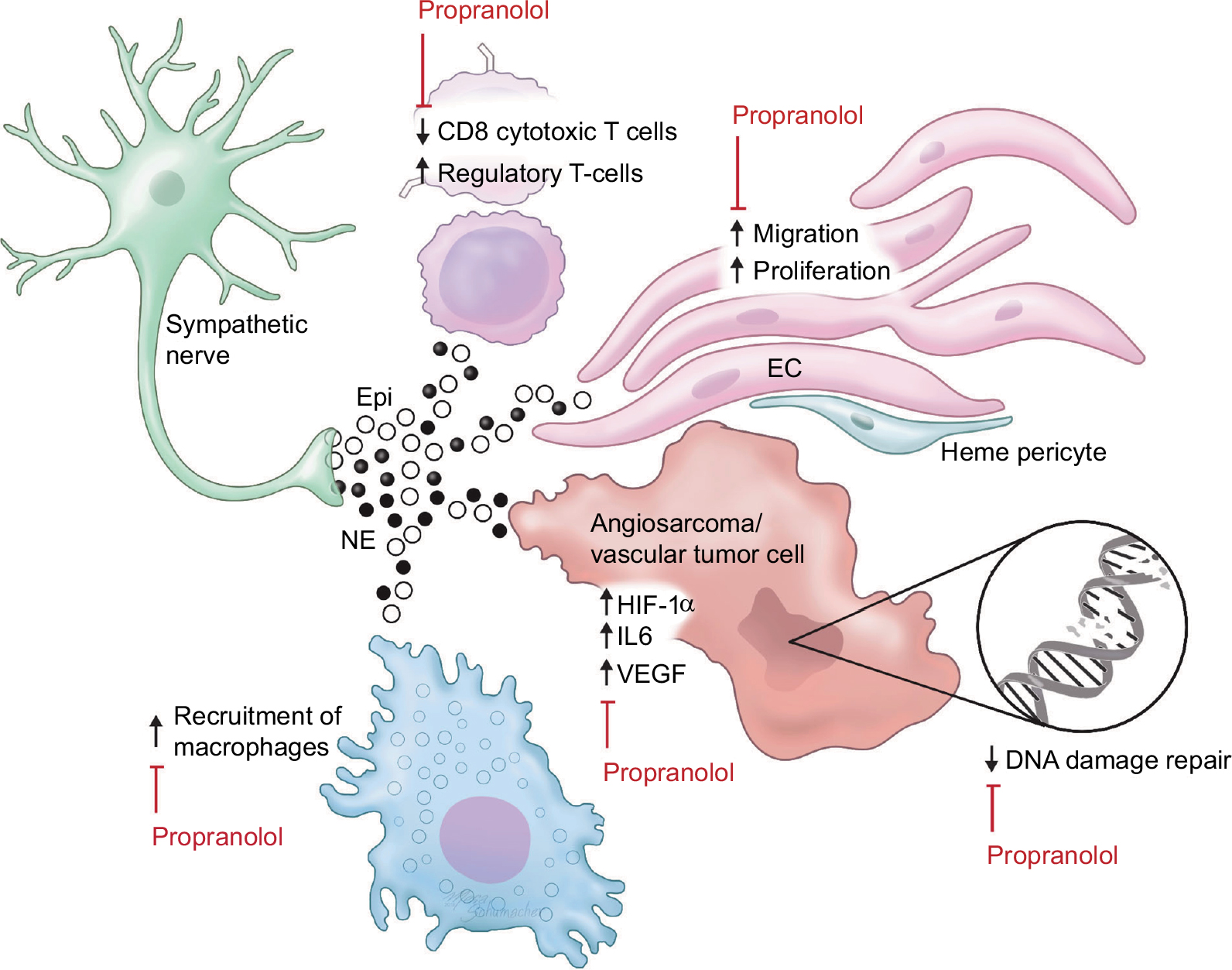 | Figure 1 Adrenergic signaling in the vascular tumor microenvironment. Notes: Epi and NE produced in sympathetic nerves act on β-adrenergic receptors present on T-cells, ECs, macrophages, and tumor cells. Activation of adrenergic receptors decreases infiltration by cytotoxic T-cells, increases the number of regulatory T-cells, and increases the recruitment and differentiation of tumor-associated macrophages. Sympathetic signaling also increases the migration and proliferation of normal ECs. In tumor cells, adrenergic signaling stimulates production of other proangiogenic and inflammatory mediators such as HIF-1α, VEGF, and IL-6 and suppresses the DNA damage response. Propranolol inhibits these oncogenic changes by blocking the β-receptors through which Epi and NE act. Abbreviations: Epi, epinephrine; NE, norepinephrine; EC, endothelial cell. |
β-adrenergic pathway modulators have direct effects on cancer cells of various subtypes in culture. Stimulation with β-agonists increases cell proliferation in a cAMP-dependent manner in lung adenocarcinoma cells.18 Activation of the β2-adrenergic pathway increases IL-6 production and inactivates the tumor suppressor LKB1 in EGFR mutant lung adenocarcinoma cells, and is a proposed mechanism for resistance to EGFR inhibitors. Indeed β-blocker use was associated with improved benefit from afatinib in the Phase III LUX-Lung3 study.19,20 Similar increases in cancer cell-specific measurements such as proliferation and invasiveness were seen in pancreatic cancer cells21 and ovarian cancer cells.22 Adrenergic stimulation led to chemoresistance in colon cancer cells23 and ovarian cancer,24 the latter by stimulating DUSP1. β-blockers are synergistic with cytotoxic chemotherapy against breast cancer25 and neuroblastoma26 cells. Although some of the effect seen in lung cancer seems to be specific for EGFR mutant-containing cells, broader pathways such as DNA damage repair pathways are also regulated in part through β-2 receptors.27
Propranolol is a small molecule nonspecific inhibitor of β-1 and β-2 adrenergic receptors, and is the focus of this review. In addition to the effects on cancer cells themselves, β-receptor inhibition with propranolol decreases proliferation, migration, and differentiation of ECs.28 Propranolol treatment inhibits angiogenesis in EC lines, but has little to no effect on vascular disruption.25 Preclinical studies in cancer models have demonstrated that increased adrenergic signaling through the β-2 receptor results in increased VEGF production in cancer cells29 and increased tumor vascularization.30 Similarly, the β-agonist isoprenaline stimulates autocrine VEGF signaling in gastric cancer cells and associated ECs via β-2 receptor-mediated signaling.31
Clinical evidence for b-receptor inhibition in cancer
Some of the first retrospective clinical data in support of β-blockers in cancer were seen in breast cancer, where β-blocker use for hypertension is associated with improved cancer-specific survival compared with patients using other types of antihypertensive medications.32 The specificity of β-receptor inhibition has an effect on survival, with a beneficial effect seen in breast cancer patients receiving the nonselective β blocker propranolol but not with the β-1 antagonist atenolol.33 Carvedilol, another nonselective β-blocker, reduces the risk of multiple cancer types with the largest effect seen in upper gastrointestinal and lung cancers in a large study from Asia.34 Additional studies show benefit of β-blocker use in patients with colorectal35 and pancreatic36 cancer. A prospective nonrandomized study of propranolol in the adjuvant setting for resected melanoma found an 80% reduction in melanoma recurrence.37 Overall, prospective clinical evidence supporting a role for propranolol in cancer treatment or prevention is limited. A summary of the largest existing clinical studies describing the impact of β-blockers on cancer incidence and outcomes is provided in Table 1.
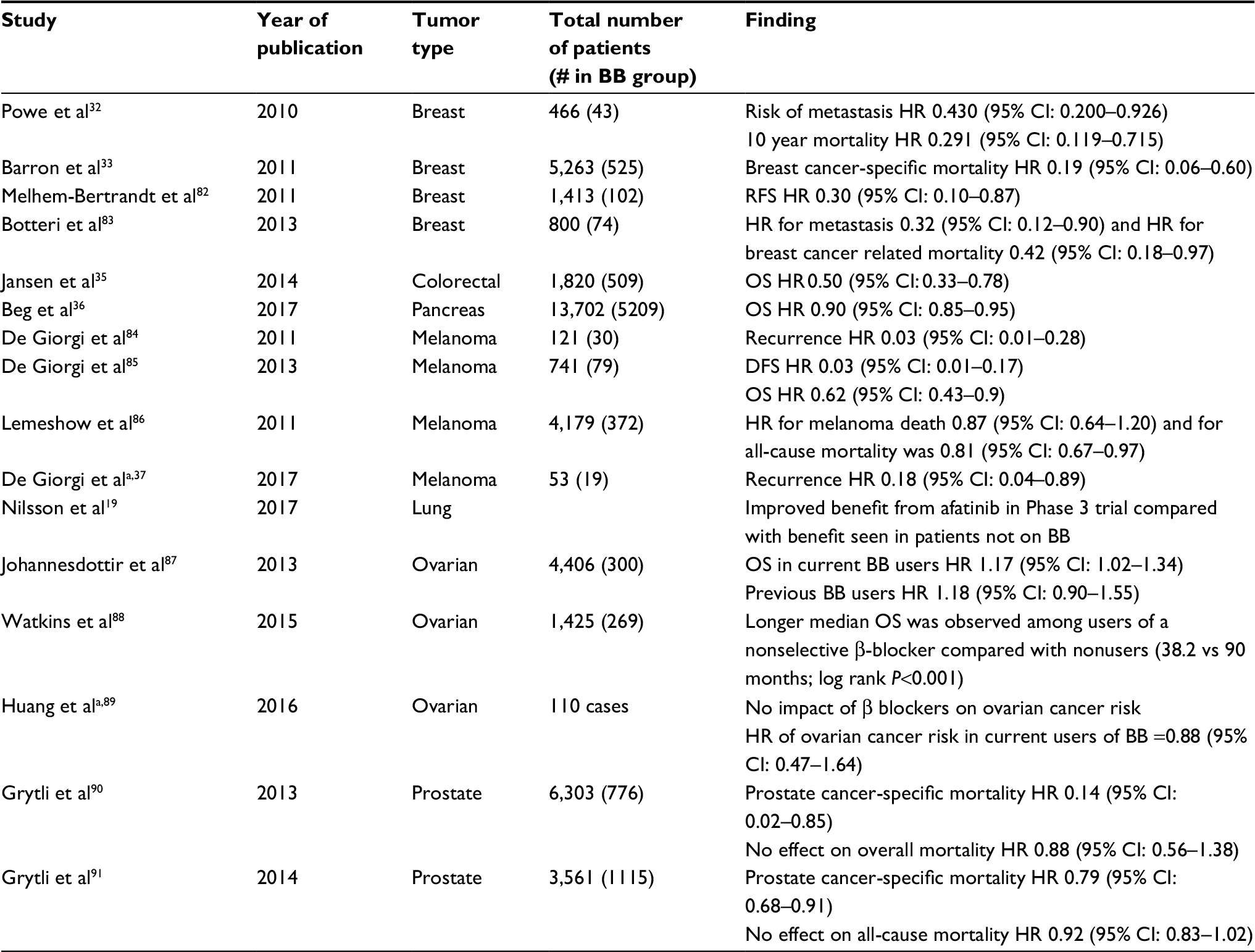 | Table 1 Clinical evaluation of β-blockers in multiple cancer subtypes Notes: aProspective studies. All others are retrospective. Abbreviations: HR, hazard ratio; RFS, relapse-free survival; DFS, disease-free survival; OS, overall survival; BB, β-blocker. |
Treatment of IH
The antiangiogenic properties of propranolol have led it to be used in vascular tumors. Indeed, propranolol has seen perhaps its greatest success in oncology in IH. There is significant controversy surrounding the cell of origin, with evidence that there is a hemangioma stem cell (HemSC) which induces proliferative changes in adjacent cells in the microenvironment.38 Despite the tendency of these tumors to first proliferate and then regress in characteristic phases, some IHs are problematic and require treatment.3 The proliferating phase of IH is characterized by VEGF-A production which stimulates hemangioma endothelial cell (HemEC) proliferation.39 Indeed, patients with IH have high increased circulating levels of VEGF-A.40,41 At the receptor level, VEGFR1 expression levels in hemangiomas are lower than those in normal ECs,42–45 consistent with its accepted role as a VEGF-A trap counteracting the stimulatory effects of VEGF-A ligand binding to VEGFR2. Decreased VEGFR1 expression levels in HemECs results in increased VEGFR2 signaling,43 and VEGFR2 knockdown in HemECs decreases cell viability and increased apoptosis, whereas VEGFR2 overexpression has the opposite effect.46 Proliferating IH has a relatively high expression of Ang2 and low expression of Ang1,47,48 as do hemangioma-derived pericytes;49 however, hemangioma-derived cell lines demonstrate increased migration and survival in response to Ang1, but not Ang2, highlighting the complicated roles these 2 ligands play.48
The tumor microenvironment also plays a critical role in hemangioma formation. Jagged1 expression on ECs and cell–cell contact between HemECs and HemSCs is required for HemSC differentiation into pericyte.44 Hemangioma-derived pericytes do not stabilize developing blood vessels as would be expected with physiologic pericytes.49 Jagged1 and Notch4 expression levels in proliferating IH are 6.5-fold and 3.2-fold higher, respectively, than those in placenta vessel control.47 Notch effector proteins HEY1, HEYL, and HES1 are highly expressed in HemSCs, whereas HEY2 is highly expressed in HemECs alone.50 Interestingly Notch1, Notch4, and Jagged1 have increased expression in involuting hemangioma ECs, and it was concluded that the involution was at least partially caused by the cells’ differentiation into a more determined EC phenotype as a result of increased Notch signaling.51
Treatment with the β-blocker propranolol at a dose of 2 mg per kg of body weight per day leads to regression of cutaneous IH lesions4 as well as potentially more life-threatening infantile hepatic hemangiomas52 and subglottic hemangiomas.53 Conversely, the use of β-2 sympathomimetic tocolytics doubled the rate of IH in preterm infants from 11% to 22% in the group studied.54 Although the mechanism remains unclear, 1 proposed mechanism by which propranolol may induce regression is by reducing the expression of HIF-1α, which in turn decreases HIF-1α-mediated signaling through the VEGF and STAT3 pathways.55 Moreover, propranolol may be targeting Hem-pericytes.49 Interestingly, GLUT1-positive cells derived from proliferating hemangiomas exhibit stem-like properties, and their growth is inhibited by mTOR inhibition but not by propranolol.56 mTOR and HIF-1a contribute to hemangioma proliferation via an autocrine VEGF signaling loop.57 Interestingly, treatment with propranolol leads to similar gene expression changes in IH and normal ECs, suggesting that the regression seen with propranolol is multifactorial, involving drug effect on multiple cell types in the microenvironment.58
Propranolol in intermediate-grade vascular sarcomas and angiosarcoma
Given their clinical success in IH, β-blockers have been studied in models of other vascular tumors. β-receptors 1, 2, and 3 are present on hemangioendothelioma and angiosarcoma by immunohistochemistry, and treatment with high doses of propranolol causes apoptosis and is synergistic with cytotoxic chemotherapy in hemangioendothelioma and angiosarcoma cell lines.59 Compared with hemangiomas, fewer of the aggressive tumors express β-2 and β-3 receptors, with about 40% of the 44 angiosarcomas in 1 series staining strongly for β-2 receptor and variable staining across the various types of hemangioendothelioma.60 mRNA expression profiling of transformed mouse ECs that behave like angiosarcoma cells revealed a broad array of differentially expressed genes after treatment with propranolol.61
The role of β-2 receptor signaling in both adaptive and innate immunity62 also makes propranolol appealing for treatment of angiosarcoma. B- and T-lymphocytes express the β-2 adrenergic receptor and are responsive to β-agonists.63 Chronic β-adrenergic receptor signaling suppresses CD8+ cytotoxic T-cells, thus reducing T-cell responses to immune checkpoint inhibitors.64 Lymphocyte egress from lymph nodes and interferon transcription is regulated by sympathetic innervation and norepinephrine.65,66 The presence of infiltrating CD8+ T-cells correlates with survival in angiosarcoma patients.67 Furthermore, β-adrenergic signaling affects myeloid cells in the microenvironment by regulating secretion of IL-6 and IL-8.68,69 Macrophage recruitment to tumors may be increased by beta adrenergic mediated secretion of chemotactic molecules by tumor cells.70 IL-6 production in angiosarcoma tumor cells increases the number of tumor promoting macrophages.71 Small reports aimed at targeting tumor-associated macrophages in angiosarcoma were promising,72,73 suggesting that the anti-inflammatory effect of propranolol will be beneficial in angiosarcoma.
Unfortunately much of what is known clinically about propranolol and its utility in treating angiosarcoma specifically are based on case reports. In 1 patient, serial biopsy before and 1 week after initiation of propranolol 40 mg twice a day resulted in a decrease in proliferative index of the tumor assessed by Ki67 staining from around 30% of positive cells to around 20%, causing a reduction in proliferation of 34%.74 As this was a single case report, and so this difference may be accounted for by sampling variance, and any determination of clinical benefit is confounded by the addition of cytotoxic chemotherapy and radiation in this patient’s treatment course.
Most reports combine propranolol with cytotoxic chemotherapy. In a preclinical model of transformed ECs, propranolol was synergistic with vinblastine, but not the chemotherapeutic agents more commonly used for angiosarcoma, such as doxorubicin or paclitaxel.75 Several patients treated with propranolol combined with metronomic vinblastine and methotrexate derived clinical benefit from this combination, though distinguishing the potential contribution of propranolol in this combination is impossible based on the described studies.75,76
Metronomic chemotherapy has been shown to have antiangiogenic effects and can improve the anticancer effects of cyclophosphamide in some settings.77 Due to the antiangiogenic effects of low-dose metronomic chemotherapy, this strategy presented a promising method for treating angiosarcoma. Treatment with metronomic trofosfamide, pioglitazone, and rofecoxib led to clinical responses in 3 of 5 angiosarcoma patients in 1 small series.78 Due its better toxicity profile, metronomic chemotherapy with cyclophosphamide was used in elderly patients with angiosarcoma with evidence of efficacy.79 Case reports combining propranolol combined with metronomic cyclophosphamide in angiosarcoma suggest promising results that warrant further investigation.80,81 The optimal dose of propranolol for angiosarcoma is not currently known. A dose finding study is currently underway in France investigating increasing dosing of propranolol combined with a stable dose of cyclophosphamide (NCT02732678).
Conclusion and future directions
The prospect of utilizing propranolol in angiosarcoma is a promising one, with hints of benefit in preclinical work and small case series and reports. However, in spite of the excitement describing the revolutionary potential of propranolol in angiosarcoma, the current role for propranolol remains an open question. Confounding factors in all of the published reports, combined with the fact that all of these studies are small case series or reports, limit the ability to make conclusive recommendations. Prospective studies with larger numbers of patients are needed. One potential study design would incorporate propranolol in the adjuvant setting to investigate a specific benefit from β-2 inhibition without the confounding impact of coadministered chemotherapy. Alternatively, incorporating propranolol into currently used systemic regimens may be preferred, but with the rarity of angiosarcoma and lack of consensus on initial management particularly for localized disease this may be difficult. Thankfully, propranolol is a relatively cheap and well-studied drug for other indications. A prospective, multicenter randomized trial should be feasible and would be able to answer once and for all if we should be including propranolol in our angiosarcoma treatment schema.
Disclosure
MJW is funded by the Conquer Cancer Foundation–ASCO Young Investigator Award and QuadW Foundation–AACR Fellowship for Clinical/Translational Sarcoma Research. The authors report no other conflicts of interest in this work.
References
Wagner MJ, Ravi V, Menter DG, Sood AK. Endothelial cell malignancies: new insights from the laboratory and clinic. NPJ Precis Oncol. 2017;1(1):11. | ||
Dickison P, Christou E, Wargon O. A prospective study of infantile hemangiomas with a focus on incidence and risk factors. Pediatr Dermatol. 2011;28(6):663–669. | ||
Chen TS, Eichenfield LF, Friedlander SF. Infantile hemangiomas: an update on pathogenesis and therapy. Pediatrics. 2013;131(1):99–108. | ||
Leaute-Labreze C, Dumas de la Roque E, Hubiche T, Boralevi F, Thambo JB, Taïeb A. Propranolol for severe hemangiomas of infancy. N Engl J Med. 2008;358(24):2649–2651. | ||
D’Angelo SP, Munhoz RR, Kuk D, et al. Outcomes of systemic therapy for patients with metastatic angiosarcoma. Oncology. 2015;89(4):205–214. | ||
Fury MG, Antonescu CR, Van Zee KJ, Brennan MF, Maki RG. A 14-year retrospective review of angiosarcoma: clinical characteristics, prognostic factors, and treatment outcomes with surgery and chemotherapy. Cancer J. 2005;11(3):241–247. | ||
Fayette J, Martin E, Piperno-Neumann S, et al. Angiosarcomas, a heterogeneous group of sarcomas with specific behavior depending on primary site: a retrospective study of 161 cases. Ann Oncol. 2007;18(12):2030–2036. | ||
Ray-Coquard IL, Domont J, Tresch-Bruneel E, et al. Paclitaxel given once per week with or without bevacizumab in patients with advanced angiosarcoma: a randomized phase II trial. J Clin Oncol. 2015;33(25):2797–2802. | ||
D’Angelo SP, Mahoney MR, Van Tine BA, et al. Alliance A091103 a phase II study of the angiopoietin 1 and 2 peptibody trebananib for the treatment of angiosarcoma. Cancer Chemother Pharmacol. 2015;75(3):629–638. | ||
Perry B, Banyard J, McLaughlin ER, et al. AKT1 overexpression in endothelial cells leads to the development of cutaneous vascular malformations in vivo. Arch Dermatol. 2007;143(4):504–506. | ||
Greenberger S, Yuan S, Walsh LA, et al. Rapamycin suppresses self-renewal and vasculogenic potential of stem cells isolated from infantile hemangioma. J Invest Dermatol. 2011;131(12):2467–2476. | ||
Antonescu CR, Yoshida A, Guo T, et al. KDR activating mutations in human angiosarcomas are sensitive to specific kinase inhibitors. Cancer Res. 2009;69(18):7175–7179. | ||
Behjati S, Tarpey PS, Sheldon H, et al. Recurrent PTPRB and PLCG1 mutations in angiosarcoma. Nat Genet. 2014;46(4):376–379. | ||
Huang SC, Zhang L, Sung YS, et al. Recurrent CIC gene abnormalities in angiosarcomas: a molecular study of 120 cases with concurrent investigation of PLCG1, KDR, MYC, and FLT4 gene alterations. Am J Surg Pathol. 2016;40(5):645–655. | ||
Guo T, Zhang L, Chang NE, Singer S, Maki RG, Antonescu CR. Consistent MYC and FLT4 gene amplification in radiation-induced angiosarcoma but not in other radiation-associated atypical vascular lesions. Genes Chromosomes Cancer. 2011;50(1):25–33. | ||
Hepler JR, Gilman AG. G proteins. Trends Biochem Sci. 1992;17(10):383–387. | ||
Cole SW, Sood AK. Molecular pathways: β-adrenergic signaling in cancer. Clin Cancer Res. 2012;18(5):1201–1206. | ||
Park PG, Merryman J, Orloff M, Schuller HM. β-adrenergic mitogenic signal transduction in peripheral lung adenocarcinoma: implications for individuals with preexisting chronic lung disease. Cancer Res. 1995;55(16):3504–3508. | ||
Nilsson MB, Sun H, Diao L, et al. Stress hormones promote EGFR inhibitor resistance in NSCLC: Implications for combinations with β-blockers. Sci Transl Med. 2017;9(415):pii: eaa4307. | ||
Sequist LV, Yang JC, Yamamoto N, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013;31(27):3327–3334. | ||
Huang XY, Wang HC, Yuan Z, Huang J, Zheng Q. Norepinephrine stimulates pancreatic cancer cell proliferation, migration and invasion via β-adrenergic receptor-dependent activation of P38/MAPK pathway. Hepatogastroenterology. 2012;59(115):889–893. | ||
Sood AK, Bhatty R, Kamat AA, et al. Stress hormone-mediated invasion of ovarian cancer cells. Clin Cancer Res. 2006;12(2):369–375. | ||
Yao H, Duan Z, Wang M, Awonuga AO, Rappolee D, Xie Y. Adrenaline induces chemoresistance in HT-29 colon adenocarcinoma cells. Cancer Genet Cytogenet. 2009;190(2):81–87. | ||
Kang Y, Nagaraja AS, Armaiz-Pena GN, et al. Adrenergic stimulation of DUSP1 impairs chemotherapy response in ovarian cancer. Clin Cancer Res. 2016;22(7):1713–1724. | ||
Pasquier E, Ciccolini J, Carre M, et al. Propranolol potentiates the anti-angiogenic effects and anti-tumor efficacy of chemotherapy agents: implication in breast cancer treatment. Oncotarget. 2011;2(10):797–809. | ||
Pasquier E, Street J, Pouchy C, et al. β-blockers increase response to chemotherapy via direct antitumour and anti-angiogenic mechanisms in neuroblastoma. Br J Cancer. 2013;108(12):2485–2494. | ||
Hara MR, Kovacs JJ, Whalen EJ, et al. A stress response pathway regulates DNA damage through β2-adrenoreceptors and β-arrestin-1. Nature. 2011;477(7364):349–353. | ||
Lamy S, Lachambre MP, Lord-Dufour S, Beliveau R. Propranolol suppresses angiogenesis in vitro: inhibition of proliferation, migration, and differentiation of endothelial cells. Vascul Pharmacol. 2010;53(5–6):200–208. | ||
Lutgendorf SK, Cole S, Costanzo E, et al. Stress-related mediators stimulate vascular endothelial growth factor secretion by two ovarian cancer cell lines. Clin Cancer Res. 2006;9(12):4514–4521. | ||
Thaker PH, Han LY, Kamat AA, et al. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat Med. 2006;12(8):939–944. | ||
Lu Y, Xu Q, Zuo Y, et al. Isoprenaline/β2-AR activates Plexin-A1/VEGFR2 signals via VEGF secretion in gastric cancer cells to promote tumor angiogenesis. BMC Cancer. 2017;17(1):875. | ||
Powe DG, Voss MJ, Zänker KS, et al. β-blocker drug therapy reduces secondary cancer formation in breast cancer and improves cancer specific survival. Oncotarget. 2010;1(7):628–638. | ||
Barron TI, Connolly RM, Sharp L, Bennett K, Visvanathan K. β blockers and breast cancer mortality: a population-based study. J Clin Oncol. 2011;29(19):2635–2644. | ||
Lin CS, Lin WS, Lin CL, Kao CH. Carvedilol use is associated with reduced cancer risk: a nationwide population-based cohort study. Int J Cardiol. 2015;184:9–13. | ||
Jansen L, Hoffmeister M, Arndt V, Chang-Claude J, Brenner H. Stage-specific associations between β blocker use and prognosis after colorectal cancer. Cancer. 2014;120(8):1178–1186. | ||
Beg MS, Gupta A, Sher D, et al. Impact of concurrent medication use on pancreatic cancer survival-SEER-medicare analysis. Am J Clin Oncol. Epub 2017 Jan 10. | ||
De Giorgi V, Grazzini M, Benemei S, et al. Propranolol for off-label treatment of patients with melanoma: results from a cohort study. JAMA Oncol. 2017;4(2):e172908. | ||
Khan ZA, Boscolo E, Picard A, et al. Multipotential stem cells recapitulate human infantile hemangioma in immunodeficient mice. J Clin Invest. 2008;118(7):2592–2599. | ||
Greenberger S, Boscolo E, Adini I, Mulliken JB, Bischoff J. Corticosteroid suppression of VEGF-A in infantile hemangioma-derived stem cells. New Engl J Med. 2010;362(11):1005–1013. | ||
Zhang L, Lin X, Wang W, et al. Circulating level of vascular endothelial growth factor in differentiating hemangioma from vascular malformation patients. Plast Reconstr Surg. 2005;116(1):200–204. | ||
Kleinman ME, Greives MR, Churgin SS, et al. Hypoxia-induced mediators of stem/progenitor cell trafficking are increased in children with hemangioma. Arterioscler Thromb Vasc Biol. 2007;27:2664–2670. | ||
Ji Y, Chen S, Li K, Li L, Xu C, Xiang B. Signaling pathways in the development of infantile hemangioma. J Hematol Oncol. 2014;7:13. | ||
Jinnin M, Medici D, Park L, et al. Suppressed NFAT-dependent VEGFR1 expression and constitutive VEGFR2 signaling in infantile hemangioma. Nat Med. 2008;14(11):1236–1246. | ||
Boscolo E, Mulliken JB, Bischoff J. VEGFR-1 mediates endothelial differentiation and formation of blood vessels in a murine model of infantile hemangioma. Am J Pathol. 2011;179(5):2266–2277. | ||
Picard A, Boscolo E, Khan ZA, et al. IGF-2 and FLT-1/VEGF-R1 mRNA levels reveal distinctions and similarities between congenital and common infantile hemangioma. Pediatr Res. 2008;63(3):263–267. | ||
Ou JM, Yu ZY, Qiu MK, et al. Knockdown of VEGFR2 inhibits proliferation and induces apoptosis in hemangioma-derived endothelial cells. Eur J Histochem. 2014;58(1):2263. | ||
Calicchio ML, Collins T, Kozakewich HP. Identification of signaling systems in proliferating and involuting phase infantile hemangiomas by genome-wide transcriptional profiling. Am J Pathol. 2009;174(5):1638–1649. | ||
Yu Y, Varughese J, Brown LF, Mulliken JB, Bischoff J. Increased Tie2 expression, enhanced response to angiopoietin-1, and dysregulated angiopoietin-2 expression in hemangioma-derived endothelial cells. Am J Pathol. 2001;159(6):2271–2280. | ||
Boscolo E, Mulliken JB, Bischoff J. Pericytes from infantile hemangioma display proangiogenic properties and dysregulated angiopoietin-1. Arterioscler Thromb Vasc Biol. 2013;33(3):501–509. | ||
Adepoju O, Wong A, Kitajewski A, et al. Expression of HES and HEY genes in infantile hemangiomas. Vasc Cell. 2013;3:19. | ||
Wu JK, Adepoju O, De Silva D, et al. A switch in Notch gene expression parallels stem cell to endothelial transition in infantile hemangioma. Angiogenesis. 2010;13(1):15–23. | ||
Mazereeuw-Hautier J, Hoeger PH, Benlahrech S, et al. Efficacy of propranolol in hepatic infantile hemangiomas with diffuse neonatal hemangiomatosis. J Pediatr. 2010;157(2):340–342. | ||
Denoyelle F, Leboulanger N, Enjolras O, Harris R, Roger G, Garabedian EN. Role of propranolol in the therapeutic strategy of infantile laryngotracheal hemangioma. Int J Pediatr Otorhinolaryngol. 2009;73(8):1168–1172. | ||
Mayer M, Minichmayr A, Klement F, et al. Tocolysis with the β-2-sympathomimetic hexoprenaline increases occurrence of infantile haemangioma in preterm infants. Arch Dis Child Fetal Neonatal Ed. 2013;98(2):F108–F111. | ||
Li P, Guo Z, Gao Y, Pan W. Propranolol represses infantile hemangioma cell growth through the β2-adrenergic receptor in a HIF-1α-dependent manner. Oncol Rep. 2015;33(6):3099–3107. | ||
Huang L, Nakayama H, Klagsbrun M, Mulliken JB, Bischoff J. Glucose transporter 1-positive endothelial cells in infantile hemangioma exhibit features of facultative stem cells. Stem Cells. 2015;33(1):133–145. | ||
Medici D, Olsen BR. Rapamycin inhibits proliferation of hemangioma endothelial cells by reducing HIF-1-dependent expression of VEGF. PLoS One. 2012;7(8):e42913. | ||
Stiles J, Amaya C, Pham R, et al. Propranolol treatment of infantile hemangioma endothelial cells: a molecular analysis. Exp Ther Med. 2012;4(4):594–604. | ||
Stiles JM, Amaya C, Rains S, et al. Targeting of β adrenergic receptors results in therapeutic efficacy against models of hemangioendothelioma and angiosarcoma. PLoS One. 2013;8(3):e60021. | ||
Chisholm KM, Chang KW, Truong MT, Kwok S, West RB, Heerema-McKenney AE. β-Adrenergic receptor expression in vascular tumors. Mod Pathol. 2012;25:1446–1451. | ||
Zhou S, Liu P, Jiang W, Zhang, H. Identification of potential target genes associated with the effect of propranolol on angiosarcoma via microarray analysis. Oncol Lett. 2017;13(6):4267–4275. | ||
Padro CJ, Sanders VM. Neuroendocrine regulation of inflammation. Semin Immunol. 2014;26(5):357–368. | ||
Pochet R, Delespesse G. β-Adrenoreceptors display different efficiency on lymphocyte subpopulations. Biochem Pharmacol. 1983;32(10):1651–1655. | ||
Nissen MD, Sloan EK, Mattarollo SR. β-adrenergic signaling impairs antitumor CD8(+) T-cell responses to B-cell lymphoma immunotherapy. Cancer Immunol Res. 2018;6(1):98–109. | ||
Kohm AP, Sanders VM. Norepinephrine and β 2-adrenergic receptor stimulation regulate CD4+ T and B lymphocyte function in vitro and in vivo. Pharmacol Rev. 2001;53(4):487–525. | ||
Nakai A, Hayano Y, Furuta F, Noda M, Suzuki K. Control of lymphocyte egress from lymph nodes through β2-adrenergic receptors. J Exp Med. 2014;211:2583–2598. | ||
D’Angelo SP, Shoushtari AN, Agaram NP, et al. Prevalence of tumor-infiltrating lymphocytes and PD-L1 expression in the soft tissue sarcoma microenvironment. Hum Pathol. 2015;46(3):357–365. | ||
Nilsson MB, Armaiz-Pena G, Takahashi R, et al. Stress hormones regulate interleukin-6 expression by human ovarian carcinoma cells through a Src-dependent mechanism. J Biol Chem. 2007;282(41):29919–29926. | ||
Shahzad MM, Arevalo JM, Armaiz-Pena GN, et al. Stress effects on FosB- and interleukin-8 (IL8)-driven ovarian cancer growth and metastasis. J Biol Chem. 2010;285(46):35462–35470. | ||
Armaiz-Pena GN, Gonzalez-Villasana V, Nagaraja AS, et al. Adrenergic regulation of monocyte chemotactic protein 1 leads to enhanced macrophage recruitment and ovarian carcinoma growth. Oncotarget. 2015;6(6):4266–4273. | ||
Yang J, Kantrow S, Sai J, et al. INK4a/ARF [corrected] inactivation with activation of the NF-κB/IL-6 pathway is sufficient to drive the development and growth of angiosarcoma. Cancer Res. 2012;72(18):4682–4695. | ||
Ishibashi M, Fujimura T, Hashimoto A, et al. Successful treatment of MMP-9-expressing angiosarcoma with low-dose docetaxel and bisphosphonate. Case Rep Dermatol. 2012;4:5–9. | ||
Fujimura T, Kambayashi Y, Furudate S, Kakizaki A, Aiba S. Immunomodulatory effect of bisphosphonate risedronate sodium on CD163+ arginase 1+ M2 macrophages: the development of a possible supportive therapy for angiosarcoma. Clin Dev Immunol. 2013;2013:325412. | ||
Chow W, Amaya CN, Rains S, Chow M, Dickerson EB, Bryan BA. Growth attenuation of cutaneous angiosarcoma with propranolol-mediated β-blockade. JAMA Dermatol. 2015;151(11):1226–1229. | ||
Pasquier E, André N, Street J, et al. Effective management of advanced angiosarcoma by the synergistic combination of propranolol and vinblastine-based metronomic chemotherapy: a bench to bedside study. EBioMedicine. 2016;6:87–95. | ||
Banavali S, Pasquier E, Andre N. Targeted therapy with propranolol and metronomic chemotherapy combination: sustained complete response of a relapsing metastatic angiosarcoma. Ecancermedicalscience. 2015;9:499. | ||
Browder T, Butterfield CE, Kräling BM, et al. Antiangiogenic scheduling of chemotherapy improves efficacy against experimental drug-resistant cancer. Cancer Res. 2000;60(7):1878–1886. | ||
Vogt T, Hafner C, Bross K, et al. Antiangiogenetic therapy with pioglitazone, rofecoxib, and metronomic trofosfamide in patients with advanced malignant vascular tumors. Cancer. 2003;98(10):2251–2256. | ||
Mir O, Domont J, Cioffi A, et al. Feasibility of metronomic oral cyclophosphamide plus prednisolone in elderly patients with inoperable or metastatic soft tissue sarcoma. Eur J Cancer. 2011;47(4):515–519. | ||
Daguze J, Saint-Jean M, Peuvrel L, et al. Visceral metastatic angiosarcoma treated effectively with oral cyclophosphamide combined with propranolol. JAAD Case Rep. 2016;2(6):497–499. | ||
Daguze J, Saint-Jean M, Dreno B. Large nose angiosarcoma treated effectively with oral cyclophosphamide combined with propranolol. J Eur Acad Dermatol Venereol. 2018;32(2):e52–e54. | ||
Melhem-Bertrandt A, Chavez-Macgregor M, Lei X, et al. β-blocker use is associated with improved relapse-free survival in patients with triple-negative breast cancer. J Clin Oncol. 2011;29(19):2645–2652. | ||
Botteri E, Munzone E, Rotmensz N, et al. Therapeutic effect of β-blockers in triple-negative breast cancer postmenopausal women. Breast Cancer Res Treat. 2013;140:567–575. | ||
De Giorgi V, Grazzini M, Gandini S, et al. Treatment with β-blockers and reduced disease progression in patients with thick melanoma. Arch Intern Med. 2011;171(8):779–781. | ||
De Giorgi V, Gandini S, Grazzini M, Benemei S, Marchionni N, Geppetti P. Effect of β-blockers and other antihypertensive drugs on the risk of melanoma recurrence and death. Mayo Clin Proc. 2013;88:1196–1203. | ||
Lemeshow S, Sørensen HT, Phillips G, et al. β-blockers and survival among Danish patients with malignant melanoma: a population-based cohort study. Cancer Epidemiol Biomarkers Prev. 2011;20:2273–2279. | ||
Johannesdottir SA, Schmidt M, Phillips G, et al. Use of β-blockers and mortality following ovarian cancer diagnosis: a population-based cohort study. BMC Cancer. 2013;13:85. | ||
Watkins JL, Thaker PH, Nick AM, et al. Clinical impact of selective and nonselective β-blockers on survival in patients with ovarian cancer. Cancer. 2015;121(19):3444–3451. | ||
Huang T, Poole EM, Eliassen AH, et al. Hypertension, use of antihypertensive medications, and risk of epithelial ovarian cancer. Int J Cancer. 2016;139(2):291–299. | ||
Grytli HH, Fagerland MW, Fossa SD, Tasken KA, Haheim LL. Use of β-blockers is associated with prostate cancer-specific survival in prostate cancer patients on androgen deprivation therapy. Prostate. 2013;73(3):250–260. | ||
Grytli HH, Fagerland MW, Fossa SD, Tasken KA. Association between use of β-blockers and prostate cancer-specific survival: a cohort study of 3,561 prostate cancer patients with high-risk or metastatic disease. Eur Urol. 2014;65(3);635–641. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.